

Significance
BDNF plays a key role in neuron development, survival, and function, with actions occurring through the stimulation of the tropomyosin-related kinase receptor B (TrkB) receptor. Whether BDNF/TrkB signaling has any physiologic role in governing myocardial function is unknown. Here we report that intact BDNF/TrkB signaling is required for the heart to fully contract and relax. These actions occur independently from and in addition to β-adrenergic influence. BDNF-induced enhancement of myocardial performance occurs via direct modulation of Ca2+ cycling in a calmodulin-dependent protein kinase II-dependent manner. Thus, BDNF/TrkB signaling represents a previously unidentified way by which the peripheral nervous system controls cardiac muscle physiology. Our study suggests that loss or alterations in BDNF/TrkB stimulation may contribute to the pathogenesis of myocardial dysfunction in acute or chronic disease conditions.
Keywords: BDNF, TrkB receptor, cardiac contractility/relaxation, CaMKII, neurotrophins
Abstract
BDNF and its associated tropomyosin-related kinase receptor B (TrkB) nurture vessels and nerves serving the heart. However, the direct effect of BDNF/TrkB signaling on the myocardium is poorly understood. Here we report that cardiac-specific TrkB knockout mice (TrkB−/−) display impaired cardiac contraction and relaxation, showing that BDNF/TrkB signaling acts constitutively to sustain in vivo myocardial performance. BDNF enhances normal cardiomyocyte Ca2+ cycling, contractility, and relaxation via Ca2+/calmodulin-dependent protein kinase II (CaMKII). Conversely, failing myocytes, which have increased truncated TrkB lacking tyrosine kinase activity and chronically activated CaMKII, are insensitive to BDNF. Thus, BDNF/TrkB signaling represents a previously unidentified pathway by which the peripheral nervous system directly and tonically influences myocardial function in parallel with β-adrenergic control. Deficits in this system are likely additional contributors to acute and chronic cardiac dysfunction.
In the brain, BDNF has a pleiotropic profile, preserving cell viability and function, preventing neuronal degeneration during stress (1), and acting as an antidepressant (2). BDNF is also expressed in various nonneuronal tissues, including smooth and skeletal muscle cells, where it enhances airway smooth muscle cell contractility (3) and acts as a metabolic enhancer (4), respectively.
In the mammalian heart, BDNF is essential for organ development because its genetic deletion leads to the thinning of cardiac chambers, microvascular leakage, and ultimately, early death in mice (5). In adult mammals, BDNF governs autonomic transmission to the heart (6) and exerts prominent angiogenic effects (7). Recent evidence also points to the presence of the BDNF receptor, tropomyosin-related kinase receptor B (TrkB), in the myocardium (8). However, the role of myocardial BDNF/TrkB signaling in cardiac physiology and myocardial response to pathologic stress, independent from its well-known trophic actions on blood vessels and autonomic efferent neurons (9), is largely unknown.
In neurons, BDNF regulates Ca2+ signaling, and calmodulin-dependent protein kinase II (CaMKII) is one of the downstream effectors of the BDNF/TrkB signaling pathway. Because CaMKII is one of the important regulators of excitation–contraction coupling, and because BDNF also enhances smooth muscle contraction by augmenting intracellular Ca2+ signaling, we first determined the effects of BDNF on cardiomyocyte contractility, relaxation, and Ca2+ dynamics. Then, to establish whether BDNF/TrkB stimulation is relevant in maintaining basal cardiac function in vivo, we generated cardiac-specific TrkB−/− mice by crossing conditional TrkB−/− mice with MHC promoter-driven Cre mice and subjected these mice and their WT littermates to pressure–volume analysis. We further investigated which signaling pathway mediates the BDNF cardiac effects, and finally, we tested whether modulation of myocyte function by BDNF is preserved or altered in myocytes from failing hearts.
Results
BDNF Directly Increases Myocyte Function, Enhancing Cardiac Ca2+ Cycling.
