

Significance
Pathogens express virulence factors to support their growth and reproduction while hosts activate various immune processes to promote pathogen clearance and minimize damage. In this study, we establish a new role for pyoverdin, an iron-binding siderophore produced by the bacterium Pseudomonas aeruginosa. In addition to promoting growth by acquiring iron, pyoverdin serves as a secreted bacterial toxin that disrupts mitochondria and iron homeostasis in Caenorhabditis elegans. We show that exposure to pyoverdin triggers mitochondrial damage and subsequent mitophagy (lysosomal degradation of damaged mitochondria). Importantly, mitophagy confers a protective effect against exposure to either pyoverdin or to a synthetic iron chelator, demonstrating a function for mitophagy in innate immunity. Finally, we show that iron chelation causes mitophagy in mammalian cells.
Keywords: mitophagy, Pseudomonas, siderophore, innate immunity, C. elegans
Abstract
In the arms race of bacterial pathogenesis, bacteria produce an array of toxins and virulence factors that disrupt core host processes. Hosts mitigate the ensuing damage by responding with immune countermeasures. The iron-binding siderophore pyoverdin is a key virulence mediator of the human pathogen Pseudomonas aeruginosa, but its pathogenic mechanism has not been established. Here we demonstrate that pyoverdin enters Caenorhabditis elegans and that it is sufficient to mediate host killing. Moreover, we show that iron chelation disrupts mitochondrial homeostasis and triggers mitophagy both in C. elegans and mammalian cells. Finally, we show that mitophagy provides protection both against the extracellular pathogen P. aeruginosa and to treatment with a xenobiotic chelator, phenanthroline, in C. elegans. Although autophagic machinery has been shown to target intracellular bacteria for degradation (a process known as xenophagy), our report establishes a role for authentic mitochondrial autophagy in the innate immune defense against P. aeruginosa.
Iron is an essential trace element used by a wide range of redox enzymes in bacteria, archaea, and eukaryotes. The requirement for iron has created an ongoing struggle between hosts and pathogens as they vie for control of this nutrient. While free iron is already stringently limited in the bloodstream of mammalian hosts, bacterial infection triggers an innate immune response that sequesters iron even further, serving as a mechanism of limiting microbial proliferation (1). Invasive microorganisms, in turn, synthesize and excrete siderophores, soluble extracellular molecules that tightly bind and help solubilize iron found in the environment. In addition, siderophores are important for acquiring iron from host iron storage proteins and the extracellular milieu, which facilitates microbial growth in this specific niche (2). Siderophores are key virulence factors in many pathosystems, including infection with Pseudomonas aeruginosa (1–4). For example, mutants of P. aeruginosa with compromised pyoverdin biosynthesis exhibit attenuated pathogenesis in both C. elegans and in mice (3–5); despite this, the virulence mechanism(s) of siderophores remains unknown. Similarly, little is known about how hosts defend themselves against siderophore exposure and subsequent loss of iron; the notable exception being secretion of a siderocalin, a protein that binds siderophores and minimizes their activity (6). Given the importance of siderophores as virulence determinants, greater insight into this defense process is needed.
P. aeruginosa is a key human nosocomial pathogen, responsible for ∼10% of hospital acquired infections and is frequently associated with adverse medical outcomes that include amputation, removal of medical devices, and death (7). P. aeruginosa also infects C. elegans, showing diverse modes of pathogenesis that are partially dependent upon the medium in which the nematodes are exposed, including intestinal infection on agar (where host death is contingent upon quorum-sensing) and a lethal intoxication in liquid that is dependent upon the P. aeruginosa siderophore pyoverdin (reviewed in ref. 8).
Results and Discussion
Pyoverdin Enters C. elegans and Is Sufficient to Mediate Host Killing.
Despite the presence of rich sources of iron within host cells, siderophores are generally assumed to scavenge iron from ferriproteins present in the extracellular milieu. However, we hypothesize that siderophores are capable of harvesting iron from intracellular sources and, consequently, function directly as toxins. We exposed worms to a pyoverdin-enriched, cell-free bacterial growth media for 24 h to determine whether detectable levels of pyoverdin could be identified within the host. After exposure, worms were washed extensively, homogenized, and subjected to centrifugation. Supernatants were assayed for the presence of pyoverdin via fluorescence spectroscopy (Materials and Methods). Significant amounts of pyoverdin were found in worm homogenates, but not in wash material (Fig. 1A), demonstrating that pyoverdin can enter the interior of C. elegans.
Fig. 1.
