

Significance
This paper describes the structure of Staphylococcus aureus TarM, an enzyme responsible for the glycosylation of wall teichoic acid that is important in pathological processes such as host immunity, phage binding, and antibiotic resistance in strains such as Methicillin-resistant S. aureus. The TarM structure is presented in an unusual ternary-like complex that features a polymeric acceptor substrate analogue and a trapped product of enzyme action, lending novel structural and mechanistic insight into the glycosylation of glycopolymers. More generally, the positioning of this product in the active site as well as the distorted conformation of its pyranose ring provide direct structural evidence for an internal substitution-like catalytic mechanism for retaining GT-B class enzymes.
Keywords: glycosyltransferase, wall teichoic acid, cell wall, crystal structure, bacterial pathogenicity
Abstract
Unique to Gram-positive bacteria, wall teichoic acids are anionic glycopolymers cross-stitched to a thick layer of peptidoglycan. The polyol phosphate subunits of these glycopolymers are decorated with GlcNAc sugars that are involved in phage binding, genetic exchange, host antibody response, resistance, and virulence. The search for the enzymes responsible for GlcNAcylation in Staphylococcus aureus has recently identified TarM and TarS with respective α- and β-(1–4) glycosyltransferase activities. The stereochemistry of the GlcNAc attachment is important in balancing biological processes, such that the interplay of TarM and TarS is likely important for bacterial pathogenicity and survival. Here we present the crystal structure of TarM in an unusual ternary-like complex consisting of a polymeric acceptor substrate analog, UDP from a hydrolyzed donor, and an α-glyceryl-GlcNAc product formed in situ. These structures support an internal nucleophilic substitution-like mechanism, lend new mechanistic insight into the glycosylation of glycopolymers, and reveal a trimerization domain with a likely role in acceptor substrate scaffolding.
A hallmark difference between Gram-negative and Gram-positive bacteria relates to the composition of their cell wall. Gram-negative bacteria possess an inner and an outer membrane, the latter of which is lined by a thin layer of peptidoglycan. Gram-positive bacteria, in contrast, lack this outer membrane, instead possessing a thick layer of peptidoglycan complemented by anionic glycopolymers known as teichoic acids (TAs) that can comprise an astonishing 60% of the dry weight of the cell wall (1). Charged with this thick cross-stitched protective matrix, Gram-positive bacteria are able to inhabit severe environments such as the gastrointestinal tract, similar to their Gram-negative counterparts.
TAs are constitutively manufactured and covalently incorporated into the cell walls of Gram-positive bacteria by attachment to peptidoglycan in the case of wall TAs (WTAs) or to the plasma membrane as with lipoteichoic acids (LTAs). TA composition varies widely depending on bacterial strain and subtype (reviewed in ref. 2). In most Staphylococcus aureus strains, WTA consists of polyribitol phosphate (polyRboP) chains of 40–60 repeats, attached via a disaccharide linkage unit to the C6 hydroxyl group of occasional N-acetylmuramic acid residues of peptidoglycan (3). LTA composition tends to be less diverse than WTA, and in S. aureus most often contains polyglycerol phosphate attached to the plasma membrane via a glycolipid anchor (reviewed in ref. 4).
Although our current understanding of the exact functions of TAs is incomplete, it is clear that bacteria invest an impressive amount of energy and resources in their construction and maintenance. Indeed, efforts to study these extraordinary polymers have yielded some important findings. For instance, it is known that, in the absence of WTA, S. aureus is viable under laboratory conditions, but that its ability to colonize and infect is greatly compromised (5, 6). Deletion of LTA leads to temperature-sensitive strains only viable at temperatures lower than 30 °C, and, interestingly, strains with deletions of both WTA and LTA are nonviable, suggesting some degree of redundancy in their respective roles (7). TAs have furthermore been implicated in resistance to antimicrobial molecules (8–11), resistance to lysozyme (11), coping with environmental stresses (7, 12), mediating interactions with receptors and biomaterials (6, 13), induction of inflammation (14–16), phage binding (17, 18), and biofilm formation (19). Faced with so many functions in an uncertain environment, TAs must remain highly adaptive, a large part of which is achieved by the d-alanylation and glycosylation of polyol hydroxyl groups. These modifications have a significant effect on the physical and interactive properties of the cell wall.
In S. aureus, C4 hydroxyls of polyRboP are heavily substituted with GlcNAc. The configuration of the glycosidic linkage varies, with certain strains of S. aureus containing WTA with almost exclusively α- or β-O-linked GlcNAc, and others displaying a mixture of anomers (20, 21). An important role of WTA sugar modification in S. aureus is that of a receptor for bacteriophage binding. This role was discovered early on, when phage-resistant mutants were observed to lack GlcNAc in their WTA (18). WTA GlcNAc residues have also long been recognized as important antigens in the host-antibody response (22–24).
Recently, Xia et al. identified a phage-resistant S. aureus RN4220 transposon mutant, whose disrupted gene was designated as TarM, a soluble cytoplasmic glycosyltransferase (GT) enzyme (25). According to the Carbohydrate-Active Enzymes database, which classifies GTs into 94 families based on sequence identity, TarM belongs to the GT4 family that includes various anomeric stereochemistry-retaining enzymes (26). On verification, recombinant TarM was shown to α-O-glycosylate WTA in vitro in a UDP-GlcNAc–dependent manner. Soon after, a second S. aureus inverting GT responsible for the β-O-GlcNAcylation of WTA was identified and designated as TarS (27). The differences in the stereochemistries of these GlcNAc glycosidic linkages appear to directly influence the biology and pathogenicity of Gram-positive bacteria, often on a strain-specific level (24, 27, 28).
Here, to our knowledge, we present the first structure of TarM in a ternary-like complex with a polyRboP acceptor substrate analog, fondaparinux, as well as a cleaved donor UDP-GlcNAc. In addition, the surprising TarM-catalyzed formation of α-glyceryl-GlcNAc and its positioning in the active site are consistent with an internal nucleophilic substitution (SNi)-like reaction mechanism.
Results
Overall Structure of TarM.
The structures presented here are of the S. aureus TarM full-length protein. The structures of TarM in the presence and absence of the nucleotide, UDP, were solved initially to 3.2 Å and 3.5 Å resolution, respectively, both forming prominent trimers. Although TarM was cocrystallized in the presence of the donor sugar UDP-GlcNAc, only the cleaved UDP product was present in the active site. Given the limited resolution of the data resulting from these native crystals, the homotrimeric interface was disrupted by mutagenesis and well diffracting crystals (2.1–2.4 Å) were obtained that were found to contain UDP and UDP-GlcNAc bound monomers in the asymmetric unit. A complex with fondaparinux, a pentameric, polyanionic synthetic heparin used clinically as an anticoagulant (trade name Arixtra; GlaxoSmithKline), was also obtained. This compound was initially tested as a ligand on the basis of the high affinity of TarM for heparin Sepharose that was exploited during purification. Binding of fondaparinux was confirmed by affinity measurements described later, and its defined length and similar physicochemical properties to those of polyRboP (both are polysaccharides with negatively charged functional groups) made it a suitable acceptor analog candidate for cocrystallization. In addition to fondaparinux, the aforementioned structure also contained product UDP as well as an α-glyceryl-GlcNAc glycoside product. Phases for the native trimeric UDP bound structure were solved by using selenomethionine single-wavelength anomalous dispersion (SAD) methods. All other structures were subsequently solved with molecular replacement by using the native structure as a search model. Refinement statistics of the final structural models are presented in Table 1.
Table 1.
Data collection and refinement statistics
| TarM SeMet | TarM apo | TarM G117R | TarM G117R fondaparinux | |
| Data collection* | ||||
| Space group | P63 | P63 | P1 | P1 |
| Cell dimensions | ||||
| a, b, c, Å | 162.3, 162.3, 228.02 | 208.60, 208.60, 120.75 | 44.07, 90.94, 94.44 | 43.47, 92.14, 96.11 |
| α, β, γ, ° | 90, 90, 120 | 90, 90, 120 | 109.27, 99.03, 101.53 | 65.89, 83.66, 84.12 |
| Wavelength, Å | 0.978 | 0.978 | 0.978 | 1.033 |
| Resolution, Å | 66.11–3.14 (3.26–3.14) | 72.33–3.4 (3.49–3.4) | 46.04–2.4 (2.48–2.4) | 87.48–2.15 (2.2–2.15) |
| Rsym or Rmerge | 0.064 (0.271) | 0.207 (0.537) | 0.031 (0.448) | 0.078 (0.505) |
| CC1/2 | 0.993 (0.811) | 0.992 (0.908) | 0.999 (0.764) | 0.997 (0.737) |
| I/σI | 7.4 (1.9) | 8.8 (3.9) | 16.3 (2.0) | 9.1 (1.6) |
| Completeness, % | 99.97 (100) | 100 (100) | 95.3 (96.5) | 97.9 (96.7) |
| Redundancy | 6.7 (6.7) | 11 (10.2) | 2.0 (2.0) | 3.9 (2.8) |
| Refinement | ||||
| Resolution, Å | 3.2 | 3.4 | 2.4 | 2.15 |
| No. reflections | 55,989 (5,588) | 41,624 (3,022) | 49,087 (4,598) | 71,890 (4,584) |
| Rwork/Rfree | 0.165/0.191 | 0.173/0.187 | 0.233/0.274 | 0.179/0.200 |
| No. atoms | ||||
| Protein | 16,132 | 8,066 | 8,058 | 8,080 |
| Ligand/ion | 75 | 80 | 64 | 272 |
| Water | 0 | 0 | 59 | 365 |
| B-factors, Å2 | ||||
| Protein | 120.8 | 98.0 | 90.6 | 58.4 |
| Ligand/ion | 123.1 | 122.0 | 69.4 | 53.9 |
| Water | NA | NA | 50.2 | 53.6 |
| rmsds | ||||
| Bond lengths, Å | 0.010 | 0.009 | 0.010 | 0.010 |
| Bond angles, ° | 1.19 | 1.12 | 1.19 | 1.11 |
NA, not applicable; SeMet, selenomethionine.
