

Significance
Catalytic hydrogenation and dehydrogenation reactions are extremely important in organic chemistry and recently for energy storage in the form of chemical bonds. Although catalysts are known which catalyze both reactions, the rates and conditions required for the two are frequently very different due to the differences associated with the bonds to be activated (C–H/O–H/N–H and C = O/C = N/H–H). The use of a bifunctional catalyst would substantially simplify the design of processes related to energy storage. In this work, organometallic complexes of iron and iridium are shown to act as catalysts for reversible dehydrogenation of alcohols to carbonyl compounds. This finding opens a pathway to the development of catalysts for direct reversible electrochemical dehydrogenation of organic fuels in energy generation and storage reactions.
Keywords: catalysis, energy storage, reversibility, hydrogenation, dehydrogenation
Abstract
Reversibility of a dehydrogenation/hydrogenation catalytic reaction has been an elusive target for homogeneous catalysis. In this report, reversible acceptorless dehydrogenation of secondary alcohols and diols on iron pincer complexes and reversible oxidative dehydrogenation of primary alcohols/reduction of aldehydes with separate transfer of protons and electrons on iridium complexes are shown. This reactivity suggests a strategy for the development of reversible fuel cell electrocatalysts for partial oxidation (dehydrogenation) of hydroxyl-containing fuels.
Hydrogenation and dehydrogenation reactions are fundamental in synthetic organic chemistry and used in a variety of large- and small-scale processes for manufacturing chemicals, pharmaceuticals, foods, and fuels. An increased need for energy storage technologies, in large part because of the recent deployment of intermittent renewable energy sources, has generated a renewed interest in hydrogen as a form of chemical energy storage. Hydrogen, which may be used in fuel cells or internal combustion engines, is best-suited for longer-term energy storage and can be stored in the form of a compressed gas or a cryogenic liquid or chemically bonded in hydrides (1). The most attractive hydrogen storage media are liquid organic hydrogen carriers (LOHCs), because they have relatively high hydrogen content and can be transported and distributed using the existing liquid fuel infrastructure (2–5).
Two strategies for coupling the chemical energy stored in these LOHCs with an energy storage device include thermal dehydrogenation to provide H2(g) for a polymer electrolyte membrane (PEM) fuel cell (expression 1) (6) and electrochemical dehydrogenation to yield protons and electrons in a direct alcohol fuel cell (expressions 2 and 3) (2, 7). In the former (acceptorless strategy), hydrogen release from LOHCs frequently requires high reaction temperatures and expensive platinum group metal (PGM) catalysts. Regeneration of the hydrogen-depleted compounds can be achieved with both PGM and less expensive non-PGM catalysts (e.g., nickel-based); however, elevated hydrogen pressures are needed (8, 9). The latter was the basis for an Energy Frontier Research Center around Electrocatalysis, Transport Phenomena, and Materials for Innovative Energy Storage funded by the Department of Energy (Acknowledgments), which assumed the use of a single electrocatalyst for dehydrogenation and hydrogenation of LOHCs that would also simplify the conventional hydrogen storage process (10). The envisioned partial electrochemical dehydrogenation of LOHCs typically involves expensive PGM catalysts (11–18), with weak bases to serve as proton scavengers in lieu of a proton exchange membrane. Reversing the applied potential in the presence of these same components provides a mechanism for regenerating the LOHCs without the need for elevated hydrogen pressure:
| [1] |
| [2] |
and
| [3] |
A thermodynamic analysis of a variety of potential LOHCs showed that cyclic hydrocarbons exhibit high hydrogen contents but that nitrogen heterocycles exhibit lower reaction enthalpies (9, 10). Nitrogen heterocycles are also more practical, because they display energy densities that are comparable with those of liquid hydrogen, and the theoretical open cell potentials of these materials are calculated to be close to or exceed the potential of the hydrogen–oxygen fuel cell (11). In addition, the overpotential of their electrooxidation is also smaller compared with cyclic hydrocarbons (12). However, basic nitrogen heterocycles are not compatible with commonly used acidic proton exchange membranes because of the formation of a nonconductive salt. One alternative class of LOHCs that does not suffer from this problem is hydroxyl-containing compounds (e.g., alcohols and diols) that feature reasonable hydrogen content and low oxidation potentials. Both mono- and polysubstituted alcohols have been proposed as hydrogen storage materials (13) and used as fuel for direct alcohol fuel cells, usually in the form of an aqueous alkaline solution (14).
Homogeneous catalysts for the acceptorless dehydrogenation of primary and secondary alcohols for the most part contain precious metals, such as Ru (19), Rh (20), and Ir (21). By comparison, the same reaction with nonprecious, earth-abundant metal catalysts is, so far, a relatively unexplored area in the literature. The first cobalt catalyst bearing a noninnocent bis(dicyclohexylphosphino)amine (PNPCy) ligand for acceptorless dehydrogenation of alcohols was reported by Hanson and coworkers (22, 23). Recently, Beller and coworkers (24) have shown hydrogen production from methanol in the presence of KOH with octahedral iron complexes (PNPiPr)Fe(H)(CO)X [X = BH4 (1) and X = Br (2)]. Remarkably, very low catalyst loadings (parts per million level) were used, and catalysis was performed at 91 °C, which suggests high thermal stability of these iron complexes and related intermediate species. However, Guan and coworkers (25) have accomplished ester hydrogenation using catalyst 1. In addition to these studies, it was recently shown that the same iron complexes can also efficiently catalyze the reversible dehydrogenation–hydrogenation of N-heterocycles (26). Yamaguchi et al. (27) have also reported Cp*Ir complexes with substituted pyridonate ligands that catalyze the reversible acceptorless dehydrogenation of 1,2,3,4-tetrahydroquinoline to quinoline in boiling xylene. Dehydrogenation is harder and routinely produced lower yields than hydrogenation, but with the 5-trifluoromethylpyridonate ligand, a quantitative yield was achieved in both directions (27). A computational analysis of the proposed catalytic cycle showed that two major pathways are possible: through a bifunctional species with coordinated pyridonate ligand or through a monomeric Cp*Ir(H)Cl complex (28).
