

Abstract
Acute lung injury (ALI) occurs frequently in patients with severe traumatic brain injury (TBI) and is associated with a poor clinical outcome. Aquaporins (AQPs), particularly AQP1 and AQP4, maintain water balances between the epithelial and microvascular domains of the lung. Since pulmonary edema (PE) usually occurs in the TBI-induced ALI patients, we investigated the effects of a thaliporphine derivative, TM-1, on the expression of AQPs and histological outcomes in the lung following TBI in rats. TM-1 administered (10 mg/kg, intraperitoneal injection) at 3 or 4 h after TBI significantly reduced the elevated mRNA expression and protein levels of AQP1 and AQP4 and diminished the wet/dry weight ratio, which reflects PE, in the lung at 8 and 24 h after TBI. Postinjury TM-1 administration also improved histopathological changes at 8 and 24 h after TBI. PE was accompanied with tissue pathological changes because a positive correlation between the lung injury score and the wet/dry weight ratio in the same animal was observed. Postinjury administration of TM-1 improved ALI and reduced PE at 8 and 24 h following TBI. The pulmonary-protective effect of TM-1 may be attributed to, at least in part, downregulation of AQP1 and AQP4 expression after TBI.
1. Introduction
Acute lung injury (ALI) commonly occurs after traumatic brain injury (TBI) [1, 2]. Respiratory failure is the most common complication of the nonneurological organ system in patients with TBI [3]. ALI has been reported for a long time, but it remains poorly understood because of the complexity of its pathophysiological mechanisms [4]. ALI is characterized by marked pulmonary vascular extravasation and intra-alveolar accumulation of protein-rich edema fluid with perivascular edema [5–7]. Water homeostasis is a crucial element of major pathophysiological processes occurring in the respiratory system. Aquaporins (AQPs), water channel proteins, play a pivotal role in this homeostasis [8–10]. Modulation of lung AQPs expression and function by using pharmacological or genetic methods may have clinical utility [11, 12]. AQPs including AQP1 and AQP4 regulate water movement across the epithelial and endothelial barriers of the lung [4, 13]. Specific deletions of AQPs in mice markedly decreased water movement across air space-capillary barriers [14]. AQP1 is expressed in pleural membranes, when pleural fluid accumulation was observed in the peritoneal cavity [14]. AQPs are also involved in lung growth and angiogenesis [15]. However, the roles of AQP1 and AQP4 expression in pulmonary edema remain controversial. Previous studies have shown that AQP1 knockout inhibits water transport [16] and reduces sublaryngeal edema [13]. Downregulation of AQP1 expression in alveolar microvessels may act as a compensatory mechanism to protect against the formation of excessive pulmonary edema (PE) in chronic heart failure [17]. However, there have also been reports suggesting that pulmonary inflammation and edema were associated with a marked reduction in the AQP1 mRNA expression level in the lung [18, 19] and the AQP1 mRNA expression level was negatively correlated with the level of the wet-to-dry weight ratio (wet/dry weight ratio) [20]. AQP4 mRNA expression is upregulated in alveolar type II (ATII) cells [21] and AQP4 is believed to regulate fluid exchange between the alveolar space and the alveolar epithelium barrier in ALI [22]. However, AQP4 deletion did not impair water transport in the airways [23]. Thaliporphine is a phenolic aporphine alkaloid obtained from Chinese herbs that possesses antioxidant and α-1 adrenoceptor antagonistic activities. A previous study showed that thaliporphine reduces the risk of cardiac muscle necrosis and arrhythmia after heart ischemia [24]. Moreover, the positive effects of thaliporphine may be attributed to the suppression of TNF-α, NO·, and O2·− production [25]. Hence, this study was aimed at investigating the effect of TM-1 on TBI-induced ALI and AQPs expression in the lung.
2. Materials and Methods
2.1. Drug Preparation
TM-1 is a derivative of (+)-thaliporphine dissolved in 100% dimethyl sulfoxide (DMSO) (30 mg in 1 mL DMSO). A dose of 10 mg/kg was used for intraperitoneal injection (i.p.).
2.2. Surgical Procedures
All animals (male Sprague-Dawley rats; body weight 250–300 g) were treated in accordance with the International Guidelines for animal research. The study design was approved by the animal ethics committee (Approval number LAC-100-0143) of Taipei Medical University. The animals were housed in groups in a controlled environment of temperature (21–25°C) and humidity (45–50%) with a 12-h light/dark cycle and ad libitum access to pellet chow and water. The CCI injury procedure was performed as described previously [26]. An impact velocity of 4 m/s and a deformation depth of 2 mm below the dura were used. The bone flap was immediately replaced and sealed, and the scalp was closed with sutures. Body temperature was monitored throughout surgery by using a rectal probe; the temperature was maintained at 37.0 ± 0.5°C using a heated pad. The rats were placed in a heated cage to maintain body temperature while recovering from anesthesia. Sham-operated rats were subjected to craniotomy as described previously but not to CCI; the impact tip was placed lightly on the dura before sealing the wound. After TBI or sham surgery, all animals were housed under the aforementioned conditions.
2.3. Time Point for TM-1 Administration
The rats received an IP TM-1 injection of 10 mg/kg (0.1 cc) or vehicle injection (0.1 cc DMSO) 3 or 4 h after TBI. The animals were sacrificed 8 or 24 h after TBI. The TM-1 administration dose was selected on the basis of pilot studies conducted in our laboratory, in which doses of 5 and 10 mg/kg were tested; the 10-mg/kg dose showed neuroprotective effects by improving behavioral deficits (unpublished results). After the CCI injury procedure, the rat tissues were processed for histological examination, wet/dry weight ratio calculation, real-time quantitative reverse transcription (RT)-PCR, western blotting, and lung injury scoring at 8 or 24 h (n = 5/time point). Five sham-operated rats were used for analysis and were administered a corresponding volume of the vehicle.