Values in parentheses are for highest-resolution shell.
TarM was observed to possess a canonical GT-B class fold (one of two structural folds identified for nucleotide sugar-dependent GTs) characterized by two β/α/β Rossmann-like domains that form distinct donor and acceptor substrate binding sites. These two flexibly linked domains face each other and form the active site within the resulting electropositive cleft, as expected for a GT4 enzyme (29). TarM also harbors a sequence-unique N-terminal domain consisting of nine antiparallel β-strands that participate in the trimerization interface (Fig. 1A). Electrostatic surface analysis furthermore reveals several prominent electropositive grooves that run along the surface of the trimer, as depicted in Fig. 1B.
Fig. 1.

Structural features of the S. aureus TarM trimer. (A) Ribbon representation of trimeric TarM in complex with UDP (displayed in stick form and colored according to heteroatom type). The separate protomers are indicated by color, and the trimerization domain and substrate donor/acceptor site Rossmann-like domains are demarcated by a dotted circle and dotted squares, respectively. The G117 residue whose mutation to arginine resulted in the monomeric protein is also marked, showing its remote position from the GT active site. (B) Electrostatic surface representation of the TarM trimer. The molecule on the left is displayed in the same orientation as A, and the molecule on the right upon 180° rotation. The trimer possesses prominent positively charged grooves along its surface, which are localized in the central trimerization site of the oligomer in the left view, and along the entire surface on the right.
The physiological relevance of the observed trimeric structure was supported by analysis of TarM via size exclusion chromatography–multiangle light scattering (SEC-MALS), with the TarM elution profile corresponding to a molecular weight of 168.7 kDa (compared with its theoretical monomer mass of 57.3 kDa), suggesting that it exists as a trimer in solution (Fig. S1). To validate the observed oligomerization, the mutant G117R, predicted to disrupt the trimerization interface (Fig. 1A), was constructed. Analysis of TarM G117R with SEC-MALS revealed an elution profile corresponding to a molecular weight of 56.8 kDa, similar to that of the theoretical monomer (Fig. S1).
The structure of the UDP-bound monomeric protein was very similar to that observed in the UDP-bound trimeric form. Brief soaking of the G117R crystals with UDP-GlcNAc resulted in a UDP and a UDP-GlcNAc bound molecule in the asymmetric unit (total two monomers in asymmetric unit), with the resulting features of each compared in Fig. S2B (ligand simulated annealing mFo-DFc OMIT maps shown in Fig. S2A). Although the relative folds of the two Rossmann-like domains are similar, the degree of opening of the active site cleft between the donor and acceptor binding domains is reduced by 12.7° in the presence of UDP-GlcNAc (from an “open” to a “closed” state), with the hinge point for rotation formed by residues 305–308 and 478–480 [analysis by DynDom (30); Movie S1]. This motion is also reflected by an overall main chain rmsd of 2.1 Å relative to the main chain rmsds of the fixed N-terminal acceptor binding/trimerization domain (0.6 Å; 3–305, 480–490) and the moving C-terminal donor binding domain (0.3 Å; 306–479). Adjacent to the hinge point, the C-terminal α-helix comprised of residues 480–493 is observed to extend from the donor binding domain to form interactions with the acceptor binding domain, effectively linking the two and allowing for rotation. This agrees with the reported observation that deletion of the seven terminal amino acids forming a large part of this α-helix results in TarM inactivation (25), presumably by disrupting proper hinge formation. Cocrystallization of the G117R mutant with UDP-GlcNAc and fondaparinux resulted in electron density defining a fully ordered fondaparinux molecule in the acceptor binding domain, and an overall structure resembling the closed state of the UDP-GlcNAc bound structure with an overall main chain rmsd of 0.3 Å (Fig. 2A). On inspection, the donor site was found to contain the cleavage products of UDP-GlcNAc, where the cleaved sugar was trapped within the active site, presumably as a result of stabilization of the enzyme’s closed state by fondaparinux. Interestingly, additional density was observed on the α-face of GlcNAc continuous with the C1 position of the cleaved sugar and in close proximity to the bound UDP. Examination of the crystal conditions identified glycerol as the most likely fit for the observed density. The bond distance between the putative primary glycerol hydroxyl and the GlcNAc anomeric carbon is ∼1.4 Å, suggesting covalent bond formation. The glycosidic glyceryl-GlcNAc linkage was confirmed by MS (Fig. S3) and NMR spectroscopy (Fig. S4), revealing an α-linkage between the C1 position of GlcNAc and a primary hydroxyl of glycerol (Fig. 2B).
Fig. 2.

Structural features of the TarM ternary-like complex. (A) Clipped-plane electrostatic surface representation of monomer TarM in complex with UDP (orange), α-glyceryl-GlcNAc (purple-blue), and fondaparinux (yellow). Substrates are shown in stick form and colored according to heteroatom type. The electrostatic representation shows that substrates occupy predominantly electropositive pockets and occupy favorable positions for GT reaction. (B) Interaction of active site residues with fondaparinux, UDP, and α-glyceryl-GlcNAc. Ligand mFo-DFc OMIT maps (contoured at 3 σ) are provided (Inset). Polar interactions between atoms are displayed by dotted lines, and relevant distances for a proposed SNi reaction mechanism given in Å. A blue dotted line along the GlcNAc molecule marks the β- and α-face as indicated.
WTA Purification and Its GlcNAcylation by TarM.
To analyze the GT activity of TarM, WTA was extracted from the cell wall of S. aureus strain RN4220. Given that WTA in this strain is largely GlcNAcylated, the extracted polymer was digested with α- and β-N-acetylglucosaminidases (NAGLUs) to release the attached GlcNAc and free acceptor sites. The digested WTA was subsequently purified on a DEAE weak anion exchange column, where UV was monitored at wavelengths of 205 nm for detection of WTA and at 280 nm for detection and removal of NAGLUs. As enzymatic assays rely on UDP detection upon TarM-catalyzed cleavage of UDP-GlcNAc (as described later), a negative control in the absence of TarM (i.e., WTA and GlcNAc only) was performed, demonstrating the absence of interfering background hydrolysis from possible trace amounts of NAGLUs. The digested, purified WTA was lyophilized and dissolved in distilled H2O to give a concentrated stock solution, and the molar concentration of constituent single RboP units in the WTA chains was determined according to the concentration of phosphate by inductively coupled plasma (ICP) MS. The integrity of the WTA chains was assessed with SEC-MALS, revealing a size distribution of 9–11 kDa, which corresponds to polyRboP polymers of ∼40–50 repeats (Fig. S5).
The activity of TarM was tested by two complementary techniques: a direct chromatography method (i.e., HPLC) and an indirect UDP fluorescence detection method. The HPLC method was used to determine the presence of activity as indicated by the release of UDP. Here, the reaction solution was filtered to remove protein and injected on a TSKgel DEAE-5PW weak anion exchange column to separate UDP-GlcNAc from the cleaved UDP, whose resolved peaks were monitored at 254 nm. The second method, based on the fluorescent monitoring of UDP release with the ADP Quest Assay kit (Profoldin), allowed for the measurement of TarM kinetic parameters (Table 2). These methods confirmed activity for the WT TarM trimer as well as the monomeric G117R mutant. TarM in both cases was capable of cleaving UDP-GlcNAc in the absence and presence of the WTA acceptor (hydrolysis being a common side reaction), which would account for our UDP bound structure upon cocrystallization with UDP-GlcNAc. In both cases, however, the GT activity in the presence of WTA was ∼20 fold greater than the hydrolysis activity in its absence. We next analyzed intrinsic enzyme processivity (or theoretical potential for processive ability Pintr) by first measuring association and dissociation rate constants (kon and koff) for the WT and G117R mutant by using biolayer inferometry. Processivity was then calculated by the approximation Pintr ∼ kcat/koff (Table 2 and Fig. S6) (31, 32). The values obtained demonstrate that the WT and the G117R monomer display similar levels of intrinsic processivity. The results suggest that the trimeric form of TarM is essential for neither donor or acceptor substrate binding, nor overall activity/processivity. The similarity in the catalytic activities of the monomer G117R mutant and the WT furthermore justified our use of the higher resolution monomeric structure for subsequent analysis of the TarM catalytic mechanism. The activity of the G117R mutant was also tested in the presence of fondaparinux, which surprisingly enhanced the hydrolysis of UDP-GlcNAc. Although fondaparinux is not glycosylated (indicated by the absence of glycosylation in the crystal structure and consistent with an unfavorable distance relative to the GlcNAc reaction center), the substrate analog does appear to stabilize the closed conformation of the enzyme, thereby orienting active site residues and substrates (in this case UDP-GlcNAc and water) for catalysis and accounting for the increased background hydrolysis. Measurement of initial rates of UDP release at varying concentrations of fondaparinux yielded an apparent Km value (0.48 ± 0.05 μM) for fondaparinux that is ∼1,000 times lower than that obtained for WTA (Fig. S7B). This is consistent with fondaparinux, as observed in our bound G117R structure, competing with WTA for the TarM acceptor binding site. The higher affinity of fondaparinux is also suggested by differential static light scattering-based thermostability measurements (Fig. S7A) (33).