As part of our ongoing effort to develop another strategy, specifically electrocatalysts for reversible partial oxidation (dehydrogenation) of alcohol-based fuels, we concentrated on the possibility of converting a known dehydrogenation catalyst to an electrocatalyst through separation of protons and electrons. We regard separately extracting protons and electrons in a catalytic dehydrogenation reaction together with the microscopic reverse (29) (i.e., separately injecting protons and electrons to effect substrate hydrogenation) as fundamental in the development of a reversible alcohol dehydrogenation–hydrogenation electrocatalyst. It was clear at the outset that the possibility of oxidation of intermediate species containing metals in low-oxidation states during dehydrogenation and the competing proton reduction to H2 in the reverse reaction substantially limited our selection of possible candidates. We recently reported electrocatalytic properties of an iridium amino-olefin complex [Ir(trop2DACH)][OTf], which is capable of catalyzing alcohol dehydrogenation with chemical oxidants as well as electrocatalytic dehydrogenation of primary alcohols with excellent faradaic efficiency (30). Two catalytic systems capable of oxidizing alcohols with a chemical oxidant (ferrocenium cation) in the presence of a base as a proton acceptor have been very recently described in literature (30, 31).
In this report, we describe catalytic systems that address both strategies outlined above as part of a unified effort to develop catalysts for reversible dehydrogenation of organic fuels in energy generation and storage reactions. These catalysts show reversible acceptorless (expression 1) and partial oxidative (expression 2) dehydrogenation of alcohols using non-PGM (iron-based) and PGM (iridium-based) catalysts, respectively.
Results and Discussion
Reversible Acceptorless Dehydrogenation of Alcohols with Iron Complexes.
We initially studied acceptorless dehydrogenation of 1-phenylethanol with catalysts iPr[PN(H)P]Fe(H)(CO)(HBH3) (1) and iPr[PN(H)P]Fe(H)(CO)(Br) (2). On investigating a variety of conditions, it was determined that the catalytic reaction was best carried out in toluene at 120 °C. Under these conditions, when 1 mol% 1 was used as the catalyst, 1-phenylethanol was quantitatively converted to acetophenone within 24 h as judged by GC-MS and NMR spectroscopy. In a closed system, only 28% of acetophenone formed after 24 h. Complex 2 was also found to be an active catalyst in the presence of a catalytic amount of KOtBu; however, a lower conversion was achieved after the same amount of time. Therefore, we focused on complex 1 to investigate the scope of substrates for the iron-catalyzed acceptorless dehydrogenation of alcohols.
As depicted in Table 1, secondary benzylic alcohols (Table 1, entries 1–4) were successfully dehydrogenated by catalyst 1, and the corresponding acetophenone derivatives were isolated in good yields (64–92%). A variety of functionalities, such as -OMe, -NO2, and -Cl, was unaffected under the catalytic conditions. Electronic influence on the dehydrogenation activity seemed to be significant, because a substrate containing an electron-withdrawing -NO2 group (Table 1, entry 3) reacted much faster than the one with an electron-donating -OMe group (Table 1, entry 2) based on respective NMR conversions after 12 h. For the -OMe derivative, 68% conversion was achieved after 12 h, whereas 100% conversion was observed for the -NO2 derivative after the same time. A more detailed kinetic analysis of the mechanism is required to rationalize these electronic effects, and these studies are currently underway. In addition to the aromatic substrates, cyclohexanol (Table 1, entry 5) was also successfully dehydrogenated to give cyclohexanone. In contrast to 1, activation of 2 with a base is required (SI Appendix, Table S1).
Table 1.
Iron-catalyzed acceptorless dehydrogenation of alcohols
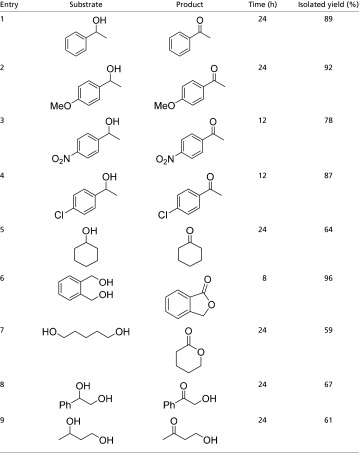 |
[1] = 0.005 M, [substrate] = 0.5 M.
Because diols can potentially release 4 eq of H to afford lactones, we investigated the reactivity of complex 1 toward several diol substrates. When 1,2-benzenedimethanol (Table 1, entry 6) was subjected to our catalytic conditions, it readily produced the corresponding lactone phthalide. Because this reaction suggested the formation of a transient aldehyde intermediate from a primary alcohol, we tried to verify its formation by performing dehydrogenation of benzyl alcohol itself. However, benzyl benzoate was isolated as the sole product as a result of an intermolecular condensation process; 1,5-pentanediol (Table 1, entry 7) was also dehydrogenated successfully to afford δ-valerolactone. Several research groups have previously reported homogeneous catalysts for the base-free oxidation of diols to lactones in the presence of a hydrogen acceptor, such as acetone (32–35). However, reports related to the acceptorless conversion of the same are extremely rare in the literature (36–39). To the best of our knowledge, only recently have a few iron-based homogeneous catalysts been reported for this transformation (40). Fe-catalyzed acceptorless alcohol dehydrogenation with a base as cocatalyst was also recently reported (41).