First, we determined the effect of exogenous application of BDNF on isolated rodent cardiomyocytes. These cells contain surface TrkB receptors (Fig. 1A), consistent with previous reports (8). Incubating mouse cardiac myocytes with BDNF (20 nM) resulted in increased cell contractility (43 ± 7.5%) and enhanced relaxation (11 ± 3%; n = 19; P < 0.01 for both) (Fig. 1B). This enhancement of function was accompanied by larger whole-cell Ca2+ transients (10 ± 4%; P < 0.01) (Fig. 1B). Ca2+ spark frequency was also increased after BDNF stimulation (1.3 ± 0.3 to 3.4 ± 0.7 sparks/100 μm/s; P < 0.05; n = 6) (Fig. 1C). We next asked whether this stimulatory action stems from an increase in sarcoplasmic reticulum (SR) Ca2+ fractional release and/or activity of L-type Ca2+ channels. BDNF markedly augmented SR Ca2+ fractional release (from 54 ± 6% to 68 ± 6%; P < 0.05; n = 11) without affecting total SR Ca2+ load, as assessed by caffeine transients (Fig. 1D). In isolated guinea pig myocytes, BDNF increased peak L-type Ca2+ current density by ∼25% compared with baseline (−3.5 ± 0.2 vs. −2.8 ± 0.4 mV; P < 0.05; n = 6) (Fig. 1E). Thus, BDNF enhances myocyte Ca2+ cycling without altering diastolic Ca2+ levels or Ca2+ SR load (Fig. 1D).
Fig. 1.
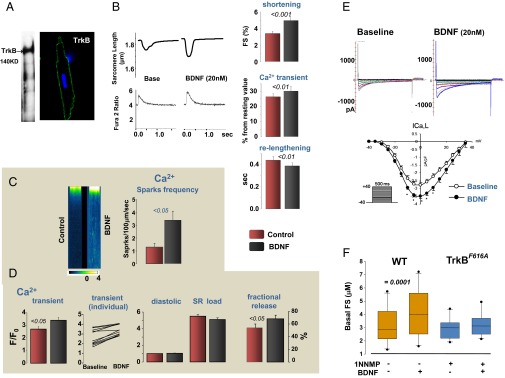
BDNF increases cardiomyocyte contractility and relaxation as a result of enhanced Ca2+ cycling, in a TrkB receptor-dependent manner. (A) TrkB protein expression is detected in isolated adult murine cardiac myocytes by Western blot (140 KD). TrkB is located on plasma membrane of cardiac myocytes by immunohistochemical study, using confocal microscopy. (B) Representative traces of sarcomere shortening and Ca2+ transients, with or without BDNF treatment. Incubating isolated murine cardiomyocytes with BDNF (20 nM) increases myocyte fractional shortening and whole-cell Ca2+ transients while accelerating myocyte relaxation; the latter is documented by decreased relaxation time (n = 19). (C) Representative confocal imaging of Ca2+ spark events in isolated rat adult cardiomyocytes: BDNF augments Ca2+ spark frequency in these cells. (D) Consistent with data in the mice, BDNF increases Ca2+ transients and SR Ca2+ fractional release in isolated rat adult cardiomyocytes without significantly affecting diastolic Ca2+ levels or SR Ca2+ load, measured by caffeine-induced total SR Ca2+ release. (E) Raw traces of BDNF’s effect on L-type Ca2+ channel activity measured in isolated guinea pig ventricular myocytes: BDNF enhances peak L-type Ca2+ channel activity from baseline (P < 0.05). (F) BDNF inotropy is abolished in cardiomyocytes isolated from TrkBF616A mice with 1-NNMP1 pretreatment.
TrkB Is Required for BDNF-Induced Enhancement of Myocyte Function.