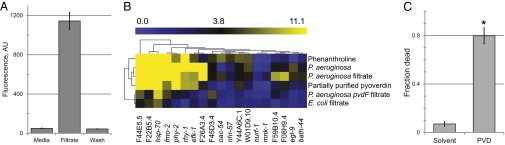
Pyoverdin enters C. elegans and causes host killing. (A) Presence of pyoverdin in worm homogenates from worms treated with uncultured media or filtrate, or in wash from worms treated with filtrate, as determined by fluorescence spectroscopy. (B) Heat map of hypoxic response gene transcription, as measured by quantitative RT-PCR (qRT-PCR). Worms were treated with the iron chelator 1,10-phenanthroline (Phe; 1 mM), exposed to P. aeruginosa, cell-free filtrates from E. coli, P. aeruginosa, or P. aeruginosa pvdF (a pyoverdin biosynthesis mutant) or partially purified pyoverdin. Expression was normalized to E. coli on plates. (C) Survival of C. elegans exposed to purified pyoverdin, compared with solvent control. n = 15,000 (A), 10,000 (B), or 100 (C) worms per replicate; error bars represent SEM. *P < 0.01, Student’s t test.
We also measured the transcriptional response in C. elegans to P. aeruginosa, partially purified pyoverdin, or to phenanthroline, a synthetic iron chelator. Under all three conditions, C. elegans exhibits a similar hypoxic crisis (Fig. 1B). Specifically, we observed up-regulation of genes (including genes that are both dependent upon and independent of HIF-1/HIF1α) that are activated in C. elegans in response to hypoxia (9). In each case, worms are subjected to iron-chelating molecules, suggesting that the removal of iron from the host is critical for pyoverdin-mediated virulence (3). Consistent with these data, exposure to E. coli, P. aeruginosa mutants with compromised pyoverdin biosynthesis, or to solvent alone was insufficient to up-regulate transcription of hypoxic response genes.
Finally, we tested whether pyoverdin was sufficient to trigger host pathology. Worms were exposed to commercially available, purified pyoverdin at concentrations equivalent to those in the liquid kill assay. Consistent with our hypothesis, exposure to purified, iron-free pyoverdin from P. aeruginosa killed C. elegans (Fig. 1C), which clearly demonstrates that pyoverdin can serve as a toxin to the host.
Pyoverdin Damages Host Mitochondria.
Mitochondria represent a rich source of cellular iron because they use a variety of heme- and iron-sulfur proteins (10). We hypothesized that siderophores might remove iron from these complexes, damaging organelles. Mitochondria exhibit characteristic quality control pathways, including constant fission and fusion events that mix mitochondrial contents, repair damage, and restore functionality (11). In the event that mitochondria were damaged by pyoverdin, mitochondrial homeostasis would be disrupted.
Mitochondrial dynamics in living worms were assessed using mitochondrially-targeted GFP in body wall muscle cells (12). Worms feeding on E. coli (on agar plates or in liquid medium) or on P. aeruginosa on agar plates exhibited normal, long-branched tubular mitochondria (Fig. 2A and Fig. S1A). In contrast, exposure to P. aeruginosa in liquid induced dramatic mitochondrial disruption in C. elegans, fragmenting the network and reducing it to large, punctate bodies (Fig. 2A and Fig. S1A). Adding phenanthroline or partially purified pyoverdin to an otherwise benign E. coli strain also caused mitochondrial fragmentation (Fig. 2A and Fig. S1A). Mitochondrial morphology correlated with killing (Fig. S1B).
Fig. 2.
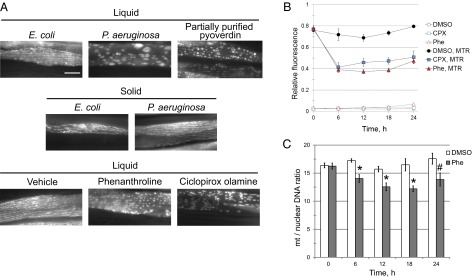
Pyoverdin disrupts mitochondrial homeostasis and triggers mitochondrial turnover. (A) Fluorescence microscopy of worms with mtGFP exposed to E. coli, P. aeruginosa, or partially purified pyoverdin in liquid; E. coli or P. aeruginosa on plates; or vehicle (DMSO), Phe (1 mM), or the iron chelator ciclopirox olamine (CPX; 1 mM) in liquid. (B) Flow vermimetry of worms exposed to Phe (1 mM), CPX (1 mM), or DMSO and labeled with MTR (4.375 μM). Fluorescence was normalized to worm size. (C) Ratio of mitochondrial to nuclear genomes for worms treated with Phe or DMSO. Three biological replicates were performed; n = 5,000 (B) or 6,000 (C) for each biological replicate. Error bars represent SEM. *P < 0.01, #P < 0.05, Student’s t test. (Scale bar: A, 20 μM.)
