

Abstract
Hexavalent chromium combines with glutathione in chloride intracellular channel carrier to form tetravalent and pentavelent chromium in plasma and organelle membranes. It also combines with NADH/NADPH to form pentavalent chromium in mitochondria. Tetravalent- and pentavalent- chromium (directly and indirectly) mediated DNA double strand breaks activate DNA damage signaling sensors: DNA-dependent-protein-kinase signals p53-dependent intrinsic mitochorndrial apoptosis, and ataxia-telangiectasia-mutated and ataxia-telangiectasia-Rad3-related signal cell-arrest for DNA repair. Tetravalent chromium may be the most potent species since it causes DNA breaks and somatic recombination, but not apoptosis. Upon further failure of apoptosis and senescence/DNA-repair, damaged cells may become immortal with loss-of-heterozygosity and genetic plasticity.
Keywords: chloride intracellular channel carrier, tetravalent chromium, pentavalent chromium, apoptosis, senescence, somatic recombination, genomic plasticity
I. The Purpose of This Review
Cr VI is a well known occupational carcinogen. Useful physico-chemical properties of chromate compounds such as color, malleability, ductility, and tensile strength, have led to its extensive use in various industries including chromate production, chromate pigment production, chrome plating, etc. Chromate production workers are exposed to both Cr(VI) and Cr(III) compounds and industrial workers in the chromate pigment industry are exposed mainly to Cr(VI). Studies have shown that lung cancer incidence has consistently increased among exposed workers in these two industries. Chrome platers exposed to Cr(VI) and other agents including nickel, also have increased risk of lung cancer (1).
In July 2008, the U.S. National Toxicology Program (NTP) published the final version of NTP Technical Report on the Toxicology and Carcinogenesis Studies of Sodium Dinchromate Dihydrate (CAS NO. 7789-12-0) in F344 Rats and B6C3F1 Mice (Drinking Water Studies). NTP (2) concluded that, under the conditions of this study, there was clear evidence of carcinogenic activity of hexavalent chromium compound sodium dichromate dihydrate in male and female F344 rats based on increased incidences of squamous cell neoplasms of the oral cavity. It also concluded that there was clear evidence of carcinogenic activity of sodium dichromate dihydrate in male and female B6C3F1 mice based on increased incidences of neoplasms in the small intestine.
The purpose of this review is to examine current available relevant data on the mechanism of Cr(VI) carcinogenesis. We examine the transport and production of reactive reductive intermediates (Cr(V) and Cr(IV)) and radicals during Cr(VI) reduction process, the genotoxic effects of these reactive chemical species, various cellular defenses against the Cr(VI) exposure (e.g. cell arrest, DNA repair and apoptosis), and the mode of action of Cr(VI) carcinogenesis. The relative importance of Cr(V) and Cr(IV) in these processes of Cr(VI) carcinogenesis is discussed.
II. Transport of Hexavalent Chromium Ion through the Chloride Phosphate Intracellular Channels
The valence state of chromium ions is the relevant physicochemical characteristic in uptake of these ions. There is a distinct difference in uptake between hexavalent and trivalent chromium in humans with a single dose of 5 mg of chromium trichloride (Cr(III)) or potassium dichromate (Cr(VI)). The bioavailability of Cr(VI) is in 2 order-of-magnitudes higher than the bioavailability of Cr(III) (6.9% vs 0.13%). The biological half-life of Cr(VI) after the cell uptake is in 1 order-of-magnitude higher than the half-life of Cr(III) (39 hr vs 10 hr) (3).
Once hexavalent chromium ion enters the body, the cells of several tissues rapidly absorb it. Cr(VI) readily crosses the cell membrane thru a concentration gradient of divalent anion in chloride phosphate anionic channel as a tetrahedral divalent CrO42− anion. In other words, there is no active transport (e.g. specific protein or ATP pump) involved (4). This anion is structurally analogous to PO43−, anions transported across cytoplasmic membranes through the chloride phosphate channel-carrier, which integrates itself onto the organelle membrane structure and passes its content into various subcellular compartments.
Channels for potassium or chloride ions are present in plasma membrane and membranes of intracellular organelles such as nucleus, mitochondria, dense core vesicles, endoplasmic reticulum, Golgi apparatus and trans-Golgi vesicles, etc. The chromate uptake is mediated by the general anion channel band-3 protein (5) now understood as the chloride intracellular channel (CLIC) carrier proteins. The CLICs play important roles in various cellular functions (6). The (CLIC) proteins are small proteins related to the omega family of glutathione S-transferases (GSTs)(7). Members of this family are capable in vitro of changing conformation from a globular, soluble state to a membrane-inserted state in which they provide chloride conductance for organelles such as the mitochondria, endoplasmic reticulum and the nucleus.
Harrop et al. (8) studied the crystal structure of the soluble form of the CLIC1 protein, a member of the CLCIs. The proteins exist in two forms: a soluble form that has a GST fold and a transmembrane form where the amino terminus is on one side of the membrane and the carboxyl terminus is on the opposite side of the membrane. CLIC1 is monomeric and structurally homologous to Ω class GST and indicated it belongs to the GST superfamily, and it has a redox-active site resembling glutaredoxin. The structure of the complex of CLIC1 with glutathione shows that glutathione occupies the redox-active site. Harrop et al. (2001) proposed that the intact glutathione binding site along with the conserved Cys24 in what is likely to be a redox-active state, and that CLIC1 activity is regulated by redox processes. In the cytoplasm, GSH is present at 1–10 mM, and the conditions are strongly reducing (9). Thus, the soluble form of CLIC1 is likely to be free of glutathione. Reactive oxygen or nitrogen species are likely to signal and alter the resting state of soluble CLIC1. We hypothesize that the hexavalent chromium will enter the cell as a divalent ion and will likely first react with glutathione in the CLIC on the cellular membrane and form the reduced species such as Cr (IV) and, possibly to a lesser extent, Cr(V).
III. Formation of Cr(VI) to Cr(V), Cr(IV), and Free Radicals
The formation and interconversion of paramagnetic chromium intermediates Cr(VI), Cr(V), Cr(IV), and Cr(III) has been reviewed by Shi et al. (10). It has been demonstrated by many studies and apparently requires no direct involvement of enzymes. GSH and NADPH, provide sufficient free energy for the redox reactions: Reduction of Cr(VI) by GHS generates Cr(IV) and Cr(V) complexes and glutathionyl radical (GS·) (10, 11, 12, 13, 14). One of the observations indicating that GSH is involved in Cr(VI) metabolism was the finding of a positive correlation between Cr(Vl)-induced DNA damage and GSH levels in cultured chick embryo hepatocytes (15,16).
One-electron reduction of Cr(VI) to Cr(V) was demonstrated under in vitro system (17, 18) and in Cr(VI) exposed living mice (19). This Cr(V) species was identified to be a Cr(V)-NADPH complex with an oxygen bond to Cr(V) (16, 20, 21 22 ). Pretreatment of the animals with ascorbate or GSH decreased the Cr(V) formation, while pretreatment with NADPH enhanced it. Metal chelators, such as 1,10-phenanthroline and diethylenetriamine pentaacetic acid (DTPA), inhibited the formation of Cr(V)–NADPH complex. These results suggest that NADPH/flavoenzymes, and not GSH or ascorbate, are the major one-electron Cr(VI) reductants in vivo. Therefore, Cr(V) can be formed thru the one-electron Cr(VI) reduction, or possibly thru Cr(IV) with reactions such as Cr(IV) + Cr(VI) →2 Cr(V), or 2Cr(IV) →Cr(V) + Cr(III) (21).
The concentration of GSH inside cells is about a 3 orders-of- magnitude higher than NADPH/NADH in the cytoplasm (23), and therefore formation of Cr(IV)-GSH is probably favored inside the cytoplasm while Cr(V)-NADPH formation occurs inside the mitochondria/endoplasmic reticulum. Both these processes follow the kinetics of the law-of-mass-action.
Since the plasma membrane has a very low permeability of Cr(III), the majority of the reduced Cr(III) from in the cells of the rats treated with potassium dichromate is reportedly sequestered in phagolysosomes in the cytoplasm for disposal. This is the cell defense mechanism to prevent the diffusion of the metal throughout the cell (24).
The Cr(VI) and transport of various Cr(VI) reduction intermediates are illustrated in Figure 1.
Figure 1. Chromium VI Reduction.

Accompany both the lower (than Cr(VI))oxidation states of Cr(IV) and Cr(V) formation, free radicals are formed via Fenton-like reactions from the interaction of chromium and cellular substance. ROS are generated from normal oxygen metabolism in cells, recognized as a list of active molecules, such as free radicals RS·, GS·, ·OH, O2·- etc. All chromium oxidation states are able to generate hydroxyl radicals with H2O2, with Cr(V) and Cr(IV) exhibiting high potency, as demonstrated in vitro by ESR spin trapping technique. Hydroxyl radical formation from Cr(III) is observed in these in vitro experiments only at non-physiological conditions at pH 10. The observed 3 order-of-magnitude difference in proton concentration under non-physiological conditions of pH 10 vs. pH 7 leads to ·OH formation. Therefore, the formation of ·OH from Cr(III) is possible but not likely (25)
Table 1 summarizes ESR experiments leading to reduction of chromium ions listed in the top row by redox reactions involving selected agents normally present in vivo. These agents are listed on the left column and include GSH, cysteine, penicilliamine, H2O2, lipid peroxide, and vitamin C. Cysteine and GSH are most commonly found intracellularly in lung and kidney, while vitamin C is present extracellularly in a high concentration (25).
Table 1. Hydroxyl Radicals Generated by Paramagnetic Chromium Radicals in ESR (25).
| Cr 6+ | Cr5+ | Cr4+ | Cr3+ | |
|---|---|---|---|---|
|
| ||||
| GSH | Cr5+ Cr4+ GS· ·OH | |||
| Cysteine | RS· O2·- RSSR· ·OH | OH | ||
| Penicillamine | OH | |||
| H2O2 | Cr6+ ·OH | Cr5+ ·OH | ·OH* | |
| Lipid peroxide | ·OH | R· RO· | ||
Vitamin C Cr5+ Cr4+ formyl·alkyl-carbonyl·
at pH 10
IV. Preparation of Cr(IV) and Cr(V) for Characterization of Mode of Action
It has been now established that mammalian exposure to Cr(VI) compounds is linked with the induction of cancer. Hexavalent chromium-mediated in vitro and in vivo reactions can cause variety of cellular injuries including nuclear transcription factor (NF)-KB activation, DNA single and double strand breaks, base modification such as 8- hydroxy-deoxyguanosine (8-OHdG) formation, and other DNA adducts (10). These types of DNA damage can lead to DNA repair, senescence, and apoptosis. Upon further failure of DNA repair or apoptosis and senescence, the chromium exposed cells with DNA damages may become immortal with further DNA changes/genetic plasticity and ultimately leading to cancer. The underlying mechanism, however, is not clear, the hypothesis of the involvement of Cr(VI) lower oxidation states (Cr(V) and Cr(IV)) directly (i.e. metal-mediated) or indirectly (i.e. radical species from the reduction of Cr(VI) has been supported by many studies.
Cr(VI) does not interact with DNA in the absence of reducing agents (26). It is believed that reduction of Cr(VI) to lower oxidation states by cellular reductants is an important step (4, 27). Relatively long-lived Cr(V) intermediates have been detected in the reduction of Cr(VI) both in vitro and in vivo (19, 27, 28. 29. 30). Cr(V) intermediate has a half-life of 37 min and are generally reactive, this chromium intermediate has been considered to be the key species in the mechanism of Cr(VI) carcinogenesis (4). This intermediate is also able to cause hydroxyl radical (·OH) generation via Fenton-like reactions (31, 32) and Haber-Weiss (33). The ·OH radicals cause cellular reactions and damage, including NF-KB activation, DNA strand breaks and 2′-deoxyguanosine (dG) hydroxylation (34).
While it appears that Cr(V) plays an important role in the mechanism of Cr(VI) carcinogenesis, studies have indicated that Cr(IV) may also be important. Cr(IV) can be generated in the reduction of Cr(VI) by cellular reductants such as glutathione (GSH) and ascorbate (16). Cr(IV) is also able to generate ·OH through a Fenton-like reaction. The reaction rate between Cr(IV) and H2O2 is faster than that between Cr(V) and H2O2, indicating that Cr(IV) may have a potential to damage cellular components, such as DNA, via free radical reactions(35, 36, 37, 38). Although Cr(IV) has been implicated in the mechanism of Cr(VI) carcinogenesis, there are only limited studies on this reactive chromium intermediate. This is because Cr(IV) is a highly reactive oxidation state of chromium and few Cr(IV)-based molecules are known that can be used as a model compound.
Liu et al (36) have synthesized a biologically related Cr(IV)—GSH compound in a solid form. The synthesis of this compound makes it possible to evaluate the role of Cr(IV) in the mechanism of Cr(IV)-induced carcinogenicity. The Cr(IV)-GSH complex was synthesized through the reaction of Cr(VI) with GSH. Its electron paramagnetic resonance (EPR) spectrum exhibits g = 1.9629 and a peak-to-peak line width of 480 G in aqueous medium as well as in the powder form. Magnetic susceptibility measurements showed that the compound has a magnetic moment of 2.53 Bohr magneton per Cr, establishing that the Cr ion has two unpaired electrons, hence its identity as Cr(IV). The Cr(IV)-GSH complex is able to generate hydroxyl radical (·OH) in the presence of molecular oxygen in aqueous medium. Catalase inhibited the ·OH radical generation while H2O2 enhanced it, indicating that the ·OH radical was generated via a Fenton-like reaction, H2O2 being generated as an intermediate in the reduction of molecular oxygen. The results imply that Cr(IV) may play an important role in the mechanism of Cr(VI) induced carcinogenesis and Cr(IV)-GSH can be used as a model compound to study the role of Cr(IV) in Cr(VI) carcinogenesis. Ramsey and Dalal (39) further discussed two stable water soluble complexes of Cr(IV) as models for Cr toxicity studies. They are Cr(dien)(O2)2·H2O (dien = diethylenetriamine) and Cr(NH3)3(O2)2.
In addition, Ramsey and Dalal (39) pointed out the availability of several Cr(V) compounds. All these compounds contain the Cr(V) ion as the peroxychromate, Cr(O2)4-3, anion with Li+, Na+ and Cs+ as the cation. (40, 41, 42). Three can be isolated as Li3Cr(O2)4·5 H2O, Na3Cr(O2)4·14 H2O and Cs3 Cr(O2)4·3H2O (42).Three of these Cr(V) compounds are highly water-soluble and could be used for comparing the relative toxicity potential for Cr(IV) and Cr(V) compounds.
V. DNA Damage Caused by Cr(VI) Reduction
DNA damages has been reported in many testing studies hexavalent chromium exposure under in vitro and in vivo testing conditions. These damages include primarily base modification and DNA strand breaks.
1. DNA Stand Break
a. Cr(VI) reduction, but not Cr(VI), causes DNA strand break
There are many in vivo and in vitro studies reported Cr(VI) exposure causes DNA strand breaks. Few examples are given below. Cr(VI) does not interact with DNA in the absence of reducing agents since Cr(VI) itself does not react readily with isolated DNA (26). Reduction of Cr(VI) to lower oxidation states can cause DNA breakage. In vivo treatment with Cr(VI) has been shown to produce persistent DNA strand breaks in chick embryo red blood cells (43). The level of Cr(VI) induced DNA strand breaks in cultured chick embryo hepatocytes was enhanced by increased intracellular concentration of GSH and was decreased by depletion of GSH (16). The level of Cr(VI)-induced DNA strand breaks in human diploid fibroblasts was decreased by superoxide dismutase and catalase but was enhanced by GSH (44).