We then tested whether the TrkB receptor is required for the inotropic/lusitropic action of BDNF. To do so, we used isolated myocytes from TrkBF616A mice (kindly provided by David D. Ginty, Department of Neurobiology, Harvard Medical School, Boston) (10). In these mice, a phenylalanine-to-alanine substitution within the kinase subdomain of the TrkB receptor renders it sensitive to specific inhibition by membrane-permeable, small-molecule protein phosphatase 1 (PP1) derivatives, including derivative of the general kinase inhibitor PP1 (1NMPP1) (11). BDNF had no effect on TrkBF616A myocytes pretreated with 1-NMMP1 (100 nM) (Fig. 1F), and 1-NMMP1 did not alter basal myocyte function: fractional shortening was 3.19 ± 0.43 (baseline) vs. 2.92 ± 0.2 (after 1NMPP1) (n = 10; P = 0.52).
Constitutive TrkB Stimulation Is Required for Optimal Cardiac Contraction and Relaxation.
To determine whether BDNF/TrkB stimulation is constitutively active in vivo and the relevance of this signaling in maintaining normal heart function, cardiac-specific TrkB knockout mice (TrkB−/−) were generated by crossing conditional TrkB−/− mice (12) (from David D. Ginty) with cardiomyocyte-specific (MHC)-Cre mice (Fig. S1). BDNF is a modulator of angiogenesis (7), and thus changes in vascularity may contribute to altering cardiac performance in TrkB−/− mice. Therefore, we first measured capillary density in TrkB−/− mice and their control littermates. The capillary density measured by counting isolectin B4-stained capillary profiles in myocardium did not differ between WT and TrkB−/− mice (Fig. S2). Next, we examined the effect of BDNF/TrkB signaling on cardiac contraction and relaxation in vivo. Pressure–volume relationships were used to distinguish intrinsic changes in myocardial contractility/relaxation from confounding effects as a result of potential changes in vascular loading conditions (i.e., preload and afterload). TrkB−/− mice displayed reduced myocardial performance, as determined by dP/dtmax (maximum time derivative of left ventricular pressure; a load-dependent index) and load-independent parameters of myocardial contractility such as ventricular elastance (Ees), dPdt/EDV (dPdt max against end-diastolic volume relationship), dPdt ip (dp/dt max divided by the instantaneous developed isovolumetric pressure), and prerecruitable stroke work (Fig. 2 and Table 1). All these indices were consistent with reduced systolic function in TrkB−/− mice, although this was not severe enough to trigger chamber dilation or reduce ejection fraction. Cardiac relaxation was also impaired, with prolongation of the relaxation time constant (Tau) from 4.0 ± 0.2 to 5.3 ± 0.3 msec (P = 0.0019) (Fig. 2B and Table 1). Total systemic vascular resistance and ventricular afterload (indexed by arterial elastance, Ea) were similar in both genotypes, so systemic load was not affected. The ventricular/arterial coupling ratio (Ees/Ea) declined solely because of the fall in contractility, which would indicate a fall in efficiency of blood transfer in TrkB−/− mice. Thus, BDNF/TrkB signaling is an important independent contributor to basal cardiac contraction and relaxation in vivo. We further examined whether TrkB−/− mice had altered responses to β-adrenergic receptor stimulation. Isoproterenol (from 10 to 40 ng/kg/min) infusion produced similar augmentation in both genotypes (n = 5 each group; Fig. 3 and Table 1). These findings indicate that a lack of BDNF/TrkB modulation does not impair β-adrenergic receptor-cAMP/PKA signaling.
Fig. 2.
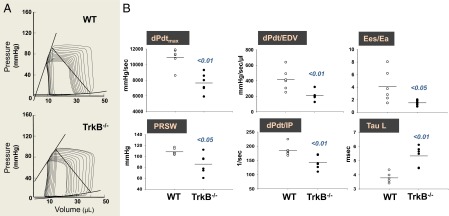
Constitutive BDNF/TrkB signaling is required for normal cardiac contraction and relaxation. (A) Representative pressure-volume loops obtained in WT littermate mice and in cardiac-specific TrkB−/− mice. (B) Cardiac contractility and relaxation are impaired in TrkB−/− mice, as indexed by dp/dtmax, dpdt/EDV, dpdt/ip, prerecruitable stroke work, Ees/Ea ratio, and Tau logistic, respectively.
Table 1.