2.4. PE Was Evaluated by the Wet/Dry Weight Ratio
The wet/dry weight ratio was used as an index of PE after TBI. To measure the index, the fresh upper right lobe of the lung was harvested (n = 5). Tissue weight was measured immediately after its excision (wet weight), and the tissue was then dried in an oven at 60°C for 5 days to attain a constant weight. The wet/dry weight ratio was calculated by dividing the wet weight by the dry weight as described previously [27].
2.5. RNA Extraction, Reverse Transcription, and Real-Time Quantitative PCR (qPCR)
Right lung tissues from the rats with TBI or sham-operated rats (n = 5) were removed after postanesthesia decapitation. Total RNA was extracted from half of the right middle lobe of the lung by using TRIzol RNA isolation reagents (Life Technologies, USA). The purity and quality of total extracted RNA were confirmed by determining the ratio of absorbance at 260 nm to that at 280 nm. Total RNA was subjected to reverse transcription by using the ReverTra Ace Set Kit according to the manufacturer's protocol (Purigo, Taiwan). The RT mixtures were diluted and used as templates in subsequent qPCR or stored at −20°C. qPCR analysis was performed using a Rotor-Gene Q detector (Qiagen, USA). For mRNA measurement, diluted cDNA was amplified using the QuantiFast SYBR Green PCR kit. Using the PCR kit, thermal cycling was initiated with a first denaturation step of 5 min at 95°C, followed by 40 cycles of 95°C for 10 s and 60°C for 30 s. Primers used for the qPCR assay were as follows: 5′-GACACCTCCTGGCTATTGACTACA-3′ (forward) and 5′-CCGCGGAGCCAAAGG-3′ (reverse) for rat AQP1, 5′-AGCCTGGGATCCACCATC-3′ (forward) and 5′-TGCAATGCTGAGTCCAAAGC-3′ (reverse) for rat AQP4, 5′-GACCCAGATCATGTTTGAGACCTTC-3′ (forward) and 5′-GAGTCCATCACAATGCCWGTGG-3′ (reverse) for rat β-actin, and 5′-GACATGCCGCCTGGAGAAAC-3′ (forward) and 5′-AGCCCAGGATGCCCTTTAGT-3′ (reverse) for rat Gapdh. The Rotor-Gene Q detector synchronized with the thermal cycler measured fluorescent emission during each extension step. Each sample was run in triplicate. Relative standard curves for the candidate gene and the rat β-actin and Gapdh gene were plotted each time the genes were analyzed. The comparative threshold cycle (Ct) value of the β-actin and Gapdh genes, which were constant irrespective of experimental conditions, was used as reference genes. All PCR products were analyzed in the geometric range of the exponential phase during PCR amplification. AQP1, AQP4, β-actin, and Gapdh mRNA levels were calculated by the Ct method [28]. The reference genes were individually β-actin and Gapdh for mRNA. The resulting change in ΔCt values (expressed as ΔΔCt) was converted to a linear form using 2(−ΔΔCt). This linear value was used in subsequent statistical analyses.
2.6. Western Blotting
Total proteins were extracted from tissues from half of the non-fixed right middle lobe of the lung of the rats with TBI or sham-operated rats by using RIPA buffer (25 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) containing protease inhibitor cocktail (complete, Mini, EDTA-free; Roche Applied Science, Germany) and homogenized using a BBX24 Bullet Blender homogenizer (Next Advance, Inc., USA). After quantitative analysis with the Bradford protein assay by using Bio-Rad Dye Reagent (500-0006, Bio-Rad, USA), the protein samples were denatured with 5x sample buffer (10% SDS, 25% beta-mercaptoethanol, 50% glycerol, 0.25 M Tris-HCl, pH 6.8, 0.01% bromophenol blue) and incubated in a water bath at 95°C for 10 min. The amounts of tissue protein lysates loaded into 10% SDS-polyacrylamide gels and resolved by standard electrophoresis (Novex, Carlsbad, CA, USA) were 30 μg for AQP1 and 100 μg for AQP4, respectively. The gels were transferred onto PVDF filters incubated with a specific primary antibody to AQP1 (1 : 1000) (Santa Cruz Biotechnology, Inc., USA) and AQP4 (1 : 1000) (Santa Cruz Biotechnology, Inc., USA). HRP conjugated secondary antibodies were goat anti-mouse IgG (1 : 1000) for β-actin and goat anti-rabbit IgG (1 : 1000) for AQP1 and AQP4. These secondary antibodies were purchased from the Jackson ImmunoRes (West Grove, PA). All blots were reported with beta-actin as an internal standard (1 : 5000) (Millipore, USA). Bands of interest were visualized using ECL reagents (PerkinElmer, Waltham, MA, USA) and quantified using the UVP BioImaging system (Biospectrum AC Imaging System, CA, USA) and ImageJ software (National Institutes of Health, USA) [28].
2.7. Analysis of Lung Injury
Fresh right lower lung lobes were immediately fixed in 10% formalin and processed using the paraffin-embedding technique. Slides were stained with hematoxylin and eosin. The lung injury score was analyzed using a semiquantitative scoring system blinded to the treatment group. The lung injury scoring system included the score of neutrophils in the alveolar space, neutrophils in the interstitial space, hyaline membrane formation, proteinaceous debris filling the air spaces, and alveolar septal thickening. These criteria were accessible to both laboratory researchers and pathologists [29]. Ten microscopic fields from each slide were analyzed. The sums of tissue slides were averaged to evaluate the severity of lung injury.