Wakeman et al (45) reported Cr(VI) exposure triggers an S-phase cell-cycle arrest in HeLa cells followed by Cr(VI) induces DNA double-strand breaks that are similar to ionizing radiation (IR). HeLa cells were mock treated or treated with 10 uM of Cr(VI) for 4 hr, or 6 Gy (IR). Double-stranded DNA breakage was confirmed by single cell gel electrophoresis (e.g., Comet assay) and positive visualization of the activated DNA damage sensor ATM and phosphorylated nuclear gamma-H2AX (both known response to DSB) by immunofluoresence microscopy.
Detection of DNA strand break by Comet assay (1st row), phosphorylation of histone H2AX (γ-H2AX) (2nd row), and with stained DAP1 to reveal nuclei (3rd row) in HeLa cells treated with 10 uM Cr(VI) for 4 hr (45).
Ha et al. (46) studied Cr(VI) exposure in normal human fibroblasts, within a toxicologically relevant dose of 1-6 uM range. Double stand breaks (DSBs) were confirmed by both Comet assay, activation of ATM and phosphorylation of histone H2AX in nucleus. Cr(VI)-induced DSBs were only observed in the S phase population, whereas no significant DSBs were observed in G1 synchronized cells. Moreover, Cr(VI)-induced gamma-H2AX foci formation was restricted to PCNA-positive cells. These results indicate that Cr(VI) - induced DSBs are S phase-dependent.
Kortenkamp et al. (47) tested the possible DNA damage induced in the course of Cr(VI) reduction. The combination of chromate and glutathione was found to cause single-strand breaks in supercoiled circular DNA of the bacteriophage PM2. Since neither Cr(VI), when administered alone, and nor Cr(III) produced any detectable DNA cleavage, the author concluded that the critical steps leading to DNA strand breaks occur in the course of the conversion of chromium(VI) to chromium(III) by GSH, the most abundant intracellular low molecular thiol.
b. Cr(V) Causes DNA Stand break
Kortenkamp et al. (47) tested the possible DNA damage of Cr(V). The green chromium(V) complex Na4(GSH)4Cr(V) ·8H2O, prepared from chromate and glutathione, cleaved supercoiled circular DNA of the bacteriophage PM2. Vitamin E and ascorbate decreased the level of Cr(VI)-induced DNA strand breaks and the levels of Cr(V) formation, whereas vitamin B2 (a diol-containing molecule) increased the level of Cr(VI)-induced DNA strand breaks and the level of Cr(V) in Chinese hamster V-79 cells (48, 49). These results suggest that the levels of Cr(VI)-induced DNA strand breaks are correlated with the levels of Cr(V) produced. The effects of oxidative DNA damage by a model Cr(V) complex, CrV)-ehba, was investigated by Sugden and Wetterhahn (34), with and without added H202. They reported that direct and H202-induced Cr(V) DNA oxidation had opposing substrate preferences, with direct Cr(V) oxidation favoring ss-DNA while H202-induced Cr(V) oxidative damage favored ds-DNA.
c. Cr(IV) Cause DNA Strand Break
Manganese(II) (Mn(II)) is a fairly specific scavenger for Cr(IV) species. Using X Hind III DNA digest, Shi et al (35 ) assessed DNA damage induced by a mixture of Cr(VI) and ascorbate with and without H202. DNA strand breaks were detected by agarose gel electrophoresis. A significant amount of DNA strand breaks occurred when the DNA was incubated with Cr(VI) and ascorbate. The amount of DNA strand breaks depended on the relative concentrations of Cr(VI) and ascorbate. Addition of H202 drastically enhanced the DNA damage. Addition of Mn(II), which can remove Cr(IV) and inhibit Cr(IV)-mediated Fenton-like reactions, inhibited DNA damage. Results from this study confirmed the similar findings reported by Luo et al. (37, 38).
The most direct evidence that Cr(IV) break DNA is that Cr(IV) model chemical Cr(IV)-GSH can cause DNA breakage. Electrophoretic mobility shift, DNA strand breakage assays and electron spin resonance (ESR) spin trapping were used to investigate the activation of DNA strand breakage by Cr(IV) as Cr(IV)–GSH complex, as well the role of free radical reactions in these processes (50). Hind III linear DNA and/or 2 mM Cr(IV) with or without 0.2% H202 were incubated in pH 7.4 phosphate buffer for various time intervals. Electrophoretic assays using A Hind III linear DNA showed that, in the presence of H202, Cr(IV) is capable of causing DNA strand breaks. Cr(IV) alone did not cause observable DNA strand breaks, nor H202 alone, Cr(IV) in the presence of H202 caused DNA strand breaks in a time-dependent manner after 6- or 8- or 24-hr of incubation. The DNA damage became more intense as incubation time increased for 8 hr and for 24 hr. Aspirin and other ·OH scavengers decreased DNA strand breaks. These results suggest Cr(IV), a very reactive chromium intermediate may play an important role in the mechanism of Cr(VI)-induced carcinogenesis.
d. The Role of Free Radical in DNA Breakage
Based on current available data, it is not clear at present the oxidation states Cr(V) and Cr(IV) or the radical species derived from the reduction of Cr(VI) ) play a major role in DNA breakage. However, the available data suggest that free radical species (e.g. ·OH) enhance the DNA breakage ability of both Cr(IV) and Cr(V).
2. DNA Base and Sugar Lesions
a. Cr(VI) Reduction Leads to DNA Base Modification
Cr(VI) reduction and its reductive intermediates Cr(V), Cr(IV) all can cause DNA oxidation. ROS such as ·OH free radicals formed via Fenton-like reactions from the interaction of chromium and cellular substance can interact most readily with guanine residues (due to the high oxidation potential of this base relative to cytosine, thymine and adenine) at several positions to generate a range of products. The most abundant and studied lesions are 8-hydroxydeoxyguanosine, 8-hydroxyguanosine, and, 7,8-dihydro-8-oxoguanine (8-oxoG), all known to be detrimental oxidation lesions for their mutagenic effect.
The formation of 8-hydroxy-deoxyguanosine (8-oxodG) is considered a marker to implicate ROS in the mechanism of toxicity and carcinogenicity of a variety of agents (51). Using high-performance liquid chromatography (HPLC) with electrochemical detection, it was found that ·OH radicals generated by Cr(V)- and Cr(IV)-mediated reactions caused 2′-deoxyguanosine (dG) hydroxylation to form 8-oxodG (12, 33).
b. Cr(V) Leads to DNA Base Modification
Sugden and Martin (52) investigate lesions generated by a model Cr(V) complex, N,N′-ethylenebis(salicylidene-animato) oxochromium(V) (Cr(V)-Salen). Reaction of Cr(V)-Salen with synthetic oligonucleotides produced guanine-specific lesions that were not predominantly 8-oxo-2′-deoxyguanosine (8-oxodG). The guanine-based lesions observed by mass spectrometry corresponded to the lesions guanidinohydantoin and spiroiminodihydantoin(Sp). Slade et al. (53) identified and quantified the base lesions formed from reaction of duplex DNA with Cr(VI) and ascorbate. These results show that Sp was present in concentrations approximately 20 times greater than that of 8-oxoG in this system. Slade et al.(54) concluded that DNA lesion 8-oxo-2′-deoxyguanosine (8-oxo-dG) can be further oxidized by the two electron oxidants Cr(V)-salen and bis(2-ethyl-2-hydroxybutyrato)oxochromium(V) (Cr(V)-ehba) at neutral pH and forms spiroiminodihydantoin by an oxo-atom transfer mechanism.
c. Cr(IV) Leads to DNA Base Modification
Shi et al., (50) reported that Cr(IV) in the form of synthesized Cr(IV)–GSH complex induces 2′-deoxyguanosine (dG) hydroxylation to generate 8-hydroxyguanosine in cultured Jurkat cells Cells were incubated with various reaction mixtures in phosphate buffer (pH 7.4) at room temperature for 24 hr under ambient air. Concentration of reaction were as follows: dG, 1 mM; Cr(IV) 1 mM; H2O2 1 mM; formate 50 mM; deferoxamine 2 mM; aspirin 2 mM
As shown in Table 2, free radical ·OH radical generated by the Cr(IV)-mediated reaction with H202 was capable of greatly enhanced the 2′-deoxyguanosine (dG) hydroxylation and 8-oxodG production. Formate, and other ·OH scavengers, decreased 8-OHdG.
Table 2. Hydroxylation of dG by Cr(IV) Reaction (50).
| Reaction | 8-OhdG/dG (pmol/ug dG ± SD (n=3) |
|---|---|
| dG | 0.2 + 0.1 |
| dG + Cr(IV) | 5.8 ± 0.7 |
| dG + Cr(IV) + H2O2 | 95.1 ± 4.2 |
| dG + H2O2 | 2.3 ± 0.4 |
| dG + Cr(IV) + H2O2 + formate | 15 ± 0.7 |
| dG + Cr(IV) + H2O2 + deferoxamine | 21 ± 0.9 |
| dG + Cr(IV) + H2O2 + aspirin | 15 ± 0.7 |
VI. DNA Damage Sensors (ATM, ATR and DNA-PK) and Activation of P53
When ionizing radiation (IR) or Cr(VI) is reductively metabolized within cells and produced various DNA damages, three known DNA damages sensors will be activated in the affected cells. These activated DNA damage sensors: DNA-dependent protein kinase (DNA-PK) (55, 56, 57), ataxia-telangiectasia mutated (ATM) and ATM –Rad3-related (ATR) (58, 59, 60).
DNA damage sensors can be directly relayed to p53 via DNA-PK or indirectly via ATM and ATR (with Chk1 or Chk2), respectively. In a normal cell p53 is inactivated for degradation by binding with its negative regulator, mdm2. Upon the signaling by the DNA damage sensors, various pathways will lead to the dissociation of the p53 and MDM2 complex. Once activated and stabilized, p53 mediates the expression of a plethora of genes involved in cell cycle control to allow DNA repair or apoptosis to discard the damage cell (61). Precisely how the nature and/or extent of DNA damage dictate differential expression of cell arrest/DNA repair versus apoptosis gene expression is not clearly understood. There is evidence that DNA damage sensor signaling upstream of p53 plays as well as the crosstalk between the sensors are important in deciding the cell fate (56, 62, 63). Figure 4 presents some general information of the signaling of the sensors in response to DNA damages caused by ionizing radiation (IR) and UV light. The specific patterns of DNA damage sensor signaling induced by Cr(VI) reduction under various exposure conditions are described in later sections of this review.
Figure 4.
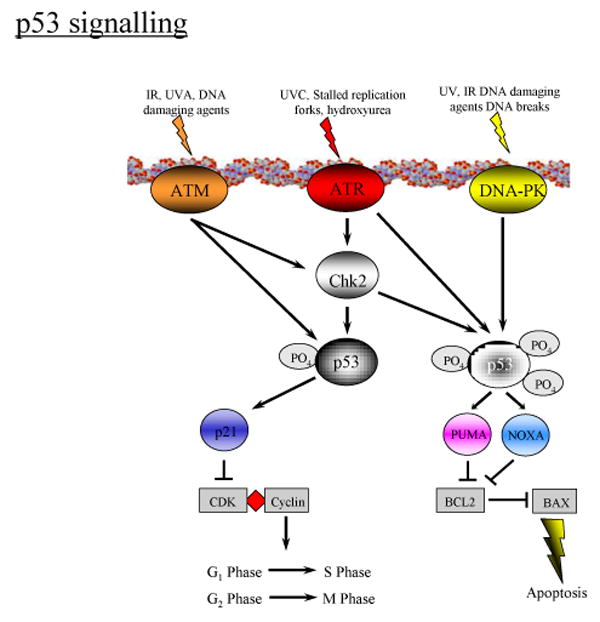
P53 Signaling of Cell Arrest/DNA Repair and Apoptosis in Response to DNA Damages by IR or UV Exposure.
VII. Cr(VI) Induced Cell Cycle Arrest
Cell cycle checkpoint pathways regulate the order and timing of cell cycle phase transition. The key cell cycle of the checkpoint pathways in response to the mammalian DNA damage are the ATM, ATR, and DNA-PK with ATM as the primary sensor of the cell arrest response to DNA double strand breaks (DSBs). The best studied function of ATM is during response to IR-induced genome damage (64). DSBs induced by IR lead to MRN (MRE11-RAD50-NBS1) recruitment. The MRE11 protein exhibits both exo- and endonuclease. RAD50 is a member of the SMC (structural maintenance of chromosomes) family, and NBS1 contains MRE11-binding domain and ATM-interacting motif. The C terminus of NBS1 recruits inactive ATM dimers to damaged DNA and this interaction leads to ATM autophosphorylation at the serine-1981 residue and dissociation into active monomer (65). The phosphorylation of H2AX histone protein at its serine-139 residue by ATM facilitates the accumulation of DSB signal mediator/transducer proteins such as 53BP1, and BRCAl. These proteins amplify the DSB response signal to downstream effectors (such as RAD51, Artemis, Chk2, and p53) triggering of G1/S and G2/M cell-arrest as well as DSB repair (66, 67, 68).
1. CR(VI) Induced G1/S Cell Cycle Arrest
G1 Checkpoint
Under normal condition, CDK inhibitor p16 inhibits CDK 4/6 to interact with Cyclin D. In growth, CyclinD/cdk4/6 complex will form and it phosphorylates the tumor suppressor retinoblastoma (Rb), this relieves the inhibition of the transcription factor E2F. E2F is then able to cause expression of cyclin E, which then interacts with CDK2 to allow for G1-S phase transition. That is the end of the first checkpoint which allows the G0-G1-S-phase transition. When DNA damages occur, the DNA damage sensor ATM/ATR will be activated. Upon ATM/ATR activation, p53 is phosphorylated. The activated p53 then up-regulates a number of genes which are also involved in the DNA damage response (MDM2, p21, p27). The accumulation and/or nucleus concentration of p21 & p27 suppress Cyclin E/Cdk2 interaction, thereby resulting in G1 arrest.
S-phase Checkpoint
The S-phase checkpoint delays DNA synthesis following DNA damage. It has been shown that there are two apparent ATM-dependent pathways control the IR-induced S-phase checkpoint (69, 70): One is to slow bulk replication by inhibiting replication origin firing and the other is to slow replication fork progression. Both pathways appear to be used. Data from IR indicates that DNA damage induces the phosphorylation of the Chk2 (ATM-dependent kinase) by ATM. Activated Chk2 then target the Cdc25A and causes destabilization of Cdc25A, prevents it from performing its normal function of removing inhibitory phosphorylations from Cdk2 (71,72). The Cdk2/CyclinE and Cdk2/CyclinA complexes remain inactive thus preventing completion of DNA synthesis. The S-phase checkpoint pathway that inhibits Cdk2/CyclinEA interaction can also be independent of Cdc25A; The alternative pathway functions through ATM-dependent phosphorylation of NBS1, BRCA1 and SMC1 (69, 73, 74). Loss of any of these proteins or mutation of the indicated phosphorylation sites results in attenuated S-phase checkpoint activation. The ATM, NBS1, BRCA1 and Cdk2 were also shown to be associated with DNA repair replication functions as described in Section VIII Repair of DNA Damages Caused by Cr(VI) Reduction. Similarly, it appears that ATR also regulate origin firing (thru ATR-dependent kinase Chk1) as well as fork progression as described above. While ATM tends to respond to DSB, ATR tends to respond to ssDNA and stabilize and repair damaged replication forks (70, 75).