Hemodynamic values in WT and TrkB−/− mice via pressure–volume relationships
| Basal hemodynamics | WT | TrkB−/− | P value | ||
| (n = 6) | (n = 5) | (n = 6) | (n = 5) | ||
| HR, min−1 | 611.7 ± 13.5 | 574 ± 25.1 | 0.22 | ||
| SBP, mm Hg | 100.1 ± 3 | 96 ± 5.2 | 0.53 | ||
| SV, μL | 23.9 ± 1.7 | 21.6 ± 1.9 | 0.39 | ||
| CO, mL/min | 14.6 ± 1 | 12.4 ± 1.1 | 0.17 | ||
| EF, % | 56.0 ± 2.3 | 56.7 ± 2.0 | 0.59 | ||
| dPdt max, mm Hg/s | 10,944.7 ± 496.3 | 7,647.2 ± 498.9 | 0.0009 | ||
| Ees, mm Hg/mL | 15.2 ± 3.2 | 6.6 ± 1.0 | 0.041 | ||
| dPdt/EDV | 417.2 ± 56.4 | 209.2 ± 25 | 0.0073 | ||
| dPdt ip, sec−1 | 185.3 ± 8.3 | 142.4 ± 9.8 | 0.0075 | ||
| Prerecruitable stroke work, mm Hg | 108.3 ± 2.4 | 85.8 ± 7.6 | 0.018 | ||
| Tau log, msec | 4.0 ± 0.2 | 5.3 ± 0.3 | 0.0019 | ||
| Ea, mm Hg/mL | 4.0 ± 0.4 | 4.3 ± 0.4 | 0.56 | ||
| Systemic vascular resistance, mm Hg⋅min⋅mL−1 | 6.5 ± 0.7 | 7.5 ± 0.6 | 0.26 | ||
| Ees/Ea | 4.1 ± 1.1 | 1.5 ± 0.6 | 0.033 | ||
| Isoproterenol, 40 ng/kg/min | |||||
| dPdt max, mm Hg/s | 13,973.7 ± 558.8 | 10,583.7 ± 655.0 | 0.005 | ||
| Percentage increase over baseline | 22.3 ± 3.0 | 38.8 ± 11.1 | 0.19 | ||
CO, cardiac output; dP/dt/EDV, dPdt max against end-diastolic volume relationship; dPdt ip, dp/dt max divided by the instantaneous developed isovolumetric pressure; dPdtmax, maximum time derivative of left ventricular pressure; Ea, arterial elastance; Ees, left ventricle chamber elastance; EF, ejection fraction %; HR, heart rate; SBP, systolic blood pressure; SV, stroke volume; Tau, relaxation time constant.
Fig. 3.
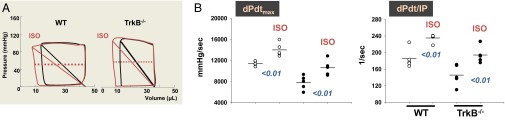
β-adrenergic response is intact in TrkB−/− mice. (A) Representative pressure-volume loops obtained before and after the infusion of the β1-β2 agonist isoproterenol (40 ng/kg/min). (B) Isoproterenol increases in vivo contractility (dP/dtmax and dPdt/IP) with the same magnitude in WT and TrkB−/− mice, despite the lower basal contractile values found in the TrkB−/− mice (n = 5 each group).
CaMKII Mediates BDNF-Induced Enhancement of Myocyte Function.
In the brain, BDNF/TrkB is mainly coupled to CaMKII (13). We found that BDNF also increased the activated and phosphorylated state of CaMKII in isolated murine ventricular myocytes. This change was paralleled by augmented phosphorylation of CaMKII-dependent sites (14, 15) on the ryanodine receptor 2 (serine 2814), and phospholamban (PLN; threonine 17) (Fig. 4A, n = 4). TrkB−/− mice displayed decreased levels of P-CaMKII and the reduction of P-CaMKII/T-CaMKII ratio (Fig. 4A). Moreover, pharmacologic inhibition of CaMKII activity by KN93 completely abolished BDNF Ca2+ transients and contractility (Fig. 4B, n = 10). Thus, CaMKII is the main mediator of BDNF/TrkB-evoked cardiac stimulatory actions that operates in parallel to β-adrenergic signaling to regulate myocardial contraction and relaxation.
