

Abstract
Paradoxically, the thymidine kinase (TK) encoded by Kaposi sarcoma-associated herpesvirus (KSHV) is an extremely inefficient nucleoside kinase, when compared to TKs from related herpesviruses. We now show that KSHV-TK, in contrast to HSV1-TK, associates with the actin cytoskeleton and induces extensive cell contraction followed by membrane blebbing. These dramatic changes in cell morphology depend on the auto-phosphorylation of tyrosines 65, 85 and 120 in the N-terminus of KSHV-TK. Phosphorylation of tyrosines 65/85 and 120 results in an interaction with Crk family proteins and the p85 regulatory subunit of PI3-Kinase, respectively. The interaction of Crk with KSHV-TK leads to tyrosine phoshorylation of this cellular adaptor. Auto-phosphorylation of KSHV-TK also induces a loss of FAK and paxillin from focal adhesions, resulting in activation of RhoA-ROCK signalling to myosin II and cell contraction. In the absence of FAK or paxillin, KSHV-TK has no effect on focal adhesion integrity or cell morphology. Our observations demonstrate that by acting as a tyrosine kinase, KSHV-TK modulates signalling and cell morphology.
Keywords: Crk/PI3-Kinase, focal adhesion, Kaposi sarcoma-associated herpesvirus, RhoA, thymidine kinase
See also: M Lagunoff (February 2015)
Introduction
Kaposi sarcoma-associated herpesvirus (KSHV) is a gammaherpesvirus that is the causative agent of several human cancers including primary effusion lymphoma (PEL), the plasmablastic variant of multicentric Castleman's disease (MCD) and Kaposi's sarcoma (KS) (Chang et al, 1994; Cesarman et al, 1995; Soulier et al, 1995; Moore et al, 1996). In pre-existing HIV infections, secondary KSHV infection induces a high incidence of Kaposi's sarcoma development, demonstrating the potent oncogenic capacity of this human virus (Martin et al, 1998). The selective inhibition of KSHV-encoded enzymes that are essential for viral replication may therefore provide good therapeutic targets to limit the impact of KSHV infection and disease progression.
Herpesviruses encode a variety of enzymes that are essential for their replication, including a thymidine kinase (TK) that overcomes a rate-limiting step during viral DNA replication in non-cycling cells by phosphorylating thymidine (Chen et al, 1979). TK-deficient herpes simplex virus (HSV) is avirulent because it no longer replicates in terminally differentiated neurons (Coen et al, 1989; Efstathiou et al, 1989; Valyi-Nagy et al, 1994). Additionally, pathogenesis studies with murid herpesvirus-4 (MuHV-4), a close relative of KSHV, have also shown that its TK is required for lytic replication in mice (Coleman et al, 2003). The fact that MuHV-4 lacking its thymidine kinase fails to infect via the upper respiratory tract argues that the TK is required for viral replication in a cell limited for nucleotides (Gill et al, 2009). The pivotal role of these viral nucleoside kinases for in vivo replication makes them good targets for anti-viral therapeutics.
The design of acyclovir, a nucleoside analogue that is selectively phosphorylated by the thymidine kinase of HSV and subsequently inhibits the viral DNA polymerase, is a prime example of how viral enzymes can be selectively targeted to prevent viral DNA replication (Elion, 1982; Smee et al, 1983; Field & Biron, 1994). Additionally, the effectiveness of nucleoside analogues such as cidofovir at reducing pathologies associated with MuHV-4 infection suggests that the TK of KSHV may represent a potential target to limit lytic replication of the virus and subsequent disease (Staskus et al, 1997; Neyts & De Clercq, 1998; Dal Canto et al, 2000; Gangappa et al, 2002).
A key point in devising anti-thymidine kinase-based therapies is to understand exactly how each enzyme functions. In contrast to other herpesviruses, the TKs encoded by gammaherpesviruses consist of a unique N-terminal domain linked to a catalytic C-terminal kinase domain (Littler et al, 1986; Littler & Arrand, 1988; Holton & Gentry, 1996). They are also very inefficient thymidine kinases (Gustafson et al, 1998, 2000). For example, the thymidine kinase of KSHV has a ∽60-fold higher Km for thymidine than that of HSV1 (Gustafson et al, 2000). Of those studied to date, the TKs encoded by gammaherpesviruses, unlike that of HSV1, are also not found in the nucleus (Degreve et al, 1998; Gill et al, 2005, 2007). Gamma2 herpesvirus TKs, such as those encoded by KSHV, rhesus monkey rhadinovirus, herpes saimiri and MuHV-4, are also phosphorylated on tyrosine residues in their N-terminal domain (Gill et al, 2005). Tyrosine phosphorylation of other viral TKs has not been observed (Gill et al, 2005). KSHV, unlike MuHV-4 and EBV, also encodes a thymidylate synthetase that can generate thymidine 5′-monophosphate via an alternative route independent of TK (Gaspar et al, 2002). The TK encoded by KSHV may therefore possess other functions that can be more effectively used by the virus during replication. Such a precedent has been reported for a number of nucleoside metabolism enzymes captured by viruses that have acquired new cellular functions (Lembo et al, 2004; Davison & Stow, 2005; Gaspar et al, 2008).
Previous observations suggest that KSHV-TK, which associates with unknown structures throughout the cytoplasm, is capable of inducing cell rounding (Gill et al, 2005). In this study, we set out to address how this viral thymidine kinase induces such morphological changes. We find that KSHV-TK is a tyrosine kinase that when auto-phosphorylated disrupts the integrity of focal adhesions and induces activation of Rho-ROCK-Myosin II-dependent cell contraction and blebbing. Moreover, our results suggest that KSHV-TK is a potential target for the development of tyrosine kinase inhibitors to limit its impact upon cell signalling pathways during lytic replication, to contribute to the treatment of virus-induced pathologies such as Kaposi sarcoma.