Wakeman et al. (45) reported Cr(VI) exposure triggers an S-phase cell-cycle arrest in HeLa cells followed by Cr(VI) induces DNA double-strand breaks that are similar to ionizing radiation (IR). HeLa cells were mock treated or treated with 10 uM of Cr(VI) for 4 hr or 6 Gy(IR). The authors reported:
Double-stranded DNA breakage and activated (phosphorylated) nuclear gamma-H2AX (known response to DSB) (see also Section V.1.a).
DNA damage sensor ATM was activated in both Cr(VI) and IR treated cells via immunoblot analysis.
Cr (VI) induced phosphorylation of SMC1 in treated Hela cells and is ATM-dependent. In addition, as demonstrated by immunoblot analysis below, there is induction of with SMC1 and phosphorylayed SMC1 by Cr (IV) treated SV40-transform human fibroblasts that are ATM-proficient (GM 637 cells, lanes 3) but not ATM-deficient (GM 9607 cells, lane 6).
Activation of the S-phase checkpoint will result in a transient decrease of DNA synthesis (76). Cr(VI) induced a time- and dose-dependent inhibition of DNA in Hela cells at tested concentrations of 5 to 80 uM. At 10 uM, 40 uM, and 80uM Cr(VI) exposure for 4 hr, the DNA synthesis was reduced to approximately 40%, 90% and 99% in comparison with the untreated cells. In cells with Cr(VI) and 500 uM caffeine co-treatment, at 10 uM, 40 uM, and 80uM Cr(VI)-caffeine co-treatment exposure for 4 hr, the DNA synthesis was approximately 120%, 60% and 50% in comparison with the untreated cells.. Caffeine is a known inhibitor of ATM and related kinases. There is no decrease of DNA synthesis in the exposed ATM-deficient (GM 9607) cells tested at 10 uM. These aforementioned data in Cr(VI) treated Hela cells and human fibroblsts concluded that the ATM-NBS1-BRACA1-SMC1 pathway is essential for activation of the chromium(VI)-induced S-phase checkpoint.
Ha et al. (46) studied Cr(VI) exposure in normal human fibroblasts, within a toxicologically relevant dose of 1-6 uM range for 1-3 hr. DSBs, activation of ATM and gamma-H2AX in nucleus were observed. Cr(VI)-induced DSBs were only observed in the S phase population, whereas no significant DSBs were observed in Cr(VI)-treated G1 synchronized cells. Moreover, Cr(VI)-induced gamma-H2AX foci formation was restricted to PCNA-positive cells. These results indicate that Cr(VI) - induced DSBs are S phase-dependent. Lal et al. (77) reported Cr(VI) induced a concentration-dependent decrease in both phospho Ser807/811 Rb (pRb) and cyclin D1 expression and the nuclear localization of p27in human diploid lung fibroblasts treated with 0-, 1-, 3- or 6- u M Cr(VI) for 24 hrs. This observation is consistent with a G1/S checkpoint arrest.
DNA damage sensor ATR works as partners to ATM in response to IR induced DNA DSB in S and G2/M cell cycle phase. The cells in S phase is vulnerable and DNA damage can hinder replication fork progression and generate replication stress. Stalled replication fork with single-strand DNA (ssDNA) can result in unstable structure and prone to collapse. Replication protein A (RPA)-coated ssDNA is recognized by ATR-ATRIP complex. Meanwhile the Rad17-Replication Factor C (RFC) complex load complex 9-1-1 (Rad9-Rad1-Hus1) to the ssDNA-5′ primer junction, and this brings TOPBP1 to the damage site and activate the ATR, downstream Chk1, and other ATR effectors to stabilize the replication fork and to slow down the DNA synthesis and provide cells time to repair DNA repair and resume DNA replication (75, 78, 79).
Wakeman and Xu (80) studied the role of ATR in regulation of S-phase checkpoint pathway of Cr(VI) exposed cells. The study reported while ATM is required for S phase arrest with SMC1 phosphrylation at 10 uM and 40 uM (to lesser extent) of Cr(VI) exposure in SV-40 transformed human fibrolasts cell line wild type GM0637, ATR causes higher extent of SMC1 phosphorylation at 40uM of Cr(VI) in GM9607 cells that are defective in ATM. In addition, the study showed that Rad17 is essential for the ATR-SMC1 S-phase cell arrest in 293T cells treated with 40 uM Cr(VI) for 4 hr.
2. Cr(VI) Induced G2/M Cell Cycle Arrest
G2 Checkpoint
The function of G2 cell cycle checkpoint is to stall the cells with DNA damages and escape the G1/S checkpoints or cells with DNA damage incurred in the G2 phase. Entry into mitosis is controlled by the activity of the cyclin dependent kinase CDK1. Upon DNA damage, the downstream kinases Chkl and Chk2 (activated by ATR and ATM, respectively) phosphorylate phosphatase Cdc25C. The phosphorylated CDC25C is sequestered in the cytoplasm thus preventing it from activating CDK1 through removal of inhibitory phosphorylations. This prevents activation of CDK1/Cyclin B complex and block mitosis. Unlike other checkpoints, the response to IR DNA damage in G2 phase is mediated primarily by ATR with ATM playing a back-up role. The maintenance phase of the G2 checkpoint probably also relies on the transcriptional regulation by BRCA1 and p53, leading to the upregulation of cell-cycle inhibitors such as p21 (81).
Cr (VI) also caused cell arrest in the G2/M phase. Zhang et al.(82) studied the Cr (VI)-induced cell growth arrest in human lung epithelial A549 cells. Treatment of the cells with Cr (VI) at 1-25 uM for 24 hrs. At 1 μM Cr(VI) already caused a growth arrest. Increase in Cr (VI) concentration enhanced the growth arrest at G2/M phase. The percentages of cells in G2/M phase are 6.53%, 10.02%, 18.10% , 26.16% and 10.88% at control, 1 uM, 5 uM, 10 uM, and 25 uM , respectively. At a concentration of 25 μM, Cr (VI)-induced apoptosis became apparent. Electron spin resonance (ESR) spin trapping measurements showed that incubation of cells with Cr (VI) generated hydroxyl radical (·OH). These results (·OH) produced by H2O2 and Cr(VI) reductive intermediates (i.e. Cr(V) and Cr(IV)) played a major role in causing G2/M phase arrest in human lung epithelial cells.
Hayashi et al. (83) treated human lymphoma U937 cells, p53 mutated cells, with 20 uM Cr(VI) for 24 h, nuclear morphological changes and DNA fragmentation were observed. The cellular response to DNA damage involves checkpoint controls that delay cell cycle progression to provide time for repair of damaged DNA, and enhances apoptotic activity. Because Cr(VI) induces DNA damage and apoptosis after a relatively long period of incubation, it is possible that the change of the cell cycle is related to Cr(V1)-induced apoptosis. The cell phase distribution after Cr(VI) treatment was analyzed by flow cytometry. The percentages of G1 phase, G2/M phase, and S phase were 47.4 ± 7.4% (Mean ± SD, n= 3), 8.9±6.1%, and 43.7 ± 5.1%, respectively, in the control group. When U937 cells were incubated in the presence of Cr(VI) for 24 h, the percentages of G2/M phase were 13.9 ± 6.4% in the group of 10 uM Cr(VI) treatment and 19.5 ± 8.4% in that of 20 uM Cr(VI) treatment, showing an increase in G2/M phase fraction compared to the control group. The results indicate that the G2 block is involved in the apoptosis induced by Cr(VI).
Based the aforementioned description of ATR/ATM activation and cell arrest via Cr(VI) exposure, it demonstrate that, Cr(VI) exposure for short duration (e.g. 4 hr) within a toxicologically relevant dose of 1-10 uM range can cause DNA double strand breaks, S1/G phase as well as the G2/M cell arrest.
VIII. Repair of DNA Damage Caused by Cr(VI) Reduction
There are several molecular mechanisms to repair DNA damages. These include base excision repair (BER) with DNA glycosylase/polymerase/ligase to replace damaged nucleotide bases caused by oxidation, nucleotide excision repair (NER) that excise and replace damaged helix-distorting lesions, and mismatch repair (MMR) to replace DNA replication errors such as mismatched nucleotides, and DNA double stand break repair (DSB Repair). There are several studies on BER of oxidative damage to DNA bases (e.g. 8-oxo-G) and DNA single strand break induced by Cr(VI), NER on Cr-DNA adducts, and involvement of MMR in Cr(VI) exposed cells. Majority of these studies are carried out under in vitro systems (84). DNA DSB is perhaps the most serious form of DNA damage because they pose problems for transcription, replication, and chromosome segregation.
1. DNA DSB Repair Pathways
Cr (VI) causes DSBs and cells with DSB DNA damage are arrested for repair in G1-, S-, G2-, and M cell cycle phase as described in Section VII. DSBs are mainly repaired by either non-homologous end joining (NHEJ) pathway or homologous recombination (HR) pathway. In the NHEJ pathway, Ku70/80 (DNA binding proteins) and DNA-PKcs (DNA-dependent protein kinase catalytic subunit) are initially recruited to the vicinity of DSBs to form DNA-PK. The activated DNA-PK can facilitate the recruitment of other repair factors (e.g. Artemis (DNA endonuclease), DNA polymerase, and DNA ligase IV/ XRCC4, XLF) in order to mediate end resection and the rejoining reaction. NHEJ repair can occur throughout the cell cycle and is more error prone due to the processing of DSB ends with nucleases followed by direct rejoining; DSB also can be repaired in an error-free manner by homologous recombination (HR) during the late S phase and G2 phase in which an undamaged sister-chromatid is available to provide repair templates. The initial step of HR is DSB end resection in order to generate a 3′-single strand DNA. This DSB end modification can be enzymatically processed by the MRN complex and possibly other nculceases. Replication protein A (RPA) the binds to the 3′- overhang to protect DNA ends. RPA is then replaced by RAD51, which mediates strand invasion and DNA synthesis using a sister-chromatid as a template. This step requires RAD51 paralogs (RAD51C, XRCC2, etc), BRCA2, and Fanconi anemia proteins. Finally, the recombination intermediate structure accompanying Holliday junctions is processed and resolved by some helicases and nucleases. It has been suggested that NHEJ and HR are both co-regulated by ATM and DNA-PK. DNA DSBs leads to MRN and ATM recruitment. Proteins involved in HR are phosphorylated by activated ATM, but some of these proteins (e.g. H2AX, P53, SMC1) can also be phosphorylated by DNA-PK. DSBs also recruit Ku heterodimer and end-binding DNA-PKcs. The DNA-PKcs protects the DNA ends until it is phosphorylated by itself and ATM to induce a conformational change that enables the DNA end processing and ligation by NHEJ enzymes (67, 85, 86).
2. Cr(VI), Cr(IV), and Possibly Cr(V) Induce Mitotic Recombination (Homologous Recombination) Due to DSB in Late S and/or G2/M Phase
As mentioned in previous sections, that Cr(VI) induces DSB, activates S-phase cell arrest via ATM-SMC1 pathway for DNA repair. Studies described below demonstrate Cr(VI) exposure induces homologous recombination to repair DNA DSB.
The somatic mutation and recombination test (SMART) D. Drosophila was developed in the 1980s. The SMART assay can detect a variety of genetic alterations, including gene mutations, deletions, aneuploidy/segmental aneuploidy and mitotic recombination. The detection of mitotic recombination is an important feature of the assay since mitotic recombination can lead to loss-of-heterozygosity (87).
There are positive results of Drosophila melanogaster SMART testing on Cr(VI) and Cr(IV); Garf and Wügler (88) reported results of SMART wing spot test and less sensitive white-ivory eye spot test in 2-3 day old larvae were fed chronically with the chromium (VI) oxide and potassium chromate. The wing spot test utilizes two loci located on the left arm of chromosome 3 − mwh (multiple wing hair) and flr3 (flare) to detect both mitotic recombination and various types of mutational events. Both loci influence development of hair growth in each adult wing blade cell. The white-ivory eye spot test makes use of the white-ivory (wi) quadruplication and detects the somatic reversion of the recessive eye color mutation wi to the wild-type (w+). Both Cr(VI) compounds have shown high mitotic recombination activities.
Katz et al (89) and Chiu et al. (90) further evaluated Cr(VI), Cr(IV) and Cr(III) for genotoxic effects in somatic tissue of D. melanogaster with SMART wing spot test. The results suggest both Cr(VI) and Cr(VI) are highly genotoxic in flies via induction of mitotic recombination. Virgin females of genotype mwh were mated to males of genotype flr3/TM3, Ser. The 3rd instar larvae were collected, washed and randomly allocated to different treatments. The larvae were treated for 6 hr with a neutral cellulose powder slurry wetted with distilled water or 20 and 40 mM concentrations K2Cr2O7 and GSH-Cr(IV). This latter acute exposure treatment was used to overcome insolubility of GSH-(IV) in water. Larvae exposed to a test substance are allowed to develop into adults, and then their wings are removed and examined microscopically for wing spots. Three endpoints are distinguished in the assay: (1) small single spots of either mwh or flr3 phenotype, which consist of just 1–2 cells; (2) large single spots of either mwh or flr3 phenotype, which consist of 3 or more cells; and (3) twin spots, which consist of adjacent mwh and flr3 spots. While small and large single spots can arise from a variety of genetic alterations, twin spots result solely from mitotic recombination.
A summary of genotoxic effects in trans-heterozygous fruit flies for Cr(VI) and Cr(IV) is presented in Table 3. Cr(VI) was a positive inducer of all endpoints at high and low dose concentrations. Similarly, Cr(IV) was found to be a positive inducer of all three endpoints at the higher concentration, and Cr(IV) was also a positive inducer of twin spots at the lower concentration. Findings that Cr(VI) and Cr(IV) induce twin spots demonstrate that chromium in these valence states induce mitotic recombination, in late S and/or G2/M cell cycle phase, due DNA double strand breaks. There was no Cr(III) induced-somatic-mutation or mitotic-recombination.
Table 3. Frequency N and Means per Wings of Induced Spots in Treated Trans-heterozygous (mwh flr+/mwh+ flr3) Flies* (89).
| Treat dose | Wings, number | Small spots | Large spots | Twin spots | |||
|---|---|---|---|---|---|---|---|
| N | per wing | N | per wing | N | per wing | ||
| H2O | 40 | 5 | 0.125 | 1 | 0.025 | 0 | 0 |
| Cr6+, Cr2Cr2O7 | |||||||
| 40 mM | 20 | 20 | 1.00 (POS) | 82 | 4.10 (POS) | 66 | 3.30 (POS) |
| 20 mM | 20 | 9 | 0.45 (POS) | 39 | 1.95 (POS) | 53 | 2.65 (POS) |
| Cr4+, GSH-Cr | |||||||
| 40 mM | 20 | 22 | 1.10 (POS) | 23 | 1.15 (POS) | 26 | 1.30 (POS) |
| 20 mM | 20 | 5 | 0.25 (INC) | 5 | 0.20 (INC) | 8 | 0.45 (POS) |
| Cr3+, CrCl3 | |||||||
| 40 mM | 20 | 3 | 0.15 (INC) | 1 | 0.05 (INC) | 0 | 0 |
Treatment with H2O as control. POS=positive result, INC=inconclusive result.