Fig. 4.
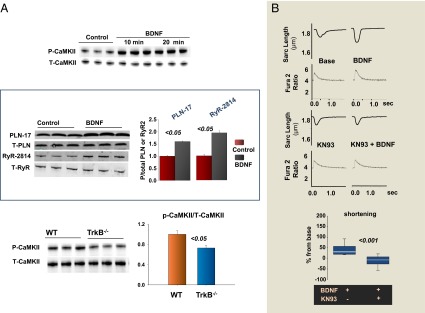
CaMKII is the major mediator of BDNF influence on myocardial mechanics. (A) BDNF induces CaMKII phosphorylation in adult murine cardiac myocytes (n = 3) and increases the phosphorylation of CaMKII-dependent sites on RyR (serine 2814) and PLN (threonine 17), respectively (n = 4); TrkB−/− mice display significantly reduced levels of constitutive phosphorylation of CaMKII, as indexed by the P-CaMKII/T-CaMKII ratio (n = 4). (B) Representative traces of BDNF’s effect on isolated mouse cardiomyocytes in the absence and the presence of the CaMKII inhibitor KN93: Pretreating cells with KN93 prevents BDNF enhancement of myocyte contraction (n = 10).
BDNF-Induced Enhancement of Myocyte Function Is Lost in Failing Hearts.
We finally tested whether BDNF-evoked cardiac enhancement is preserved in failing myocytes, using cardiomyocytes isolated from Gαq overexpressing (Gαq OE) mice that display progressive cardiac dilation and reduced ejection fraction (16). Enhanced Gαq signaling is a common pathway mediating maladaptive cardiac hypertrophy and adverse remodeling. In response to pathologic stress, Gαq signaling is activated by α-adrenergic agonist, angiotensin II, or endothelin, and so on, promoting cardiac growth, apoptosis, and fibrosis, and ultimately resulting in cardiac dysfunction (16). Therefore, Gαq OE mice mimic this pathologic response and are often used as a heart failure model. In the present study, GαqOE myocytes did not respond to BDNF (20 nM) (Fig. 5A, n = 19 for control and n = 24 for Gαq myocytes); therefore, we sought to determine the mechanisms underlying this insensitivity. First, we examined the expression of TrkB in GαqOE hearts. Intriguingly, the expression of full-length TrkB was unchanged, but the truncated form of TrkB (TrkB-T1), which lacks tyrosine kinase activity (17), was markedly increased (Fig. 5B, n = 4). TrkB-T1 is one of the TrkB splicing variants, and it often acts as a dominant negative form that suppresses BDNF/TrkB signaling; increased TrkB-T1 levels have been implicated in neurologic disorders (18, 19). Next, we further examined the CaMKII signaling cascade in GαqOE hearts. We found that these hearts display elevated P-CaMKII and total T-CaMKII at baseline (Fig. 5C, n = 4), consistent with previous reports (20). The expression of total PLN was unchanged; however, a decreased level of SERCA2a was evident (Fig. 5C). Moreover, the phosphorylation at Threonine 17 of PLN was markedly reduced (Fig. 5C), despite chronic activation of CaMKII. Thus, chronically activated CaMKII and/or altered CaMKII downstream targets can also account for loss in BDNF/TrkB stimulatory action in Gαq OE myocytes. These findings were recapitulated in mice with pressure overload-induced heart failure via transverse aortic constriction. Transverse aortic constriction hearts displayed higher expression of the TrkB-T1 and chronically activated CaMKII signaling pathway (Fig. S3), demonstrating that altered TrkB receptor and chronically activated CaMKII signaling pathway are not peculiar to a transgenic mouse model, but also pertain to other heart failure models generated via chronic hemodynamic stress.
Fig. 5.