Table 2.
Kinetic parameters of WT and G117R trimerization mutant TarM
| Construct | Hydrolysis reaction* | GT reaction† | |||
| Km, μM | kcat, min−1 | Km, μM | kcat, min−1 | Pintr | |
| WT | 65 ± 5 | 6.0 ± 0.6 | 65 ± 10 (UDP-GlcNAc) | 126 ± 10 | 11,700 ± 1,200 |
| 390 ± 30 (WTA) | |||||
| G117R | 55 ± 5 | 7.0 ± 0.4 | 30 ± 5 (UDP-GlcNAc) | 146 ± 10 | 12,800 ± 1,100 |
| 540 ± 50 (WTA) | |||||
Catalytic mutants with activities below the reliable detection limit of the assay were studied by the HPLC-based UDP-detection method as presented in Fig. 3B.
Parameters were determined by titration with increasing concentrations of UDP-GlcNAc.
Values of kcat and Km were determined in the presence of and absence of 500 μM WTA with increasing concentrations of UDP-GlcNAc. Km for WTA was determined in the presence of 1 mM UDP-GlcNAc with increasing concentrations of WTA. The concentration of WTA reflected the measured phosphate concentration (by ICP-MS) pertaining to single RboP units in the WTA polymer chain.
Investigation of the Catalytic Mechanism of TarM.
TarM is a predicted member of the Carbohydrate-Active Enzymes database GT4 enzyme family (26), consisting of a GT-B catalytic domain that is typical of that family and an additional N-terminal antiparallel β-sheet domain that facilitates trimerization. According to earlier studies (25), TarM is a retaining GT that catalyzes the α-GlcNAcylation of polyRboP. GT-B class retaining enzymes often use a metal cation-independent catalytic mechanism, instead using active site residues to stabilize charges on the leaving group UDP of the donor substrate. Accordingly, addition of EDTA or MgCl2 had no influence on TarM activity as measured by direct HPLC-based assay. The absence of metal cation was also supported by the lack of observed peaks in the anomalous difference map of Mn2+-soaked crystals (collected at 1.55 Å), as well as by the lack of detected metal in ICP-MS experiments.
We next analyzed ligand coordination in TarM structures to predict putative active site residues involved in catalysis, and, on this basis, created selected mutants H249A, R326A, K331A, E403A, and E411A with which to study the roles of these residues. The thermostabilities of all mutants with respect to WT were measured by differential static light scattering in the presence and absence of UDP-GlcNAc (Fig. 3A). The hydrolysis and GT activities of WT TarM and the monomeric mutant G117R were measured by fluorescence-based (Table 2) and HPLC-based assays (Fig. 3B) as already described. For activities outside reliable detection limits by the fluorescence-based method, the HPLC-based assay provided a direct and comparative measure of residual activity. Two of the mutants, K331A and R326A, were completely inactive, whereas activities of mutants H249A, E403A, and E411A were severely reduced by comparison with the WT enzyme (Fig. 3B). These results, together with the positioning of the residues in the TarM active site, suggest the following catalytic involvement of active site residues, as illustrated in Fig. 2B and Figs. S2B and S8. Here, UDP is positioned such that it is stabilized by offset π-π stacking interactions between the uracil ring and Y382, and via hydrogen bonding of the ribose hydroxyls with the side chain of E411. The side chains of R326 and K331 appear to stabilize negative charges on the UDP phosphates, compensating for the absence of a stabilizing cation that is commonly present in other GTs. The observation that UDP-GlcNAc provides little thermostability to the K331A mutant compared with WT and other mutants suggests a significant role for this residue in donor substrate binding (Fig. 3A). The E403 side chain interacts with the GlcNAc C3 hydroxyl, as well as the nearby side chain of K331, thereby likely establishing the correct orientation of K331 for stabilization of the UDP leaving group. The positioning of the H249 and N304 side chains suggests hydrogen bonding with the C6 hydroxyl of GlcNAc. In addition, sequence alignment of S. aureus TarM with similar proteins in various organisms reveals high-level conservation of the aforementioned residues, further highlighting their importance in catalysis (Fig. S9).
Fig. 3.
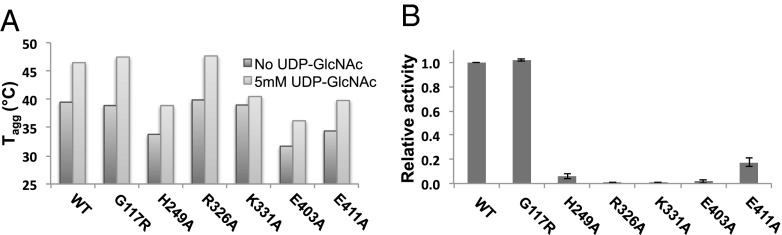
Comparison of thermostability and activity of TarM mutants. (A) Thermostability (i.e., Tagg) of WT TarM and various mutants in the absence and presence of 5 mM UDP-GlcNAc. Thermostability was analyzed by differential static light scattering (StarGazer) as a measure of Tagg upon thermodenaturation. (B) Relative activity of WT TarM and various mutants by HPLC-based UDP detection. For comparative purposes, relative activity is given as a fraction of the WT activity whose value was adjusted to 1.0. In all cases, TarM was incubated with 1 mM UDP-GlcNAc for 1 h preceding measurement.
Because of inherent difficulties in obtaining a homogenous acceptor substrate as a result of the heterogeneity in WTA polymer length, we instead crystallized TarM in the presence of fondaparinux, a synthetic heparin pentasaccharide. This compound was selected based on its defined length and similar physicochemical properties to polyRboP (Fig. 4B). Fondaparinux binds TarM in the N-terminal GT-B acceptor substrate binding domain within a dominant and solvent-exposed electropositive groove, positioning it in close proximity to the active site with bound UDP and α-glyceryl-GlcNAc molecules (Fig. 2A). Fondaparinux is stabilized by a number of interactions, with five of its eight sulfate moieties forming direct interactions along the positively charged binding groove (Fig. S7C). Notably, the 2-sulfoamino group of the nonreducing terminal saccharide closest to the putative GT reaction center is within hydrogen bonding distance to R326, whose side chain interacts with the UDP β-phosphate as well as the glycosidic oxygen of α-glyceryl-GlcNAc and appears in this way to bridge the substrates (Fig. 2B). Based on its proximity to the active site and similar molecular properties, we propose that fondaparinux is an acceptor analog that binds TarM in a similar manner to the physiological polyRboP acceptor. In further support of this hypothesis, a series of five ordered sulfate ions were observed in the apo TarM structure, crystallized in the presence of ammonium sulfate, along the continuous positively charged groove extending from the active site. Two of these ions overlap with interacting sulfate groups in fondaparinux and the remaining three extend across the trimerization interface into the N-terminal β-insertion domain of a neighboring protomer. These sulfate ions are spaced in good agreement with the interphosphate distances in polyRboP, and we propose that they highlight the path in which part of the natural extended WTA substrate (∼40–50 units) wraps across the surface of the novel trimerization interface, as modeled for illustration in Fig. 4.
Fig. 4.

Binding of modeled polyRboP to TarM. (A) Model illustrating the proposed binding of an extended polyRboP polymer (cyan) to the positively charged binding groove that extends from the active site along the acceptor binding domain and across to the trimerization domain of a neighboring protomer. Location of ordered sulfate residues in the apo structure are shown in green (Fo-Fc difference map contoured at 4 σ) and the bound fondaparinux (shown for comparison) in yellow. (B) Comparison of polyRboP (Top) and fondaparinux (Bottom) chemical structures. SO4/PO4 groups of respective fondaparinux and modeled polyRboP that overlap with ordered sulfates are displayed in green, and groups that overlap between the two molecules (but not the ordered sulfates) are shown in orange. A nonoverlapping fondaparinux SO4 group that interacts with the electropositive groove in the acceptor binding domain is displayed in purple. (C) Model of the binding of a PRboP unit (cyan) to the active site of TarM. The model places the PRboP C4 hydroxyl (circled) 3.5 Å away from the anomeric carbon (indicated with dashed line), and 3.6 Å from the leaving group phosphate. The PRboP unit is predicted to bind in a similar manner to fondaparinux (yellow), sharing the same phosphate binding pocket as the 6-O-sulfate group of the terminal saccharide of fondaparinux (circled), with the other phosphate coordinated by R326 (green) similar to the fondaparinux sulfoamino group. (D) Same TarM trimer view shown in A but with surface removed to highlight path of polyRboP binding (cyan) across the trimerization interface.