In a series of reactions, if the concentration of intermediate chemical species, e.g. in this case Cr(IV) markedly increases above the concentration of the initial chemical reactant species (Cr(VI)), there will be a corresponding faster and more concentrated buildup of the terminal product (somatic recombination) compared to the initial reactants. The total number of wing spots induced by Cr(VI) and Cr(IV) treatments at 20 and 40 mM in trans-heterozygous flies are summarized in Table 4. It shows that the ratio of spots formation at 40 mM vs 20 mM for Cr(VI) and Cr(IV) are 1.66 (8.40/5.05) and 3.94 (3.55/0.90), respectively. The conservation of higher ratio of response associated with C(IV) over Cr(VI) with a doubling of mutagen concentration treatments and Cr(IV) ability to increase twin spots or somatic recombination (25 ).
Table 4. Total wing spots in trans-heterozygous flies (mwh flr+/mwh+ flr3) treated with Cr6+ and Cr4+ (89).
| Treatment | Total number spots | Mean number |
|---|---|---|
| 40 mM Cr6+ | 168 | 8.40 |
| 20 mM Cr6+ | 101 | 5.50 |
| 40 mM Cr4+ | 71 | 3.55 |
| 20 mM Cr4+ | 18 | 0.90 |
Independently, Spanó et al. (90) determined the recombinagenic and mutagenic activities for potassium chromate with D. melanogaster SMART wing spot test. Two different crosses involving the wing cell markers mwh and flr3 were used: the standard cross and a high bioactivation cross. The high bioactivation cross is characterized by a high constitutive level of cytochromes P450 and therefore enhance the Cr(VI) reduction to Cr(V). Three-day-old larvae derived from both crosses were treated chronically with the oxidizing agent potassium chromate. The oxidizing agent potassium chromate was equally and highly genotoxic in both crosses.
Studies above shown that Cr(IV), Cr(IV), and possibly Cr(V) are highly genotoxic in flies via induction of mitotic recombination. Mitotic recombination leads to a loss-of-heterozygosity in an organism, which is suspected of playing a major role in the development of human cancers (91, 92, 93, 94, 95).
The pathways (established with current available data) of G1 Phase-, S Phase-, and G2/M Phase-cell cycle arrest as well as DNA repair caused by Cr(VI) exposure are presented in Figure 7.
Figure 7.

Cell Arrest and DNA Repair Resulted from Double Strand DNA Breaks Due to Cr(VI) Exposure. Diagram Based on Available in in vivo/in vitro Experimental Data.
IX. Apoptosis Caused by Cr(VI) Reduction
1. Intrinsic-Mitochondrial Apoptosis Pathway and Extrinsic Death Factor pathway
a. Intrinsic Mitochondrial Apoptosis Pathway
Cell death, apoptosis, occurs in at least two pathways: the intrinsic mitochondria-mediated pathway and extrinsic death-receptor pathway; Reactive oxygen species (ROS), glutathione depletion or DNA damage can stimulate apoptotic signaling which causes mitochondrial membrane permeabilization (MMP). MMP can be induced via different pathways. For example, MMP can be resulted via mitochondrial permeability transition (MPT) pore formed across inner membranes when Ca2+ reaches a critical threshold or via mitochondrial outer membrane pores formed by regulation of p 53–dependent proapoptic (e.g. Bax, Bak) and antiapoptotic (e.g. Bcl2, Bc1-X1) proteins. MMP leads to the release of apoptogenic proteins such as cytochrome c and other apoptosis-inducing factors that form a molecular platform APOPTOSOME in cytoplasm for activation of caspase 9 which in turn to produce cascade of proteins and effectors (caspase 3, caspase 6, and caspase 7) for the morphological and biochemical changes of apoptosis (96, 97, 98, 99, 100).
b. P53-Dependent Intrinsic-Mitochondrial Apoptosis Pathway
In response to DNA damage, the tumor suppressor gene p53 regulates the expression of proapoptotic members of the Bcl-2 family (e.g. Bax, Bak) at the outer mitochondrial membrane to induce MOMP (mitochondrial outer membrane permeabilization). A subgroup of the Bcl-2 family, BH3 (Bcl-2 homology 3)-only proteins, such as PUMA were also activated primarily by p53 and act indirectly on the Bcl-2 family members Bax and/or Bak by relieving the inhibition imposed by antiapoptotic members such as Bcl-2. In addition, nuclear caspase-2 activated via p53-inducible nuclear death-domain (PIDDosome) platform was reportedly to be required and act in conjunction with Bax/Bak for p53-mediated apoptosis by act upstream of MOMP (101, 102, 103, 104, 105, 106, 107, 108, 109, 111, 112).
c. Extrinsic Apoptosis Pathway
The extrinsic death receptor signaling is mediated by the binding of ligand, such as FasL (Fas ligand), to its member receptor and, together with the Fas-associated death domain (FADD), forms the death-inducing signaling complex (DISC). Subsequent activation of the initiator caspase-8, and possibly caspase-10, the DISC (death inducing signaling complex) recruits and activates initiator caspase-8 and 10, results in (i) direct activation effector caspases such as executor caspase-3,-6, and -7 or (ii) engagement of mitochondrial apoptotic signaling through cleavage of the pro-apoptotic protein Bid (BH3 only pro-apoptotic protein) to tBid (truncated form of Bid). It was reported that Caspase 2 may be also required for cleavage of Bid, which is necessary for MMP and cytochrome c release from mitochondria (97, 113, 114, 115).
The current view of apoptosis pathways and their intiator- and executor- caspases are summarized as follows:
Mitochondrial intrinsic pathway via APOPTOSOME platform with initator caspase-9 and effectors caspase-3, -6, -7.,
P53-dependent mitochondrial intrinsic pathway via PIDDSOME platform with initator caspase-2 and amplification loop with effectors caspase-3, and other unkown factors.,
Extrinsic apoptosis pathway via DISC platform with initiator caspase-8, -10 and effector caspase-3, -6, -7.
2. Cr(VI) Induced Apoptosis via Both p53 -Dependent and p53-Independent Intrinsic Mitochondrial Pathways
Ye et al. (116) treated human lung tumor cell line A549 cells Cr(VI) at a final concentration of 75-500 uM for 3 h and then cultured in fresh cell medium and examined at different time intervals. Cr(VI) caused apoptosis in the exposed cells as measured by DNA fragmentation, mitochondria damage, and cell morphology. The induction of apoptosis raised with time of the exposure and peaked at 24 hours followed by decline. The extent of the apoptosis is in dose-dependent manner with Cr(VI) concentration from 75 to 300 uM, saturated at a dose above 300 uM, and decreased slightly at 500 uM. At 300 uM, Cr(VI)-induced ROS generation occurred within a few minutes after Cr(VI) treatment of the cells, whereas p53 induction took at least 5 h. Both reactive oxygen species (ROS) O2·- and H2O2 were visualized in cytoplasm, and O2·- were also visualized in nucleus with intracellular staining. The level of Cr(VI)-induced apoptosis was similar in both p53-positive cells and p53-negative cells independent of p53 status in the early stage (0–2 h) of Cr(VI) treatment. However, at the later stage (3–24 h), the level of the apoptosis is about twice higher in p53-positive cells than in p53-negative cells. These results suggest that ROS generated through Cr(VI) reduction is responsible to the early stage of apoptosis, whereas p53 contributes to the late stage of apoptosis and enhance the Cr(VI)-induced apoptosis. In contrary to strong induction apopotosis in Cr(VI) treated cells, Ye et al. (1999) reported that there is very little observed apoptosis of cells treated with 300 uM of Cr(IV) for 3 hr under the same condition.
Hayashi et al. (117) further investigated the apoptotic mechanism induced by Cr(VI). When human lymphoma U937 cells, p53 mutated cells, were treated with 20 uM Cr(VI) for 24 h, nuclear morphological changes and DNA fragmentation were observed. Production of hydroxyl radicals (by electron paramagnetic resonance (EPR)-spin trapping), increased lipid peroxidation, and increased intracellular calcium ion concentration (monitored by digital imaging) were observed in Cr(VI)-treated cells. An intracellular Ca2+chelator, BAPTA-AM, and calpain inhibitors suppressed the Cr(VI)-induced DNA fragmentation by 60% and 20%, respectively; This suggested strongly a Ca+2 –dependent pathway is involved in Cr(VI) induced apoptosis via Ca2+-calpain pathway and it is also possible Ca2+ is able to activate other targets to trigger apoptosis, such as Ca2+- Mg2+ -dependent nuclease; The number of treated cells also showed low mitochondrial membrane potential (control 4.61+/-1.85% vs Cr(VI) treated 26.55 +/-5.12 %), high level of superoxide anion radicals O2·- (2.98+/- 1.24% vs 35.605+/- 5.67%), and high activity of caspase-3 (5.03+/- 0.31% vs 58.33+/-1.85%). These are indicators of mitochondria-caspase-dependent pathway; No increase of the expressions of Fas and phosphorylated JNK was observed after Cr(VI) treatment indicates the extrinsic apoptosis in not involved in Cr(VI) induced apoptosis under the experimental condition. The authors concluded that ROS play an important role in both Cr(VI) induced P53 independent apoptosis thru Ca+2 –dependent pathway and p53-dependent mitochondrial-caspase pathway. Cell cycle analysis revealed that the fraction of G2/M phase tended to increase after 24 h of treatment, suggesting that Cr(VI) induced apoptosis is related to the G2 block.
3. Cr (VI) and Cr(V), but not Cr(IV), Induced p53-Dependent Intrinsic Mitochondrial Apoptosis
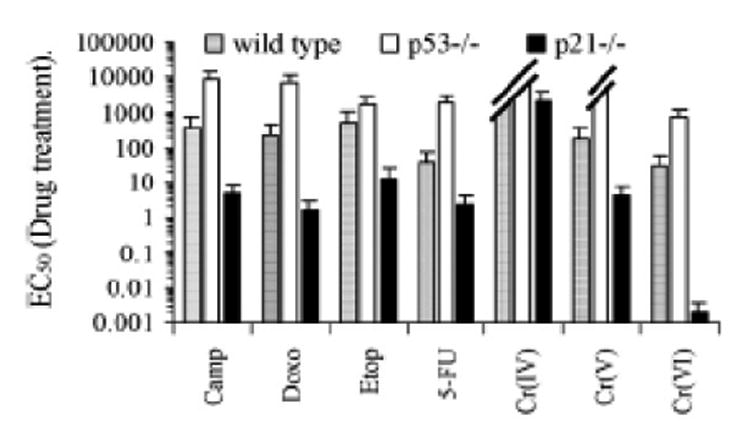
Hill et al. (118) studied the apoptosis induced by of Cr(VI) in wild type, p53-/- and p21-/- human colon carcinoma HCT116 cells treated with 30 uM of Cr(VI) for 24 hr or with Cr(V) or Cr(IV) or other DNA damage agent for 72 hr. Cell line sensitivity was then assessed by MTS assay (n = 3) and EC50 was determined for each cell line tested. As shown in Figure 8, the Cr(IV) failed to induce cell death in either wild type or p53-/- cells, consequently an EC50 value could not be determined at the 72 hr time point. This finding is consistent with the conclusion of Ye et al. (116) that Cr(IV) does not cause apoptosis. While Cr(V) and Cr(VI) both induced cell death, Cr(V) failed to induce cell death in p53-/- cells (an EC50 value could not be determined), and Cr(VI)exhibited increased EC50 in p53-/- cells in comparison to the wild type. The elevated EC50 value in Cr(VI) in P53-/- cells may be due to the ROS induction of p53-independent apoptosis. These results suggest that: i) Cr(IV) does not induce apoptosis, ii) Cr(V) induces mainly p53-dependent apoptosis, and iii) Cr(VI) can induce both p53-dependent and p-53-independent intrinsic mitochondrial apoptosis. These results are consistent with the findings of Ye et al. (116) and Hayashi et al. (117). For each treatment, p21-/-HCT116 cells demonstrate significantly reduced viability. This suggests that p21 is important for replicative stress response/cell arrest, and its absence allows more DNA damage leading to more apoptosis.
Figure 8. EC50 of Wild Type, p53-/-, p21-/- HCT116 Cells Treated with 30 uM Cr(VI) for 24 hr or Cr(V) or Cr(IV) or Other DNA Damage Agent for 72 hr (118).
4. DNA-Dependent Protein Kinase Mediated Cr(VI) Induced p53-Dependent Intrinsic Mitochondrial Apoptosis
Hill et al. (119) further studied which DNA-damager sensor is responsible for the Cr(VI) induced p53-dependent intrinsic mitochondrial apoptosis in treated wild type p53-/-, p21-/- HCT116 cells . When p53+/+ HCT116 cells were pre-incubated for 1 hr with DNA-dependent protein kinase (DNA-PK)-inhibitor at 150 uM concentration followed by 30 uM Cr(VI) exposure for 12 hr, Cr(VI)-induced p53 accumulation as well as its phosphorylation at ser15 and ser46 in p53+/+ cells were efficiently blocked. The DNA-PK inhibitor did not affect Cr(VI)-induced ATR or ATM activation as it did not block Chk1 phosphorylation at ser345 by ATR or Chk2 phosphorylation at thr68 by ATM. In contrast, pretreatment for 1 hr with 10 mM caffeine, which effectively blocked Cr(VI)-induced Chk1 and Chk2 phosphorylation had no effect on the high levels of induced p53 or ser15 and ser46 phosphorylation. When p53+/+ HCT116 cells were pre-incubated with DNA-PK inhibitor and then treated with Cr(VI) there was a significant reduction of cellular sensitivity to Cr(VI). This reduced sensitivity was not observed when cells were pre-incubated with caffeine prior to Cr(VI) exposure. Relative to p53+/+ cells, the p53-/- cells were highly resistant to Cr(VI)-induced cell death, and were immune to the effects of the DNA-PK inhibitor or caffeine. There are very little effects of DNA-PK inhibitor I or caffeine on p53 stabilization/phspohorylation and apoptosis in p21-/- HCT116 cells. Based on the aforementioned observation, the authors concluded that Cr(VI) induced p53-dependent apoptosis is mediated via sensor DNA-PK.
5. Increased p53/PUMA/BAX, and Decreased P21/Bcl-2 during Cr (VI) Induced p53-Dependent Intrinsic Mitochondrial Apoptosis
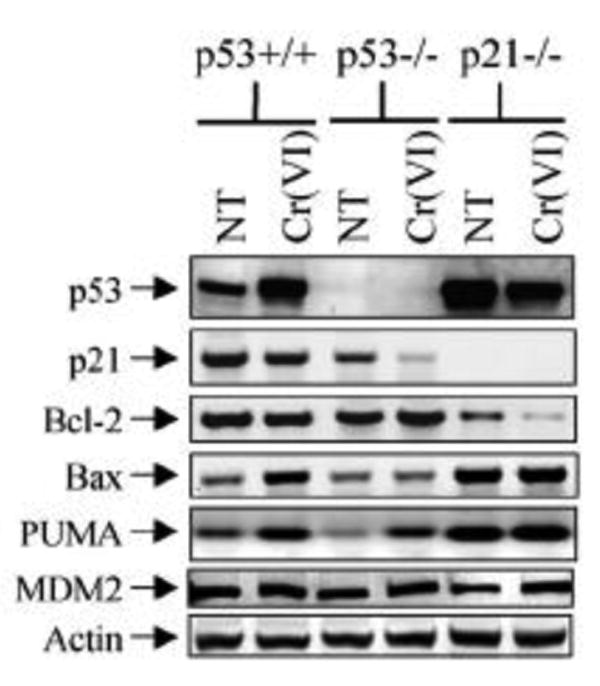
Consistent with the molecular pathway of p53-dependent intrinsic mitochondrial apoptosis, increased Bax, PUMA, and MDM2 and decreased Bcl-2 and p21 were observed in wild type cells, but not inP53-/- and P21-/- cells, following 30 uM of Cr(VI) treatment for 12 hr. Note under non-DNA damage conditions, the p53, Bax, PUMA level in p21-/- cells were already significantly higher, and Bcl-2 significantly lower than in wild-type cells (Figure 10).