Generation of ATP via oxidative phosphorylation is one of the most important roles for mitochondria in metazoans. This function requires the development and maintenance of an electric potential across the mitochondrial membrane (ΔψM). Loss of this membrane potential can also change mitochondrial dynamics. Therefore, ΔψM was assayed by MitoTracker Red (MTR) staining in worms treated with 1 mM phenanthroline, 0.5 mM ciclopirox olamine, or DMSO. Fluorescence was markedly reduced in worms exposed to either chelator (Fig. 2B), showing that iron sequestration disrupts ΔψM.
When mitochondrial fusion fails to restore homeostasis, excessively damaged mitochondria are sequestered from further fusion with the remainder of the cellular mitochondria and are targeted for turnover via autophagic degradation, a process known as mitophagy (11, 13). Mitochondrial turnover was assayed by staining with 10-nonyl acridine orange (NAO), a mitochondrial dye relatively insensitive to ΔψM (14, 15). Staining decreased substantially after iron depletion (Fig. S2), suggesting a decrease in mitochondrial mass. To confirm mitochondrial degradation, the ratio of mitochondrial and nuclear genomes was assayed via quantitative PCR (qPCR). This ratio also decreased after iron chelation (Fig. 2C). Combined, these data demonstrate that iron chelation reduces mitochondrial number. Comparing the magnitude of the decreases in MTR staining (∼50% reduction) and in the mitochondrial:nuclear genome ratio (∼20% reduction) supports the conclusion that decreased MTR staining is at least partially due to decreased ΔψM and not only a consequence of a decrease in the number of mitochondria.
Mitophagy Is Involved in the Innate Immune Response Against P. aeruginosa.
Mitophagy uses a well-characterized sequence of events that are shared with generalized autophagy, including the development of an isolation membrane and maturation of the autophagosome. We used translational fusions of two proteins that are critical for this process, BEC-1/Beclin1 and LGG-1/LC3, to monitor intracellular events after exposure to chelators. Treatment with phenanthroline or P. aeruginosa triggered coalescence of the constitutively expressed BEC-1/BECN1::RFP and mCherry::LGG-1/LC3 proteins from a diffuse, cytoplasmic pattern into discrete punctae that likely represent autophagosomes, demonstrating activation of autophagy (Fig. 3 A and B). Mitophagy also requires at least two specific regulators (PINK-1/PINK1 and PDR-1/Parkin) to target particular mitochondria for recycling. PINK-1/PINK1 is constitutively expressed, but under healthy conditions, is imported into mitochondria and degraded by matrix-resident proteases. Exposure to phenanthroline or P. aeruginosa inhibited mitochondrial import and degradation of PINK-1/PINK1::GFP, increasing protein levels (Fig. 3C). These data demonstrate that iron sequestration triggers a cascade that begins with loss of ΔψM and concludes with mitophagy.
Fig. 3.
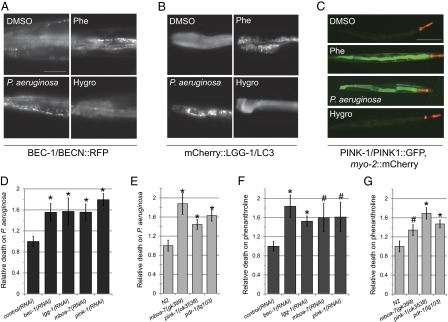
Mitophagy confers resistance to P. aeruginosa. (A–C) Fluorescence microscopy of (A) BEC-1/BECN::RFP, (B) mCherry::LGG-1/LC3, or (C) myo-2::mCherry; PINK-1/PINK1::GFP worms treated with DMSO, Phe (1 mM), P. aeruginosa, or hygromycin (80 µg/mL). (D and E) Pyoverdin-mediated killing by P. aeruginosa and (F and G) Phe toxicity are enhanced by RNAi knockdown (D and F) or mutation (E and G) of autophagy pathway genes bec-1/BECN1, lgg-1/LC3, mboa-7/MBOAT7, or the mitophagic regulators pink-1/PINK1 and pdr-1/PARK2. Survival was normalized to empty plasmid (D and F) or wild-type (E and G) controls. Three biological replicates were used for each experiment; n = 850 (D and E), n = 360 (F and G) per replicate. Error bars represent SEM. *P < 0.01, #P < 0.05 (Student’s t test). (Scale bars: A and B, 50 μm; C, 200 μm.)