As mentioned, there is no evidence that fondaparinux is GlcNAcylated, as observed in the crystal structure and by the unfavorable positioning of its terminal saccharide relative to the reaction center. Instead, in addition to UDP, we observe a TarM-catalyzed α-GlcNAcylated glycerol product (confirmed by MS and NMR, as described earlier) trapped in the active site as a result of crystal packing and/or fondaparinux stabilization of the closed state of the enzyme. Increasing concentrations of glycerol furthermore enhanced TarM activity in the presence and absence of fondaparinux (Fig. S10). The major alternative form of WTA is composed of glycerol-3-phosphate units, such that the trapped α-glyceryl-GlcNAc molecule (with the glyceryl hydroxyl continuous with the adjacent fondaparinux by way of hydrogen bonding) likely reflects the position of the native GlcNAcylated polyRboP product. Interestingly, the leaving group UDP β-phosphate O3B is within close hydrogen bonding distance (2.4 Å; Fig. 2B) to the α-glyceryl-GlcNAc glycosidic oxygen. This is consistent with the role of UDP in the orientation and base-activation of the glycerol hydroxyl in a front face SNi-like catalytic mechanism (Fig. S8B), where nucleophilic attack occurs on the same face as leaving group departure, and where the leaving group itself is responsible for base activation of the incoming nucleophile.
To illustrate the active site binding conformation of the native acceptor, we modeled the binding of a single phosphate-ribitol-phosphate (PRboP) monomer with AutoDock Vina using a highly exhaustive search protocol. Analysis of the 20 top scoring poses shows the PRboP ligand forms two dominant clusters partially overlapping with the terminal saccharide group of fondaparinux. The lowest energy pose shares a common phosphate binding site with the fondaparinux terminal saccharide 6-O-sulfate group, whereas the ribitol and other phosphate are oriented closer to the donor binding pocket than the fondaparinux terminal sugar, with the phosphate interacting with R326 (analogous to the 2-sulfoamino group of fondaparinux; Fig. 4C). The model places the acceptor in a favorable position for leaving group UDP-mediated general base deprotonation of the ribitol 4C hydroxyl and subsequent nucleophilic attack at the GlcNAc anomeric reaction center.
Discussion
The GlcNAc residues that decorate the WTA of the highly prevalent clinical pathogen S. aureus serve important functions in drug resistance, elicitation of host immune response, and phage interaction. The configuration of the GlcNAc linkage furthermore appears to define and modulate these processes. TarM is a retaining GT-B responsible for decoration of WTA with α-GlcNAc in S. aureus. Here, we have presented various structural snapshots of TarM, showing a native trimer and an unexpected ternary-like complex including a product formed by TarM itself. Together with kinetic and mutational data, these structures further the understanding of the catalytic mechanism and potential inhibition of WTA α-GlcNAcylation and, also more generally, support the proposed SNi-like catalytic mechanism for anomeric retention in retaining GT-B family members.
The GT-B fold consists of two flexibly linked Rossmann-like domains that face each other (rather than abut as with the GT-A fold), whose architecture forms distinct donor and acceptor sites at the resulting cleft. There is no distinct correlation between protein fold and the stereochemical outcome of the product: GT-As and GT-Bs are capable of catalyzing the transfer of a glycosyl group to an acceptor with inversion or retention of configuration at the anomeric center. As expected, the TarM GT-B domain bears close structural resemblance to other GT-B class enzymes, including the structures of the first two GT4 family members, AviGT4 and WaaG (Escherichia coli) (34). To study the catalytic mechanism of TarM, we generated several structure-directed active site mutants, all at sites that were highly conserved (Fig. S9), permitting us to identify key residues involved in catalysis. Comparison with structures of other retaining GT-Bs reveals several similarities in active site composition. For instance, as with other retaining GT-Bs [MalP (GT35; E. coli) (35), AGT (GT78; Enterobacteria phage T4) (36), WaaG (GT4; E. coli) (34), MshA (GT4; Corynebacterium glutamicum) (37), OtsA (GT20; E. coli) (38)], a pair of conserved lysine/arginine residues (here, K331 and R326) are found within hydrogen bonding distance to a donor sugar UDP phosphate. These likely take on a charge stabilization role in the absence of a divalent cation. A nearby glutamate residue (here E403; substituted by aspartate in OtsA) appears necessary for the proper orientation of K331. Other similarities include a histidine (here H249; in WaaG, an aspartate assumes this role) with a side chain that hydrogen bonds with the C6 hydroxyl of GlcNAc and a main chain carbonyl that may possibly help to stabilize a proposed oxocarbenium-like transition state.
We next studied the acceptor substrate binding site for insight into GT transfer. The acceptor binding domains of GTs are more variable than are the donor binding domains, and generally less well-characterized as a result of the relative paucity of structural data on GTs with bound acceptor molecules. We were able to obtain a ternary-like complex containing a pentasaccharide acceptor analog (fondaparinux), along with a cleaved UDP product and a trapped α-glyceryl-GlcNAc glycoside that we propose mimics the native GlcNAcylated ribitol product. The chemical relevance of α-glyceryl-GlcNAc in the active site is supported by the observation that various Gram-positive bacterial strains such as Bacillus subtilis have a WTA composed of polyglycerol phosphate. To the best of our knowledge, this represents the first enzyme-catalyzed product complex for a retaining GT-B and provides further support for a front-face mechanism of anomeric retention. In contrast to the single displacement reaction mechanisms of inverting GTs, the catalytic mechanism used by retaining GTs has been the subject of debate. A double displacement mechanism was initially proposed whereby catalysis proceeds via the formation of a covalent anomerically inverted glycosyl-enzyme intermediate, followed by opposite face attack by the activated acceptor with overall retention of glycosidic configuration. As more structures of retaining GTs became available, the lack of suitable active site nucleophiles or observed intermediates led to the proposal of a front face SNi-like reaction mechanism whereby the nucleophile attacks from the same face as leaving group departure, and the leaving group itself is responsible for the base activation of the incoming nucleophile (39). A significant contribution came from studies on OtsA, whose active site bears a close resemblance to that of TarM, in which a ternary complex of UDP and a bisubstrate inhibitor (validoxylamine A 6′-O-phosphate; VA6P) identified a putative transition state-like arrangement (38). The structure showed that hydrogen bonding existed between the leaving group oxygen of UDP and the nucleophile mimic of the sugar moiety, suggestive of an SNi-like reaction mechanism. Kinetic isotope effects and linear free energy relationship experiments (40), along with computational studies (41) on the inhibitor complex, confirmed that VA6P and UDP acted together as synergistic transition state mimics, supporting front-face nucleophilic attack involving hydrogen bonding between leaving group and nucleophile. Structural comparison with our TarM ternary-like complex reveals a very similar active site composition with good structural agreement between UDP and donor sugar glycoside products (Fig. 5). Notably, the glycosidic oxygen of the α-glyceryl-GlcNAc in TarM is positioned similarly to the glycosidic nitrogen of VA6P in OtsA, with similar bonding distances and geometries to the UDP product. A strong 2.4-Å hydrogen bond between the leaving group phosphate O3B and the glycosidic oxygen (similar to 2.6 Å in OtsA) suggests that the leaving group phosphate oxygen could contact the incoming acceptor nucleophile, consistent with the phosphate acting as a base to deprotonate the acceptor. In addition, the distance between the anomeric carbon and interacting UDP is closer in the TarM complex at 3.0 Å compared with 3.5 Å in the OtsA structure. One potentially important mechanistic observation is the strained conformation of the sugar ring. In the OtsA complex, the cyclohexenyl ring of VA6P has flattened geometry with an approximate E3 conformation, proposed to mimic the oxocarbenium ion-like character of the SNi transition state. Although not distorted to the same degree, the pyranose ring of the α-glyceryl-GlcNAc in TarM is flattened along O5-C1-C2-C3 with a 4H5 conformation [Fig. 5; analysis by Platon (42)]. This small distortion, with the axial α-bond moving into a pseudoequatorial conformation, could alleviate the crowding under the sugar where, according to the SNi mechanism, there is one group leaving and another attacking in the same space. Thus, the binding geometry of this in situ product provides further support for the SNi-like mechanism proposed based on the structure of the VA6P inhibitor complex and subsequent analyses.
Fig. 5.

Comparison of TarM and OtsA ternary complexes. TarM (blue) in complex with UDP and enzyme catalyzed α-glyceryl-GlcNAc is superimposed with OtsA (yellow) in complex with UDP and bisubstrate inhibitor VA6P. Residues are displayed in stick form and colored according to heteroatom type. The UDP leaving group phosphate O3B to glycosidic O/N (TarM/OtsA) or anomeric carbon distances are labeled. Residues are identified according to TarM numbering and residue differences denoted in parenthesis. Cremer–Pople parameters for ring conformation are shown below.