Results
KSHV-TK induces cell contraction and blebbing
Previous work reveals that KSHV-TK induces cell rounding and detachment of adherent cells (Gill et al, 2005). To obtain additional insights into these morphological changes, we analysed the effects of expressing GFP-tagged KSHV-TK (GFP-KSHV-TK) on cell morphology using time-lapse microscopy. Cells expressing GFP-KSHV-TK initially had a flat epithelial morphology but were observed to undergo a dramatic contraction ∽16 h post-transfection (Fig1A; Supplementary Movies S1 and S2). Expression of GFP-KSHV-TK in COS-7 and BHK-21 cells resulted in a similar phenotype (Supplementary Fig S1A). In contrast, expression of GFP-tagged HSV1-TK had no effect on cell shape or obvious changes in focal adhesion integrity as discussed later (Supplementary Fig S1B).
Figure 1.

- Live cell imaging reveals that expression of GFP-KSHV-TK, but not GFP induces contraction of HeLa cells (see Supplementary Movies S1 and S2). The time from the start of imaging is indicated above each panel, and the scale bars represent 18 μm. Quantification of GFP-KSHV-TK-induced cell contraction. The graphs show cell area (μm2) of cells expressing the indicated protein, and error bars represent SEM from three independent experiments in which n = 224 for GFP and n = 203 for GFP-KSHV-TK. ***P < 0.001.
- Phase images taken from Supplementary Movie S3 illustrating GFP-KSHV-TK induced blebbing of contracted cells. The time from the start of imaging is indicated above each panel, and the scale bars represent 18 μm.
- Scanning electron micrographs of HeLa cells expressing GFP or GFP-KSHV-TK.
- Immunofluorescence images illustrating the association of GFP-KSHV-TK, but not GFP, with actin stress fibres. Scale bar, 10 μm.
Closer examination reveals that GFP-KSHV-TK-expressing cells that had contracted underwent extensive membrane blebbing (Fig1B; Supplementary Movie S3). The morphological changes observed in time-lapse studies were also evident in the scanning electron microscope (Fig1C). GFP-KSHV-TK-induced membrane blebbing was not due to apoptosis as cells remained viable as they excluded trypan blue. They also showed none of the hallmarks of apoptosis when examined in the transmission electron microscope (Supplementary Fig S1C). Consistent with this, the apoptosis inhibitor Z-ZAD-FMK did not prevent GFP-KSHV-TK-induced cell contraction (Supplementary Fig S1D).
KSHV-TK-induced contraction is dependent on RhoA, ROCK and myosin II
Prior to cell contraction, GFP-KSHV-TK forms an extensive array of filaments, a proportion of which align with a subset of actin stress fibres (Fig1D). It was also noticeable that GFP-KSHV-TK also promoted an increase in the number of central actin stress fibres in spread cells before they contracted (Fig1D and Supplementary Fig S1E). To examine how KSHV-TK induces actin stress fibre formation, we examined the consequences of inhibiting RhoA signalling via ROCK to myosin II, the major signalling pathway involved in their formation (Pellegrin & Mellor, 2007). We found that co-expression of the dominant negative GFP-RhoN19 mutant partially inhibited the ability of RFP-KSHV-TK to promote cell contraction (Fig2A). Treatment of cells with Y-27632 or blebbistatin, which inhibit ROCK and myosin II, respectively, also abrogated KSHV-TK-induced stress fibre formation and cell contraction (Fig2B). Treatment of cells with Y-27632 or blebbistatin, in the absence of KSHV-TK, did not alter the integrity of focal adhesions (Supplementary Fig S2A). Consistent with these observations, we found that the level of GTP-bound RhoA is higher in GFP-KSHV-TK expressing cells (Fig2C).
Figure 2.

- Co-expression of GFP-RhoN19 but not GFP inhibits RFP-KSHV-TK-induced cell contraction. Graphs represent cell area (μm2) of KSHV-TK, RhoN19 or KSHV-TK and RhoN19 expressing cells. Error bars represent SEM from 3 independent experiments, n = 204 for KSHV-TK, n = 223 for RhoN19 and n = 189 for KSHV-TK and RhoN19.
- GFP-KSHV-TK-induced cell contraction does not occur in cells treated with the ROCK and myosin II inhibitors Y-27632 and blebbistatin, respectively. Error bars represent SEM from three independent experiments, n = 213 for DMSO, n = 189 for Y-27632 and n = 245. ***P < 0.001.
- Immunoblot analysis of rhotekin pull-down (P.D.) assays reveals that GFP-KSHV-TK increases the level of GTP-bound RhoA (GTP-RhoA). Actin is included as a loading control, and the input lysates are indicated.
- GFP-KSHV-TK but not GFP induces the loss of phosphotyrosine, phospho-FAK Y925 and phospho-paxillin Y31 epitopes from focal adhesions.