Reaction of diols with both primary and secondary hydroxyl groups with catalyst 1 results in similar yield and chemoselectivity (exclusive dehydrogenation of the secondary alcohol moiety) for compounds with different acidity of the methine C–H bond (Table 1, entry 8 compared with entry 9). This preference can be explained by the difference in thermodynamics for dehydrogenation of primary and secondary hydroxyl groups. For example, the Gibbs free energy of dehydrogenation for 1-butanol is >30 kJ/mol more positive than that for 2-butanol [+33.5 vs. +2.5 kJ/mol at 120 °C based on values from the National Institute of Standards and Technology database (42)].
To show the possibility of using these iron catalysts for the reverse reaction of alcohol dehydrogenation, we examined their activity toward the hydrogenation of a corresponding ketone (Table 2). Although Guan and coworkers (25) have shown that complex 1 reduces ester functionalities with 10 atm H2, hydrogenation of a simple ketone has not been reported so far. In our investigation, we found that 1 mol% complex 1 catalyzes quantitative hydrogenation of 4-methoxyacetophenone with 5.4 atm H2 pressure in toluene at room temperature (Table 2, entry 1). Both complexes 2 and iPr[PNP]Fe(H)(CO) (3) also served as equally effective catalysts in this hydrogenation reaction (Table 2, entries 2 and 3).
Table 2.
Iron-catalyzed hydrogenation of 4'-methoxyacetophenone
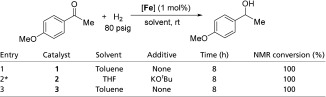 |
10 mol% KOtBu was added. [Catalyst] = 0.005 M, [KOtBu] = 0.05 M, [4′-methoxyacetophenone] = 0.5 M.
A plausible mechanism for the iron-catalyzed alcohol dehydrogenation is outlined in Scheme 1. Based on our current understanding of the dehydrogenation of N-heterocycles (26), we propose that the amido-iron complex 3 is the active catalyst in the alcohol dehydrogenation. Consistent with this hypothesis, catalytic hydrogenation of 4-methoxyacetophenone (Table 2, entry 3) and dehydrogenation of 1-phenylethanol (in toluene at 120 °C) were quantitative when 1 mol% 3 was directly used as the catalyst. The other iron species that is possibly involved in the catalytic cycle is a trans-dihydride complex (4). The existence of this species, which tends to lose H2 even at room temperature and thereby, regenerates the active dehydrogenation intermediate 3, was previously supported by NMR spectroscopy and trapping experiments (26). A computational analysis using density functional theory (DFT) suggests that the dehydrogenation of alcohol, a key elementary step, occurs in a concerted fashion through metal–ligand cooperation (43). A stepwise pathway involving an iron-alkoxide intermediate (shown in Scheme 1) is also possible, although less likely, because it would require higher activation energy (DFT calculations are shown in ref. 43). Catalytic results mentioned in this report indicate that iron complexes 1–3 represent a rare class of homogeneous catalysts for reversible dehydrogenation/hydrogenation of alcohols and ketones.
Scheme 1.

Proposed pathway for the alcohol dehydrogenation on iron PNP complexes.
Reversible Oxidative Dehydrogenation of Primary Alcohols with Iridium Complexes.
To drive a dehydrogenation reaction toward separate generation of protons and electrons requires a stoichiometric oxidant combined with a weak base as a proton sink, and for the reverse reaction (catalytic hydrogenation), a stoichiometric amount of reductant is necessary combined with a (weak) conjugate acid as a proton source. Herein, we studied two iridium(I) complexes, [Ir(trop2DACH)][OTf] (5), a highly efficient catalyst used in the dehydrogenation of a broad range of primary alcohols to aldehydes (44), and the related [Ir(trop2DAD)][OTf] (6) (45), which features an unsaturated and sterically less encumbered ligand framework, as reversible alcohol dehydrogenation–hydrogenation catalysts (Scheme 2).
Scheme 2.

Reversible oxidative dehydrogenation of 4-methoxybenzyl alcohol catalyzed by complexes 5 and 6.
Dehydrogenation of benzylic and allylic alcohols proceeds at room temperature with very low loadings of 5 (0.01 mol%) and requires catalytic amounts of strong base (e.g., KOtBu; 0.03 mol%) and stoichiometric 1,4-benzoquinone (BQ) as a hydrogen (H2) scavenger (44, 46). We now report our findings with 5 and 6 for alcohol dehydrogenation in terms of separately extracting protons and electrons through chemical catalysis results that used stoichiometric oxidants (e.g., ferrocenium) combined with a weak base (phenolate). We targeted separation of proton and electron transfer processes (instead of simultaneous transfer of protons and electrons to the same molecule, such as in BQ) to show the first necessary step in a broader strategy to transition Grützmacher’s system to an electrocatalytic (i.e., electrode-driven) dehydrogenation and use such complexes as electrocatalysts (30). To verify the possibility of operating these catalysts in both directions, we have also studied the reverse reaction, separately injecting protons and electrons for aldehyde hydrogenation using 5 and 6 combined with stoichiometric reductants (e.g., cobaltocene) and weak conjugate acid (phenol).