It has been observed that different strains of S. aureus display α, β, or a mixture of the two GlcNAc-WTA linkages. Interestingly, however, enzyme preparations from several different strains appear capable of achieving α- and β-O-GlcNAcylation of WTA to various extents, regardless of the strains tendency to do so natively (24). Furthermore, it has been reported that WTA from S. aureus strain H displays varied content of α-O-linked GlcNAc upon slight variations in its preparation (43). These observations together would suggest underlying regulation in the expression or activity of the WTA GTs. The additional finding that in S. aureus Copenhagen, displaying 15% α- and 85% β-O-linkages, there exist two distinct polymers of TA with exclusive α- or β-O-GlcNAc (22), may suggest that TarM and TarS are processive enzymes. Here, we have demonstrated that TarM is indeed processive. In addition, the complex of TarM with fondaparinux, which we propose acts as an acceptor mimic, and analysis of the surface electrostatics potentially help explain the basis of this processivity. Such analysis reveals large positively charged grooves snaking across the surface of the TarM trimer (Fig. 1). Notably, a prominent basic groove extends along the N-terminal acceptor binding GT-B domain from the active site and across the trimerization interface into the β-insertion domain of a neighboring protomer. Our crystal structures revealed the presence of fondaparinux within this groove, as well as a series of five ordered sulfate ions that lie along the length of the positively charged path extending from the trimerization domain of one protomer onto that of its neighbor (Fig. 4 A and D). We propose this binding groove provides a means for TarM to scaffold the extended electronegative WTA acceptor across its surface, facilitating glycosylation along the WTA chain before release. Interestingly, the G117R monomeric mutant displayed a similar level of intrinsic processivity compared with the native trimer, suggesting that the WTA chain can maintain analogous intrasubunit interactions within its own trimerization domain with little effect on scaffolding or processivity. Trimerization, however, could act to increase the local concentration of the enzyme at the membrane, and could possibly play a role in other associations in the WTA pathway. In addition, the different open and closed states of the respective structures with bound UDP vs. UDP-GlcNAc or fondaparinux (with UDP and α-glyceryl-GlcNAc) may, in addition to bringing the donor and acceptor substrates together for the GT reaction, create a motion that extends along the acceptor binding path, such that the movement could be involved in enzyme processivity.
An interesting feature of the TarM structure is the N-terminal β-trimerization domain (residues 69–199), which belongs to Pfam database (44) family domain of unidentified function (DUF) 1975. This DUF is predicted (by Pfam) to be present in the N termini of various prokaryotic α-GTs including the species presented in the sequence alignment (Fig. S9). Recently, the crystal structure of the GT-B O-GlcNAc transferase GtfA of Streptococcus pneumoniae, involved in protein glycosylation, has revealed a similar domain organization to TarM (overall rmsd 2.2 Å), with a β-inserted DUF1975 domain that is proposed to participate in complex formation with its GtfB coactivator (45). The domain also appeared to be involved in acceptor polypeptide binding according to molecular docking simulations. Our structure demonstrates that this domain participates in protein–protein interactions (via trimerization vs. coactivator binding in the GtfA monomer) and, in addition, provides evidence that it is also involved in the binding of the polymeric acceptor substrates. Because of its prevalence and apparent versatility, this domain could present a general feature for protein–protein interaction and acceptor binding in harboring enzymes. Like TarM, GtfA is also involved in the glycosylation of surface bound polymers (serine-rich repeat glycoproteins in the case of GtfA). These polymers, as with WTA, are implicated in adhesion, immune evasion, colonization, biofilm formation, and virulence in Gram-positive bacteria (reviewed in ref. 46), which presents an interesting parallel between the two cell surface polymer decorating systems. By contrast, TarS (an inverting GT-A) is not predicted to contain domain DUF1975.
The anomeric configuration of WTA GlcNAc sugars has been shown to be highly influential in S. aureus pathobiology. The α- vs. β-linkage of WTA GlcNAc residues, for example, appears to be important in the host antibody response. In laboratory S. aureus strains as well as a community-associated Methicillin-resistant S. aureus (MRSA) strain, ΔTarS mutants (with intact α-O-GlcNAc) escape host anti–WTA-IgG mediated opsonophagocytosis, whereas ΔTarM mutants (with intact β-O-GlcNAc) retain susceptibility and are engulfed (28). However, the antibody specificity could be adaptive given that the recognition of α- vs. β-linkage, as studied thus far, appears to depend on the predominant anomeric configuration of the sugar within a given strain (24), as well as on the previous pathogenic exposure of the host serum (22). As such, the modification of the type of linkage produced by the bacteria may be advantageous for its survival. It was also recently observed that TarS deletion resulted in the sensitization of MRSA to β-lactam antibiotics, whereas TarM deletion did not alter the resistance phenotype (27). These authors propose that β-O-GlcNAc may act as a scaffold for recruitment of PBP2a, a protein largely responsible for MRSA, based on the recent finding that PBP2a directly binds WTA in vitro (47). As such, the preliminary result that fondaparinux shows higher affinity binding to TarM/TarS compared with its native substrate, WTA, may present an interesting avenue for drug design aimed at MRSA resensitization.
It has been observed that, in S. aureus strains possessing WTA with exclusive α- or β-O-linked GlcNAc, the presence of GlcNAc, regardless of its anomeric form, was important for phage binding (17). It has, however, recently been shown that the α-O-GlcNAc of WTA serves as an adsorption receptor for serogroup A, B, and F phage binding in K6 (TarM disrupted mutant) RN4220 S. aureus (48). Nevertheless, the later finding that the K6 mutant also contains a truncation in the TarS gene (27) would warrant further studies of specificity with strict ΔTarM or ΔTarS mutants. As phage therapy is gaining increasing attention as a result of the continued pattern of broad-spectrum antibiotic resistance (49), the study of TarM and TarS will be of great importance in this reemerging field. Adding to this is a recent finding that shows binding of helper phages, such as certain temperate phages of serogroup B to GlcNAc residues of S. aureus WTA, allow for horizontal transfer of mobile genetic elements such as those typically encompassing antibiotic resistance genes over long phylogenic distances extending to genera (50). We are indeed only beginning to appreciate the importance of the WTA “glycocode” in shaping the evolution and resistance profile of this and other Gram-positive bacteria. As such, TarM and TarS are important players in many aspects of bacterial biology and pathogenicity and will likely prove lucrative targets for novel therapeutic agents in light of an ever-decreasing antibiotic arsenal.
Materials and Methods
Cloning and Protein Synthesis.
The full-length ORF (amino acids 1–493) encoding S. aureus TarM (SACOL1043) was cloned into the expression vector pET41b with a thrombin cleavable C-terminal 8xHis tag. Mutagenic TarM constructs were produced with the Quick Change mutagenesis kit (Qiagen). Constructs were transformed into Rosetta (DE3) E. coli. Cells were grown in Luria–Bertani broth (supplemented with 35 µg/mL kanamycin) to an OD at 600 nm of 0.6–0.8, at which point IPTG was added at a final concentration of 1 mM. Protein expression was carried out overnight at room temperature. Cells were pelleted and stored at −80 °C until required. Selenomethionine-labeled protein was produced by using the auxotrophic E. coli strain B834 and grown in M9 minimal media containing 100 mg/L of selenomethionine according to the protocol of Leahy et al. (51).
Protein Purification.
Cell pellets were resuspended in buffer A (20 mM NaH2PO4, pH 7.3, 500 mM NaCl, 5% glycerol). A complete protease inhibitor tablet at 1× final concentration (Roche) and DNase 1 at 1 μg/mL final concentration were added, and cells were lysed at 12,000 psi by using a French press (Thermo Electron). Cell debris was pelleted by centrifugation at 20,000 × g for 30 min. The resulting supernatant was loaded onto a 1-mL HisTrap HP cartridge (GE Lifesciences) and eluted over a 30-mL linear gradient to 100% buffer B (500 mM imidazole, 20 mM NaH2PO4, pH 7.3, 500 mM NaCl, 5% glycerol). Fractions containing the purest protein were pooled and the His-tag cleaved by overnight incubation with thrombin (10 U/mg protein) at 4 °C. The protein was dialyzed into buffer A and passed over a 1-mL His-trap HP cartridge (GE Lifesciences), and the flow through, containing the non–His-tagged protein, was collected. The sample was exchanged into buffer C (20 mM Hepes, pH 7.3, 300 mM NaCl, 5% glycerol), loaded onto a 5-mL HiTrap Heparin cartridge (GE Lifesciences), and eluted over a 30-mL linear gradient to 100% buffer D (20 mM Hepes, pH 7.3, 2 M NaCl, 5% glycerol). Fractions containing the purest protein were pooled, concentrated, and loaded on a Superdex 200 column (GE Lifesciences) equilibrated in buffer E (20 mM Hepes, pH 7.5, 500 mM NaCl, 5% glycerol), and the fractions were collected and concentrated. The protein was frozen in liquid N2 and stored at −80 °C until required.
Metal Binding Analysis.
The metal content of TarM was measured by using an ICP mass spectrometer (NexION 300D ICP-MS; PerkinElmer Life Sciences), and the data were analyzed with NexION software. A calibration standard (cat. no. IV-STOCK-4; Inorganic Ventures) containing metals of interest (Mg2+, Mn2+, Co2+, Ni2+, Cu2+, Zn2+) was diluted with an internal standard solution containing 10 μg/L Sc and 1% nitric acid (cat. no. IV-ICPMS-71D; Inorganic Ventures), and this was used to generate standard curves spanning 1–100 μg/L for each metal. Protein samples were appropriately diluted with internal standard solution to adjust metal concentrations within the range of the standard curve. To confirm absence of cation, the protein sample was spiked with metals of interest and measured as a positive control.
WTA Isolation and Purification.