KSHV-TK disassembles focal adhesions
The dramatic changes in morphology induced by KSHV-TK are indicative of a possible alteration in the interaction of the cell with the extracellular substrate. To explore this possibility, we examine whether KSHV-TK induces changes in the composition and/or integrity of focal adhesions. We found that there was a reduction in the normal phosphotyrosine staining of focal adhesions in cells expressing GFP-KSHV-TK (Fig2D). The reduction in phosphotyrosine signal at focal adhesions, however, was not always readily apparent as it was frequently masked by a strong phosphotyrosine signal associated with KSHV-TK (Fig2D). To overcome this problem, we used phospho-specific antibodies to examine the level of tyrosine-phosphorylated FAK and paxillin at focal adhesions in cells expressing KSHV-TK (Fig2D and Supplementary Fig S2B). Immunofluorescence analysis with antibodies against phosphorylated tyrosines 397, 861 and 925 of FAK reveals that level of activated phospho-FAK associated with focal adhesions is lower in cells expressing GFP-KSHV-TK (Fig2D and Supplementary Fig S2B). A similar reduction in the level of phospho-paxillin was observed using antibodies directed at phosphorylated tyrosines 31 and 118 of paxillin (Fig2D and Supplementary Fig S2B). Quantification reveals that in cells expressing GFP-KSHV-TK, the phospho-paxillin-Y31 and phospho-FAK-Y925 signal associated with focal adhesions were reduced by 84.4 and 67.23%, respectively, compared to GFP alone (Supplementary Fig S2C). Consistent with its inability to induce cell contraction, expression of GFP-HSV1-TK did not induce a loss in the phosphotyrosine or phospho-paxillin signal at focal adhesions or stimulate actin stress fibre formation (Supplementary Fig S1).
Not only did KSHV-TK induce the loss of the phospho-FAK and paxillin signal associated with focal adhesions, it also altered the cellular distribution of total FAK and paxillin (Supplementary Fig S3A). Staining for other focal adhesion components including parvin, VASP, vinculin and zyxin revealed a similar reduction at focal adhesions together with an increase in the cytoplasmic level of the protein (Supplementary Fig S3A and B). The ability of KSHV-TK to induce loss of focal adhesion integrity was identical regardless of whether the protein was tagged with GFP, RFP or not (Supplementary Fig S3C).
Virally expressed KSHV-TK also induces disassembly of focal adhesions
To confirm our observations in transfected cells, we sought to analyse the effect of KSHV-TK expression on focal adhesion integrity during lytic viral replication. As it is still not possible to assess the role of specific KSHV-encoded proteins during lytic replication using the KSHV BAC mutagenesis system, we generated a recombinant MuHV-4 virus in which KSHV-TK was substituted for the TK of this closely related gammaherpesvirus (Fig3A). This is possible as the MuHV-4 TK is essential for in vivo replication, but is dispensable for replication in cell culture (Coleman et al, 2003). The resulting recombinant virus is denoted MuHV-4 [g-KSHV-TK] (Fig3A). Cells infected with MuHV-4 [g-KSHV-TK] expressed GFP-KSHV-TK, which became tyrosine-phosphorylated (Fig3B). The recombinant virus also had similar early and late lytic protein expression as the parental MuHV-4 (Supplementary Fig S4A). Infection with MuHV-4 [g-KSHV-TK] induced a similar loss of the phospho-FAK Y925 and phospho-paxillin Y31 signal associated with focal adhesions as observed in cells expressing GFP-KSHV-TK (Fig3C). In contrast, infection with MuHV-4 had no effect on focal adhesion integrity or distribution of phospho-FAK or phospho-paxillin (Fig3C). The cell rounding observed in cells infected with the recombinant MuHV-4 (MuHV-4, g-KSHV-TK), expressing KSHV-TK, could be blocked by the inclusion of the ROCK inhibitor Y-27632 during viral replication (Fig3D).
Figure 3.

- A schematic illustrating the BAC strategy used to generate a recombinant MuHV-4 virus (MuHV-4 [g-KSHV-TK]) in which the endogenous MuHV-4 TK is exchanged for GFP-KSHV-TK.
- Immunoblot analysis of BHK cells infected with MuHV-4 and MuHV-4 [g-KSHV-TK] or uninfected cells using antibodies against phosphotyrosine (p-Tyr), GFP, MuHV-4 TK, ORF17 (capsid) and glycoprotein N.
- Immunofluorescence analysis of MuHV-4- and MuHV-4 [g-KSHV-TK]-infected cells with the indicated antibodies reveals GFP-KSHV-TK expressed in the context of lytic infection (MuHV-4 [g-KSHV-TK]) induces the loss of phospho-FAK Y925 and paxillin Y31 from focal adhesions. White arrowheads indicate examples of infected cells expressing GFP-KSHV-TK or GFP (indicating WT MuHV-4 infection, BAC+ virus).
- Immunofluorescence analysis reveals that GFP-KSHV-TK expressed in the context of lytic infection (MuHV-4 [g-KSHV-TK]) does not alter focal adhesion integrity (paxillin) in the presence of the ROCK inhibitor Y-27632.
- Immunofluorescence analysis of paxillin in the lytically induced rKSHV.219/Vero cell line in the presence or absence of Y-27632.
In addition, although lytic replication is very inefficient in culture, we took advantage of two available systems to examine the impact of lytic KSHV replication on cell morphology. In one approach, Vero cells containing latent KSHV (rKSHV.219) (GFP+ cells) were induced to promote lytic replication, which can be observed by the expression of RFP that is driven by a pan-lytic KSHV promoter (Vieira & O'Hearn, 2004). Cells in which lytic replication was visible (GFP+/RFP+) were clearly contracted, compared to cells with latent virus (GFP+) which retained a more flattened morphology (Fig3E). The cell rounding observed during KSHV lytic replication could also be blocked by the incubation of Y-27632 (Fig3E). In the second experimental approach, virus was generated from a recombinant KSHV-Lyt virus that constitutively expresses the main lytic switch regulator, RTA (Budt et al, 2011). Filtered supernatants containing KSHV-lyt were then used to infect HeLa cells in the absence or presence of Y-27632. In the absence of Y-27632, infected cells (GFP+) underwent cell contraction and had a strong phosphotyrosine staining (Supplementary Fig S4B). In the presence of Y-27632, the infected cells maintained a more flattened morphology (Supplementary Fig S4B). Taken together, our data clearly show that the ROCK-specific inhibitor Y-27632 is sufficient to block the cell rounding induced by KSHV-TK in overexpression and viral contexts, including KSHV lytic replication.