To determine whether mitophagy promotes survival in the face of P. aeruginosa exposure, we measured survival of worms with disruptions in genes required for autophagy or mitophagic regulators that were exposed to P. aeruginosa or to phenanthroline. Mitophagy is important for organismal defense against iron removal, because disruption of conserved genes with roles in autophagy (bec-1/BECN1, lgg-1/LC3, mboa-7/MBOAT7) or the mitophagic regulators (pink-1/PINK1 and pdr-1/PARK2) increased lethality of treatment with P. aeruginosa or phenanthroline (Fig. 3 D–G). Sensitivity to iron sequestration correlated with decreased mitophagy, as determined by increased MTR staining and mitochondrial:nuclear genome ratio (Fig. S3). Iron sequestration-induced mitochondrial fragmentation was also strongly diminished when autophagic or mitophagic genes were disrupted (Fig. S4). This protective effect was specific to liquid context and to iron removal, as disruption of autophagy or mitophagy did not generally increase sensitivity to P. aeruginosa infection on plates or to treatment with the translational inhibitors hygromycin or G418 (Fig. S5). A notable exception was lgg-1/LC3(RNAi) worms, which did exhibit enhanced sensitivity.
Damaged mitochondria often produce increased amounts of reactive oxygen species (ROS), which can cause nonspecific pathology to the organism. We ruled out this explanation for the protective effect that we observed, because treatment with phenanthroline at a concentration sufficient to trigger mitophagy and killing did not increase ROS (Fig. S6). The protective mechanism of mitophagy remains to be determined.
C. elegans Uses a Multifaceted Innate Immune Response to Liquid Killing.
Within the past 15 y, the innate immune system of C. elegans has been subjected to intensive research, and several pathways crucial for resisting bacterial infection have been identified; the most central of these is the PMK-1/p38 MAPK pathway, which is indispensible for the hosts’ immune response to infection with most bacterial species (16, 17). Interestingly, compromising the p38 MAPK cascade, including by mutations in tir-1/SARM, nsy-1/MAP3K, sek-1/MAPKK, and pmk-1/p38 or by RNAi-mediated knockdown of PMK-1/p38 pathway members caused decreased susceptibility to P. aeruginosa in liquid (Fig. 4 A and B). Hyperactivation of this pathway by RNAi-mediated knockdown of the inhibitory phosphatase vhp-1/MKP strongly increased sensitivity to P. aeruginosa, likely due to the effect of vhp-1/MKP on the PMK-1/p38 pathway, because inhibition of kgb-1/JNK (the other known kinase target of vhp-1/MKP) had no impact on host survival. In addition, a PMK-1/p38-dependent reporter was not activated in P. aeruginosa liquid intoxication, although liquid conditions did not preclude activation of the reporter (Fig. S7A), and genes transcriptionally activated by P. aeruginosa on plates were not activated when worms were exposed to P. aeruginosa in liquid (Fig. S7 B and C). One possible explanation for PMK-1/p38 activity reducing survival in liquid killing is that PMK-1/p38-dependent immune effectors monopolize resources (e.g., chaperones that ameliorate endoplasmic reticulum stress) (18) required for pyoverdin-elicited mitophagy. Alternatively, PMK-1/p38 may induce host pathology by initiating inflammation-like processes.
Fig. 4.

PMK-1/p38 activity is deleterious for survival in liquid killing, but ZIP-2 is required. P. aeruginosa pathogenesis of worms with RNAi (A) or mutations (B and C) targeting PMK-1/p38 pathway members (A and B) or zip-2 (C). Also shown is PMK-1/p38 hyperactivation by knockdown of the negative regulatory phosphatase vhp-1/MKP. Disruption of kgb-1/JNK, the other target of vhp-1/MKP, did not alter sensitivity. Normalized to (A) control RNAi or to (B and C) wild type. (D) Gene expression data for ZIP-2–dependent genes in both N2 and glp-4 backgrounds, as determined by qRT-PCR. Three biological replicates were performed; n = 850 (A–C), n = 10,000 (D) per replicate. Error bars represent SEM. *P < 0.01, #P < 0.05 (Student’s t test).