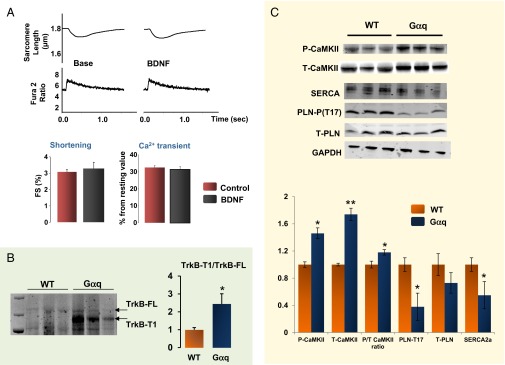
BDNF-evoked enhancement of cardiac contractility is lost in failing cardiomyocytes that display increased truncated TrkB and altered CaMKII signaling/targets. (A) Myocytes isolated from Gαq mice are insensitive to BDNF (20 nM) (control cells = 19; Galphaq myocytes = 24): raw traces and cumulative data for sarcomere shortening and whole-cell Ca2+ transients. (B) The expression of full-length TrkB is unchanged in Gαq OE hearts; however, the truncated TrkB (Trk-T1) is markedly increased. (C) In Gαq OE mice hearts, CaMKII phosphorylation is constitutively up-regulated, as shown by the increased P-CaMKII/T-CaMKII ratio. The expression of SERCA2a is markedly decreased in Gαq hearts; this change is coupled to unchanged expression of total PLN, and to reduced levels of PLN phosphorylation levels at the T17 residue (*P < 0.05, **P < 0.01).
Discussion
During its lifetime, the heart is under the constant influence of the autonomic nervous system. Sympathetic efferent fiber activation is designed to release cardioactive neurotransmitters, which is crucial to adjusting cardiac performance to increased workload. This on-demand mechanism is essential during exercise, and at least initially, it maintains adequate cardiac output in the presence of chronic hemodynamic stress such as hypertension. However, autonomic fibers also contain and release neurotrophins, but our understanding of their influence on myocardial function has been mostly confined to their ability to exert trophic actions on autonomic efferents and vessels serving the heart.
Here, we report that endogenous BDNF, via stimulation of sarcolemmal TrkB receptors and CaMKII-associated signaling, establishes a tonic control on basal cardiac contractility and relaxation. Thus, our study reveals a previously unidentified role for BDNF/TrkB signaling as a direct modulator of myocardial mechanical function. Several physiologic and pathophysiologic implications arise from these findings. For example, BDNF/TrkB signaling could contribute to enhanced cardiac performance during exercise. In fact, exercise augments BDNF levels in the brain, the skeletal muscle, and plasma, enhancing function and improving energy metabolism in these organs (21, 22). Our present findings may also provide a potential explanation for recent clinical reports showing that in heart failure patients, a correlation exists between low circulating levels of BDNF and worsening of symptoms (23, 24). Although it is known that growth factors such as insulin-like growth factor 1 and vascular endothelial growth factor have pharmacologic actions to modulate cardiac contractility (25, 26), no studies have established their relevance in vivo or their signaling in this respect. Here we report that a tyrosine kinase-associated receptor such as TrkB, and its endogenous ligand BDNF, directly control the cardiac excitation–contraction coupling process, independently and in parallel to G protein-coupled receptor signaling. Thus, disruption of tyrosine kinase-based signaling and consequent perturbations in cardiac mechanical work can largely contribute to loss in ventricular systolic function that accompanies the use of tyrosine kinase inhibitors during anticancer therapies (27).
In the present study, we also found the BDNF-induced enhancement of contraction was abolished in failing myocytes. The loss of efficacy is probably a result of the increased truncated form of TrkB, which is the dominant negative form of BDNF/TrkB signaling. The underlying mechanism of elevated TrkB-T1 remains to be elucidated. In contrast, CaMKII is chronically activated, despite the diminished TrkB activation. Indeed, CaMKII is also a key downstream signal pathway of both α- and β-adrenergic receptors. In heart failure, this kinase is constitutively activated by the neurohormone overdrive that characterizes this syndrome and that contributes to the development of adverse cardiac hypertrophy and remodeling (28). Thus, in addition to the decreased TrkB activity, the constitutive activation of CaMKII presented in failing hearts can be another major reason accounting for the loss in efficacy of BDNF/TrkB signaling in failing hearts.