Cell contraction is dependent on auto-phosphorylation of KSHV-TK
Immunofluorescence and Western blot analysis reveals that KSHV-TK is clearly tyrosine-phosphorylated (Figs2D and 3A). To examine whether the catalytic activity of KSHV-TK is responsible for its auto-phosphorylation and whether this modification is required to induce cell contraction, we generated a kinase-dead mutant (KSHV-TK-DEAD) by changing three essential glycine residues in the ATP binding motif of the kinase domain to valine (Saraste et al, 1990). Immunoblot analysis reveals that KSHV-TK-DEAD has a dramatic reduction in its level of tyrosine phosphorylation (Fig4A). Moreover, in contrast to the wild-type protein, KSHV-TK-DEAD is diffuse in the cytoplasm and does not form a reticular network, nor does it induce cell contraction or blebbing (Fig4A and B). Immunofluorescence analysis of the distribution of FAK, parvin, paxillin, VASP, vinculin and zyxin also indicated there were no obvious changes in the integrity of focal adhesions (compare Fig4C and D and Supplementary Fig S5 with Fig2D and Supplementary Figs S2B and S3A).
Figure 4.

- Cellular localisation and immunoblot analysis of phosphotyrosine (upper panel) and GFP (lower panel) in cells expressing wild-type or kinase-dead GFP-KSHV-TK.
- Scanning electron micrographs of HeLa cells expressing GFP or GFP-KSHV-TK-DEAD.
- Immunofluorescence analysis of HeLa cells expressing GFP-KSHV-TK-DEAD (white arrowheads) with the indicated phospho antibodies.
- Immunofluorescence images of the localisation of FAK and zyxin in HeLa cells expressing GFP-KSHV-TK-DEAD (white arrowheads).
- Purified recombinant GST-EBV-TK, GST-MuHV-4 and GST-KSHV-TK immunoblotted for phosphotyrosine and GST following a sequential dephosphorylation and kinase reaction.
- Purified recombinant GST-KSHV-TK and GST-KSHV-TK-KINASE-DEAD immunoblotted for phosphotyrosine and GST.
- Immunoblot analysis demonstrating that the N-terminus of KSHV-TK is only auto-phosphorylated by the kinase domain in cis.
To further validate the ability of KSHV-TK to act as a tyrosine kinase and auto-phosphorylate itself, we expressed and purified the protein from E. coli. The EBV and MuHV-4-related gammaherpesvirus TKs were used as controls. Immunoblot analysis reveals that in contrast to the TKs from EBV and MuHV-4, KSHV-TK is tyrosine-phosphorylated when purified from E. coli (Fig4E). The ability of purified recombinant KSHV-TK to undergo auto-phosphorylation in the presence of ATP after treatment with protein tyrosine phosphatase 1B (PTP1B) further confirmed its tyrosine kinase activity (Fig4E). Furthermore, the auto-phosphorylation of KSHV-TK was abolished when the kinase-dead mutant was purified from E. coli (Fig4F). By expressing the individual domains of KSHV-TK in mammalian cells, we could also demonstrate that the N-terminal region of KSHV-TK is only phosphorylated when attached to its C-terminal kinase domain (Fig4G).
To investigate whether the loss of focal adhesion integrity is due to phosphorylation of KSHV-TK or phosphorylation of cellular targets, we set out to define which tyrosine residues are auto-phosphorylated in KSHV-TK. Western blot analysis of a series of N-terminal deletion mutants reveals that auto-phosphorylation of KSHV-TK is lost when the first 100 amino acids of this protein are removed (Fig5A). Motif searches in the first 100 amino acids with the Scansite server (scansite.mit.edu/motifscan_seq.phtml) predict that when phosphorylated, tyrosine residues 65 and 85 of KSHV-TK are potential SH2 binding sites for a number of proteins. Given this, we mutated each tyrosine residue to phenylalanine, alone or in combination to examine their contribution to KSHV-TK-induced focal adhesion disassembly. Western blot analysis reveals that the single tyrosine mutants (Y65F and Y85F) are still phosphorylated (Supplementary Fig S6A). The single mutants also induced loss of focal adhesion integrity and cell rounding that was indistinguishable from the wild-type protein (Supplementary Fig S6B). In contrast, the double mutant (denoted as Y2F) had significantly reduced auto-phosphorylation and did not induce cell rounding or changes in focal adhesion integrity (Supplementary Fig S6B and C). These results suggest that auto-phosphorylation of tyrosines 65 and 85 of KSHV-TK is sufficient to disrupt focal adhesion integrity and induce cell contraction. To confirm that KSHV-TK auto-phosphorylates tyrosines 65 and 85, we performed Mass spec analysis of KSHV-TK purified from E. coli (Supplementary Fig S7). This analysis confirmed tyrosines 65 and 85 are phosphorylated and also revealed that tyrosine 120 is additionally modified (Supplementary Fig S7). Phosphorylation of tyrosine Y120 of KSHV-TK is also consistent with the partial phosphorylation of the Y2F mutant (Fig5B and Supplementary Fig S6A). Mutation of tyrosine 120 in conjugation with Y65 and Y85 completely abolished tyrosine phosphorylation of KSHV-TK (Fig5B). Cells expressing the triple mutant Y65/85/120F (denoted as Y3F) were indistinguishable from controls as they had a flat morphology and prominent focal adhesions (Fig5C–E). Consistent with this, the KSHV-TK-DEAD, Y2F and Y3F mutants failed to increase the level of GTP-bound RhoA induced by the wild-type protein (Fig5F). In order to examine the impact of expressing the KSHV-TK-Y3F during lytic viral replication, we generated a recombinant MuHV-4 virus. In contrast to the cell rounding induced by KSHV-TK, the MuHV-4-infected cells expressing KSHV-TK-Y3F retained a flat morphology (Fig5G).