The bZIP transcription factor ZIP-2 also plays a key role in defense against P. aeruginosa infection (19). ZIP-2 is also known to be activated by damage-associated molecular patterns, including translational inhibition and mitochondrial disruption (20, 21), which suggests that ZIP-2 may bridge the divide between immunity and stress response genes. Unlike PMK-1/p38 pathway members, mutation of ZIP-2 caused significant sensitivity to pyoverdin intoxication (Fig. 4C). Consistent with this, ZIP-2–dependent genes (19) were up-regulated in liquid killing (Fig. 4D).
Inhibition of the insulin signaling pathway by mutation of daf-2/IGFR results in resistance to a wide variety of abiotic and biological insults (17, 22–25). To determine whether the insulin signaling pathway is involved in the host response to liquid killing, we used RNAi to knock down a panel of daf-2/IGFR pathway genes. Disruption of daf-2/IGFR, age-1/PIP2K, and akt-1/AKT1 increased survival in liquid killing (Fig. S8A). Importantly, knockdown of daf-16/FOXO by RNAi increased host sensitivity to P. aeruginosa, demonstrating that the resistance conferred is biologically relevant. Moreover, overexpression of the FOXO transcription factor DAF-16 increased resistance (Fig. S7A). Liquid killing conditions drove a DAF-16/FOXO-GFP translational fusion construct into the nuclei of cells (Fig. S8 B–E). However, we also observed that DAF-16/FOXO-GFP translocates to the nucleus in the absence of P. aeruginosa in liquid conditions (Fig. S8D). We assayed the biological importance of DAF-16/FOXO translocation by comparing the longitudinal survival of worms reared on control and daf-16/FOXO(RNAi) of worms in liquid conditions with E. coli. Consistent with this, daf-16/FOXO(RNAi) mutants survived a significantly shorter time than controls (Fig. S7F), as was previously observed (26). Although it is difficult to assess what fraction of daf-16/FOXO(RNAi) mutants’ increased sensitivity is specific (as a result of the pathogen exposure) and what portion is generic (as a result of liquid exposure), activation of DAF-16/FOXO is important for host survival.
Iron Chelators Trigger Mitochondrial Turnover in Mammalian Cells.
Because P. aeruginosa is an important human pathogen, we tested whether iron sequestration triggered pathology in mammalian cells. MTR staining after treatment with either ciclopirox olamine or phenanthroline reduced ΔψM and increased mitochondrial fragmentation (Fig. 5A), much as was observed in C. elegans. Acute iron sequestration also activated mitochondrial turnover (Fig. 5B), potentially by the same mitophagic pathways that confer resistance to pyoverdin in C. elegans.
Fig. 5.

Mitochondrial turnover after chelator treatment is evolutionarily conserved. (A) Confocal laser scanning microscopy of HEK293T cells treated with 0.3 mM Phe, 0.1 mM CPX, or vehicle (DMSO) for 12 h and stained with MTR CMXRos, 200 nM. (B) Mitochondrial (nd1, nd4) to nuclear (TUB) genome ratio after treatment with either 0.5 mM Phe or 0.1 mM CPX for 12 h. Ratios are normalized to DMSO controls. (C) qRT-PCR of PPARGC1A (PGC-1α) after treatment with either 0.5 mM Phe or 0.1 mM CPX for 12 h. Expression was normalized to DMSO control. (D) Immunoblotting for NRF1, phosphorylated NRF2, or β-tubulin (loading control) in total protein extracts after lysis of HEK293T cells treated with 0.5 mM Phe, 0.1 mM CPX, or vehicle (DMSO) for 12 h. For all experiments, three biological replicates were performed, and a representative replicate is shown (A and D). Statistical significance was determined via Student’s t test, *P < 0.01. Error bars represent SEM.
Mitochondria are constantly generated and recycled, even in healthy cells; as such, decreased mitochondrial biogenesis may also account for our observations. To rule out this explanation, we assayed expression of PPARGC1A (PGC-1α), a master regulator of mitochondrial biogenesis, after chelator treatment (Fig. 5C). Treatment significantly increased expression of PGC-1α. We also assayed NRF1 and NRF2, key transcription factors in mitochondrial biogenesis, and discovered that levels of NRF1 and phosphorylation of NRF2 were essentially indistinguishable after treatment with phenanthroline or ciclopirox olamine (Fig. 5D). Combined, these data support our conclusion that mitochondria are being turned over and that biogenesis is not likely to be compromised.