Considering the high efficiency reported in the original system using 5 with stoichiometric BQ (example, TOF greater than 150,000 h−1 for benzyl alcohol dehydrogenation in chlorobenzene at 90 °C) (44, 46), we used phenolate anion as a substitute base combined with various chemical oxidants for the catalytic dehydrogenation of 4-methoxybenzyl alcohol to p-anisaldehyde (Table 3). Oxidations were carried out anaerobically at room temperature with 0.03 mol% 5 or 0.1 mol% 6 and required 2 eq each phenolate and oxidant relative to 4-methoxybenzyl alcohol. Oxidants were surveyed with each catalyst over a relatively narrow range of chemical potentials in o-dichlorobenzene (o-DCB). All were greater than −0.13 V vs. the ferrocenium/ferrocene couple (Fc+/0), which is the peak oxidation potential (observed to be pseudoreversible at scan rates ≥ 1 V/s) that we reported for monodeprotonated 5, namely the neutral Ir-amido-amine complex [Ir(trop2DACH-1H)]0 (30).
Table 3.
Dehydrogenation of 4-methoxybenzyl alcohol with chemical oxidants in the presence of sodium phenolate as base in o-DCB using 5 (0.03 mol%) and 6 (0.1 mol%) as catalysts
| Entry | Catalyst | Oxidant | E1/2 (V) | Time (h) | GC yield (%) |
| 1 | 5 | [Fe(η5-C5H4Me)2]BF4 | −0.11 | 2 | 75 |
| 2 | 5 | [FeCp2]PF6 | 0.00 | 2 | 70 |
| 3 | 5 | [Fe(η5-C5H4C(O)Me)Cp]BF4 | 0.25 | 2 | 11 |
| 4 | 5 | AgOTf | 0.39* | 2 | 94 |
| 5 | 5 | [Fe(η5-C5H4C(O)Me)2]BF4 | 0.46 | 2 | 4 |
| 6 | 6 | [Fe(η5-C5H4Me)2]BF4 | −0.11 | 4 | 75 |
| 7 | 6 | [FeCp2]PF6 | 0.00 | 4 | 76 |
| 8 | 6 | [Fe(η5-C5H4C(O)Me)Cp]BF4 | 0.25 | 4 | 17 |
| 9 | 6 | AgOTf | 0.39* | 4 | 63 |
| 10 | 6 | [Fe(η5-C5H4C(O)Me)2]BF4 | 0.46 | 4 | 16 |
Reported here as the observed peak potential and not formal E1/2.
From Table 3, we found 5 to be more active and efficient for the dehydrogenation of 4-methoxybenzyl alcohol compared with 6, exhibiting higher rates with lower catalyst loadings while maintaining comparable moderate-to-high conversions to aldehyde. We observed a 94% yield of p-anisaldehyde after 2 h when the reaction was carried out with 5, phenolate, and AgOTf (Table 3, entry 4), whereas the same reaction with 6 gave only 63% yield after 4 h (Table 3, entry 9). Oxidant solubility played an important role in catalysis—acetylated ferrocenium oxidants ([Fe(η5-C5H4C(O)Me)Cp]+ and [Fe(η5-C5H4C(O)Me)2]+ in Table 3, entries 3, 5, 8, and 10) showed very poor solubility and performance in o-DCB compared with [FeCp2]+ and [Fe(η5-C5H4Me)2]+ oxidants. Of significant note was that, by separating proton and electron transfer events in 4-methoxybenzyl alcohol dehydrogenation, no penalties were observed in terms of reaction rate or percentage conversion to aldehyde (for Table 3, entry 4 specifically) relative to the concerted process (i.e., Grützmacher’s 5-BQ system exhibited a TOF of 625 h−1 at 25 °C in chlorobenzene for oxidation of 4-methoxybenzyl alcohol).
Electrochemical studies of 5 and related complexes ([Ir(trop2DACH-1H)]0 and [Ir(trop2DACH-2H)]−) (46) allowed for refinement of our understanding around catalyst activation and determination of a thermochemical cycle (details in SI Appendix). For example, calculation of H atom removal from 5 (homolytic bond dissociation) to form [Ir(trop2DACH-1H)]+ (Scheme 3, Center) was determined to require 79.2 kcal/mol, with comparable energy necessary to repeat the deprotonation/oxidation step (77.6 kcal/mol). (Note that [Ir(trop2DACH-2H)]+ may also be considered as an Ir(III)-bis-amide complex.) By derivation of additional thermochemical data, the pKa of a monodeprotonated monooxidized species, [Ir(trop2DACH-1H)]+, was calculated to be 9.2 (in DMSO), which is actually lower than the pKa1 of 10.5 measured for 5 (46). Together, these data are consistent with activation of 5 beyond simple monodeprotonation and oxidation under our catalysis conditions. Likely, a second deprotonation by phenolate base occurs to engender [Ir(trop2DACH-2H)]0—an intermediate also proposed by Grützmacher in the original KOtBu-initiated BQ-mediated system. Additional oxidation of [Ir(trop2DACH-2H)]0 to [Ir(trop2DACH-2H)]+ is reasonable given that the potentials of chemical oxidants in Table 3 are much higher than the E1/2 measured for interconversion between these two species (−0.20 V vs. Fc+/0) (SI Appendix).
Scheme 3.