2.8. Statistical Analyses
All data are presented as mean ± SEM. Between-group comparisons were made by one-way analysis of variance with the nonparametric Kruskal-Wallis test with Dunn's multiple comparison posttest. Instat 3 software (GraphPad Software Inc.; San Diego, CA, USA) was used for all analyses. P values of <0.05 were considered statistically significant. Pearson's regression analysis was conducted to analyze the correlation between the wet/dry weight ratio and the lung injury score in SAS version 7.1.
3. Results
3.1. TM-1 Administration 3 or 4 h after Injury Reduced Pulmonary Edema Developed at 8 or 24 h after TBI
The wet/dry weight ratio, which reflects edema formation, of lung samples harvested from the rats with TBI dramatically increased at 8 and 24 h after injury compared with that of lung samples harvested from the sham-operated rats. Following TM-1 administration (10 mg/kg, i.p.) at 3 or 4 h after injury, the lung wet/dry weight ratio significantly decreased at 8 h or 24 h after injury in the TM-1 treated group compared with the TBI + veh group. Significant difference was observed between the rats administered TM-1 (10 mg/kg) 3 or 4 h after injury and sacrificed 24 h after injury (Figure 1).
Figure 1.
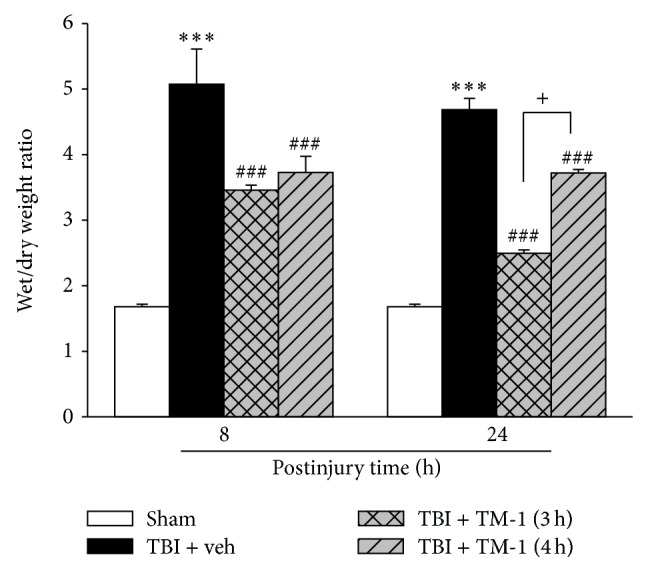
TM-1 administered 3 or 4 h after injury reduced the TBI-induced lung wet/dry weight ratio measured 8 and 24 h after TBI. Decrease in the wet/dry weight ratio at 3 or 4 h after injury with TM-1 administration (10 mg/kg) when the rats were sacrificed at 8 or 24 h after injury, respectively. Data are expressed as mean ± SD (n = 5/group). *** P < 0.001 TBI + veh group compared with the sham-operated group; ### P < 0.001 TM-1 treated groups compared with the TBI + veh groups; + P < 0.05, comparison between wet/dry weight ratios of rats sacrificed at 24 h following TM-1 administration (10 mg/kg) at 3 and 4 h after injury.
3.2. TM-1 Administration at 3 or 4 h after Injury Diminished the mRNA Expression of AQP1 in Pulmonary Injury at 8 or 24 h after TBI
Eight and 24 h after injury, the mRNA expression of AQP1 notably increased in the TBI + veh group (TBI treated with vehicle) compared with the sham-operated group. This increase in the mRNA expression was significantly attenuated by TM-1 administration at 3 or 4 h after injury and sacrifice at 8 or 24 h, respectively. Significant difference was observed between the rats administered TM-1 (10 mg/kg) at 3 or 4 h after injury. Similar measured patterns in AQP1 mRNA expression were observed using the two reference genes, β-actin and Gapdh (Figures 2(a) and 2(b)).
Figure 2.

TM-1 administered 3 or 4 h after injury reduced the TBI-induced AQP1 mRNA expression measured 8 and 24 h after TBI. Relative AQP1 mRNA expression was measured by reverse transcription Q-PCR using β-actin (a) and Gapdh (b) as reference genes. AQP1 mRNA expression significantly decreased following TM-1 administration (10 mg/kg) at 3 and 4 h after injury in the rats sacrificed 8 or 24 h after injury, respectively. Compared with the TBI + veh groups, the TM-1 treated groups exhibited significantly decreased mRNA expression. Data are expressed as mean ± SD (n = 5/group). *** P < 0.001 TBI + veh group compared with the sham-operated group; # P < 0.05 and ### P < 0.0001 TM-1 treated group compared with the TBI + veh groups; +++ P < 0.001, comparison between mRNA expression following TM-1 administration (10 mg/kg) 3 and 4 h after injury in the rats sacrificed 8 or 24 h after TBI.
3.3. TM-1 Administration 3 or 4 h after Injury Diminished the Protein Expression of AQP1 in Pulmonary Injury 8 or 24 h after TBI
Eight and 24 h after injury, the protein level expression of AQP1 notably increased in the TBI + veh group compared with the sham-operated group (Figure 3). A significant reduction in AQP1 protein expression was attenuated by TM-1 administration at 3 or 4 h after TBI and AQP1 protein levels examined when animals were sacrificed at 8 h or 24 h after TBI. There was no significant difference in the TM-1 administration group whether treatment was given at 3 or 4 h post injury for animals subsequently sacrificed at either 8 or 24 h after TBI.
Figure 3.