Figure 5.
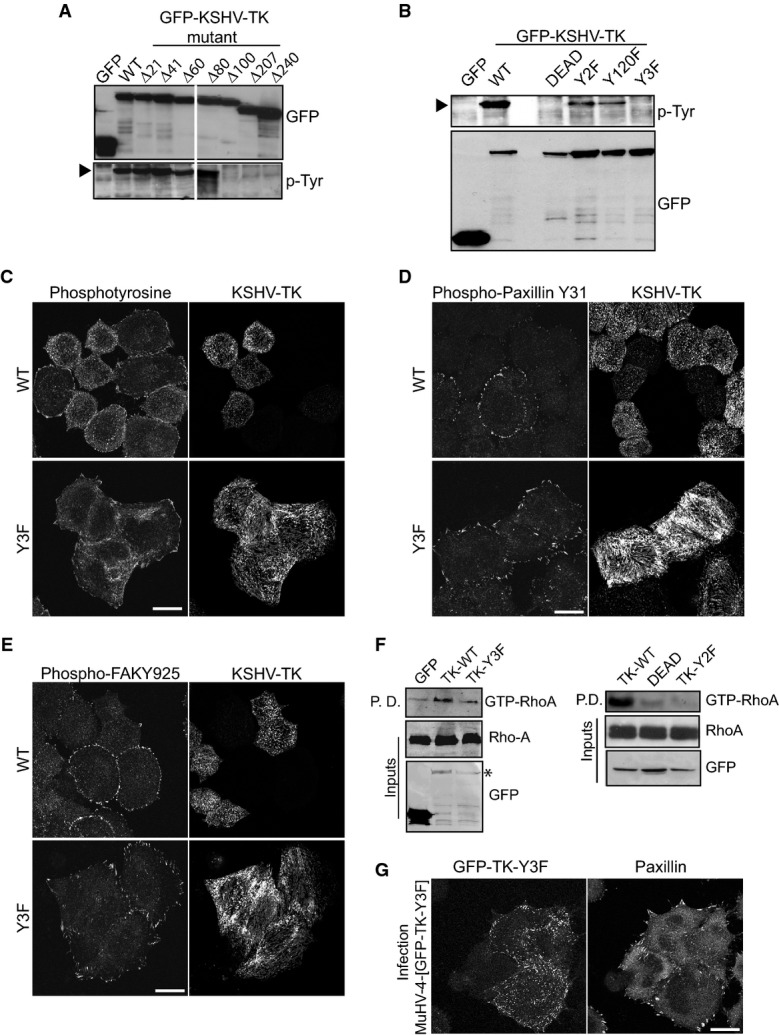
- Immunoblot analysis of N-terminal deletions of KSHV-TK demonstrates that the first 100 amino acids are required for tyrosine phosphorylation.
- Immunoblot analysis reveals that mutation of tyrosines 65, 85 and 120 ablates tyrosine phosphorylation of the KSHV-TK.
- Immunofluorescence analysis with the indicated antibodies reveals that mutation of tyrosines 65, 85 and 120 to phenylalanine abrogates the ability of GFP-KSHV-TK to induce loss of phosphotyrosine epitopes, phospho-paxillin and phospho-FAK from focal adhesions.
- Immunoblot analysis of level of GTP-bound and total RhoA in cells expressing the indicated GFP-tagged KSHV-TK proteins.
- Immunofluorescence analysis reveals that localisation of paxillin at focal adhesions is unaffected by KSHV-TK-Y3F expressed in the context of lytic infection.
The kinase activity of KSHV-TK is required for its association with FAK
Expression of KSHV-TK leads to a loss of phospho-paxillin at focal adhesions (Fig2D). Phosphorylation of paxillin at focal adhesions is mediated by a complex of FAK–Src (Glenney & Zokas, 1989; Schaller & Parsons, 1995; Frame, 2004; Schlaepfer & Mitra, 2004). It is possible that the KSHV-TK kinase activity disrupts the ability of FAK-Src to phosphorylate paxillin. Consistent with this, the ability of KSHV-TK to associate with FAK was dependent upon its kinase activity, as the KSHV-TK-DEAD but not the Y2F or Y3F mutants failed to associate with FAK (Fig6A). To further define the contribution of FAK and paxillin in the KSHV-TK-induced phenotypes, we examined the effects of expressing GFP-KSHV-TK on the localisation of paxillin and zyxin in FAK−/− or paxillin-depleted cells. In FAK−/− but not FAK+/+ cells, both paxillin and zyxin were readily detected in peripheral focal adhesions, and the cells did not undergo contraction (Fig6B and Supplementary Fig S6D). Immunofluorescence analysis reveals that RNAi-mediated depletion of paxillin did not inhibit tyrosine phosphorylation of GFP-KSHV-TK, which still forms an extensive array of filaments throughout the cell (Fig6C). The loss of paxillin, however, did prevent GFP-KSHV-TK-induced disassembly of focal adhesions and cell contraction (Fig6C). Our observations clearly demonstrate that KSHV-TK-mediated focal adhesion disassembly and cell contraction involve FAK and paxillin.