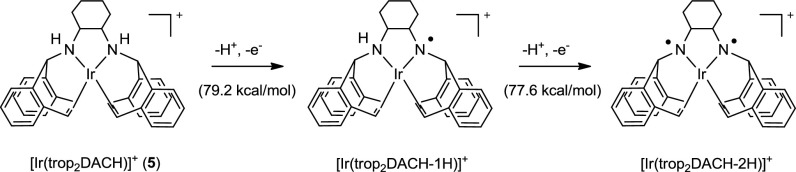
Products of [Ir(trop2DACH)] oxidation.
Although aldehyde yields for 5 (Table 3, entries 1 and 2) and 6 (Table 3, entries 6 and 7) are comparable, albeit under slightly different conditions, our understanding around activation of 6 as a dehydrogenation catalyst is relatively limited. Accommodation of a fifth ligand (i.e., phenolate) by 6 is expected given that the thiophenolate [Ir(trop2DAD)(SPh)]0 is known and has been crystallographically characterized (47). Cyclic voltammetry (CV) of 1 mM 6 in o-DCB/0.08 M n-Bu4NPF6 electrolyte shows two one-electron (quasi)reversible waves (E1/21 = −0.63 V and E1/22 = −1.35 V vs. Fc+/0), which can be attributed to consecutive reduction of the diazabutadiene ligand and is consistent with prior observations in THF (45). Addition of 4 eq phenolate base to 6 results in the appearance of two additional one-electron (quasi)reversible waves at more positive potentials (E1/23 = −0.33 V and E1/24 = −0.06 V vs. Fc+/Fc) (SI Appendix). It is presumed that activation of 6 for dehydrogenation catalysis involves formation of [Ir(trop2DAD)(OPh)]0 followed by oxidation of the coordinated phenolate complex. (Note that 4 mM phenolate does not show electrochemical activity up to +0.6 V vs. Fc+/0, which is well above any potential used in these experiments.)
We also studied the reverse reaction (catalytic hydrogenation of p-anisaldehyde to 4-methoxybenzyl alcohol) using 5 or 6 combined with phenol as a proton source and stoichiometric reductants (Table 4). It was necessary to run hydrogenations with excess phenol (acid), because we observed formation of large amounts of a precipitate, which was identified by 1H NMR, CV, HPLC-TOF/MS, and thermogravimetric analysis (TGA) coupled FTIR as an adduct of cobaltocenium phenolate with phenol ([CoCp2][OPh]·HOPh). Therefore, hydrogenation reactions were carried out with 1 mol% 5 or 6, 5 eq phenol, and 2.5 eq reductant relative to p-anisaldehyde to achieve high yields. Reductants were surveyed with each catalyst over a relatively wide range of redox potentials in o-DCB (Table 4), most being strong enough to activate (reduce) 5 and 6 by one and two electrons, respectively. Reduction of 5 to metalloradical [Ir(trop2DACH)] was reported previously in DMSO (46), and in o-DCB/0.08 M n-Bu4NPF6 electrolyte, the peak potential observed by CV at −1.36 V vs. Fc+/0 indicates that its formation should be possible by [CoCp2], [Co(η5-C5H4Et)2], and [CoCp*2]. Based on CV studies of 6 under the same electrochemical conditions, reductants [FeCp*2] and [Cr(η6-C6H6)2] listed in Table 4 are not strong enough to reduce both C = N bonds in 6.
Table 4.
Hydrogenation of p-anisaldehyde with chemical reductants in the presence of phenol as a proton source in o-DCB using 5 and 6 (1 mol%) as catalysts
| Entry | Catalyst | Reductant | E1/2 (V)* | Time (h) | Yield (%) |
| 1 | 5 | [FeCp*2] | −0.54 | 72 | 0 |
| 2 | 5 | [Cr(η6-C6H6)2] | −1.19 | 72 | 10 |
| 3 | 5 | [CoCp2] | −1.33 | 72 | 60 |
| 4 | 5 | [Co(η5-C5H4Et)2] | −1.44 | 72 | 25 |
| 5 | 5 | [CoCp*2] | −1.95 | 72 | 36 |
| 6 | 6 | [FeCp*2] | −0.54 | 24 | 0 |
| 7 | 6 | [Cr(η6-C6H6)2] | −1.19 | 24 | 3 |
| 8 | 6 | [CoCp2] | −1.33 | 24 | 89 |
| 9 | 6 | [Co(η5-C5H4Et)2] | −1.44 | 24 | 49 |
| 10 | 6 | [CoCp*2] | −1.95 | 24 | 0 |
Relative to Cp2Fe+/Cp2Fe.
Data from Table 4 support our expectations that [FeCp*2] was not strong enough to activate either 5 or 6 for catalytic aldehyde hydrogenation. Likewise, [Cr(η6-C6H6)2] was not expected or observed to activate both 5 and 6. Our initial hydrogenation attempts that used 2.5 eq each phenol and [CoCp2] provided low yields of p-anisaldehyde (48% and 27% with 6 and 5, respectively) and produced large amounts of precipitate that indicated acid-starved conditions of this reaction. To avoid this acid-starved condition, all hydrogenation reactions in Table 4 were run as a 2:1 ratio of phenol to reductant. We identified a definitive sweet spot at the [CoCp2] redox potential and found 6 to be more effective and active compared with 5. An 89% yield of 4-methoxybenzyl alcohol was observed after 24 h at room temperature with 6, phenol, and [CoCp2] (Table 4, entry 8), whereas the same reaction with 5 afforded a 60% yield after 72 h (Table 4, entry 3). Such as was observed in the dehydrogenation reaction, solubility of the reductants used played an important role in the effectiveness of catalytic hydrogenation. [CoCp2*] has a reduction potential sufficient to reduce 5 and 6; however, very poor solubility in o-DCB rendered it largely ineffective with 5 (despite stirring for 3 d) and incapable of reducing 6. (Note that [CoCp2*] remained in a reduced state at the end of the reaction and did not consume/reduce phenol to any significant extent.) Conversely, [Co(η5-C5H4Et)2] is a liquid, and hydrogenations were observed to be homogeneous with near-complete consumption of reductant. With high o-DCB solubility, excess phenol consumed [Co(η5-C5H4Et)2], and low conversions resulted (despite negligible side products).