Figure 10. Western blot of p53, p21, Bcl-2, PUMA, and MDM2 in Vr(VI) Treated HCT116 cells with 30 uM Cr(VI) for 12 hr.
6. Cr(VI) and Cr(V), but not Cr(IV) Induce Caspases
a. Cr(V), but not Cr(IV), Induces p53 Dependent Caspase 2 Activity
Caspase 2 is located in nucleus and has been identified as a part of p53 mediated MOMP apoptosis. It has been demonstrated that caspase 2 is required for p53 dependent activation of Bax and “caspase2-caspase3” amplification loop. (101, 120). However, no experiment has been previously reported on caspase 2 activity in Cr(VI) induced apoptosis. Caspase 2 &3 activity analyses were therefore performed on Cr(V) or Cr(IV) exposed Neo-(as the control) and Bcl-2 tranfected wild-type Jurkat T-lymphocytes (clones E6.1 and A3) cells. Apoptosis was induced with Cr(V) or Cr(IV ) at a final concentration of 10 uM for 6 or 24 hours. Cells (5 × 105) were pelleted and washed once with ice-cold phosphate-buffered saline (PBS) for caspase activities as described previously by Robertson et al. (121).
Activaties of caspase 2 (Figure 11a) are significantly increased upon exposure to Cr(V) in time-dependent manner, but Cr(IV) did not increase caspase 2 activity. Bcl-2 (apoptosis inhibitor) effectively blocked the Cr(V) ability to activate caspase 2. These results confirmed that Cr(VI) and Cr(V), but not Cr(IV), induce intrinsic mitochondrial apoptosis through the activation of p53 and caspase 2.
Figure 11a. Activation of Caspase 2 in Jurkat T-Lymhocytes after 10 uM Cr(V) or Cr(IV) Exposure for 24 hr.

b. Cr(VI) & Cr(V), but not Cr(IV), Induces p53 Dependent Caspase 3 Activity
Caspase-3 is an execution caspase in the late stage of apoptosis. Caspase-3 is activated in the apoptotic cell both by extrinsic (death ligand) and intrinsic (mitochondrial) pathways (97). In addition, it has been suggested that “caspase-2 – caspase 3 amplification loop” promotes the activation of Bax during p53-dependent mitochondrial apoptosis.
P53-dependent activation of caspase-3 by Cr(VI) has been demonstrated by Hill et al. (118). When exposed to Cr(VI) at 30 uM concentration for 12 hours, wild type HCT116 cells showed increase of caspase 3 and caspase 3 cleavage, whereas p53-/- HCT116 cells showed less increase of caspase-3 and minimal caspase 3 cleavage. The exposed p21-/- cells demonstrated caspase 3 cleavage significantly faster than wild type or p53-/- cells. Collectively, these results indicate that caspase 3 activation is p53 dependent via Cr(VI) exposure. The enhanced cell death and caspase 3 observed in p21-/- HCT116 cells implies that caspase activation is p53-dependent event and is indirectly controlled (via caspase 2) by p21.
The finding of increased caspse 3 activity in Cr(VI) by Hill et al. (118) has confirmed the result by Hayashi (117) as described in Section IX.2 and Ha et al (122). Ha et al (122) reported that the percentage of apoptotic cells of normal fibroblast treated with Cr(VI) for 24 hours at 0-, 3- and 6-uM are 2%, 2.6-4.3% and 40%, respectively. The caspase-3 and cleaved caspase-3 also increased in a dose-dependent manner. The amount of cleave caspase 3 is 8 fold higher in cells treated with Cr(VI) for 24 hr than the control.
Caspase-3 activity analyses were performed on 10 uM Cr(V) or Cr(IV) exposed Neo-(as the control) and Bcl-2 transfected wild-type Jurkat T-lymphocytes (clones E6.1 and A3) cells as described by Robertson et al. (121 2002). Activaties of caspase-3 are significantly increased upon exposure to Cr(V) in time-dependent manner, but Cr(IV) did not increase caspase 3 activity. Bcl-2 (apoptosis inhibitor) effectively blocked the Cr(V) ability to activate caspase 3.
In summary, the results of the reported apoptosis and its upstream regulators and caspase activation collectively suggest the following:
Cr(VI) induces apoptosis in exposed cell via p53-dependent and p53-indepednetmitochondrial intrinsic pathway. ROS plays an important role in both apoptosis pathways.
Cr(V) causes apoptosis primarily via p53-depedent mitochondria intrinsic pathway.
Cr (IV) does not cause apoptosis.,
DNA-PK is the primary DNA damage sensor controls the Cr(VI) induced p53-dependent mitochondrial apoptosis that resulted in p53 upregulate the expression of proapoptotic Bax and Puma mediated MOMP.
Caspase 2 in nucleus downregulated by p53/p21 is required for Cr(VI) and Cr(V) induced p53-dependent mitochondrial apoptosis by act upstream of MOMP.
The resulting MOMP activated downstream effector caspases such as caspase-3 then mediates the final steps of Cr(VI) and Cr(V) induced apoptosis.
p21 (suppressor of Cyclin E/Cdk2 interaction, thereby resulting in cell arrest) has an important role in the cellular response to normal replicative stress and its absence leads to a “chronic DNA damage” state that primes the cell for p53-dependent apoptosis.
Apoptosis pathways resulted from Cr(VI) exposure based on in vitro and in vivo experimental data are presented in Figure 12.
Figure 12.

Apoptosis Resulted from Double Strand DNA Breaks Due to Cr(VI) Exposure. Diagram Based on Available InVitro/InVivo Experimental Data.
X. Cr(VI) Carcinogenesis and Conclusions
1. The Balance of Cell Arrest and Apoptosis Induced by Cr(VI) Exposure
As described in previous sections, (VI) is reductively metabolized within cells and produces DNA double strand breaks during the reductive process. Three known DNA damages sensors ATM, ATR and DNA-PK will be then activated in the affected cells. These DNA damage signals are relayed to the master p53 directly via (DNA-PK) or indirectly (by Chk1 or Chk2) via ATM and related (ATR). In a normal cell, p53 is inactivated for degradation by binding with its negative regulator, mdm2. Upon the signaling by the DNA damage sensors, various pathways will lead to the dissociation of the p53 and mdm2 complex. Once activated and stabilized, p53 mediates the expression of a plethora of genes involved in cell respectively cycle control to allow DNA repair or apoptosis to discard the damage cell. Cr(VI) exposure induced DNA-dependent protein kinase (DNA-PK)-mediated apoptotic response (118, 119) as well as ATM/ATR-SMC1 dependent S-phase arrest. (45, 80) and G2/M-phase cell arrest (116, 117). (See Figures 7 and 12 for cell cycle arrest and apoptosis, respectively). These events induced by DNA DSB from Cr(VI) exposure are similar to those caused by IR exposure. Precisely how the nature and/or extent of DNA damage dictate differential expression of cell arrest/DNA repair versus apoptosis gene expression is not clearly understood and under research. There is evidence that DNA damage sensor signaling upstream of p53 also plays an important role in deciding cell fate. Limited crosstalk exists between the DNA-PK, ATM and ATR kinase pathways, as illustrated by experiments where signaling through one pathway is partially or totally deficient (56, 62, 63). Woo et al. (57) suggest the function of DNA-PK activity in regard to p53-activation is to promote apoptosis and has no significant effect on IR-induced ATM-dependent cell cycle arrest. Therefore it appears that while DNA-PK, ATR and ATM function as upstream p53 activators in response to DNA damage, they play antagonistic role. On the other hand, recent study by Shi et al (123) indicates that DNA-PK, in DNAPKcs-PIDDosome, may be also involved G2/M DNA damage checkpoint and NHEJ DNA repair in γ-radiation exposed mammalian cells. DNAPKcs-PIDDosome is a large nuclear protein complex consists of DNA-PK, Caspase 2, and PIDD (p53-inducible death-domain). It has been reported that Cr(VI) exposure induces both G1/S- and G2/M cell cycle arrest. It is possible that DNA-PK is the first sensor to detect DNA damage prior to the activation of ATM/ATR. And ATM/ATR is responsible to Cr(IV) induced G1/S cell arrest while DNA-PK may be involved in Cr(VI) induced G2/M cell arrest. Recent studies also identified a significant level of sophistication and interplay between DNA damage, p53 activation, recruitment of the basal transcription apparatus and RNA polymerase II (RNA Pol II) machinery modification after various forms of DNA damage, suggesting that p53-dependent discriminatory transcription of downstream targets is a also major determinant of cell fate upon DNA damage (124, 125,126,127).
2. Cr(VI) Can Cause Cancer under Very Low Concentration and Chronic Exposure Condition
Cr(VI) does not interact with DNA in the absence of reducing agents (26). Cr(VI) reduction intermediates Cr(V) and Cr(IV) cause DNA DSB with isolated DNA (50). The DBS is further increased with added H2O2. Based on these observations, one can conclude that reductive intermediates (i.e., Cr(V) and Cr(IV)) cause DNA DBS and the free radicals produced during the reduction process further enhanced the DNA breakages.
Cells respond to DNA DSB due to exposure to Cr(VI) reduction with cell arrest (i.e. p53/ATM/ATR-SMC1 dependent S-phase arrest and G2/M phase), DNA repair (e.g. somatic recombination) as well as apoptosis (i.e. p53/DNA-PK/caspase-2 dependent intrinsic mitochondrial apoptosis via preferential expression of pro-apoptotic genes PUMA and BAX). Cell cycle arrest is favored at low dose for shorter exposure (e.g. 1-6 uM for 30 min to 4 hr) (Wakeman et al. 2004; Ha et al. 2004). On the other hand, apoptosis is favored at shorter exposure at high Cr(VI) concentrations (e.g. 75uM for 3 hr) (116) or longer time with lower concentrations (e.g. 25-30 uM for 12hr or 1-3 uM for 24 hrs) (82, 118, 119, 117, 122). Ha et al. (2003) reported the clonogenic survival of normal fibroblast treated with Cr(VI) for 24 hr at 0.1 uM, 0.5 uM and 1 uM are 98%, 78% and 51-59%, respectively. In addition, cells with Cr(VI) induced apoptosis triggers p21 (a p53 downstream cell arrest promoter) degradation.
Question shall be raised among all the changes in genetic characters in the cancer cell such as avoidance of apoptosis, senescence/cell-cycle-arrest, endocrine-independence, genetic plasticity and distant metastasis, which characteristic is primary event that initiates the carcinogenic process. (128, 129, 130, 131). From the studies of apoptosis and senescence in targeted knockout myc-, ras- and p53- mice during cancer development, apoptosis appears to be the premier mechanism in the control of cell number and the maintenance of stem cell in the cellular population. Apoptosis will persist to threaten subsequent cell generations from the damaged cells that survived the first cell-cycle. And a DNA-damaging chemical that allows the continuous ability for this malignant-progenitor-cell to escape from cell death and to proliferate is the character that a carcinogen is expected to possess (132, 133, 134,135, 136, 137, 138, 139).
In a parallel study of the progression of early premalignant and malignant lesions in human urinary bladder and lung, the DNA damage response such as DNA DSB-induced apoptosis precedes the activation of ATM/Chk2/H2AX and followed by the increase in DNA replication and cell proliferation (58). Following this model, the carcinogenic process Cr(VI) exposure is expected to occur in cycles of DNA damage response→ apoptosis→cell-cycle-arrest/DNA repair, followed by an increase in unregulated DNA-replication and cell-division and cancer development.
It is likely that long-term and very low concentration exposure to Cr(VI) is sufficient to cause DNA damage, p53 activation, p53-dependent apoptosis and p21 degradation (prevents cell arrest). Cells under this condition will undergo apoptosis. However, damaged cells with mutated p53 or other connected regulatory genes (e.g. M2D2, H2AX), thus compromised PUMA and BAX expression, and would not be able to undergo apoptosis under chronic low-dose Cr(VI) exposure. Meanwhile these cells also would not be able to undergo cell growth arrest due to the suppression of p21 (or other relayed cell arrest genes such as SMC1,CDK2, NBS) expression. These cells with DNA damages will not be arrested and continue to proliferate, leading to the emergence of tumor.
3. Cr(IV), a Reductive Intermediate of Cr(VI), Is likely the Most Potent Carcinogenic Chromium Species
In vivo and in vitro treatment with Cr(VI) has been shown to produce persistent DNA strand breaks in cells (43, 44). The level of Cr(VI) induced DNA strand breaks enhanced by increased intracellular concentration of GSH and was decreased by depletion of GSH (16). The formation of Cr(IV)-GSH from Cr(VI) via two-electron transfer is probably favored inside the cytoplasm while Cr(V)-NADPH formation via one-electron transfer occurs inside the mitochondria/endoplasmic reticulum. The concentration of GSH inside cells is about a 3 order of magnitude higher than NADPH/NADH in the cytoplasm (23). These argue that Cr(IV) is more abundant and potent carcinogen than Cr(V) to cause DNA DSB (25).
Exposure to Cr(IV) or Cr(V) or Cr(VI) all induce DNA DSB and DNA base modification. In addition, Cr(IV) appears to be more efficient than Cr(VI) to produce somatic recombination in D. Drosophila SMART test (25, 89). The cells with DNA DSB and increased somatic recombination will have increased chance of loss-of –heterozygosity. However, unlike Cr(VI), Cr(IV) don't kill cells. Both Cr(VI) and Cr(V), but not Cr(IV), cause p53-dependent and p53-independent intrinsic mitochondrial apoptosis (118, 119). Both Cr(V) and Cr(V), but not Cr(IV), induce caspase-2 (required for p53-dependent intrinsic mitochondrial apoptosis) and caspase-3 (required for both p-53-dependent and p53-independent intrinsic mitochondrial apoptosis). This implies that Cr(IV) can provide added character to the exposed cells with DNA damages to survive and propagate from one generation to the next.
It is also worth to mention that Cr(VI) and Cr(IV), but not Cr(III), are able to induce (50) NF-KB activation in Jurkat cells treated with 2 or 4 uM Cr(IV) in dose-dependent manner. NF-KB is considered a primary oxidative stress response transcription factor that functions to enhance the transcription of a variety of genes. NF-KB binding sites serve as an enhancer element in the c-myc oncogene (140). It is possible that Cr(VI) & Cr(IV) could induce over-expression of c-myc proto-oncogene via NF-KB activation. Since over-expression of this oncogene is associated with carcinogenesis, NF-KB activation and a subsequent proto-oncogene expression may added to the mechanism for induction of neoplastic transformation by Cr(VI) as well as Cr(IV).
Based on the relative abundance of Cr(IV) species during Cr(VI) reduction, the increased chance of DNA damages (e.g. increased DNA DSB and somatic recombination) and the fact Cr(IV) does not cause cell apoptosis, we suspect that Cr(IV) is likely the most potent carcinogenic Cr species.