Figure 6.

- Immunoblot analysis reveals that GFP-KSHV-TK WT, Y2F and Y3F but not GFP-KSHV-TK-DEAD associate with FAK.
- GFP-KSHV-TK does not displace paxillin or zyxin from focal adhesions (white arrowheads) or induce contraction in the absence of FAK. Scale bar, 20 μm.
- GFP-KSHV-TK does not induce loss of zyxin and p-Tyr from focal adhesions or contraction of HeLa cells depleted of paxillin. Error bars represent SEM from three independent experiments, n = 196 for mock siRNA and n = 229 for paxillin siRNA; ***P < 0.001. Scale bar, 20 μm.
KSHV-TK interacts with Crk family members and the p85 subunit of PI3-Kinase
We set out to identify cellular binding partners for phosphorylated tyrosines 65 and 85 given their absolute requirement in KSHV-TK-induced cell contraction. Upon closer examination, we realised that Y65 (pYDVP) and Y85 (pYATP) matched the preferred Crk SH2 binding motif of pY-x-x-P (Fig7A) reviewed in Birge et al (2009). Immunoprecipitation of GFP-KSHV-TK and its Y2F mutant reveals that endogeneous CrkI, CrkII and CrkL interact with the wild-type protein but not with the Y2F mutant lacking tyrosines 65 and 85 (Fig7B). Immunofluorescence analysis also reveals that endogeneous CrkII associates with KSHV-TK but not the Y2F mutant (Fig7C). To confirm KSHV-TK interacts with the SH2 domain of CrkL and CrkII, we performed pull-down assays on cells co-expressing different GFP-tagged CrkL and CrkII domains. We found that RFP-KSHV-TK only interacts with the SH2 domain of CrkL and CrkII (Fig7D). In addition, the co-expression of the SH2 of either CrkII or CrkL was sufficient to block RFP-KSHV-TK-induced cell rounding (Fig7E).
Figure 7.

- A schematic representation of the Crk SH2 consensus binding motifs in KSHV-TK.
- Immunoblot analysis of GFP antibody immunoprecipitates reveals that GFP-KSHV-TK but not Y2F or GFP associates with CrkI, II and L.
- Immunofluorescence analysis reveals CrkII associates with GFP-KSHV-TK but not the Y2F mutant. Scale bars, 20 μm.
- Immunoblot analysis of RFP antibody immunoprecipitates reveals that RFP-KSHV-TK specifically associates with the SH2 domain of CrkII and CrkL.
- Immunofluorescence analysis of phosphotyrosine epitopes in cells expressing GFP-CrkII SH2, GFP-CrkL SH2, RFP-KSHV-TK or a combination of CrkII/LSH2 and RFP-KSHV-TK. Arrowheads represent contracted KSHV-TK-expressing cells. The SH2 domains of CrkII or L block KSHV-TK-induced cell rounding. Scale bars, 20 μm.
Interestingly, tyrosine 120 of KSHV-TK conforms to the preferred PI3-Kinase SH2 binding motif of pY-x-x-M (Fig8A) (Songyang et al, 1993). Immunoblot analysis of GFP immunoprecipitations reveals that in contrast to the wild-type protein and the Y2F mutant, KSHV-TK-Y120F has a significantly reduced interaction with endogeneous p85 regulatory subunit of PI3K (Fig8B). Furthermore, immunoprecipitations demonstrate that CrkII and/or CrkL are unable to interact with KSHV-TK via an association with PI3K (Fig8C and D). This suggests that the two pathways downstream of phosphorylated tyrosines 65/85 and 120 are operating independently from each other.
Figure 8.
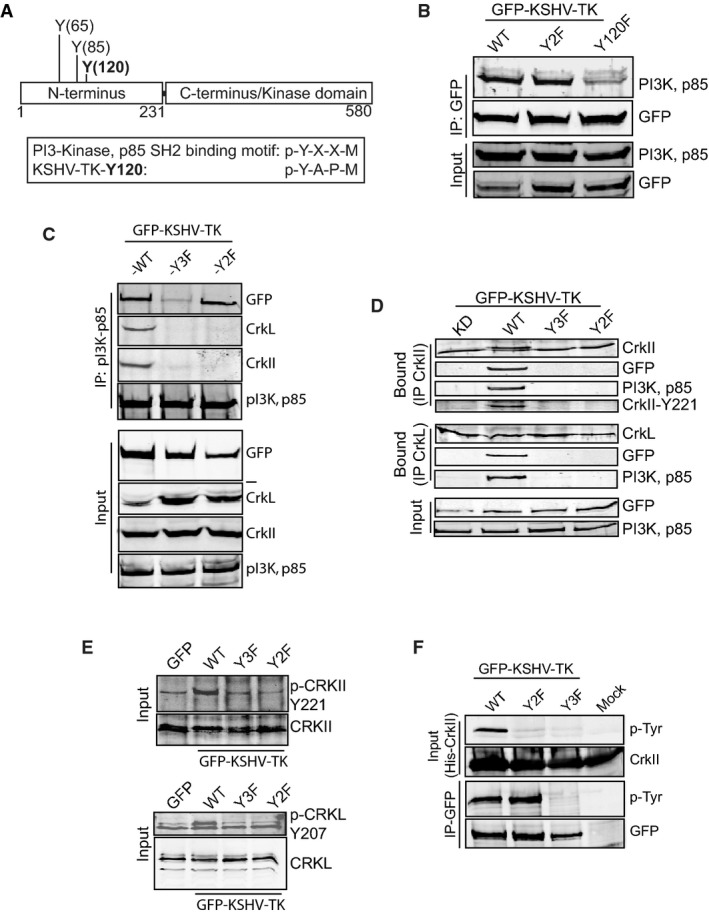
- A schematic representation of the p85 regulatory subunit SH2 consensus binding motif of PI3-Kinase in KSHV-TK.