Summary
In conclusion, we have developed a rare class of homogeneous iron catalysts (1–3) for efficient reversible acceptorless dehydrogenation–hydrogenation of alcohols and diols under mild conditions. Moreover, we showed that it is possible to decouple proton and electron transfer processes in both directions through use of metallocene-based chemical oxidants/reductants, presumably as surrogates for catalytic electrode-driven (de)hydrogenation reactions. This work further illustrates the importance of noninnocent ligands in enabling reversible (de)hydrogenation catalysis. In all cases, catalytically active metal complexes bear such ligands, with nitrogen atoms (capable of forming/breaking N–H bonds) acting as a proton sink/source. The obtained results are of importance for the development of reversible dehydrogenation electrocatalysts and ultimately, the application of such catalysts in regenerative liquid fuel cells using chemically bound hydrogen for energy storage.
Methods
All organometallic compounds were prepared and handled under a nitrogen atmosphere using standard Schlenk and glove box techniques. E1/2 values are reported relative to Cp2Fe+/Cp2Fe and were measured in o-DCB/0.08 M n-Bu4NPF6 as electrolyte at 0.1 V/s with Pt working and counter electrodes and Ag wire/10 mM AgNO3 solution as reference electrode in CH3CN/0.1 M n-Bu4NPF6 electrolyte. Experimental details are in SI Appendix.
General Procedure for the Iron-Catalyzed Dehydrogenation of Alcohols.
In a glove box, a 50-mL flame-dried Schlenk flask equipped with a condenser was charged with an iron catalyst (25 µmol), an alcohol substrate (2.5 or 25 mmol), and 5 mL toluene. The solution was stirred at 120 °C for a specific time under a constant N2 flow. After the reaction, the solution was allowed to cool to room temperature, filtered through a short silica gel column, and eluted with THF. The resulting filtrate was evaporated under vacuum to afford the pure product. 1H and 13C{1H} NMR spectra of the products were recorded in CDCl3 and matched with the chemical shifts reported in the literature. The results are summarized in Table 1.
General Procedure for the Iron-Catalyzed Hydrogenation of 4′-Methoxyacetophenone.
In a glove box, a 25-mL stainless steel Parr pressure reactor was loaded with an iron complex (25 µmol), KOtBu (if required), 4′-methoxyacetophenone (375 mg, 2.5 mmol), and 5 mL toluene (or THF). The reactor was sealed, flushed with H2 three times, and finally, placed under 80 psig H2 pressure. The solution was then stirred at room temperature for 8 h. After the reaction, the solution was filtered through a short silica gel column and eluted with THF. The resulting filtrate was evaporated to dryness to afford the pure hydrogenation product. 1H and 13C{1H} NMR spectra of the product were recorded in CDCl3 and matched with the reported spectra in the literature. For all of the reactions, quantitative conversions were achieved (Table 2).
General Procedure for the Iridium-Catalyzed Oxidative Dehydrogenation of Alcohols.
Dehydrogenation reactions were carried out with 0.03 mol% (5) or 0.1 mol% (6) catalyst; however, both used 1.5 mmol alcohol, 3 mmol oxidant, and 3 mmol sodium phenolate as the base at room temperature in o-DCB (reaction volume ∼ 5 mL). 1H NMR yields were determined against hexamethylbenzene as an internal standard based on peak integration of either methoxy or aromatic protons.
General Procedure for the Iridium-Catalyzed Reductive Hydrogenation of Alcohols.
All hydrogenation reactions used 1 mol% catalyst (5 or 6), 1.25 mmol aldehyde, 3.13 mmol reductant, and 6.26 mmol phenol as acid at room temperature in o-DCB (reaction volume ∼ 10 mL). 1H NMR yields were determined against hexamethylbenzene as an internal standard based on peak integration of either methoxy or aromatic protons.
Supplementary Material
Acknowledgments
This material is based on work supported as part of the Center for Electrocatalysis, Transport Phenomena, and Materials for Innovative Energy Storage (an Energy Frontier Research Center funded by Department of Energy, Office of Science, Office of Basic Energy Sciences Award DE-SC0001055), and funding was provided by a grant from Empire State Development.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1420199112/-/DCSupplemental.
References
- 1.Weidenthaler C, Felderhoff M. Solid-state hydrogen storage for mobile applications: Quo vadis? Energy Environ Sci. 2011;4(7):2495–2502. [Google Scholar]
- 2.Crabtree RH. Hydrogen storage in liquid organic heterocycles. Energy Environ Sci. 2008;1(1):134–138. [Google Scholar]
- 3.Ichikawa M. Organic liquid carriers for hydrogen storage. In: Walker G, editor. Solid-State Hydrogen Storage: Materials and Chemistry. CRC; Boca Raton, FL: 2008. pp. 500–532. [Google Scholar]
- 4.Teichmann D, Arlt W, Wasserscheid P, Freymann R. A future energy supply based on liquid organic hydrogen carriers (LOHC) Energy Environ Sci. 2011;4(8):2767–2773. [Google Scholar]
- 5.Yadav M, Xu Q. Liquid-phase chemical hydrogen storage materials. Energy Environ Sci. 2012;5(12):9698–9725. [Google Scholar]
- 6.Pez GP, Scott AR, Cooper AC, Cheng H. 2008. US Patent 7429372.