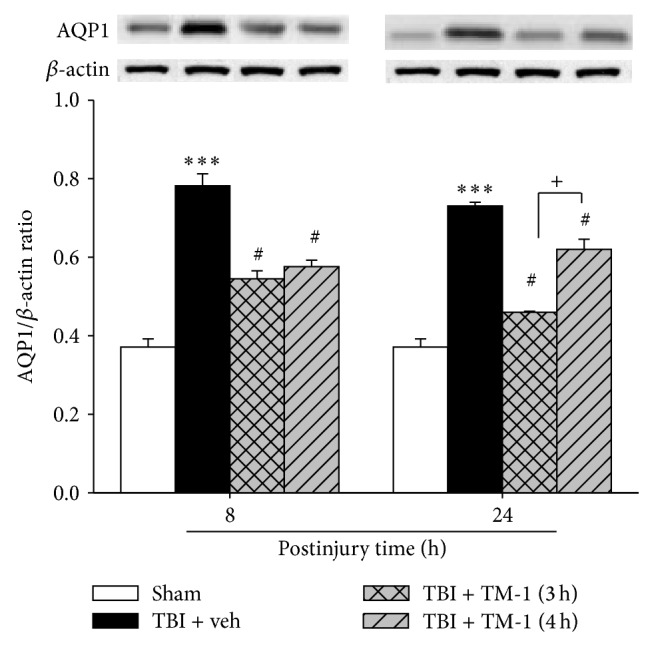
TM-1 administered 3 or 4 h after injury reduced TBI-induced lung AQP1 protein expression levels measured 8 and 24 h after TBI. The AQP1 protein level showed a marked decrease following TM-1 administration 3 or 4 h after injury (10 mg/kg), respectively. Rats were sacrificed 8 or 24 h after TBI. Data are expressed as mean ± SD (n = 5/group). *** P < 0.001 compared with the sham-operated group; # P < 0.05 compared with the untreated groups; +++ P < 0.001, comparison between mRNA expression following TM-1 administration (10 mg/kg) 3 and 4 h after injury in the rats sacrificed 8 and 24 h after TBI; + P < 0.05, comparison between protein levels following TM-1 administration (10 mg/kg) 3 and 4 h after injury in the rats sacrificed 24 h after TBI.
3.4. TM-1 Administration at 3 or 4 h after Injury Diminished the mRNA Expression of AQP4 in Pulmonary Injury at 8 and 24 h after TBI with β-actin and Gapdh Reference Genes
Three and 4 h after injury, TM-1 administration substantially reduced AQP4 mRNA expression in the treated group 8 and 24 h after injury compared with the TBI + veh group (Figures 4(a) and 4(b)). Significant difference was observed between the rats administered TM-1 (10 mg/kg) at 3 or 4 h after injury and sacrificed at 24 h after injury (Figures 4(a) and 4(b)).
Figure 4.

TM-1 administered 3 or 4 h after injury reduced the TBI-induced AQP4 mRNA expression measured 8 and 24 h after TBI. (a) β-actin and (b) Gapdh as reference genes. AQP4 mRNA expression significantly decreased following TM-1 administration (10 mg/kg) 3 and 4 h after injury in the rats sacrificed 8 or 24 h after injury, respectively. Compared with the TBI + veh groups, the TM-1 treated groups exhibited significantly decreased mRNA expression. Data are expressed as mean ± SD (n = 5/group). *** P < 0.001 TBI + veh group compared with the sham-operated group; ## P < 0.01 and ### P < 0.001 compared with the TBI + veh groups; + P < 0.05, comparison between mRNA expression following TM-1 administration (10 mg/kg) 3 and 4 h after injury in the rats sacrificed 8 or 24 h after TBI.
3.5. TM-1 Administration at 3 or 4 h after Injury Diminished the mRNA and Protein Expression of AQP4 in Pulmonary Injury 8 and 24 h after TBI
Three and 4 h after injury, TM-1 administration substantially reduced AQP4 protein level expression in the TM-1 treated group 8 and 24 h after injury compared with the TBI + veh group. Significant difference was observed between the rats administered TM-1 (10 mg/kg) 3 or 4 h after injury and sacrificed 24 h after injury (Figure 5).
Figure 5.
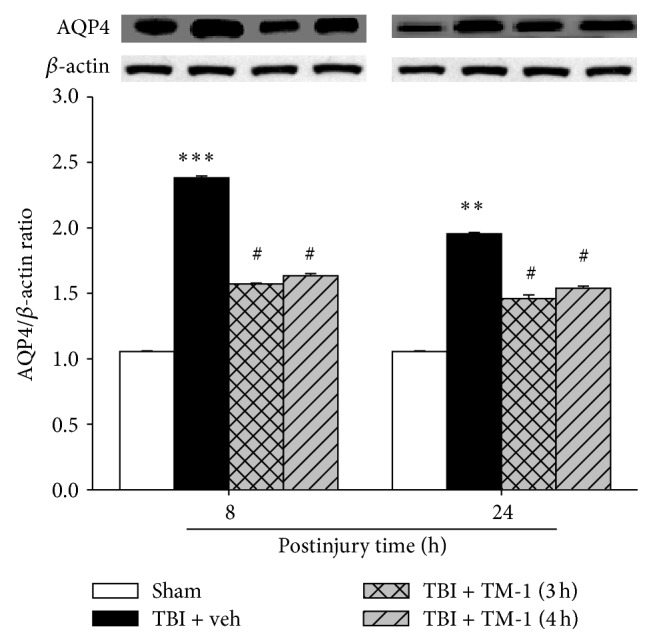
TM-1 administered 3 or 4 h after injury reduced the TBI-induced AQP4 protein level measured 8 and 24 h after TBI. The AQP4 protein level showed a marked decrease following TM-1 administration (10 mg/kg) 3 and 4 h after injury, respectively. Rats were sacrificed 8 or 24 h after TBI. Data are expressed as mean ± SD (n = 5/group). ** P < 0.01 and *** P < 0.001 TBI + veh group compared with the sham-operated group; # P < 0.05 TM-1 treated groups compared with the TBI + veh groups; no significance in AQP4 protein expression following TM-1 administration (10 mg/kg) at 3 and 4 h after injury in the rats sacrificed at 8 or 24 h after TBI.