4. Cr(VI) Causes Cancer via Multistage-Multipath Mechanism
Multistage and multiple pathway mechanisms of carcinogenesis have long been identified and reported in both rodents and humans. Multiple pathways in multistage carcinogenesis were first demonstrated by Leslie Fould, based on his life-long studies of murine mammary adenocarcinoma (141). The biological counterparts of this conceptual multi-stage multi-path model can best be explained by our molecular understanding of the regulation of various cellular functions (e.g. cell arrest, DNA repair, apopotosis, etc) by inter-connected oncogenes. Changes in any of these regulatory genes can deter the affected cell to maintain its normal condition. These genetic changes can be due to DNA damages and errors in DNA repair (e.g. somatic recombination and loss of heterozygosity). With the current understanding of the various elaborated cellular responses (such as cell arrest, DNA repair and apoptosis described in Figure 7 and Figure 12) to Cr(VI) induced DNA DSB, it is reasonable to assume the multi-stage and multi-path mode of action of Cr(VI) carcinogenesis.
In summary, hexavalent chromium (Cr(VI)) enters the cells along the concentration gradient of divalent anion through the intracellular chloride-phosphate anionic channel.. Cr(VI) can combines with glutathione/glutathione synthetase to form possibly predominantly tetravalent chromium (Cr(IV)), as well as some pentavalent chromium (Cr(V)), in chloride intracellular channels (CLIC) in the plasma and subcellular organelle membrane. Once these reductive intermediates are formed, they diffuse to cellular compartments including mitochondria and nucleus and can damage the DNA in both organelles. In mitochondria and endoplasmic reticulum, the Cr(VI) will combine with NADH/NADPH to from pentavalent chromium (Cr(V). Accompany both the Cr(IV) and Cr(V) formation, free radicals are formed via Fenton-like reactions. Cr(V) and Cr(IV) causes metal- and/or free radical-mediated breakage of DNA phosphodiester bond and DNA base pair modification. Three known DNA damages sensors ATM, ATR and DNA-PK are activated in the affected cells. The DNA damage signals are relayed to the master p53 directly via (DNA-PK) or indirectly (by Chk1 or Chk2) via ATM and related (ATR). Once activated and stabilized, p53 mediates the expression of a plethora of genes involved in a) cell cycle arrest to allow DNA repair or b) apoptosis to discard the damage cell. Cr(VI) exposure activates ATM/ATR to signal the cells with damaged DNA to cell-arrest. Cr(VI) & Cr(V), but not Cr(IV), induces apoptosis via DNA-PK sensor signaled p53-dependent intrinsic mitochondrial apoptosis. Cr(VI) and Cr(V), but not Cr(IV), induce activities of caspase -2 and –3. Caspase 2 in nucleus is downregulated by p53/p21, and is required for p53-mediated apoptosis by act upstream on BAX and Puma to cause mitochondria outer membrane permeabilization (MOMP). Cytochrome C is then release and execution caspases such as caspase 3 is activated for final apoptosis execution; Cr(VI) also induce p53-independent, but Ca+2 –dependent, intrinsic mitochondrial apoptosis. ROS plays an important role in both apoptosis pathways. The effective concentration of Cr(VI) for DNA breakage/cell arrest/apoptosis is very low, especially under chronic exposure condition. Upon failure of apoptosis and cell arrest, the Cr(VI) exposed cells with unrepaired/misrepaired DNA damages may become immortal with further DNA changes and genetic plasticity. We hypothesize that Cr(IV) may be the most potent pen-ultimate carcinogen by causing DNA DSBs, somatic recombination and loss-of-heterozygosity, but not apoptosis. The current data indicate that pathways of cell defenses (e.g. cell cycle arrest, somatic recombination, apoptosis) against Cr(VI) induced DNA DSB are elaborated/interconnected. This supports the conclusion that Cr(VI) causes cancer via multistage- multipath pathways.
Figure 2. Detection of DNA Strand Breaks in Cr(VI) Exposed Hela Cells (45).
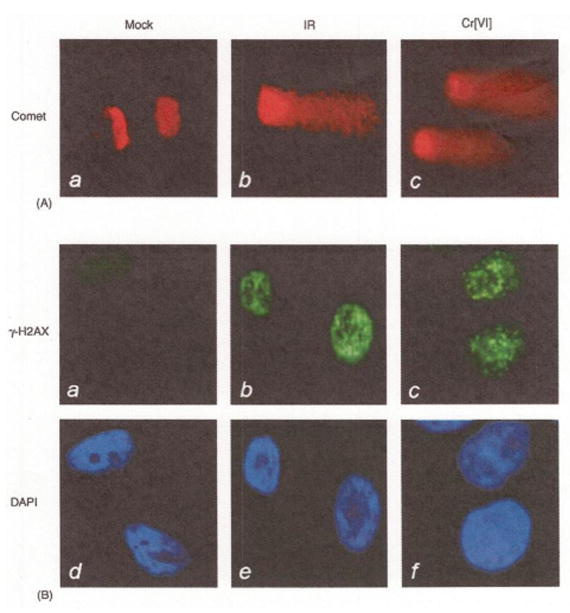
Figure 3. DNA Strand Breaks by Cr (IV) Reactions* (50).
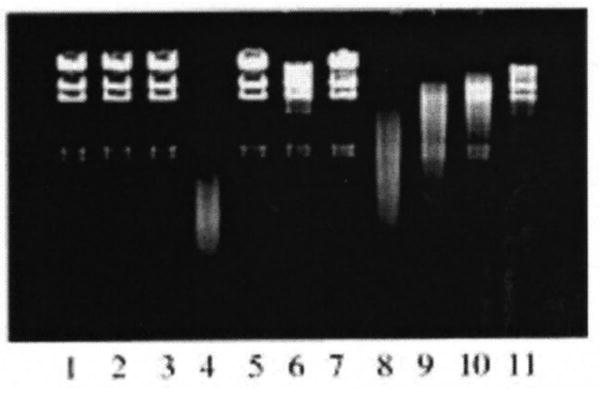
*Lane 1, untreated control A Hind III-digested DNA in pH 7.4 phosphate buffer; lane 2, same as lane 1 but with 2 mM Cr(IV) added; lane 3, same as lane I but with 0.2%H202 added; lane 4, incubation mixture containing DNA, 2 mM Cr(IV) and 0.2% H20,; lane 5, same as lane 4, but with 5000 waits/m1 catalase added; lane 6, same as lane 4, but with 2 mM deferoxamine added; lane 7, same as lane 4 but with 50 mM sodium formate added; lanes 8-11, same as lane 4, but with 0.1, 1, 2, and 5 mM aspirin added. The samples were incubated for 8 hr.
Figure 5.
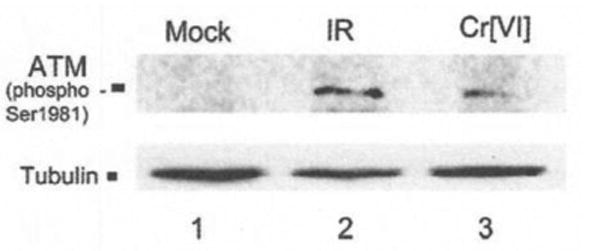
Immunoblot analysis of ATM phosphorylation in cells treated with 10 uM Cr(VI) or irradiation (6Gy) (45).
Figure 6.
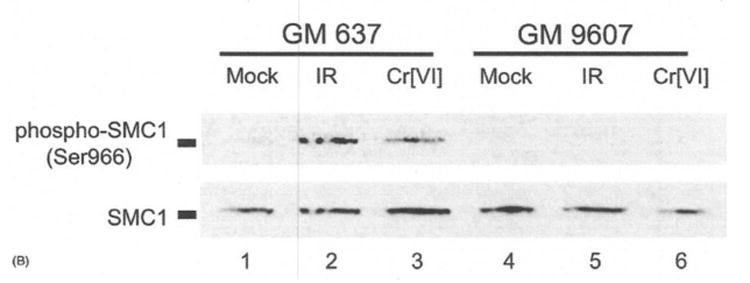
Immonoblot analysis of SMC1 phosphorylation of ATM proficient GM 637 cells or ATM-deficient GM 9607 cells treated with 10 uM Cr(VI) for 4 hr or 6 Gy ionizing radiation (45).
Figure 9. Effects of PK Inhibitor I or Caffeine on the EC50 Value of Cr(VI) Treated with 30uM Cr(VI) for 12 hr (119 ).
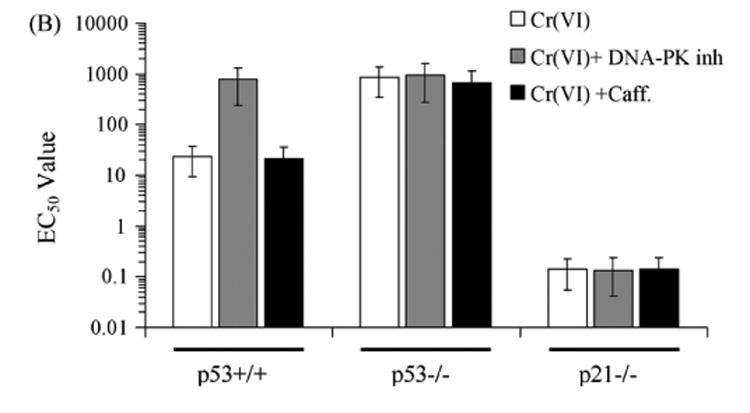
Figure 11b. Activation of Caspase 3 in Jurkat T-Lymhocytes after 10 uM Cr(V) or Cr(IV) Exposure for 24 hr.

Footnotes
Disclaimer: The opinions and conclusions in this paper are those of the authors and do not necessarily reflect their affiliated institutions.
References
- 1.IARC. Vol. 49. Lyon, France: 1990. IARC Monograph on the Evaluation of Carcinogenic Risks to Humans: Chromium, Nickle and Welding; pp. 389–401. [PMC free article] [PubMed] [Google Scholar]
- 2.NTP Technical Report on the Toxicology and Carcinogenesis Studies of Sodium Dinchromate Dihydrate (CAS NO. 7789-12-0) in F344 Rats and B6C3F1 Mice (Drinking Water Studies) National Toxicology Program July 2008 NTP TR 546 Research Triangle Park, NC 27709. [PubMed] [Google Scholar]
- 3.Kerger BD, Paustenbach DJ, Corbett GE, Finley BL. Absorption and elimination of trivalent and hexavalent chromium in humans following ingestion of a bolus dose in drinking water. Toxicol Appl Pharmacol. 19961;41:145–158. doi: 10.1006/taap.1996.0271. [DOI] [PubMed] [Google Scholar]
- 4.Connett PH, Wetterhahn KE. Metabolism of the carcinogenic chromate by cellular constituents. Struct Bend. 1983;54:93–124. [Google Scholar]
- 5.Buttner B, Beyersmann D. Modification of erythrocyte anion carrier by chromate. Xenobiotica. 1985;15:735–741. doi: 10.3109/00498258509047435. [DOI] [PubMed] [Google Scholar]
- 6.Ulmasov B, Bruno J, Woost PG, Edwards JC. Tissue and subcellular distribution of CLCI1. BMC Cell Biol. 2007 Feb 27;8:8. doi: 10.1186/1471-2121-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cromer BA, Mottoa CJ, Board PG, Parker MW. From glutathione transferase to pore in a CLIC. Eur Biophys J. 2002:356–364. doi: 10.1007/s00249-002-0219-1. [DOI] [PubMed] [Google Scholar]
- 8.Harrop SJ, DeMaerez MZ, Fairlie WD, Reztsova T, Valen-zuela SA, Mazzanti M, Tonini R, Qiu MR, Jankova L, Warton K, Bauskin AR, Wu WM, Pankurst S, Campell TJ, Breit SN, Curmi PMG. Crystal structure of a soluble form of the intracellular chloride ion channel CLIC (NCC27) at 1.4 A° resolution. J Biol Chem. 2001;276(48):44993–45000. doi: 10.1074/jbc.M107804200. [DOI] [PubMed] [Google Scholar]
- 9.Gilbert HF. Adv Enzymol Relat Areas Mol Biol. 1990;63:69–172. doi: 10.1002/9780470123096.ch2. [DOI] [PubMed] [Google Scholar]
- 10.Shi X, Chiu A, Chen CT, Halliwell B, Castranova V, Vallyathan V. Reduction of chromium(VI) and its relationship to carcinogenesis. J Toxicol Environ Health B Crit Rev. 1999 Jan-Mar;2(1):87–104. doi: 10.1080/109374099281241. [DOI] [PubMed] [Google Scholar]
- 11.Dalal NS, Shi X. ESR evidence for the glutathionyl radical in die reduction of chromium-(VI) by glutathione. In: Hayaishi O, Niki E, Konodo M, Yoshikawa T, editors. Medical, biomedical and chemical aspects of free radicals. Amsterdam: Elsevier; 1989. pp. 547–550. [Google Scholar]
- 12.Shi X, Dalai NS. On the mechanism of the chromate reduction by glutathione: ESR evidence for the glutathionyl radical and err isolable Cr(V) intermediate. Biochem Biophys Res Common. 1988;156:137–142. doi: 10.1016/s0006-291x(88)80815-5. [DOI] [PubMed] [Google Scholar]
- 13.Bose RN, Moghaddas S, Gelerinter E. Long-lived chromium(V) and chromium(V) metabolites in the chromium(V1)-glutathione reaction: NMR, ESR, HPLC, and kinetic characterization. Inorg Chem. 1992;31:1987–1994. [Google Scholar]
- 14.Liu KJ, Shi X, Dalal NS. Synthesis of Cr(IV)-GSH, its identification and its free hydroxyl generation: a model compound for Cr(VI) carcinogenicity. Biochem Biophys Res Commun. 1997;235:54–58. doi: 10.1006/bbrc.1997.6277. [DOI] [PubMed] [Google Scholar]
- 15.Cupo DY, Wetterhahn KE. Binding of chromium to chromatin and DNA from liver and kidney of rats treated with sodium dichromate and chromiurn (III) chloride in vivo. Cancer Res. 1985;45:1146–1151. [PubMed] [Google Scholar]
- 16.Cupo DY, Wetterhahn KE. Modification of chromium(VI)-induced DNA drainage by glutathione and cytochromes P-450 in chick embryo hepatocytes. Proc Natl Acad Sci USA. 1985;82:6755–6759. doi: 10.1073/pnas.82.20.6755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liebross RH, Wetterhahn KE. In vivo formation of chromium(V) in chick embryo red blood cells. Chem Res, Toxicol. 1990;3:401–403. doi: 10.1021/tx00017a002. [DOI] [PubMed] [Google Scholar]
- 18.Liebross RH, Wetterhahn KE. In vivo formation of chromium(V) in chick embryo liver and red blood cells. Carcinogenesis. 1992;13:2111–2120. doi: 10.1093/carcin/13.11.2113. [DOI] [PubMed] [Google Scholar]
- 19.Liu KJ, Jiang J, Swartz HM, Shi X. Low-frequency EPR detection of chromium(V) formation by chromium(VI) reduction in whole live mice. Arch Biochem Biophys. 1994 Sep;313(2):248–52. doi: 10.1006/abbi.1994.1384. [DOI] [PubMed] [Google Scholar]
- 20.Rossi SC, Gorman N, Wetterhahn KE. Mitochondrial reduction of the carcinogen chromate: formation of chromium(V) Chem Res Toxicol. 1988;1(2):101–7. doi: 10.1021/tx00002a003. [DOI] [PubMed] [Google Scholar]
- 21.Liu K, Shi X, Jiang JJ, Goda F, Dalal NS, Swath HM. Chromate-induced chromium(V) formation in live mice and its control by cellular antioxidants: An L-band EPR study. Arch, Biochem, BioPhys. 1995;323:33–39. doi: 10.1006/abbi.1995.0006. [DOI] [PubMed] [Google Scholar]
- 22.Liu KJ, Shi X, Jiang JI, Docia F, Dalal NS, Swartz HM. Low frequency electron paramagnetic resonance investigation on metabolism of chromium(V1) by whole mice. Annu Clin Lab Sci. 1996;26:176–184. [PubMed] [Google Scholar]
- 23.Meister A, Anderson ME. Glutathione. Ann Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 24.Berry JP, Hourdry J, Galle P, Lagrue G. Chromium concentration by proximal renal tubule cells: and intrastructural, microanalytical and cytochemical study. The J of Histochemistry and cytochemistry. 1978;26(8):651–657. doi: 10.1177/26.8.690404. [DOI] [PubMed] [Google Scholar]
- 25.Chiu A, Katz AJ, Beaubier J, Chiu N, Shi X. Genetic and cellular mechanisms in chromium and nickel carcinogenesis considering epidemiologic findings. Mol Cell Biochem. 2004;255:181–94. doi: 10.1023/b:mcbi.0000007274.25052.82. [DOI] [PubMed] [Google Scholar]
- 26.Tsapakos MJ, Wetterhahn KE. The interaction of chromium with nucleic acids. Chem Biol Interact. 1983;46(2):265–277. doi: 10.1016/0009-2797(83)90034-0. 1983. [DOI] [PubMed] [Google Scholar]
- 27.Jennette KE. Microsomal reduction of the carcinogen chromate produce chromium(V) J Am Chem Soc. 1982;104:874–875. [Google Scholar]
- 28.Shi X, Dalal Z, Gannett P. Chromate-mediated free radical generation from cysteine, penicillamine, hydrogen peroxide, and lipid hydroperoxides. Biochim Biophys Acta. 1994;1226:65–72. doi: 10.1016/0925-4439(94)90060-4. [DOI] [PubMed] [Google Scholar]
- 29.Liu KJ, Shi X, Jiang JJ, Goda F, Dalal N, Swartz HM. Chromate-Induced Chromium(V) Formation in live mice and its control by cellular antioxidants: An L-band Electron Paramagnetic Resonance Study. Arch Biochem Biophys. 1995;323:33–39. doi: 10.1006/abbi.1995.0006. [DOI] [PubMed] [Google Scholar]
- 30.Shi XG, Dalal NS. On the hydroxyl radical formation in the reaction between hydrogen peroxide and biologically generated chromium(V) species. Arch Biochem Biophys. 1990a Mar;277(2):342–50. doi: 10.1016/0003-9861(90)90589-q. [DOI] [PubMed] [Google Scholar]