S. aureus WTA was isolated and purified according to modified previously established protocols (52, 53). S. aureus RN4220 cells were grown in a culture of 20 mL tryptic soy broth overnight at 37 °C, and the cells were collected at 2,000 × g for 10 min. The cells were washed once in 30 mL of buffer 1 (50 mM MES, pH 6.5), resuspended in buffer 2 (4% SDS, 50 mM MES, pH 6.5), and boiled in a water bath for 1 h. The cell debris was collected at 10,000 × g for 10 min, resuspended in 2 mL of buffer 2, and sedimented at 14,000 × g for 10 min. The pellet was washed in subsequent 1-mL volumes of buffer 2, buffer 3 (2% NaCl, 50 mM MES, pH 6.5), and buffer 1. The pellet was resuspended in 1 mL of buffer containing proteinase K (20 mM Tris⋅HCl, pH 8.0, 0.5% SDS, 20 μg proteinase K) and digested at 50 °C for 4 h. The sample was pelleted at 14,000 × g for 10 min and washed once in buffer 3 and three times with distilled H2O. The sample was then resuspended in 1 mL of 0.1 M NaOH and shaken at room temperature for 16 h. The remaining insoluble cell wall debris was removed by centrifugation at 14,000 × g for 10 min and the supernatant containing the hydrolyzed crude WTA was neutralized with addition of HCl to a final concentration of 0.1 M. The sample was dialyzed against distilled H2O by using a 3-kDa molecular weight cut-off (MWCO) membrane. For digestion of attached GlcNAc, the sample was exchanged into buffer (100 mM sodium citrate, pH 4.5, 250 mM NaCl) over a PD10 desalting column (GE Lifesciences) and incubated with 0.5 mg/mL α-NAGLU (R&D Systems) and 0.2 mg/mL β-NAGLU (New England Biolabs) overnight at 37 °C. For purification, the sample was exchanged into buffer A (20 mM Tris⋅HCl, pH 7.2) over a PD10 desalting column, applied on a 5-mL DEAE FF cartridge (GE Lifesciences), and eluted over a 30-mL linear gradient to 100% buffer B (20 mM Tris⋅HCl, pH 7.2, 1 M NaCl), with UV monitored at 205 nm. Peaks with the highest 205-nm readings were pooled and dialyzed against distilled H2O using a 3-kDa MWCO membrane. The sample was frozen, lyophilized, and resuspended in distilled H2O. WTA concentration was measured according to the concentration of phosphorus detected by ICP-MS, whereby a phosphorus calibration curve spanning 1–100 μg/L was created by using ribitol-5-phosphate diluted with internal standard solution (Metal Binding Analysis). WTA samples were then diluted with internal standard solution to fall within the range of the standard curve and measured in triplicate. WTA polymer size was assessed by SEC-MALS by using a Superdex 200 10/300 GC column (GE Healthcare) eluted with a flow rate of 0.2 mL/min in 20 mM Hepes, pH 7.3, 500 mM NaCl, 5% glycerol buffer, using an Agilent 1100 series HPLC (Agilent Technologies), coupled in line to a Dawn HeleosII 18-angle MALS light scattering detector and Optilab T-rEX differential refractometer monitor (Wyatt Technology), and using a refractive index increment dn/dc of 0.143 mL/g.
In Vitro Activity Assay.
TarM hydrolysis activity was studied by using the ADP Quest Assay kit (DiscoverRx) according to the manufacturer’s protocol and performed in a 384-well black assay plate with a total volume of 10 μL. Various concentrations of TarM and mutant proteins were incubated with 1 mM UDP-GlcNAc and assay kit reagents. Upon cleavage of UDP-GlcNAc by TarM, UDP is released, resulting in a fluorescence signal detected continuously at 530 nm excitation and 590 nm emission wavelengths using a Synergy H4 multimode plate reader (BioTek). Km and kcat values were determined by using optimal protein concentrations and varying concentrations of UDP-GlcNAc, WTA, or fondaparinux while using UDP for the standard curve. TarM hydrolysis of UDP-GlcNAc was further verified by chromatography. Here, 10 µM TarM was incubated with 1 mM UDP-GlcNAc, after which aliquots of the reaction mixture were separated at indicated times and filtered through a 3-kDa MWCO filtration unit to remove protein. Fractions (10 μL) of the filtrate were injected onto a TSKgel DEAE-5PW weak anion exchange column (TOSOH Biosciences), and the separated UDP-GlcNAc and UDP peak areas were monitored and quantified at a UV wavelength of 254 nm using an ÄKTA Micro purification system (GE Lifesciences). TarM GT activity was analyzed as described earlier by using 1 mM UDP-GlcNAc in the presence of increasing concentrations of WTA (purified and GlcNAc digested as described).
Determination of Association/Dissociation Rate Constants.
Biolayer inferometry was performed by using an Octet Red instrument (FortéBio) with streptavidin sensors (FortéBio). TarM was biotinylated by using EZ-Link NHS-PEG4-Biotin (Thermo Scientific). Biolayer inferometry was performed at 25 °C in a 96-well plate (Greiner Bio-One) and a 200-μL well volume. After a brief equilibration of the sensors in assay buffer (20 mM Hepes, pH 7, 500 mM NaCl), TarM_WT or TarM_G117R was loaded onto the sensors for 5 min at 300 nM, followed by the blocking of unbound streptavidin with 15 μg/mL EZ-Link Biocytin (Thermo Scientific) in Superblock Blocking Buffer in PBS solution (Thermo Scientific). Next, a baseline was acquired for 3 min, followed by the association of WTA for 5 min (kon) and dissociation for 15 min (koff) in assay buffer (20 mM Hepes, pH 7, 500 mM NaCl). Various optimal concentrations of WTA (0.31 mM, 0.62 mM, 1.25 mM, 2.5 mM, and 5 mM) were titrated with double referencing to rule out nonspecific binding to sensors, and the KD was calculated based on kon and koff rates fitted to a heterogeneous ligand model by using the FortéBio data analysis software.
Thermostability Analysis.
TarM thermostability was measured as a function of its temperature dependent aggregation by differential static light scattering (StarGazer-2; Harbinger Biotechnology and Engineering) according to the method of Vedadi et al. (33). Briefly, 50 μL of 0.4 mg/mL protein under various conditions was heated from 25 °C to 85 °C at a rate of 1 °C/min in individual wells of a clear-bottom 384-well plate (Nunc). Protein aggregation, as a measure of the intensity of scattered light, was scanned every 30 s with a CCD camera. The integrated intensities were plotted against temperature, with the inflection point of each fitted curve, using a Boltzmann regression, defined as the aggregation temperature, Tagg.
Crystallization, Structure Solution, and Modeling.
Native TarM (∼20 mg/mL) was crystallized by sitting-drop vapor diffusion in the presence of 2 mM UDP-GlcNAc and 2 mM MgCl2 using a reservoir solution of 8–10% wt/vol PEG 3350, 200 mM NaCl, 200 mM sodium acetate (pH 8.0), 100 mM Hepes (pH 7.0), and 20% ethylene glycol. Before data collection, crystals were flash-cooled directly from the drop. Crystals belonged to space group P63 with unit cell dimensions a = b = 162.3 Å, c = 228.0 Å. For phasing, selenomethionine derivative crystals were obtained and a single-wavelength SAD experiment was carried out. Data were processed with iMosflm (54), XDS (55), and Aimless (56). Phasing was carried out with SHARP (57) using SHELX (58) to determine the heavy atom substructure. Model building was performed with Buccaneer (59) and refined using Refmac (60), Buster (61), Phenix (62), and Coot (63) using TLS parameters in the later stages. The final model has good stereochemistry, with 94.40% of residues in the favored region of the Ramachandran plot and 0.41% outliers (Table 1). Four molecules are found in the asymmetric unit with crystal symmetry generating four trimers. Crystals for native TarM in the absence of donor were obtained by sitting-drop vapor diffusion by using a reservoir solution of 200 mM Na/K tartrate, 100 mM trisodium citrate, pH 6.5, and 2.1 M ammonium sulfate. Crystals were cryoprotected with a reservoir solution containing 30% glycerol and were subsequently flash-cooled. Crystals belonged to space group P63 with unit cell dimensions a = b = 208.6 Å, c = 120.8 Å. The structure was solved with molecular replacement using Phaser (64) with the donor bound structure as a search model, finding two molecules in the asymmetric unit and refined as described earlier. The final model has good stereochemistry, with 93.58% of residues in the favored region of the Ramachandran plot and 0.41% outliers. The G117R mutant was crystallized at ∼20 mg/mL by sitting-drop vapor diffusion in the presence of 2 mM UDP-GlcNAc, 2 mM MgCl2, and 1 mM fondaparinux sodium [methyl O-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-d-glucopyranosyl-(1→4)-O-β-d-glucopyranuronosyl-(1→4)-O-2-deoxy-3,6-di-O-sulfo-2-(sulfoamino)-α-d-glucopyranosyl-(1→4)-O-2-O-sulfo-α-l-idopyranuronosyl-(1→4)-2-deoxy-6-O-sulfo-2-(sulfoamino)-α-d-glucopyranoside, decasodium salt; GlaxoSmitheKline] using 0.1 Mes, pH 6.5, 10–14% wt/vol PEG 20000 as the reservoir solution. Crystals were cryoprotected with reservoir solution containing 40% glycerol and were subsequently flash-cooled. UDP/UDP-GlcNAc bound crystals diffracted X-rays to ∼2.4 Å resolution, and UDP/fondaparinux bound crystals diffracted X-rays to ∼2.1 Å. Both of these crystals belonged to space group P1 with two monomers in the asymmetric unit. All data were collected at the Canadian Light Source (beam lines 08ID-1 and 08B1-1). The structure was solved with molecular replacement by using Phaser (64) with the native structure as a search model. The structures were refined as before. Both models have good stereochemistry, with 95.42% Ramachandran favored, 0.41% outliers for the UDP/UDP-GlcNAc structure and 96.33% favored, 0.78% outliers for the fondaparinux complex (Table 1). The CCP4 package (65) was used where applicable. Structural validation was carried out using MolProbity (66). Data and coordinates have been deposited to the Protein Data Bank with accession codes 4X6L, 4X7M, 4X7P, and 4X7R.