3.6. TM-1 Administration 3 or 4 h after Injury Improved Pathological Changes 8 and 24 h after TBI
Sham-operated rats (vehicle-treated group) showed a normal alveolar morphology (Figure 6). Eight and 24 h after TBI, infiltration of numerous neutrophils in interstitial spaces, hemorrhage, and marked swelling of alveolar walls were observed in the rats with ALI. Extensive interstitial edema and infiltration of inflammatory cells were the same as these which have been previously described [30]. These pathological changes were attenuated in the rats administered TM-1 (10 mg/kg) at 3 or 4 h after injury and sacrificed at 8 or 24 h after TBI (Figure 6, middle and lower panels). No significant pathological changes were observed in the vehicle-treated sham-operated group.
Figure 6.

TM-1 administered 3 or 4 h after injury reduced alveolar swelling with infiltration of inflammatory cells 8 and 24 h after TBI by H&E staining. The vehicle-treated sham-operated group showed a normal alveolar morphology. Alveolar swelling and infiltration of inflammatory cells increased 8 and 24 h in vehicle-treated TBI rats. Alveolar swelling and infiltration of inflammatory cells decreased following TM-1 administration 3 and 4 h after injury. Rats were sacrificed 8 or 24 h after TBI. Bar = 25 μm.
3.7. TM-1 Administration 3 or 4 h after Injury Improved the Lung Injury Score 8 and 24 h after TBI
In Figure 7, the results illustrated a higher lung injury score in the TBI groups than in the sham-treated group (in which lung injury score was almost zero). These lung injury scores were significantly lower in the TM-1 treated groups (3 or 4 h after injury) than in the TBI + veh group. When the rats were sacrificed at 8 h or 24 h after TBI, the lung injury score was markedly lower in the TM-1 treated groups (3 or 4 h after injury) than in the TBI + veh groups. No significance was found between the rats administered TM-1 (10 mg/kg) at 3 or 4 h after injury and sacrificed at 8 or 24 h after TBI.
Figure 7.
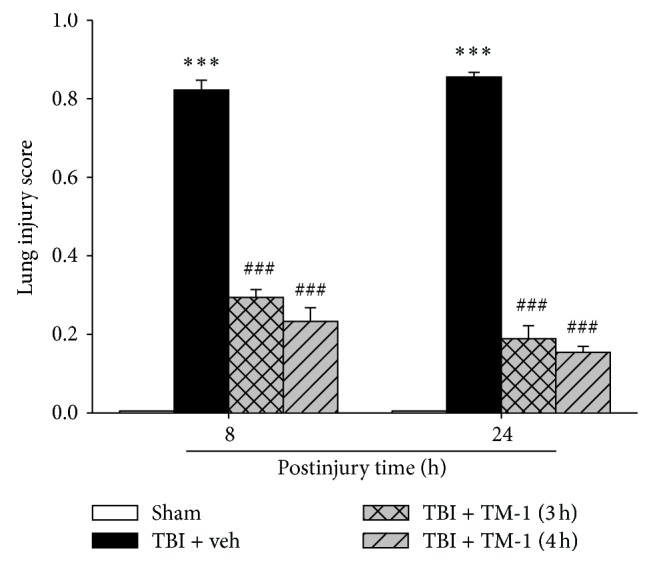
TM-1 administered 3 or 4 h after injury reduced the TBI-induced lung injury score 8 and 24 h after TBI. Significant difference was observed between the sham-operated and TBI groups. The lung injury score decreased with TM-1 administration (10 mg/kg) 3 and 4 h after injury. Rats were sacrificed 24 h after TBI. The lung injury score decreased following TM-1 administration (10 mg/kg) 3 and 4 h after injury. Rats were sacrificed 24 h after TBI. Data are expressed as mean ± SD (n = 5/group). *** P < 0.001 compared with the sham-operated group; ### P < 0.001 compared with the untreated groups.
3.8. TM-1 Administration at 3 or 4 h after Injury Showed a Positive Correlation between the Wet/Dry Weight Ratio and the Lung Injury Score at 8 and 24 h after TBI
Pearson's regression analysis showed that a positive correlation existed between the wet/dry weight ratio and the lung injury score. In Figure 8(a), lung injury score and lung wet/dry weight ratio showed a positive correlation (df = 13, r = 0.7224) and a significant difference (P < 0.05) at 3 or 4 h after injury and sacrificed at 24 h after TBI. Similarly, in Figure 8(b), and lung injury scores and lung wet/dry weight ratio were examined when animals were sacrifice at 24 h after TBI.
Figure 8.

Rats administered TM-1 3 or 4 h after injury showed a positive correlation between the wet/dry weight ratio and the lung injury score. (a) The wet/dry weight ratio and the lung injury score in rats sacrificed at 8 h after TBI exhibited a positive correlation (df = 13, r = 0.72224) and a significant difference (P < 0.01) in the three groups. (b) The wet/dry weight ratio and the lung injury score in rats sacrificed at 24 h after TBI exhibited a positive correlation (df = 13, r = 0.72633) and a significant difference (P < 0.01) in the three groups.