- Immunoblot analysis reveals that GFP-KSHV-TK WT and Y2F but not Y120F associate with p85 of PI3-Kinase.
- Immunoblot analysis of p85 PI3-Kinase antibody immunoprecipitates reveals that GFP-KSHV-TK and its Y2F mutant but not Y120F associate with p85 of PI3-Kinase.
- Immunoblot analysis of CrkII and CrkL antibody immunoprecipitates reveals that GFP-KSHV-TK and the p85 PI3-Kinase associate with CrkII and CrkL. No association is observed with the Y2F, Y3F or kinase-dead TK.
- Tyrosine phosphorylation of CrkII and CrkL is only upregulated in cells expressing GFP-KSHV-TK.
- Immunoblot analysis reveals that recombinant His-CrkII binds to and is phosphorylated by KSHV-TK but not the Y2F or Y3F mutants.
KSHV-TK induces phosphorylation of Crk bound to tyrosines 65 and 85
Given that tyrosine phosphorylation of Crk family proteins impacts on their cellular function (reviewed in Birge et al, 2009), we decided to see whether KSHV-TK induces phosphorylation of CrkII and CrkL on tyrosines 221 and 207, respectively (Fig7D). Immunoblot analysis with phospho-specific antibodies on endogeneous CrkII and CrkL immunoprecipitated from cells expressing GFP-KSHV-TK reveals that both proteins become phosphorylated in the presence of the viral kinase (Fig8E). Increased tyrosine phosphorylation of CrkII and CrkL was not observed in cells expressing the GFP-KSHV-TK Y2F or Y3F mutants (Fig8E). This suggests that the interaction of CrkII and CrkL with tyrosine 65/85 of KSHV-TK is required for their phosphorylation. The Y2F result also provides further evidence that the two signalling pathways act independently, as the presence of PI3K bound to KSHV-TK does not enhance CrkII or CrkL phosphorylation. To confirm this is the case, we examined the ability of recombinant His-CrkII to bind and be phosphorylated by GFP-KSHV-TK. We found that the wild-type but not the Y2F or Y3F mutants could bind and phosphorylate His-CrkII (Fig8F). Furthermore, the interaction of His-CrkII is specific for phosphorylated tyrosine 65/85 as in the presence of phosphorylated tyrosine 120 in the Y2F mutant, there was still no binding (Fig8F). Taken together, our results indicate that Crk family members and PI3K are acting independently of each other downstream of phosphotyrosine 65/85 and 120, respectively.
Discussion
One of the most successful anti-viral therapeutics used to limit herpesvirus replication has been the design of inhibitory nucleoside analogues, such as acyclovir and ganciclovir, that are specifically phosphorylated by the virus encoded thymidine kinase and block viral DNA replication (Elion, 1982, 1993; Smee et al, 1985; Reardon & Spector, 1989; Sia & Patel, 2000; Galarraga et al, 2005). Based on the conservation of their kinase domains, one would assume that the TK expressed by the gammaherpesvirus KSHV would represent a favourable target for nucleoside analogue directed therapies. However, previous studies have shown that the thymidine kinase encoded by KSHV is an extremely inefficient enzyme compared to related herpesvirus TKs (Gustafson et al, 2000). Moreover, the ability of the TK from KSHV but not that of HSV1 to induce cell rounding when expressed in mammalian cells suggested that the two kinases may have very different functions during infection (Gill et al, 2005). In trying to understand how KSHV-TK induces cell contraction, we have now uncovered that the protein is actually a tyrosine kinase that auto-phosphorylates tyrosines 65, 85 and 120 in its N-terminal domain. Phosphorylation of tyrosines 65 and 85 of KSHV-TK ultimately leads to the loss of focal adhesion integrity and the induction of cell contraction followed by blebbing. These KSHV-TK-induced morphological changes are dependent on FAK and paxillin as well as the activity of RhoA-ROCK signalling to myosin II.
It is well established that paxillin is a pivotal player in the formation of focal adhesions (Turner, 2000). Once recruited to the leading edge of migrating cells, paxillin becomes tyrosine phosphorylated by a complex of FAK–SRC and acts as a scaffolding protein to recruit both structural and signalling proteins to the developing focal adhesion (Burridge et al, 1992; Nakamura et al, 2000; Nikolopoulos & Turner, 2000; Laukaitis et al, 2001). In this study, we have demonstrated that KSHV-TK leads to a loss of paxillin phosphorylated on tyrosines 31 and 118 at focal adhesions. This presumably results from the ability of catalytically active KSHV-TK to bind to and interfere with the normal cellular functionality of FAK-SRC. Phosphorylated tyrosines 31 and 118 of paxillin bind directly to the SH2 domain of p120RasGAP preventing its interaction with p190RhoGAP, which in turn leads to the suppression of RhoA signalling at focal adhesions (Iwasaki et al, 2002; Tsubouchi et al, 2002). Our observations suggest that KSHV-TK induces cell contraction and blebbing by indirectly activating RhoA signalling to myosin II by disrupting the ability of FAK-Src to phosphorylate tyrosine residues 31 and 118. Consistent with this, FAK-deficient cells expressing KSHV-TK do not undergo contraction and focal adhesion disassembly. However, the ability of FAK to still interact with the phosphorylation-deficient Y3F KSHV-TK mutant suggests an additional cellular protein(s), presumably upstream of FAK or in addition to FAK, that enables KSHV-TK to induce focal adhesion disassembly and cell contraction.