Acceptor binding was modeled using Autodock Vina (67) by using the TarM G117R:UDP/fondaparinux structure as the receptor and a phosphate-ribitol phosphate monomer as the ligand.
Analysis of Quaternary Structure.
Purified protein was applied to a Superdex 75 HR 10/30 column (GE Healthcare) equilibrated in 30 mM Hepes, pH 7.3, 500 mM NaCl, 5% glycerol buffer, using an Agilent 1100 series HPLC (Agilent Technologies), coupled in line to a Dawn HeleosII 18-angle MALS light scattering detector and Optilab T-rEX differential refractometer monitor (Wyatt Technology). Monomeric BSA (Sigma-Aldrich) was used to normalize the light scattering detectors. Data were collected and analyzed with the Astra 6 software package provided by the manufacturer (Wyatt Technology). The protein molar mass was calculated, assuming a refractive index increment (dn/dc) value of 0.186 mL⋅g−1.
LCMS and NMR Analysis of the Glyceryl-GlcNAc Glycoside After Acetylation.
TarM 100 μM was reacted with 10 mM UDP-GlcNAc in the presence and absence of 1 mM fondaparinux and 40% glycerol overnight. The enzyme was subsequently removed from the reaction mixture by using a 3-kDa MWCO filtration device, the filtrate was lyophilized and then dissolved in pyridine (2.5 mL), acetic anhydride (2.5 mL) was slowly added, and the reaction mixture was stirred at room temperature (Ac2O/pyridine 1:1, 5 mL total volume). After 20 h, the mixture was cooled in an ice bath and MeOH (7 mL) was slowly dropped into the solution. The reaction mixture was concentrated and coevaporated with toluene several times until only a sticky oil remained, which was dissolved in dichloromethane (20 mL). Silica gel (5 g) was added to this solution, dichloromethane removed by evaporation, and the remaining solid transferred to a silica gel column. Glycerol-tri-O-acetate was eluted with petroleum ether/ethyl acetate 1:1, and the residual compounds were eluted with ethyl acetate. All compounds that eluted with ethyl acetate were pooled, reduced, and further purified by C18 chromatography (Zorbax Eclipse XDB C18, 9.4 × 250 mm, 4 mL/min, 25% acetonitrile) to give two compounds of almost identical retention time and mass (recorded on a Waters ZQ2000 LC MS attached to a Waters 2695 separation module with flow injection analysis; spectra analyzed with Masslynx 4.0; electrospray ionization positive mode: 528.4 [M+Na]+), which were identified by NMR (Bruker Avance II, 400 MHz; assignment was based on 1H COSY-heteronuclear single quantum coherence spectra analyzed with Topspin 3.2) as the l and d glycero isomers of α-glyceryl-GlcNAc.
Supplementary Material
Acknowledgments
The authors thank Gideon J. Davies for valuable advice on sugar conformational analysis and reading the manuscript, the Centro de Biologia Estructural del Mercosur/CCP4 workshop (Montevideo, Uruguay) for support in training (S.S.), and the staff at the Canadian Light Source beam line CMCF-1 for assistance with data collection. This work was supported by the Canadian Institutes of Health Research [S.S. (Banting Fellow), L.J.W., S.G.W., and N.C.J.S.], Michael Smith Foundation for Health Research Fellowship program (S.S. and R.J.G.), Howard Hughes Medical Institute International Scholar program (N.C.J.S.), British Columbia Knowledge Development Fund (N.C.J.S.), Cystic Fibrosis Canada (E.L.), FWF Austrian Science Fund (M.B.), and Deutsche Forschungsgemeinschaft (L.B.). N.C.J.S. and S.G.W. are Tier 1 Canada Research Chairs.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4X6L, 4X7M, 4X7P, and 4X7R).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1418084112/-/DCSupplemental.
References
- 1.Neuhaus FC, Baddiley J. A continuum of anionic charge: Structures and functions of D-alanyl-teichoic acids in gram-positive bacteria. Microbiol Mol Biol Rev. 2003;67(4):686–723. doi: 10.1128/MMBR.67.4.686-723.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weidenmaier C, Peschel A. Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat Rev Microbiol. 2008;6(4):276–287. doi: 10.1038/nrmicro1861. [DOI] [PubMed] [Google Scholar]
- 3.Kojima N, Araki Y, Ito E. Structure of the linkage units between ribitol teichoic acids and peptidoglycan. J Bacteriol. 1985;161(1):299–306. doi: 10.1128/jb.161.1.299-306.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown S, Santa Maria JP, Jr, Walker S. Wall teichoic acids of gram-positive bacteria. Annu Rev Microbiol. 2013;67:313–336. doi: 10.1146/annurev-micro-092412-155620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weidenmaier C, et al. Role of teichoic acids in Staphylococcus aureus nasal colonization, a major risk factor in nosocomial infections. Nat Med. 2004;10(3):243–245. doi: 10.1038/nm991. [DOI] [PubMed] [Google Scholar]
- 6.Weidenmaier C, et al. Lack of wall teichoic acids in Staphylococcus aureus leads to reduced interactions with endothelial cells and to attenuated virulence in a rabbit model of endocarditis. J Infect Dis. 2005;191(10):1771–1777. doi: 10.1086/429692. [DOI] [PubMed] [Google Scholar]
- 7.Oku Y, et al. Pleiotropic roles of polyglycerolphosphate synthase of lipoteichoic acid in growth of Staphylococcus aureus cells. J Bacteriol. 2009;191(1):141–151. doi: 10.1128/JB.01221-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peschel A, Vuong C, Otto M, Götz F. The D-alanine residues of Staphylococcus aureus teichoic acids alter the susceptibility to vancomycin and the activity of autolytic enzymes. Antimicrob Agents Chemother. 2000;44(10):2845–2847. doi: 10.1128/aac.44.10.2845-2847.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kohler T, Weidenmaier C, Peschel A. Wall teichoic acid protects Staphylococcus aureus against antimicrobial fatty acids from human skin. J Bacteriol. 2009;191(13):4482–4484. doi: 10.1128/JB.00221-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peschel A, et al. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J Biol Chem. 1999;274(13):8405–8410. doi: 10.1074/jbc.274.13.8405. [DOI] [PubMed] [Google Scholar]
- 11.Bera A, et al. Influence of wall teichoic acid on lysozyme resistance in Staphylococcus aureus. J Bacteriol. 2007;189(1):280–283. doi: 10.1128/JB.01221-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoover DG, Gray RJ. Function of cell wall teichoic acid in thermally injured Staphylococcus aureus. J Bacteriol. 1977;131(2):477–485. doi: 10.1128/jb.131.2.477-485.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aly R, Shinefield HR, Litz C, Maibach HI. Role of teichoic acid in the binding of Staphylococcus aureus to nasal epithelial cells. J Infect Dis. 1980;141(4):463–465. doi: 10.1093/infdis/141.4.463. [DOI] [PubMed] [Google Scholar]
- 14.Morath S, Geyer A, Hartung T. Structure-function relationship of cytokine induction by lipoteichoic acid from Staphylococcus aureus. J Exp Med. 2001;193(3):393–397. doi: 10.1084/jem.193.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoebe K, et al. CD36 is a sensor of diacylglycerides. Nature. 2005;433(7025):523–527. doi: 10.1038/nature03253. [DOI] [PubMed] [Google Scholar]
- 16.Draing C, et al. Cytokine induction by Gram-positive bacteria. Immunobiology. 2008;213(3-4):285–296. doi: 10.1016/j.imbio.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 17.Chatterjee AN. Use of bacteriophage-resistant mutants to study the nature of the bacteriophage receptor site of Staphylococcus aureus. J Bacteriol. 1969;98(2):519–527. doi: 10.1128/jb.98.2.519-527.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park JT, Shaw DR, Chatterjee AN, Mirelman D, Wu T. Mutants of staphylococci with altered cell walls. Ann N Y Acad Sci. 1974;236(0):54–62. doi: 10.1111/j.1749-6632.1974.tb41481.x. [DOI] [PubMed] [Google Scholar]
- 19.Gross M, Cramton SE, Götz F, Peschel A. Key role of teichoic acid net charge in Staphylococcus aureus colonization of artificial surfaces. Infect Immun. 2001;69(5):3423–3426. doi: 10.1128/IAI.69.5.3423-3426.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baddiley J. Teichoic acids in walls and cells of grampositive bacteria. Fed Proc. 1962;21:1084–1088. [PubMed] [Google Scholar]
- 21.Sanderson AR, Strominger JL, Nathenson SG. Chemical structure of teichoic acid from Staphylococcus aureus, strain Copenhagen. J Biol Chem. 1962;237:3603–3613. [PubMed] [Google Scholar]
- 22.Torii M, Kabat EA, Bezer AE. Separation of teichoic acid of Staphylococcus aureus into two immunologically distinct specific polysaccharides with alpha- and beta-n-acetylglucosaminyl linkages respectively. Antigenicity of theichoic acids in man. J Exp Med. 1964;120:13–29. doi: 10.1084/jem.120.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nathenson SG, Strominger JL. Enzymatic synthesis and immunochemistry of N-acetylglucosaminylribitol linkages in the teichoic acids of Staphylococcus aureus stains. J Biol Chem. 1962;237:3839–3841. [PubMed] [Google Scholar]