4. Discussion
Our study showed that, compared with the TBI + veh groups, a decrease in the wet/dry weight ratio was observed in the TM-1 treated groups. Histological results showed that extensive interstitial edema and infiltration of inflammatory cells occurred in injured lungs in the TBI + veh groups, but little interstitial edema and infiltration of inflammatory cells were observed in the TM-1 treated groups. These results are consistent with those of other studies [30]. The lung injury score revealed the severity of lung injury. TM-1 administered at 3 or 4 h after injury reduced the TBI-induced lung injury score measured at 8 and 24 h after TBI. To our knowledge, our study is the first to illustrate a positive correlation between the lung injury score and the wet/dry weight ratio in the same animal. Tissue edema was uniformly synchronized with tissue pathological changes. The mRNA and protein expression of AQP1 and AQP4 measured in TBI-induced lung injury was increased and attenuated by TM-1 administered (10 mg/kg) 3 or 4 h after injury, and the rats were sacrificed 8 and 24 h after TBI. The previous study demonstrated that moderate PE appeared after 8 h and peaked after 16 h of hypoxia. In addition, in lung tissue, the mRNA expression of inflammatory cytokines was upregulated after 8 h of hypoxia, and the mRNA expression of collagens was significantly increased after 8–16 h [31]. Our results are similar to those observed in the aforementioned study. Our data showed that TM-1 decreased extensive interstitial edema, infiltration of inflammatory cells, and the lung injury score. Adrenergic mechanisms may underlie this decrease. Norepinephrine and other adrenoceptor agonists are known to induce the activation of proinflammatory cytokines, such as TNF-α, interleukin (IL)-1, and IL-6. These cytokines are involved in the pathogenesis of PE. PE is typically accompanied by inflammation and followed by pulmonary vascular hypertrophy, permeability, and fibrosis [32]. Thaliporphine is a phenolic aporphine alkaloid obtained from Chinese herbs that possesses antioxidant and α-1 adrenoceptor antagonistic activity. In the previous study, thaliporphine is shown to be a partial Ca2+ channel agonist with strong Na+ and K+ channel-blocking activities and is attributed to the suppression of inflammatory cytokines and the production of free radicals [25]. In addition, thaliporphine is an antioxidant and α-1 adrenoceptor antagonist that decreased the risk of cardiac muscle necrosis [33–35]. We therefore assume that the antiadrenergic mechanisms of TM-1 may have a major role in the treatment of pulmonary injuries characterized by edema and inflammation after TBI. Chiao et al. [25] illustrated that the thaliporphine was evaluated for its antitoxic properties against toxemia induced by the bacterial endotoxin lipopolysaccharide; cardiovascular and hemodynamic changes were monitored. The authors showed that thaliporphine treatment is not associated with any significant changes in mean arterial blood pressure and heart rate or vascular hyporeactivity in response to epinephrine. We believe therefore that cardiovascular parameters in our study were unlikely affected by the thaliporphine. So we did not monitor the cardiovascular and hemodynamic changes in our study. Downregulation of AQP1 expression prevented the formation of excessive PE in chronic heart failure [17]. AQP1 knockout inhibits water transport through pulmonary capillaries and reduced high-pressure PE [16]. Furthermore, AQP1 expression increased in tuberculous pleural effusion [36]. In another study, ALI resulted in the upregulation of AQP4 mRNA expression in rat ATII cells [21]. These results are consistent with those of our study. Our study also showed the downregulation of mRNA and protein expression of AQP1 and AQP4 as well as the positive effect of improving PE with TM-1 administration after TBI. These results may be, in part, explained by the partial Ca2+ channel agonist with strong Na+ and K+ channel-blocking activities of thaliporphine, which have been shown by previous studies. AQP4 mRNA expression was upregulated in the ATII cell membrane to regulate fluid exchange between the alveolar space and the alveolar epithelium barrier and facilitated pulmonary liquid clearance in case of sodium pump transport damage during ALI [22]. Pulmonary inflammation and PE were associated with a marked reduction in AQP1 mRNA levels in the lung [18, 19]. In addition, AQP1 mRNA expression negatively correlated with the level of the wet/dry weight ratio [20]. In the airways, AQP4 deletion did not impair water transport [23]. Experimental designs, therapeutic agents, animals, or cell types in the previous studies and the present study were different. However, our data imply that the mRNA and protein expression of AQP1 and AQP4 are affected by TM-1 administration and that AQPs not only regulate water balance but also regulate inflammation in lung injury after TBI. AQP5 is the main subtype in lung and expressed in type I alveolar epithelium cells [10, 37]. There is no correlation between AQP5 immunohistochemistry and alveolar septal sickness, peribronchial/vascular cuffing, perivascular edema, alveolar lipid containing macrophage, medial hyperplasia, and mast cell to perivascular edema [38]. There were some studies that demonstrated that AQP5 were not required for the physiological clearance of lung water or for accumulation of extravascular lung water in the injured lung [14, 23, 39]. Therefore, the thaliporphine derivative TM-1 could be a novel agent for reducing ALI and PE after TBI. However, further studies are required to identify the underlying mechanisms of modulation in water balance between AQPs and inflammation in the lung after TBI.
5. Conclusions
TM-1 simultaneously diminished the lung injury score and the wet/dry weight ratio in rats with TBI. In addition, TM-1 reduced TBI-induced PE by downregulating AQP1 and AQP4 expression. Our data suggest that the decreased expression of AQP1 and AQP4 contributes, at least in part, to the beneficial effects of TM-1 on TBI-induced PE.