In conclusion, by binding to TrkB receptors and triggering a CaMKII-dependent signaling cascade, endogenous BDNF acts in parallel with the β-adrenergic system to maintain or enhance myocardial Ca2+ cycling, and thus cardiac contraction and relaxation. Our study suggests that loss or decreases in circulating BDNF levels and alterations in TrkB receptor structure/function, along with abnormal CaMKII signaling, could further contribute to cardiac dysfunction under acute and chronic diseased conditions.
Materials and Methods
Detailed methods are available in the SI Materials and Methods.
Statistics.
Results are expressed as means ± SEM. Significance was estimated by one-way repeated measures ANOVA, Student's t test for paired observations, or Mann–Whitney U test, as appropriate; P ≤ 0.05 was considered significant.
Studies Approval.
All animal protocols were approved by the Animal Care and Use Committee of the Johns Hopkins University in Baltimore, MD, and the Loyola University in Chicago, IL, following established NIH guidelines.
Supplementary Material
Acknowledgments
We are grateful to Dr. David D. Ginty for providing us with TrkBF616A and TrkB conditional knockout mice. We also thank Dr. Brian O’Rourke for valuable scientific discussion. This work was supported by American Heart Association Grant-in-Aid GRNT17070027, Johns Hopkins University Magic That Matters Funds, and NIH T32 Training Grant T32HL-0227.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1417949112/-/DCSupplemental.
References
- 1.Lu B, Nagappan G, Guan X, Nathan PJ, Wren P. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat Rev Neurosci. 2013;14(6):401–416. doi: 10.1038/nrn3505. [DOI] [PubMed] [Google Scholar]
- 2.Berton O, Nestler EJ. New approaches to antidepressant drug discovery: Beyond monoamines. Nat Rev Neurosci. 2006;7(2):137–151. doi: 10.1038/nrn1846. [DOI] [PubMed] [Google Scholar]
- 3.Abcejo AJ, et al. Brain-derived neurotrophic factor enhances calcium regulatory mechanisms in human airway smooth muscle. PLoS ONE. 2012;7(8):e44343. doi: 10.1371/journal.pone.0044343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marosi K, Mattson MP. BDNF mediates adaptive brain and body responses to energetic challenges. Trends Endocrinol Metab. 2014;25(2):89–98. doi: 10.1016/j.tem.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donovan MJ, et al. Brain derived neurotrophic factor is an endothelial cell survival factor required for intramyocardial vessel stabilization. Development. 2000;127(21):4531–4540. doi: 10.1242/dev.127.21.4531. [DOI] [PubMed] [Google Scholar]
- 6.Yang B, Slonimsky JD, Birren SJ. A rapid switch in sympathetic neurotransmitter release properties mediated by the p75 receptor. Nat Neurosci. 2002;5(6):539–545. doi: 10.1038/nn0602-853. [DOI] [PubMed] [Google Scholar]
- 7.Kermani P, Hempstead B. Brain-derived neurotrophic factor: A newly described mediator of angiogenesis. Trends Cardiovasc Med. 2007;17(4):140–143. doi: 10.1016/j.tcm.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okada S, et al. Brain-derived neurotrophic factor protects against cardiac dysfunction after myocardial infarction via a central nervous system-mediated pathway. Arterioscler Thromb Vasc Biol. 2012;32(8):1902–1909. doi: 10.1161/ATVBAHA.112.248930. [DOI] [PubMed] [Google Scholar]