Our results extend in several key ways previous work (27, 28) showing that iron chelation triggers mitochondrial degradation. For example, we demonstrate involvement of mitophagic regulators in the response to iron sequestration. Striking physiological differences have been observed after chelator exposure [e.g., the involvement (this study)—or lack thereof (27)—of the canonical mitophagic regulators PINK1 and Parkin, the presence (28) or absence (27) of mitochondrial damage, etc.]; this may suggest that various chelators cause mitochondrial destruction but that the mechanisms may differ and may include nonmitophagic degradation pathways like mitoptosis (29). In addition, we show that mitophagy is necessary for organismal resistance to pyoverdin, a key virulence factor that kills C. elegans.
To our knowledge, this is the first report of authentic autophagy (i.e., the turnover of host mitochondria promoting resistance to intoxication with pyoverdin or the extracellular pathogen, P. aeruginosa) serving a role in innate immunity. Although there are reports of autophagy conferring resistance to many intracellular pathogens (e.g., Listeria monocytogenes or Mycobacterium tuberculosis) (30, 31), this occurs via autophagic machinery targeting foreign, invasive microbes for degradation. Finally, our data suggest a mechanism for pyoverdin-mediated virulence for P. aeruginosa in acute mammalian infection models (4, 5), and suggest that pyoverdin and host mitophagy may be potential targets for the development of novel anti-P. aeruginosa therapies.
Materials and Methods
C. elegans and Pseudomonas Strains.
All C. elegans strains were maintained on nematode growth medium (NGM) seeded with E. coli strain OP50 as a food source and were maintained at 15 °C (32), unless otherwise noted.
P. aeruginosa strain PA14 is a previously described clinical isolate (33). PA14 pyoverdin mutants (pvdF and pvdP) were obtained from a transposon insertion library (34) and verified by DNA sequencing.
Mammalian Cell Culture.
HEK293T cells were grown at 37 °C in a humidified, 5% CO2 incubator in DMEM (Invitrogen) supplemented with 10% FBS (HyClone) and penicillin/streptomycin mix (Invitrogen). Cells were split when they reached >95% confluency.
Partial Purification of Pyoverdin.
P. aeruginosa was inoculated in modified M9 media (3) and grown for 24 h. Bacteria were pelleted at 20,000 × g. Supernatants were filtered twice through 0.22-μm filters. Filtrate was then boiled for 30 min and centrifuged at 207,000 × g for 4 h to denature and precipitate bulk protein contaminants. Supernatants were collected from centrifuge tubes by side puncture. Contamination with other proteins was assessed by Coomassie staining. The presence of pyoverdin in each fraction was verified by fluorescence spectrometry (excitation maximum 405 nm, emission maximum 460 nm). Samples from P. aeruginosa pvdF, a pyoverdin biosynthetic mutant, were prepared in parallel.
Flow Vermimetry.
All experiments were performed with a COPAS large-particle biosorter. Before fluorescence measurement, synchronized young adult worms were washed from NGM plates seeded with E. coli OP50 and exposed to 1 mM 1,10-phenanthroline (Sigma; dissolved in DMSO (Sigma), final DMSO concentration was 1% vol/vol), 0.5 mM ciclopirox olamine (Sigma, dissolved in DMSO, final DMSO concentration was 1% vol/vol), or vehicle (DMSO, 1% vol/vol) for varying lengths of time. After exposure, worms were stained with either 10 µM 10-nonyl acridine orange bromide (NAO; Invitrogen) or 4.375 μM MitoTracker Red CMXRos (MTR; Invitrogen) in S basal. Samples were also collected from worms that were not stained. Gating was adjusted to limit measurement acquisition from nonadult worms or debris; fluorescence was normalized to worm size as based on time of flight. Fluorescence measurement was collected by a long-pass filter with excitation at 488 nm (NAO or no dye control) or 562 nm (MTR or no dye control). Each biological replicate consisted of five technical replicates, each of ∼1,000 worms.
For determination of ROS, worms were collected and processed as above, except that they were stained with either 250 μM dichlorofluorescein diacetate (DCF-DA; Sigma) or with 4.375 μM MitoTracker Red CM-H2Xros (Invitrogen). Additional samples had 1 mM H2O2 (Sigma) added as a positive control for ROS before staining. Fluorescence measurement was collected by a long-pass filter with excitation at 488 nm (DCF-DA) or 562 nm (MTR-CMH2Xros). Each biological replicate consisted of five technical replicates, each of ∼1,000 worms.
C. elegans Pathogenesis and Chemical Exposure Assays.
Slow kill assays were performed essentially as previously described (35), except that synchronized young adult worms were used. Liquid kill assays were performed essentially as previously described (36). To determine phenanthroline sensitivity, the liquid kill assay was modified as follows: first, P. aeruginosa PA14 was substituted with E. coli OP50 at a final density of OD600 = 0.03. Second, 1,10-phenanthroline was added at a final concentration of 1 mM.