Acknowledgment
This study was financially supported in part by Grants from the Ministry of Science and Technology (MOST 103-2321-B-038-002 and NSC 102-2321-B-038-003 to Jia-Yi Wang).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Baumann A., Audibert G., McDonnell J., Mertes P. M. Neurogenic pulmonary edema. Acta Anaesthesiologica Scandinavica. 2007;51(4):447–455. doi: 10.1111/j.1399-6576.2007.01276.x. [DOI] [PubMed] [Google Scholar]
- 2.Gajic O., Manno E. M. Neurogenic pulmonary edema: another multiple-hit model of acute lung injury. Critical Care Medicine. 2007;35(8):1979–1980. doi: 10.1097/01.CCM.0000277254.12230.7D. [DOI] [PubMed] [Google Scholar]
- 3.Zygun D. A., Kortbeek J. B., Fick G. H., Laupland K. B., Doig C. J. Non-neurologic organ dysfunction in severe traumatic brain injury. Critical Care Medicine. 2005;33(3):654–660. doi: 10.1097/01.CCM.0000155911.01844.54. [DOI] [PubMed] [Google Scholar]
- 4.Martin G. S., Brigham K. L. Fluid flux and clearance in acute lung injury. Comprehensive Physiology. 2012;2(4):2471–2480. doi: 10.1002/cphy.c100050. [DOI] [PubMed] [Google Scholar]
- 5.Kandatsu N., Nan Y.-S., Feng Q.-G., et al. Opposing effects of isoflurane and sevoflurane on neurogenic pulmonary edema development in an animal model. Anesthesiology. 2005;102(6):1182–1189. doi: 10.1097/00000542-200506000-00018. [DOI] [PubMed] [Google Scholar]
- 6.Leal Filho M. B., Morandin R. C., de Almeida A. R., et al. Hemodynamic parameters and neurogenic pulmonary edema following spinal cord injury: an experimental model. Arquivos de Neuro-Psiquiatria. 2005;63(4):990–996. doi: 10.1590/S0004-282X2005000600016. [DOI] [PubMed] [Google Scholar]
- 7.Leal Filho M. B., Morandin R. C., de Almeida A. R., et al. Importance of anesthesia for the genesis of neurogenic pulmonary edema in spinal cord injury. Neuroscience Letters. 2005;373(2):165–170. doi: 10.1016/j.neulet.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 8.Agre P., King L. S., Yasui M., et al. Aquaporin water channels—from atomic structure to clinical medicine. The Journal of Physiology. 2002;542(1):3–16. doi: 10.1113/jphysiol.2002.020818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nielsen S., King L. S., Christensen B. M., Acre P. Aquaporins in complex tissues. II. Subcellular distribution in respiratory and glandular tissues of rat. American Journal of Physiology—Cell Physiology. 1997;273(5):C1549–C1561. doi: 10.1152/ajpcell.1997.273.5.C1549. [DOI] [PubMed] [Google Scholar]
- 10.Kreda S. M., Gynn M. C., Fenstermacher D. A., Boucher R. C., Gabriel S. E. Expression and localization of epithelial aquaporins in the adult human lung. The American Journal of Respiratory Cell and Molecular Biology. 2001;24(3):224–234. doi: 10.1165/ajrcmb.24.3.4367. [DOI] [PubMed] [Google Scholar]
- 11.Flynn G. A., Migliati E. R., Ritter L. S., Yool A. J. Patent. US20110172195 A1. Google Patents; 2011. Aquaporin modulators and methods of using them for the treatment of edema and fluid imbalance. [Google Scholar]
- 12.Wang Q., Ishikawa T., Michiue T., Zhu B.-L., Guan D.-W., Maeda H. Intrapulmonary aquaporin-5 expression as a possible biomarker for discriminating smothering and choking from sudden cardiac death: a pilot study. Forensic Science International. 2012;220(1-3):154–157. doi: 10.1016/j.forsciint.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y., Xi H., Xing W., Gu J. Aquaporin changes in compound 48/80 induced inflammatory sublaryngeal edema in rat. Journal of Voice. 2012;26(6):e815–e823. doi: 10.1016/j.jvoice.2012.04.007. [DOI] [PubMed] [Google Scholar]
- 14.Song Y., Yang B., Matthay M. A., Ma T., Verkman A. S. Role of aquaporin water channels in pleural fluid dynamics. American Journal of Physiology: Cell Physiology. 2000;279(6):C1744–C1750. doi: 10.1152/ajpcell.2000.279.6.C1744. [DOI] [PubMed] [Google Scholar]
- 15.Verkman A. S. Physiological importance of aquaporin water channels. Annals of Medicine. 2002;34(3):192–200. doi: 10.1080/713782138. [DOI] [PubMed] [Google Scholar]
- 16.Bai C., Chen Z., Song Y. Experimental study for lung fluid transport by epithelial cells and aquaporins. Zhonghua Jie He He Hu Xi Za Zhi. 2001;24(2):105–108. [PubMed] [Google Scholar]
- 17.Müllertz K. M., Strøm C., Trautner S., et al. Downregulation of aquaporin-1 in alveolar microvessels in lungs adapted to chronic heart failure. Lung. 2011;189(2):157–166. doi: 10.1007/s00408-010-9276-x. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y. B., Li S. X., Chen X. P., et al. Autophagy is activated and might protect neurons from degeneration after traumatic brain injury. Neuroscience Bulletin. 2008;24(3):143–149. doi: 10.1007/s12264-008-1108-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mascia L. Acute lung injury in patients with severe brain injury: a double hit model. Neurocritical Care. 2009;11(3):417–426. doi: 10.1007/s12028-009-9242-8. [DOI] [PubMed] [Google Scholar]