- 31.Shi X, Dalal NS. ESR spin trapping detection of hydroxyl radicals in the reaction of Cr(V) complexes with hydrogen peroxide. Free radical res Commun. 1990b;10:17–26. doi: 10.3109/10715769009145929. [DOI] [PubMed] [Google Scholar]
- 32.Shi X, Dalal NS. Evidence for a Fenton-type mechanism for the generation of OH radicals in the reduction of Cr(VI) in cellular media. Archim Biochem Biophys. 1990c;277:342–350. doi: 10.1016/0003-9861(90)90417-w. [DOI] [PubMed] [Google Scholar]
- 33.Shi X, Dalal NS. The role of Cr(V), The role of superoxide radical in chromium(VI) generated hydroxyl radical: The Cr(VI) Haber-Weiss. cycle Arch Biochim Biophy. 1992;292:323–327. doi: 10.1016/0003-9861(92)90085-b. [DOI] [PubMed] [Google Scholar]
- 34.Sugden KD, Wetterhahn KE. Direct and hydrogen peroxide-induced chromium(V) oxidation of deoxyribose in single-stranded and double-stranded calf thymus DNA. Chem Res Toxicol. 1997;10(12):1397–406. doi: 10.1021/tx970135r. [DOI] [PubMed] [Google Scholar]
- 35.Shi X, Mao Y, Knapton AD, Ding M, Rojanasakut Y, Gannett PM, Dalal NS, Liu K. Reaction of Cr(VI) with ascorbate and hydrogen peroxide generates hydroxyl radicals and cause DNA damage: Role of Cr(IV)-mediated Fenton-like reaction. Carcinogenesis. 1994;15:2475–2478. doi: 10.1093/carcin/15.11.2475. [DOI] [PubMed] [Google Scholar]
- 36.Liu KJ, Shi X, Dalal NS. Synthesis of Cr(IV)-GSH, its identification and its free hydroxyl generation: a model compound for Cr(VI) carcinogenicity. Biochem Biophys Res Commun. 1997;235:54–58. doi: 10.1006/bbrc.1997.6277. [DOI] [PubMed] [Google Scholar]
- 37.Luo H, Lu YD, Mao Y, Shi X, Dalal N. Role of chromium(IV) in the chromium(VI)-related free radical formation, dG hydroxylation and DNA damage. J Inorg Biochem. 1996;64:25–35. doi: 10.1016/0162-0134(95)00241-3. [DOI] [PubMed] [Google Scholar]
- 38.Luo H, Lu Y, Shi X, Dalal NS. Chromium(IV)/H2O2 Fenton-like reaction cause DNA damage: implication to chromate's genotoxicity. Ann Clin Lab Sci. 1996;26:185–191. [PubMed] [Google Scholar]
- 39.Ramsey CM, Dalal NS. Crystalline and water soluble Cr(4+) and Cr(5+)model compounds for chromium toxicity studies. Molecular and Cellular Biochemistry. 2004;255:113–118. doi: 10.1023/b:mcbi.0000007267.97472.da. [DOI] [PubMed] [Google Scholar]
- 40.Dalal NS, Milar JM, Jagadeesh MS, Seehra MS. Paramagnetic resonance and susceptibility of K3CrO8. J Chem Phys. 1981;74:19, 16–1923. [Google Scholar]
- 41.Cage B, Dalal NS. Unhydrated Cr(V) peroxychromates M3CrO8 (M = Na,K, Rb): Low-dimensional antiferromagnets exhibiting large specific heats at mK to 5 K temperatures. Chem Mat. 2001;13:880–890. [Google Scholar]
- 42.Cage B, Geyer W, Abboud KA, Dalal NS. Hydrated Cr(V) peroxychromates M3Cr O8. X H2O (M = Li, Na, Cs): Model 3d1 systems exhibiting linear chain behavior and antiferromagnetic interactions. Chem Mat. 2001;13:871–879. [Google Scholar]
- 43.Hamilton JW, Wetterhahn KF. Chromium(VI)-induced DNA- damage in check embryo liver and blood cells in vivo, Carcinogenesis. 1986;7:2085–2088. doi: 10.1093/carcin/7.12.2085. [DOI] [PubMed] [Google Scholar]
- 44.Snyder RU. Role of active species in metal-induced DNA strand breakage in human diploid fibroblasts. Mutat Res. 1988;193:237–246. doi: 10.1016/0167-8817(88)90034-x. [DOI] [PubMed] [Google Scholar]
- 45.Wakeman TP, Kim WJ, Callens S, Chiu A, Brown KD, Xu B. The ATM-SMC1 pathway is essential for activation of the chromium[VI] -induced S-phase checkpoint. Mutation Research. 2004;554:241–251. doi: 10.1016/j.mrfmmm.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 46.Ha L, Ceryak S, Patierno SR. Generation of S phase-dependent DNA double-strand break by Cr(VI) exposure: involvement of ATM in Cr(VI) induction of gamma-H2AX. Carcinogenesis. 2004;25:2265–2274. doi: 10.1093/carcin/bgh242. [DOI] [PubMed] [Google Scholar]
- 47.Kortenkamp A, Ozolins Z, Beyersmann D, O'Brien P. Generation of PM2 DNA breaks in the course of reduction of chromium(VI) by glutathione. Muni Res. 1989;216(1):19–26. doi: 10.1016/0165-1161(89)90019-8. [DOI] [PubMed] [Google Scholar]
- 48.Sugiyama M. Effects of vitamin E and vitamin 82 on chromate-induced DNA lesions. Biol Trace Elem Res. 1989;21:399–404. doi: 10.1007/BF02917281. [DOI] [PubMed] [Google Scholar]
- 49.Sugiyama M, Tsuzuki K, Ogura R. Effects of ascorbic acid on DNA damage, cytotoxicity, glutathione reductase, and formation of paramagnetic chromate in Chinese hamster V-79 cells treated with sodium(VI) I Biol Chem. 1991;266:3383–3386. [PubMed] [Google Scholar]
- 50.Shi X, Ding M, Ye J, Wang S, Leonard SS, Zang L, Castranova V, Vallyathan V, Chiu A, Dalal N, Liu K. Cr(IV) causes activation of nuclear transcription factor-kappa B, DNA strand breaks and dG hydroxylation via free radical reactions. J Inorg Biochim. 1999;75(1):37–44. doi: 10.1016/S0162-0134(99)00030-6. [DOI] [PubMed] [Google Scholar]
- 51.Dizadaroglu M. Chemical determination of free radical-induced damage to DNA. Free Radical Biol Med. 1991;10:225–242. doi: 10.1016/0891-5849(91)90080-m. [DOI] [PubMed] [Google Scholar]
- 52.Sugden KD, Martin BD. Guanine and 7,8-dihydro-8-oxo-guanine-specific oxidation in DNA by chromium(V) Environ Health Perspect. 2002;110(Suppl 5):725–8. doi: 10.1289/ehp.02110s5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Slade PG, Hailer MK, Martin D, Sugden KD. Guanine-specific oxidation of double-stranded DNA by Cr(VI) and ascorbic acid forms spiroiminodihydantoin and 8-oxo-2′-deoxyguanosine. Chem Res Toxicol. 2005;18(7):1140–1149. doi: 10.1021/tx050033y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Slade PG, Priestley ND, Sugden KD. Spiroiminodihydantoin as an oxo-atom transfer product of 8-oxo-2′-deoxyguanosine oxidation by chromium(V) Org Lett. 2007;9:4411–4414. doi: 10.1021/ol701667t. [DOI] [PubMed] [Google Scholar]
- 55.Achanta G, Pelicano H, Feng L, Plunkett W, Huang P. Interaction of p53 and DNA-PK in response to nucleoside analogues: potential role as a sensor complex for DNA damage. Cancer Res. 2001;61:8723–8729. [PubMed] [Google Scholar]
- 56.Rinaldo C, Prodosmo A, Mancini F, Iacovelli S, Sacchi A, Moretti F, Soddu S. MDM2-regulated degradation of HIPK2 prevents p53Ser46 phosphorylation and DNA damage-induced apoptosis. Mol Cell. 2007;25:739–750. doi: 10.1016/j.molcel.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 57.Woo RA, McLure KG, Lees-Miller SP, Rancourt DE, Lee PW. DNA-dependent protein kinase acts upstream of p53 in response to DNA damage. Nature. 1998;394:700–704. doi: 10.1038/29343. [DOI] [PubMed] [Google Scholar]
- 58.Bartek J, Bartkova J, Lukas J. DNA damage signaling guards against activated oncogenes and tumour progression. Oncogene. 2007;26(56):7773–9. doi: 10.1038/sj.onc.1210881. [DOI] [PubMed] [Google Scholar]
- 59.Vaziri C, Saxena S, Jeon Y, Lee C, Murata K, Machida Y, Wagle N, Hwang DS, Dutta A. A p53-dependent checkpoint pathway prevents rereplication. Mol Cell. 2003;11:997–1008. doi: 10.1016/s1097-2765(03)00099-6. [DOI] [PubMed] [Google Scholar]
- 60.Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene. 2001;20:1803–1815. doi: 10.1038/sj.onc.1204252. [DOI] [PubMed] [Google Scholar]
- 61.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 62.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 63.Lees-Miller SP, Chen YR, Anderson CW. Human cells contain a DNA-activated protein kinase that phosphorylates simian virus 40 T antigen, mouse p53, and the human Ku autoantigen. Mol Cell Biol. 1990;10:6472–6481. doi: 10.1128/mcb.10.12.6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shiloh Y. ATM and ATR: networking cellular responses to DNA damage. Curr Opin, Genet Dev. 2001;11:71–77. doi: 10.1016/s0959-437x(00)00159-3. [DOI] [PubMed] [Google Scholar]
- 65.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 66.Shiloh Y. ATM: from phenotype to functional genomics-and back. Ernst Schering Res Found Workshop. 2002:5 1–70. doi: 10.1007/978-3-662-04667-8_4. Review. [DOI] [PubMed] [Google Scholar]
- 67.Iijima K, Ohara M, Seki R, Tauchi H. Dancing on damaged chromatin: functions of ATM and the RAD50/MRE11/NBS1 complex in cellular responses to DNA damage. J Radiat Res (Tokyo) 2008;49(5):451–64. doi: 10.1269/jrr.08065. Review. [DOI] [PubMed] [Google Scholar]
- 68.Satyanarayana A, Kaldis P. Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene. 2009;28:2925–2939. doi: 10.1038/onc.2009.170. [DOI] [PubMed] [Google Scholar]
- 69.Zhao S, Weng YC, Yuan SS, Lin YT, Hsu HC, Lin SC, Gerbino E, Song MH, Zdzienicka MZ, Gatti RA, Shay JW, Ziv Y, Shiloh Y, Lee EY. Functional link between ataxia-telangiectasia and Nijmegen breakage syndrome gene products. Nature. 2000;405:473–477. doi: 10.1038/35013083. [DOI] [PubMed] [Google Scholar]
- 70.Willis N, Rhind N. Regulation of DNA replication by the S-phase damage checkpoint. Cell Division. 2009;4:13. doi: 10.1186/1747-1028-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Falck J, Mailand N, Syljuasen RG, Bartek J, Likasm J. The ATM-Chk2-Cc1c25A checkpoint pathway guards against radioresistant DNA synthesis. Nature. 2001;410:842–847. doi: 10.1038/35071124. [DOI] [PubMed] [Google Scholar]
- 72.Bartek J, Falck J, Lukas J. CHK2 kinase--a busy messenger. Nat Rev Mol Cell Biol. 2001;2(12):877–86. doi: 10.1038/35103059. [DOI] [PubMed] [Google Scholar]
- 73.Kim ST, Xu B, Kastan MB. Involvement of the cohesin protein, Smcl, in Atm-dependent and independent responses to DNA damage. Genes Dev. 2002;16:560–570. doi: 10.1101/gad.970602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yazdi PT, Wang Y, Zhao S, Patel N, Lee EY, Qin J. SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev. 2002;16:571–582. doi: 10.1101/gad.970702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cimprich KA, Cortez D. ATR: An essential regulator of genomic integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kastan MB, Lim DS. The many substrates and functions of ATM. Mol Cell Biol Review. 2000;1(3):179–186. doi: 10.1038/35043058. [DOI] [PubMed] [Google Scholar]
- 77.Lal M, Bae D, Camilli TC, Patierno SR, Ceryak S. AKT1 mediates bypass of the G1/S checkpoint after genotoxic stress in normal human cells. Cell Cycle. 2009;8(10):1589–1602. doi: 10.4161/cc.8.10.8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Adams KE, Medhurst AL, Dart DA, Lakin ND. Recruitment of ATR to sites of ionising radiation-induced DNA damage requires ATM and components of MRN protein complex. Oncogene. 2006;25:3894–3904. doi: 10.1038/sj.onc.1209426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Myers JS, Cortez D. Rapid activation of ATR by ionizing radiation requires ATM and Mre11. J Biol Chem. 2006;281:9346–9350. doi: 10.1074/jbc.M513265200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wakeman TP, Xu B. ATR regulates hexavalent chromium-induced S-phase checkpoint through phosphorylation of SMC1. Mutat Res. 2006;610:14–20. doi: 10.1016/j.mrgentox.2006.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kastan MB, Bartek J. Cell cycle checkpoints and cancer. Nature. 2004;432(7015):316–23. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 82.Zhang Z, Leonard SS, Wang S, Vallyathan V, Castranova V, Shi X. Cr (VI) induces cell growth arrest through hydrogen peroxide-mediated reactions. Molecular and Cellular Biochemistry. 2001;222:77–83. [PubMed] [Google Scholar]
- 83.Hayashi Y, Kondo T, Zhao QL, Ogawa R, Cui ZG, Feril LB, Jr, Teranishi H, Kasuya M. Signal transduction of p53-independent apoptotic pathway induced by hexavalent chromium in U937 cells. Toxicology and Applied Pharmacology. 2004;197:96–106. doi: 10.1016/j.taap.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 84.Wise SS, Holmes AL, Wise JP., Sr Hexavalent chromium-induced DNA damage and repair mechanisms. Rev Environ Health. 2008;23:39–57. doi: 10.1515/reveh.2008.23.1.39. [DOI] [PubMed] [Google Scholar]
- 85.Weterings E, Chen DJ. DNA-dependent protein kinase in non-homologous end joining: a lock with multiple keys? Mini-review Journal of Cell Biology. 2007;179:183–186. doi: 10.1083/jcb.200705106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shrivastav M, De Haro LP, Nickloff JA. Regulation of DNA double-strand break repair pathway choice. Review Cell Research. 2008;18:134–147. doi: 10.1038/cr.2007.111. [DOI] [PubMed] [Google Scholar]
- 87.Graf U, Frei H, Kagi A, Katz AJ. Würgler FE: Thirty compounds tested in the Drosophila wing spot test. Mutat Res. 1989;22:359–373. doi: 10.1016/0165-1218(89)90112-2. [DOI] [PubMed] [Google Scholar]