- 24.Nathenson SG, Ishimoto N, Anderson JS, Strominger JL. Enzymatic synthesis and immunochemistry of alpha- and beta-N-acetylglucosaminylribitol linkages in teichoic acids from several strains of Staphylococcus aureus. J Biol Chem. 1966;241(3):651–658. [PubMed] [Google Scholar]
- 25.Xia G, et al. Glycosylation of wall teichoic acid in Staphylococcus aureus by TarM. J Biol Chem. 2010;285(18):13405–13415. doi: 10.1074/jbc.M109.096172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42(database issue):D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown S, et al. Methicillin resistance in Staphylococcus aureus requires glycosylated wall teichoic acids. Proc Natl Acad Sci USA. 2012;109(46):18909–18914. doi: 10.1073/pnas.1209126109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kurokawa K, et al. Glycoepitopes of staphylococcal wall teichoic acid govern complement-mediated opsonophagocytosis via human serum antibody and mannose-binding lectin. J Biol Chem. 2013;288(43):30956–30968. doi: 10.1074/jbc.M113.509893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lairson LL, Henrissat B, Davies GJ, Withers SG. Glycosyltransferases: Structures, functions, and mechanisms. Annu Rev Biochem. 2008;77:521–555. doi: 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- 30.Poornam GP, Matsumoto A, Ishida H, Hayward S. A method for the analysis of domain movements in large biomolecular complexes. Proteins. 2009;76(1):201–212. doi: 10.1002/prot.22339. [DOI] [PubMed] [Google Scholar]
- 31.Horn SJ, Sørlie M, Vårum KM, Väljamäe P, Eijsink VG. Measuring processivity. Methods Enzymol. 2012;510:69–95. doi: 10.1016/B978-0-12-415931-0.00005-7. [DOI] [PubMed] [Google Scholar]
- 32.Kurasin M, Väljamäe P. Processivity of cellobiohydrolases is limited by the substrate. J Biol Chem. 2011;286(1):169–177. doi: 10.1074/jbc.M110.161059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vedadi M, et al. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc Natl Acad Sci USA. 2006;103(43):15835–15840. doi: 10.1073/pnas.0605224103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinez-Fleites C, et al. Insights into the synthesis of lipopolysaccharide and antibiotics through the structures of two retaining glycosyltransferases from family GT4. Chem Biol. 2006;13(11):1143–1152. doi: 10.1016/j.chembiol.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 35.Geremia S, Campagnolo M, Schinzel R, Johnson LN. Enzymatic catalysis in crystals of Escherichia coli maltodextrin phosphorylase. J Mol Biol. 2002;322(2):413–423. doi: 10.1016/s0022-2836(02)00771-4. [DOI] [PubMed] [Google Scholar]
- 36.Larivière L, Sommer N, Moréra S. Structural evidence of a passive base-flipping mechanism for AGT, an unusual GT-B glycosyltransferase. J Mol Biol. 2005;352(1):139–150. doi: 10.1016/j.jmb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 37.Vetting MW, Frantom PA, Blanchard JS. Structural and enzymatic analysis of MshA from Corynebacterium glutamicum: Substrate-assisted catalysis. J Biol Chem. 2008;283(23):15834–15844. doi: 10.1074/jbc.M801017200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Errey JC, et al. Mechanistic insight into enzymatic glycosyl transfer with retention of configuration through analysis of glycomimetic inhibitors. Angew Chem Int Ed Engl. 2010;49(7):1234–1237. doi: 10.1002/anie.200905096. [DOI] [PubMed] [Google Scholar]
- 39.Sinnott ML, Jencks WP. Solvolysis of D-glucopyranosyl derivatives in mixtures of ethanol and 2,2,2-trifluoroethanol. J Am Chem Soc. 1980;102(6):2026–2032. [Google Scholar]
- 40.Lee SS, et al. Mechanistic evidence for a front-side, SNi-type reaction in a retaining glycosyltransferase. Nat Chem Biol. 2011;7(9):631–638. doi: 10.1038/nchembio.628. [DOI] [PubMed] [Google Scholar]
- 41.Ardèvol A, Rovira C. The molecular mechanism of enzymatic glycosyl transfer with retention of configuration: Evidence for a short-lived oxocarbenium-like species. Angew Chem Int Ed Engl. 2011;50(46):10897–10901. doi: 10.1002/anie.201104623. [DOI] [PubMed] [Google Scholar]
- 42.Spek AL. Structure validation in chemical crystallography. Acta Crystallogr D Biol Crystallogr. 2009;65(pt 2):148–155. doi: 10.1107/S090744490804362X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haukenes G, Ellwood DC, Baddiley J, Oeding P. Serological cross-reactivity between polysaccharide A and teichoic acid of Staphylococcus aureus. Biochim Biophys Acta. 1961;53:425–426. doi: 10.1016/0006-3002(61)90461-9. [DOI] [PubMed] [Google Scholar]
- 44.Finn RD, et al. Pfam: The protein families database. Nucleic Acids Res. 2014;42(database issue):D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi WW, et al. Structure of a novel O-linked N-acetyl-D-glucosamine (O-GlcNAc) transferase, GtfA, reveals insights into the glycosylation of pneumococcal serine-rich repeat adhesins. J Biol Chem. 2014;289(30):20898–20907. doi: 10.1074/jbc.M114.581934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lizcano A, Sanchez CJ, Orihuela CJ. A role for glycosylated serine-rich repeat proteins in gram-positive bacterial pathogenesis. Mol Oral Microbiol. 2012;27(4):257–269. doi: 10.1111/j.2041-1014.2012.00653.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qamar A, Golemi-Kotra D. Dual roles of FmtA in Staphylococcus aureus cell wall biosynthesis and autolysis. Antimicrob Agents Chemother. 2012;56(7):3797–3805. doi: 10.1128/AAC.00187-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xia G, et al. Wall teichoic Acid-dependent adsorption of staphylococcal siphovirus and myovirus. J Bacteriol. 2011;193(15):4006–4009. doi: 10.1128/JB.01412-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chan BK, Abedon ST, Loc-Carrillo C. Phage cocktails and the future of phage therapy. Future Microbiol. 2013;8(6):769–783. doi: 10.2217/fmb.13.47. [DOI] [PubMed] [Google Scholar]
- 50.Winstel V, et al. Wall teichoic acid structure governs horizontal gene transfer between major bacterial pathogens. Nat Commun. 2013;4:2345. doi: 10.1038/ncomms3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leahy DJ, Hendrickson WA, Aukhil I, Erickson HP. Structure of a fibronectin type III domain from tenascin phased by MAD analysis of the selenomethionyl protein. Science. 1992;258(5084):987–991. doi: 10.1126/science.1279805. [DOI] [PubMed] [Google Scholar]
- 52.Meredith TC, Swoboda JG, Walker S. Late-stage polyribitol phosphate wall teichoic acid biosynthesis in Staphylococcus aureus. J Bacteriol. 2008;190(8):3046–3056. doi: 10.1128/JB.01880-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Endl J, Seidl PH, Fiedler F, Schleifer KH. Determination of cell wall teichoic acid structure of staphylococci by rapid chemical and serological screening methods. Arch Microbiol. 1984;137(3):272–280. doi: 10.1007/BF00414557. [DOI] [PubMed] [Google Scholar]
- 54.Leslie AW, Powell H. Processing diffraction data with mosflm. In: Read R, Sussman J, editors. Evolving Methods for Macromolecular Crystallography, NATO Science Series. Vol 245. Springer; Dordrecht, The Netherlands: 2007. pp. 41–51. [Google Scholar]
- 55.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66(pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr. 2013;69(pt 7):1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vonrhein C, Blanc E, Roversi P, Bricogne G. Automated structure solution with autoSHARP. Methods Mol Biol. 2007;364:215–230. doi: 10.1385/1-59745-266-1:215. [DOI] [PubMed] [Google Scholar]
- 58.Sheldrick GM. Experimental phasing with SHELXC/D/E: Combining chain tracing with density modification. Acta Crystallogr D Biol Crystallogr. 2010;66(pt 4):479–485. doi: 10.1107/S0907444909038360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr D Biol Crystallogr. 2006;62(pt 9):1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 60.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(pt 3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 61.Tronrud DE. TNT refinement package. Methods Enzymol. 1997;277:306–319. doi: 10.1016/s0076-6879(97)77017-4. [DOI] [PubMed] [Google Scholar]
- 62.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40(pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67(pt 4):235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen VB, et al. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(pt 1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.