- 20.Cao C.-S., Yin Q., Huang L., Zhan Z., Yang J.-B., Xiong H.-W. Effect of angiotensin II on the expression of aquaporin 1 in lung of rats following acute lung injury. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue. 2010;22(7):426–429. doi: 10.3760/cma.j.issn.1003-0603.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 21.Chen C.-L., Li T.-P., Zhu L.-H. Effect of MAPK signal transduction pathway inhibitor U0126 on aquaporin 4 expression in alveolar type II cells in rats with oleic acid-induced acute lung injury. Nan Fang Yi Ke Da Xue Xue Bao. 2009;29(8):1525–1528. [PubMed] [Google Scholar]
- 22.Zhu L.-H., Li T.-P., He L. Role of AQP-4 in pulmonary water metabolism in rats in early stage of oleic acid-induced acute lung injury. Nan Fang Yi Ke Da Xue Xue Bao. 2008;28(5):707–711. [PubMed] [Google Scholar]
- 23.Borok Z., Verkman A. S. Lung edema clearance: 20 years of progress invited review: role of aquaporin water channels in fluid transport in lung and airways. Journal of Applied Physiology. 2002;93(6):2199–2206. doi: 10.1152/japplphysiol.01171.2001. [DOI] [PubMed] [Google Scholar]
- 24.Hung L. M., Lee S. S., Chen J. K., Huang S. S., Su M. J. Thaliporphine protects ischemic and ischemic-reperfused rat hearts via an NO-dependent mechanism. Drug Development Research. 2001;52(3):446–453. doi: 10.1002/ddr.1146. [DOI] [Google Scholar]
- 25.Chiao C.-W., Lee S.-S., Wu C.-C., Su M.-J. Thaliporphine increases survival rate and attenuates multiple organ injury in LPS-induced endotoxaemia. Naunyn-Schmiedeberg’s Archives of Pharmacology. 2005;371(1):34–43. doi: 10.1007/s00210-004-1014-6. [DOI] [PubMed] [Google Scholar]
- 26.Chen S., Pickard J. D., Harris N. G. Time course of cellular pathology after controlled cortical impact injury. Experimental Neurology. 2003;182(1):87–102. doi: 10.1016/S0014-4886(03)00002-5. [DOI] [PubMed] [Google Scholar]
- 27.Kitamura Y., Hashimoto S., Mizuta N., et al. Fas/FasL-dependent apoptosis of alveolar cells after lipopolysaccharide-induced lung injury in mice. American Journal of Respiratory and Critical Care Medicine. 2001;163(3):762–769. doi: 10.1164/ajrccm.163.3.2003065. [DOI] [PubMed] [Google Scholar]
- 28.Wen L. L., Chiu C. T., Huang Y. N., Chang C. F., Wang J. Y. Rapid glia expression and release of proinflammatory cytokines in experimental Klebsiella pneumoniae meningoencephalitis. Experimental Neurology. 2007;205(1):270–278. doi: 10.1016/j.expneurol.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 29.Matute-Bello G., Downey G., Moore B. B., et al. An official american thoracic society workshop report: features and measurements of experimental acute lung injury in animals. American Journal of Respiratory Cell and Molecular Biology. 2011;44(5):725–738. doi: 10.1165/rcmb.2009-0210ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamashita T., Kawashima S., Ohashi Y., et al. Resistance to endotoxin shock in transgenic mice overexpressing endothelial nitric oxide synthase. Circulation. 2000;101(8):931–937. doi: 10.1161/01.CIR.101.8.931. [DOI] [PubMed] [Google Scholar]
- 31.Rassler B., Marx G., Reissig C., et al. Time course of hypoxia-induced lung injury in rats. Respiratory Physiology & Neurobiology. 2007;159(1):45–54. doi: 10.1016/j.resp.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 32.Rassler B. Role of α- and β-adrenergic mechanisms in the pathogenesis of pulmonary injuries characterized by edema, inflammation and fibrosis. Cardiovascular and Hematological Disorders-Drug Targets. 2013;13(3):197–207. doi: 10.2174/1871529X1303140129154602. [DOI] [PubMed] [Google Scholar]
- 33.Yu S. M. Thaliporphine selectively inhibits expression of the inducible, but not the constitutive, nitric oxide synthase. Biochemical Journal. 1994;303(1):289–294. doi: 10.1042/bj3030289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Su M.-J., Chang Y.-M., Chi J.-F., Lee S.-S. Thaliporphine, a positive inotropic agent with a negative chronotropic action. European Journal of Pharmacology. 1994;254(1-2):141–150. doi: 10.1016/0014-2999(94)90381-6. [DOI] [PubMed] [Google Scholar]
- 35.Chang W.-L., Lee S.-S., Su M.-J. Attenuation of post-ischemia reperfusion injury by thaliporphine and morphine in rat hearts. Journal of Biomedical Science. 2005;12(4):611–619. doi: 10.1007/s11373-005-7401-2. [DOI] [PubMed] [Google Scholar]
- 36.Du H., Xie C., He Q., Deng X. Increased expression of aquaporin-1 on the pleura of rats with a tuberculous pleural effusion. Lung. 2007;185(6):325–336. doi: 10.1007/s00408-007-9023-0. [DOI] [PubMed] [Google Scholar]
- 37.King L. S., Nielsen S., Agre P. Aquaporins in complex tissues. I. Developmental patterns in respiratory and glandular tissues of rat. American Journal of Physiology—Cell Physiology. 1997;273(5):C1541–C1548. doi: 10.1152/ajpcell.1997.273.5.C1541. [DOI] [PubMed] [Google Scholar]
- 38.Singha O., Kengkoom K., Chaimongkolnukul K., et al. Pulmonary edema due to oral gavage in a toxicological study related to aquaporin-1, -4 and -5 expression. Journal of Toxicologic Pathology. 2013;26(3):283–291. doi: 10.1293/tox.26.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Verkman A. S., Matthay M. A., Song Y. Aquaporin water channels and lung physiology. American Journal of Physiology—Lung Cellular and Molecular Physiology. 2000;278(5):L867–L879. doi: 10.1152/ajplung.2000.278.5.L867. [DOI] [PubMed] [Google Scholar]