- 88.Graf U, Würgler FE. The somatic white-ivory eye spot test does not detect the same spectrum of genotoxic events as the wing somatic mutation and recombination test in Drosophila melanogaster. Environ Mol Mutagen. 1996;27:219–26. doi: 10.1002/(SICI)1098-2280(1996)27:3<219::AID-EM7>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 89.Katz AJ, Chiu A, Beaubier J, Shi X. Combining Drosophila melanogaster somatic-mutation-recombination and electron-spin-resonance-spectroscopy data to interpret epidemiologic observations on chromium carcinogenicity. Mol Cell Biochem. 2001;222:61–8. doi: 10.1023/a:1017959222379. [DOI] [PubMed] [Google Scholar]
- 90.Spanó MA, Frei H, Würgler FE, Graf U. Recombinagenic activity of four compounds in the standard and high bioactivation crosses of Drosophila melanogaster in the wing spot test. Mutagenesis. 2001;16(5):385–94. doi: 10.1093/mutage/16.5.385. [DOI] [PubMed] [Google Scholar]
- 91.Sengstag C. The role of mitotic recombination in carcinogenesis. Crit Rev Toxicol. 1994;24:323–353. doi: 10.3109/10408449409017922. [DOI] [PubMed] [Google Scholar]
- 92.Shammas MA, Reis RJS. Recombination and its role in DNA repair, cellular immortalization and cancer. Age. 1999;22:71–88. doi: 10.1007/s11357-999-0009-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cavenee WK, White RL. The genetic basis of cancer. Sci Am. 1995;272:72–79. doi: 10.1038/scientificamerican0395-72. [DOI] [PubMed] [Google Scholar]
- 94.Tischfield JA. Loss of heterozygosity or: How I learned to stop worrying and love mitotic recombination. Am J Hum Genet. 1997;61:995–999. doi: 10.1086/301617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shao C, Deng L, Henegariu O, Liang L, Stambrook PJ, Tischfield JA. Chromosomal instability contributes to loss_of_heterozygosity in mice lacking p53. Proc Natl Acad Sci USA. 2000;97:7405–7410. doi: 10.1073/pnas.97.13.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kroemer G. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nature Med. 1997;36:614–620. doi: 10.1038/nm0697-614. [DOI] [PubMed] [Google Scholar]
- 97.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 98.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Sci. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 99.Jimi S, Uchiyama M, Takaki A, Suzumiya J, Syuji H. Mechanism of cell death induced by cadmium and arsenic. Ann NY Acad Sci USA. 2004;1011:325–331. doi: 10.1007/978-3-662-41088-2_32. [DOI] [PubMed] [Google Scholar]
- 100.Yoon YS, Cho HS, Lee H, Yoon G. Mitochondrial dysfunction via disruption of complex-II activity during iron chelation-induced senescence-like growth arrest of Chang cell. Ann NY Acad Sci. 2004;1011:123–132. doi: 10.1007/978-3-662-41088-2_13. [DOI] [PubMed] [Google Scholar]
- 101.Baptiste-Okoh N, Barsotti AM, Prives C. Casepase 2 is both required for p53-mediated apoptosis and downregulated by p53 in a p21-dependent manner. Cell Cycle. 2008;7(9):1133–1138. doi: 10.4161/cc.7.9.5805. [DOI] [PubMed] [Google Scholar]
- 102.Vousden KH. p53 and PUMA: A deadly duo. Sci. 2005;309:1685–1686. doi: 10.1126/science.1118232. [DOI] [PubMed] [Google Scholar]; Tomita Y, Marchenko N, Nemajerova A, Dehner A, Klein C, Pan HG, Kessler H, Pancoska P, Moll UM. WTp53, but not tumor derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization. J Biol Chem. 2006;281(13):8600–8606. doi: 10.1074/jbc.M507611200. [DOI] [PubMed] [Google Scholar]
- 103.Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11:577–590. doi: 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- 104.Mioll UM, Wolff S, Spiedel D, Deppert W. Transcription independent pro-apoptotic functions of p53. Curr Opinion Cell Bio. 2005;l17:631–636. doi: 10.1016/j.ceb.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 105.Tomita Y, Marchenko N, Nemajerova A, Dehner A, Klein C, Pan HG, Kessler H, Pancoska P, Moll UM. WTp53, but not tumor derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization. J Biol Chem. 2006;281(13):8600–8606. doi: 10.1074/jbc.M507611200. [DOI] [PubMed] [Google Scholar]
- 106.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DCS. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 107.Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, green DR, Newmeuyer DD. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17:525–535. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 108.Susin SA, Zamzami N, Castedo M, Hirsch T, Marchetti P, Macho A, Daugas E, Geuskens M, Kroemer Bcl-2 inhibits the mitochondrial release of an apoptogenic process. J Exp Med. 1996;184:1331–1341. doi: 10.1084/jem.184.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Eskes R, Desagher S, Antonsson B, Martinou JC. Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol Cell Biol. 2000;20(3):929–935. doi: 10.1128/mcb.20.3.929-935.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Baptiste N, Prives C. p53 in the cytoplasm: a question of overkill. Cell. 2004;116(4):487–489. doi: 10.1016/s0092-8674(04)00164-3. [DOI] [PubMed] [Google Scholar]
- 111.Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Sci. 2003;303:1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 112.Chipuki JE, Bouchier –Hayes L, Kuwana T, Newmeyer DD, Green DR. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Sci. 2005;309:1732–1735. doi: 10.1126/science.1114297. [DOI] [PubMed] [Google Scholar]
- 113.Guo Y, Srinivasula SM, Druilhe A, Fernandes-Alnemri T, Alnemri ES. Caspase-2 induces apoptosis by releasing proapoptotic proteins from mitochondria. J Biol Chem. 2002;277:13430–13437. doi: 10.1074/jbc.M108029200. [DOI] [PubMed] [Google Scholar]
- 114.Gao Z, Shao Y, Jiang X. Essential roles of the Bcl-2 family of proteins in caspase-2-induced apoptosis. J Biol Chem. 2005;280:38271–38275. doi: 10.1074/jbc.M506488200. [DOI] [PubMed] [Google Scholar]
- 115.Wagner KW, Engels IH, Deveraux QL. Caspase-2 can function upstream of bid cleavage in the TRAIL apoptosis pathway. J Biol Chem. 2004;279:35047–35052. doi: 10.1074/jbc.M400708200. [DOI] [PubMed] [Google Scholar]
- 116.Ye J, Wang S, Leonard SS, Sun Y, Butterworth L, Antonini J, Ding M, Rojanasakul Y, Vallyathan V, Castranova V, Shi X. Role of Reactive Oxygen Species and p53 in Chromium(VI)-induced Apoptosis. J Biol Chem. 1999;274(49):34974–34980. doi: 10.1074/jbc.274.49.34974. [DOI] [PubMed] [Google Scholar]
- 117.Hayashi Y, Kondo T, Zhao QL, Ogawa R, Cui ZG, Feril LB, Jr, Teranishi H, Kasuya M. Signal transduction of p53-independent apoptotic pathway induced by hexavalent chromium in U937 cells. Toxicology and Applied Pharmacology. 2004;197:96–106. doi: 10.1016/j.taap.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 118.Hill R, Leidal AM, Madureira PA, Gillis LD, Cochrane HK, Waisman DM, Chiu A, Lee PW. Hypersensitivity to chromium-induced DNA damage correlates with constitutive deregulation of upstream p53 kinases in p21-/- HCT116 colon cancer cells. DNA Repair. 2008;7(2):239–52. doi: 10.1016/j.dnarep.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 119.Hill R, Leidal AM, Madureira PA, Gillis LD, Waisman DM, Chiu A, Lee PW. Chromium-mediated apoptosis: Involvement of DNA-dependent protein kinase (DNA-PK) and differential induction of p53 target genes. DNA Repair. 2008;7(9):1484–99. doi: 10.1016/j.dnarep.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 120.Zhivotovsky B, Orrenius S. Caspase-2 function in response to DNA damage. Biochem Biophys Res Commun. 2005;331:859–867. doi: 10.1016/j.bbrc.2005.03.191. [DOI] [PubMed] [Google Scholar]
- 121.Robertson JD, Enoksson M, Suomela M, Zhivotovsky B, Orrenius S. Caspase-2 acts upstream of mitochondria to promote cytochrome c release during etoposide-induced apoptosis. J Biol Chem. 2002;277:29803–29809. doi: 10.1074/jbc.M204185200. [DOI] [PubMed] [Google Scholar]
- 122.Ha L, Ceryak S, Patierno SR. Chromium (VI) activates ataxia telangiectasia mutated (ATM) protein. Request of ATM for both apoptosis & recovery from terminal growth arrest. J Biol Cheem. 2003;278:17885–17894. doi: 10.1074/jbc.M210560200. [DOI] [PubMed] [Google Scholar]
- 123.Shi M, Vivian CJ, Lee KJ, Ge C, Morotomi-Yano K, Manzl C, Bock F, Sato S, Tomomori-Sato C, Zhu R, Haug JS, Swanson SK, Washburn MP, Chen DJ, Chen BP, Villunger A, Florens L, Du C. DNA-PKcs-PIDDosome: a nuclear caspase-2-activating complex with role in G2/M checkpoint maintenance. Cell. 2009;136(3):508–20. doi: 10.1016/j.cell.2008.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 124.Tanaka T, Ohkubo S, Tatsuno I, Prives C. hCAS/CSE1L associates with chromatin and regulates expression of select p53 target genes. Cell. 2007;130:638–650. doi: 10.1016/j.cell.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 125.Mattia M, Gottifredi V, McKinney K, Prives C. p53-dependent p21 mRNA elongation is impaired when DNA replication is stalled. Mol Cell Biol. 2006:1309–1320. doi: 10.1128/MCB.01520-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Espinosa JM, Emerson BM. Transcriptional regulation by p53 through intrinsic DNA/chromatin binding and site-directed cofactor recruitment. Mol Cell. 2001;8:57–69. doi: 10.1016/s1097-2765(01)00283-0. [DOI] [PubMed] [Google Scholar]
- 127.Espinosa JM, Verdun RE, Emerson BM. p53 functions through stress- and promoter-specific recruitment of transcription initiation components before and after DNA damage. Mol Cell. 2003;12:1015–1027. doi: 10.1016/s1097-2765(03)00359-9. [DOI] [PubMed] [Google Scholar]
- 128.Nowell PC. The clonal evolution of tumor cell populations. Sci. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 129.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 130.Loeb LA, Loeb KR, Anderson JP. Multiple mutations and cancer. Proc Acad Sci USA. 2003;110:776–781. doi: 10.1073/pnas.0334858100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, Simpson JT, Stebbings LA, Leroy C, Edkins S, Mudie LJ, Greenman CD, Jia M, Latimer C, Teague JW, Lau KW, Burton J, Quail MA, Swerdlow H, Churcher C, Natrajan R, Sieuwerts AM, Martens JW, Silver DP, Langerød A, Russnes HE, Foekens JA, Reis-Filho JS, van't Veer L, Richardson AL, Børresen-Dale AL, Campbell PJ, Futreal PA, Stratton MR. Complex landscape of somatic rearrangement in human breast cancer genome. Nature. 2009;462:1005–1010. doi: 10.1038/nature08645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Harris AW, Pinkert CA, Crawford M, Langdon WY, Brinster RL, Adams JM. The Eμ-myc transgenic mouse. A model for high incidence spontaneous lymphoma and leukemia of early. B cells J Exp Med. 1988;167:353–371. doi: 10.1084/jem.167.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dang CV, O'Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-myc target gene network Seminars in Cancer Biology. 2006;16:253–264. doi: 10.1016/j.semcancer.2006.07.014. Review. [DOI] [PubMed] [Google Scholar]
- 134.Nair SK, Burley SK. X-ray structure of myc-max and mad-max recognizing DNA: molecular basis of regulation by proto-oncogene transcription factors. Cell. 2003;112:193–205. doi: 10.1016/s0092-8674(02)01284-9. [DOI] [PubMed] [Google Scholar]
- 135.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–348. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- 136.Herbst A, Hemann MT, Tworkowski KA, Salghetti SE, Lowe SW, Tansey WP. A conserved element in myc that negatively regulates its proapoptotic activity. EMBO Report. 2005;6(2):177–183. doi: 10.1038/sj.embor.7400333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Strasser A, Harris AW, Beth ML, Cory S. Novel primitive lymphoid tumors induced transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–333. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- 138.Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes hematopoetic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;355:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- 139.Felsher DW, Bishop JM. Reversible tumorigenesis by myc in hematopoetic linkages. Mol Cell. 1999;4:199–207. doi: 10.1016/s1097-2765(00)80367-6. [DOI] [PubMed] [Google Scholar]
- 140.Ji L, Arcinas M, Boxer LM. NF-kappa B sites function as positive regulators of expression of the translocated c-myc allele in Burkitt's lymphoma. Mol Cell Biol. 1994;14(12):7967–74. doi: 10.1128/mcb.14.12.7967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Flouds L. The histological analysis of tumors. A critical review. Am J Cancer. 1940;39:1–24. [Google Scholar]