- 7.Soloveichik GL, Zhao J-C. 2008. US Patent Appl 20080248345.
- 8.Biniwale RB, Rayalu S, Devotta S, Ichikawa M. Chemical hydrides: A solution to high capacity hydrogen storage and supply. Int J Hydrogen Energy. 2008;33(1):360–365. [Google Scholar]
- 9.Bond GC. Metal-Catalysed Reactions of Hydrocarbons. Springer; Berlin: 2006. [Google Scholar]
- 10.Soloveichik GL, Lemmon JP, Zhao J-C. 2008. US Patent Appl 2008/0248339.
- 11.Rodríguez M, et al. Synthesis, structure, and redox and catalytic properties of a new family of ruthenium complexes containing the tridentate bpea ligand. Inorg Chem. 2001;40(17):4150–4156. doi: 10.1021/ic010064q. [DOI] [PubMed] [Google Scholar]
- 12.Hino T, Wada T, Fujihara T, Tanaka K. Redox behavior of new Ru-dioxolene and -ammine complexes and catalytic activity toward electrochemical oxidation of alcohol under mild conditions. Chem Lett. 2004;33(12):1596–1597. [Google Scholar]
- 13.Cheung K-C, Wong W-L, Ma D-L, Lai T-S, Wong K-Y. Transition metal complexes as electrocatalysts—development and applications in electro-oxidation reactions. Coord Chem Rev. 2007;251(17-20):2367–2385. [Google Scholar]
- 14.Serra D, Correia MC, McElwee-White L. Iron and ruthenium heterobimetallic carbonyl complexes as electrocatalysts for alcohol oxidation: Electrochemical and mechanistic studies. Organometallics. 2011;30(21):5568–5577. [Google Scholar]
- 15.Ozawa H, Hino T, Ohtsu H, Wada T, Tanaka K. A new type of electrochemical oxidation of alcohols mediated with a ruthenium–dioxolene–amine complex in neutral water. Inorg Chim Acta. 2011;366(1):298–302. [Google Scholar]
- 16.Yamazaki S, et al. Electrocatalytic oxidation of alcohols by a carbon-supported Rh porphyrin. Chem Commun (Camb) 2012;48(36):4353–4355. doi: 10.1039/c2cc30888f. [DOI] [PubMed] [Google Scholar]
- 17.Vannucci AK, et al. Water oxidation intermediates applied to catalysis: Benzyl alcohol oxidation. J Am Chem Soc. 2012;134(9):3972–3975. doi: 10.1021/ja210718u. [DOI] [PubMed] [Google Scholar]
- 18.Hull JF, et al. Reversible hydrogen storage using CO2 and a proton-switchable iridium catalyst in aqueous media under mild temperatures and pressures. Nat Chem. 2012;4(5):383–388. doi: 10.1038/nchem.1295. [DOI] [PubMed] [Google Scholar]
- 19.Muthaiah S, Hong SH. Acceptorless and base-free dehydrogenation of alcohols and amines using ruthenium-hydride complexes. Adv Synth Catal. 2012;354(16):3045–3053. [Google Scholar]
- 20.Trincado M, Banerjee D, Grutzmacher H. Molecular catalysts for hydrogen production from alcohols. Energy Environ Sci. 2014;7(8):2464–2503. [Google Scholar]
- 21.Zeng G, Sakaki S, Fujita K-i, Sano H, Yamaguchi R. Efficient catalyst for acceptorless alcohol dehydrogenation: Interplay of theoretical and experimental studies. ACS Catal. 2014;4(3):1010–1020. [Google Scholar]
- 22.Zhang G, Hanson SK. Cobalt-catalyzed acceptorless alcohol dehydrogenation: Synthesis of imines from alcohols and amines. Org Lett. 2013;15(3):650–653. doi: 10.1021/ol303479f. [DOI] [PubMed] [Google Scholar]
- 23.Zhang G, Vasudevan KV, Scott BL, Hanson SK. Understanding the mechanisms of cobalt-catalyzed hydrogenation and dehydrogenation reactions. J Am Chem Soc. 2013;135(23):8668–8681. doi: 10.1021/ja402679a. [DOI] [PubMed] [Google Scholar]
- 24.Alberico E, et al. Selective hydrogen production from methanol with a defined iron pincer catalyst under mild conditions. Angew Chem Int Ed Engl. 2013;52(52):14162–14166. doi: 10.1002/anie.201307224. [DOI] [PubMed] [Google Scholar]
- 25.Chakraborty S, et al. Iron-based catalysts for the hydrogenation of esters to alcohols. J Am Chem Soc. 2014;136(22):7869–7872. doi: 10.1021/ja504034q. [DOI] [PubMed] [Google Scholar]
- 26.Chakraborty S, Brennessel WW, Jones WD. A molecular iron catalyst for the acceptorless dehydrogenation and hydrogenation of N-heterocycles. J Am Chem Soc. 2014;136(24):8564–8567. doi: 10.1021/ja504523b. [DOI] [PubMed] [Google Scholar]
- 27.Yamaguchi R, Ikeda C, Takahashi Y, Fujita K. Homogeneous catalytic system for reversible dehydrogenation-hydrogenation reactions of nitrogen heterocycles with reversible interconversion of catalytic species. J Am Chem Soc. 2009;131(24):8410–8412. doi: 10.1021/ja9022623. [DOI] [PubMed] [Google Scholar]