For filtrate pathogenesis experiments, pyoverdin-rich filtrates were produced as previously described (3). Filtrates comprised 50% (vol/vol) of final volume. E. coli OP50 was added to a final density of OD600 = 0.03. Killing was scored as for liquid kill assays.
For chemical killing assays, synchronized L1 larvae were spotted onto NGM media supplemented with carbenicillin and 1 mM IPTG and spotted with E. coli containing RNAi constructs. Young adults were washed off of plates and dropped onto freshly made NGM plates spotted with E. coli OP50 that was supplemented with 80 µg/mL hygromycin B (Sigma) or 500 µg/mL G418 (Invitrogen).
Before exposure to antimycin A, rotenone, or hygromycin B, synchronized young adult worms were washed from NGM plates seeded with OP50, and resuspended in S basal supplemented with 50 µM antimycin A (Sigma), 50 µM rotenone (Sigma), or 80 µg/mL hygromycin B in the presence of OP50. Worms were exposed for 8 h, and RNA was then harvested (see below).
At least three biological replicates were performed for each experiment. P values were determined via Student’s t test.
Supplementary Material
Acknowledgments
The authors thank Dr. David Fay for providing the WY753 and W756 strains, Dr. Vamsi Mootha for providing the HEK293T cell line, and Dr. Daniel Kirienko for technical assistance. This work was supported by a Massachusetts Biomedical Research Corporation Tosteson Postdoctoral Fellowship Award (to N.V.K.); Ruth L. Kirschstein National Research Service Award F32 AI-100501 (to N.V.K.); and National Institutes of Health Grants R01 AI-085581 and P30 DK040561 (to F.M.A.) and R01 AG16636 (to G.R.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1424954112/-/DCSupplemental.
References
- 1.Cassat JE, Skaar EP. Iron in infection and immunity. Cell Host Microbe. 2013;13(5):509–519. doi: 10.1016/j.chom.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hammer ND, Skaar EP. Molecular mechanisms of Staphylococcus aureus iron acquisition. Annu Rev Microbiol. 2011;65:129–147. doi: 10.1146/annurev-micro-090110-102851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kirienko NV, et al. Pseudomonas aeruginosa disrupts Caenorhabditis elegans iron homeostasis, causing a hypoxic response and death. Cell Host Microbe. 2013;13(4):406–416. doi: 10.1016/j.chom.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meyer JM, Neely A, Stintzi A, Georges C, Holder IA. Pyoverdin is essential for virulence of Pseudomonas aeruginosa. Infect Immun. 1996;64(2):518–523. doi: 10.1128/iai.64.2.518-523.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takase H, Nitanai H, Hoshino K, Otani T. Impact of siderophore production on Pseudomonas aeruginosa infections in immunosuppressed mice. Infect Immun. 2000;68(4):1834–1839. doi: 10.1128/iai.68.4.1834-1839.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sia AK, Allred BE, Raymond KN. Siderocalins: Siderophore binding proteins evolved for primary pathogen host defense. Curr Opin Chem Biol. 2013;17(2):150–157. doi: 10.1016/j.cbpa.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aloush V, Navon-Venezia S, Seigman-Igra Y, Cabili S, Carmeli Y. Multidrug-resistant Pseudomonas aeruginosa: Risk factors and clinical impact. Antimicrob Agents Chemother. 2006;50(1):43–48. doi: 10.1128/AAC.50.1.43-48.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Utari PD, Quax WJ. Caenorhabditis elegans reveals novel Pseudomonas aeruginosa virulence mechanism. Trends Microbiol. 2013;21(7):315–316. doi: 10.1016/j.tim.2013.04.006. [DOI] [PubMed] [Google Scholar]
- 9.Jiang H, Guo R, Powell-Coffman JA. The Caenorhabditis elegans hif-1 gene encodes a bHLH-PAS protein that is required for adaptation to hypoxia. Proc Natl Acad Sci USA. 2001;98(14):7916–7921. doi: 10.1073/pnas.141234698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang ML, Lane DJ, Richardson DR. Mitochondrial mayhem: The mitochondrion as a modulator of iron metabolism and its role in disease. Antioxid Redox Signal. 2011;15(12):3003–3019. doi: 10.1089/ars.2011.3921. [DOI] [PubMed] [Google Scholar]