- 9.Caporali A, Emanueli C. Cardiovascular actions of neurotrophins. Physiol Rev. 2009;89(1):279–308. doi: 10.1152/physrev.00007.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen X, et al. A chemical-genetic approach to studying neurotrophin signaling. Neuron. 2005;46(1):13–21. doi: 10.1016/j.neuron.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 11.Bishop AC, et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407(6802):395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- 12.Liu Y, et al. Sexually dimorphic BDNF signaling directs sensory innervation of the mammary gland. Science. 2012;338(6112):1357–1360. doi: 10.1126/science.1228258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chao MV. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4(4):299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- 14.Huke S, Bers DM. Ryanodine receptor phosphorylation at Serine 2030, 2808 and 2814 in rat cardiomyocytes. Biochem Biophys Res Commun. 2008;376(1):80–85. doi: 10.1016/j.bbrc.2008.08.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mattiazzi A, Kranias EG. CaMKII regulation of phospholamban and SR Ca2+ load. Heart Rhythm. 2011;8(5):784–787. doi: 10.1016/j.hrthm.2010.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adams JW, et al. Enhanced Galphaq signaling: A common pathway mediates cardiac hypertrophy and apoptotic heart failure. Proc Natl Acad Sci USA. 1998;95(17):10140–10145. doi: 10.1073/pnas.95.17.10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eide FF, et al. Naturally occurring truncated trkB receptors have dominant inhibitory effects on brain-derived neurotrophic factor signaling. J Neurosci. 1996;16(10):3123–3129. doi: 10.1523/JNEUROSCI.16-10-03123.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vidaurre OG, et al. Imbalance of neurotrophin receptor isoforms TrkB-FL/TrkB-T1 induces neuronal death in excitotoxicity. Cell Death Dis. 2012;3:e256. doi: 10.1038/cddis.2011.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong J, Rothmond DA, Webster MJ, Weickert CS. Increases in two truncated TrkB isoforms in the prefrontal cortex of people with schizophrenia. Schizophr Bull. 2013;39(1):130–140. doi: 10.1093/schbul/sbr070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mishra S, et al. Cardiac hypertrophy and heart failure development through Gq and CaM kinase II signaling. J Cardiovasc Pharmacol. 2010;56(6):598–603. doi: 10.1097/FJC.0b013e3181e1d263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pedersen BK, Febbraio MA. Muscles, exercise and obesity: Skeletal muscle as a secretory organ. Nat Rev Endocrinol. 2012;8(8):457–465. doi: 10.1038/nrendo.2012.49. [DOI] [PubMed] [Google Scholar]
- 22.Pedersen BK, et al. Role of exercise-induced brain-derived neurotrophic factor production in the regulation of energy homeostasis in mammals. Exp Physiol. 2009;94(12):1153–1160. doi: 10.1113/expphysiol.2009.048561. [DOI] [PubMed] [Google Scholar]
- 23.Takashio S, et al. Abstract 12400: Significant Association between Decreased Plasma Levels of Brain-derived Neurotrophic Factor in Heart Failure. Circulation. 2013;128(Supplement 22):A12400. [Google Scholar]
- 24.Fukushima A, et al. Decreased serum brain-derived neurotrophic factor levels are correlated with exercise intolerance in patients with heart failure. Int J Cardiol. 2013;168(5):e142–e144. doi: 10.1016/j.ijcard.2013.08.073. [DOI] [PubMed] [Google Scholar]
- 25.Cittadini A, et al. Insulin-like growth factor-1 but not growth hormone augments mammalian myocardial contractility by sensitizing the myofilament to Ca2+ through a wortmannin-sensitive pathway: Studies in rat and ferret isolated muscles. Circ Res. 1998;83(1):50–59. doi: 10.1161/01.res.83.1.50. [DOI] [PubMed] [Google Scholar]
- 26.Rottbauer W, et al. VEGF-PLCgamma1 pathway controls cardiac contractility in the embryonic heart. Genes Dev. 2005;19(13):1624–1634. doi: 10.1101/gad.1319405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Force T, Kolaja KL. Cardiotoxicity of kinase inhibitors: The prediction and translation of preclinical models to clinical outcomes. Nat Rev Drug Discov. 2011;10(2):111–126. doi: 10.1038/nrd3252. [DOI] [PubMed] [Google Scholar]
- 28.Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol. 2011;51(4):468–473. doi: 10.1016/j.yjmcc.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.