- 28.Li H, Jiang J, Lu G, Huang F, Wang Z-X. On the “reverse gear” mechanism of the reversible dehydrogenation/hydrogenation of a nitrogen heterocycle catalyzed by a Cp*Ir Complex: A computational study. Organometallics. 2011;30(11):3131–3141. [Google Scholar]
- 29.Tolman RC. The Principles of Statistical Mechanics. Dover; New York: 1938. [Google Scholar]
- 30.Bonitatibus PJ, Jr, Rainka MP, Peters AJ, Simone DL, Doherty MD. Highly selective electrocatalytic dehydrogenation at low applied potential catalyzed by an Ir organometallic complex. Chem Commun (Camb) 2013;49(90):10581–10583. doi: 10.1039/c3cc46051g. [DOI] [PubMed] [Google Scholar]
- 31.Weiss CJ, Das P, Miller DL, Helm ML, Appel AM. Catalytic oxidation of alcohol via nickel phosphine complexes with pendant amines. ACS Catal. 2014;4(9):2951–2958. [Google Scholar]
- 32.Lin Y, Zhu X, Zhou Y. A convenient lactonization of diols to γ- and δ-lactones catalyzed by transition metal polyhydrides. J Organomet Chem. 1992;429(2):269–274. [Google Scholar]
- 33.Endo Y, Bäckvall J-E. Aerobic lactonization of diols by biomimetic oxidation. Chemistry. 2011;17(45):12596–12601. doi: 10.1002/chem.201102168. [DOI] [PubMed] [Google Scholar]
- 34.Buntara T, et al. Caprolactam from renewable resources: Catalytic conversion of 5-hydroxymethylfurfural into caprolactone. Angew Chem Int Ed Engl. 2011;50(31):7083–7087. doi: 10.1002/anie.201102156. [DOI] [PubMed] [Google Scholar]
- 35.Murahashi S, Naota T, Ito K, Maeda Y, Taki H. Ruthenium-catalyzed oxidative transformation of alcohols and aldehydes to esters and lactones. J Org Chem. 1987;52(19):4319–4327. [Google Scholar]
- 36.Zhang J, Balaraman E, Leitus G, Milstein D. Electron-rich PNP- and PNN-type ruthenium(II) hydrido borohydride pincer complexes. Synthesis, structure, and catalytic dehydrogenation of alcohols and hydrogenation of esters. Organometallics. 2011;30(21):5716–5724. [Google Scholar]
- 37.Zhao J, Hartwig JF. Acceptorless, neat, ruthenium-catalyzed dehydrogenative cyclization of diols to lactones. Organometallics. 2005;24(10):2441–2446. [Google Scholar]
- 38.Muñiz K. Bifunctional metal-ligand catalysis: Hydrogenations and new reactions within the metal-(di)amine scaffold. Angew Chem Int Ed Engl. 2005;44(41):6622–6627. doi: 10.1002/anie.200501787. [DOI] [PubMed] [Google Scholar]
- 39.Tseng K-NT, Kampf JW, Szymczak NK. Base-free, acceptorless, and chemoselective alcohol dehydrogenation catalyzed by an amide-derived NNN-ruthenium(II) hydride complex. Organometallics. 2013;32(7):2046–2049. [Google Scholar]
- 40.Kamitani M, Ito M, Itazaki M, Nakazawa H. Effective dehydrogenation of 2-pyridylmethanol derivatives catalyzed by an iron complex. Chem Commun (Camb) 2014;50(59):7941–7944. doi: 10.1039/c4cc01498g. [DOI] [PubMed] [Google Scholar]
- 41.Song H, Kang B, Hong SH. Fe-catalyzed acceptorless dehydrogenation of secondary benzylic alcohols. ACS Catal. 2014;4(9):2889–2895. [Google Scholar]
- 42.NIST NIST Chemistry WebBook. 2005. Available at webbook.nist.gov/chemistry. Accessed January 2, 2015.
- 43.Chakraborty S, et al. Well-defined iron catalysts for the acceptorless reversible dehydrogenation-hydrogenation of alcohols and ketones. ACS Catal. 2014;4(11):3994–4003. [Google Scholar]
- 44.Königsmann M, et al. Metalloenzyme-inspired catalysis: Selective oxidation of primary alcohols with an iridium-aminyl-radical complex. Angew Chem Int Ed Engl. 2007;46(19):3567–3570. doi: 10.1002/anie.200605170. [DOI] [PubMed] [Google Scholar]
- 45.Breher F, et al. Tropad: A new ligand for the synthesis of water-stable paramagnetic [16+1]-electron rhodium and iridium complexes. Chemistry. 2003;9(16):3859–3866. doi: 10.1002/chem.200204700. [DOI] [PubMed] [Google Scholar]
- 46.Donati N, Königsmann M, Stein D, Udino L, Grützmacher H. Iridium aminyl radical complexes as catalysts for the catalytic dehydrogenation of primary hydroxyl functions in natural products. C R Chim. 2007;10(8):721–730. [Google Scholar]
- 47.Häbe K. Chemie von “Non-Innocent” Liganden, Rhodium und Iridium in Stickstoff-Olefinkomplexen. Doctor of Science. ETH Zürich; Zurich: 2006. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.