- 11.Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013;17(4):491–506. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benedetti C, Haynes CM, Yang Y, Harding HP, Ron D. Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics. 2006;174(1):229–239. doi: 10.1534/genetics.106.061580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12(1):9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keij JF, Bell-Prince C, Steinkamp JA. Staining of mitochondrial membranes with 10-nonyl acridine orange, MitoFluor Green, and MitoTracker Green is affected by mitochondrial membrane potential altering drugs. Cytometry. 2000;39(3):203–210. doi: 10.1002/(sici)1097-0320(20000301)39:3<203::aid-cyto5>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 15.Zhang H, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283(16):10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Kim DH, et al. A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science. 2002;297(5581):623–626. doi: 10.1126/science.1073759. [DOI] [PubMed] [Google Scholar]
- 17.Shivers RP, Youngman MJ, Kim DH. Transcriptional responses to pathogens in Caenorhabditis elegans. Curr Opin Microbiol. 2008;11(3):251–256. doi: 10.1016/j.mib.2008.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Richardson CE, Kooistra T, Kim DH. An essential role for XBP-1 in host protection against immune activation in C. elegans. Nature. 2010;463(7284):1092–1095. doi: 10.1038/nature08762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Estes KA, Dunbar TL, Powell JR, Ausubel FM, Troemel ER. bZIP transcription factor zip-2 mediates an early response to Pseudomonas aeruginosa infection in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2010;107(5):2153–2158. doi: 10.1073/pnas.0914643107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dunbar TL, Yan Z, Balla KM, Smelkinson MG, Troemel ER. C. elegans detects pathogen-induced translational inhibition to activate immune signaling. Cell Host Microbe. 2012;11(4):375–386. doi: 10.1016/j.chom.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McEwan DL, Kirienko NV, Ausubel FM. Host translational inhibition by Pseudomonas aeruginosa exotoxin A triggers an immune response in Caenorhabditis elegans. Cell Host Microbe. 2012;11(4):364–374. doi: 10.1016/j.chom.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsu AL, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300(5622):1142–1145. doi: 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- 23.Henderson ST, Johnson TE. daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr Biol. 2001;11(24):1975–1980. doi: 10.1016/s0960-9822(01)00594-2. [DOI] [PubMed] [Google Scholar]
- 24.Murakami S, Johnson TE. A genetic pathway conferring life extension and resistance to UV stress in Caenorhabditis elegans. Genetics. 1996;143(3):1207–1218. doi: 10.1093/genetics/143.3.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lamitina ST, Strange K. Transcriptional targets of DAF-16 insulin signaling pathway protect C. elegans from extreme hypertonic stress. Am J Physiol Cell Physiol. 2005;288(2):C467–C474. doi: 10.1152/ajpcell.00451.2004. [DOI] [PubMed] [Google Scholar]
- 26.Henderson ST, Bonafè M, Johnson TE. daf-16 protects the nematode Caenorhabditis elegans during food deprivation. J Gerontol A Biol Sci Med Sci. 2006;61(5):444–460. doi: 10.1093/gerona/61.5.444. [DOI] [PubMed] [Google Scholar]
- 27.Allen GF, Toth R, James J, Ganley IG. Loss of iron triggers PINK1/Parkin-independent mitophagy. EMBO Rep. 2013;14(12):1127–1135. doi: 10.1038/embor.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park SJ, et al. Mitochondrial fragmentation caused by phenanthroline promotes mitophagy. FEBS Lett. 2012;586(24):4303–4310. doi: 10.1016/j.febslet.2012.10.035. [DOI] [PubMed] [Google Scholar]
- 29.Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta. 2013;1833(12):3448–3459. doi: 10.1016/j.bbamcr.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 30.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13(10):722–737. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stiernagle T. Maintenance of C. elegans. WormBook. 2006;11:1–11. doi: 10.1895/wormbook.1.101.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rahme LG, et al. Common virulence factors for bacterial pathogenicity in plants and animals. Science. 1995;268(5219):1899–1902. doi: 10.1126/science.7604262. [DOI] [PubMed] [Google Scholar]
- 34.Liberati NT, et al. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc Natl Acad Sci USA. 2006;103(8):2833–2838. doi: 10.1073/pnas.0511100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan MW, Mahajan-Miklos S, Ausubel FM. Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc Natl Acad Sci USA. 1999;96(2):715–720. doi: 10.1073/pnas.96.2.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kirienko NV, Cezairliyan BO, Ausubel FM, Powell JR. Pseudomonas aeruginosa PA14 pathogenesis in Caenorhabditis elegans. Methods Mol Biol. 2014;1149:653–669. doi: 10.1007/978-1-4939-0473-0_50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.