We have now found that when phosphorylated tyrosines 65 and 85 of KSHV-TK interact with the SH2 domain of Crk family proteins. Crk family members are potent oncogenic SH2/SH3 adaptor proteins that play a critical role in regulating cell adhesion through the formation of a diverse array of signalling complexes (Birge et al, 2009). Crk family members bind to phosphorylated paxillin and p130Cas via their SH2 domain and in doing so partially protect these proteins from dephosphorylation by cellular phosphatases (Schaller & Parsons, 1995; Iwasaki et al, 2002; Takino et al, 2003). In the case of paxillin, phosphorylation of Y31 and Y118 is sufficient to prevent RhoA activation (Tsubouchi et al, 2002). Crk family members also bind to Rho-GTPase exchange factors DOCK180, SOS1 and C3G via their N-terminal SH3 domain (Birge et al, 2009). The formation of Cas-Crk-C3G complex promotes cell adhesion and spreading resulting from Rap1 activation (Sakakibara et al, 2002). In contrast, paxillin-Crk-DOCK180 and paxillin-CrkII-GIT2/PKL-β-PIX complexes promote Rac1 activation and cell spreading (Ichiba et al, 1999; Lamorte et al, 2003; Valles et al, 2004). The strong association of CrkII with KSHV-TK filaments suggests that the viral kinase is sequestering Crk family proteins away from their normal cellular binding partners including phosphorylated paxillin. The sequestering activity immediately suggests KSHV-TK induces activation of RhoA and cell contraction by blocking the formation of Crk signalling complexes involved in promoting or maintaining cell spreading. In addition, KSHV-TK also increased the phosphorylation of CrkII and CrkL on tyrosine residues 221 and 207, respectively, a modification that promotes an auto-inhibited conformation for both proteins (reviewed in Birge et al, 2009). In its auto-inhibited state, the Crk N-terminal SH3 domain is also unable to interact with GEFs including DOCK180 and C3G. Failure of C3G to bind Crk inactivates Rap1 and consequently stimulates RhoA activation (Huang et al, 2008), a state reminiscent of that induced by KSHV-TK.
Phosphorylated tyrosine 120 of KSHV-TK binds to the p85 regulatory subunit of PI3-Kinase, although the biological significance of this interaction remains to be defined. Intriguingly, we do not see any association between Crk family members and the p85 subunit of PI3K in the absence of KSHV-TK expression. The ability of phosphorylated KSHV-TK to form a complex with both Crk and PI3K p85 is reminiscent of that observed when tyrosine-phosphorylated Cbl forms a complex with both Crk and PI3K p85 in activated T cells (Gelkop et al, 2001). In activated T lymphocytes, C3G is believed displaced from the Crk-Cbl-C3G complex enabling the formation of the Crk-Cbl-PI3K p85 complex (Gelkop et al, 2001). C3G is known to activate Rap1, block Ras-dependent signalling and in doing so promote immune cell anergy (Moodie et al, 1993; Boussiotis et al, 1997). The ability of KSHV-TK to simultaneously bind to both Crk and PI3K p85 could also block C3G binding to Crk and represent a platform for further Crk and PI3K p85 signalling.
Unfortunately, in the absence of an animal model, it is currently not possible to examine the cellular consequences of KSHV-TK expression in vivo or its contribution to KSHV pathogenesis. We have found that expression of KSHV-TK alone or in the context of infection with a recombinant MuHV-4 induces cell contraction and persistent blebbing. Lytically replicating KSHV was also capable of promoting ROCK-dependent cell rounding. However, understanding the precise contribution of KSHV-TK phosphorylation to viral replication and spread will only be possible once a better lytic KSHV replication system is available that permits the generation of site-specific mutants, which at present are currently unavailable.
In recent years, it has become clear that cell blebbing is an important aspect driving the migration of embryonic and tumour cells in 3D environments (Sahai & Marshall, 2003; Charras & Paluch, 2008; Fackler & Grosse, 2008; Lammermann & Sixt, 2009; Paluch & Raz, 2013). As in the case of KSHV-TK, these cellular blebs are induced via the activation of RhoA-ROCK signalling to myosin II. Bleb-induced matrix deformation followed by reattachment enables cancer cells to move in an amoeboid-like fashion through the extra-cellular matrix without the requirement for proteases (Pinner & Sahai, 2008; Tozluoglu et al, 2013). Based on our observations, we suggest that expression of KSHV-TK early during lytic replication will induce changes in the interaction of infected cells with both their non-infected neighbours and the extracellular matrix. It is also possible that KSHV-TK promotes infected cell migration in a manner analogous to that observed for tumour cells potentially by modulating the functionality of Crk family members and/or FAK-Src signalling via paxillin. Inducing migration of infected cells may provide a selective advantage during viral pathogenesis, as it will enhance the spread of infection independently of virus shedding. Moreover, endothelial cells, which are believed to be the source of Kaposi's sarcoma spindle cells (Hong et al, 2004), are also capable of moving via cell blebbing (Norman et al, 2010). It is possible that TK-mediated activation of RhoA during lytic virus replication, potentially via paxillin dephosphorylation and Crk phosphorylation, promotes the movement of these cells. The latter would assist in seeding KSHV-infected cells to new anatomical sites for establishment and replication, consistent with the multifocal vascular nature of Kaposi's sarcoma. The use of RhoA activation to promote cell movement contrasts the situation for vaccinia virus, where the viral protein F11 enhances the spread of infection by inducing cell migration and modulating the integrity of the cortical actin by binding RhoA to inhibit its downstream signalling (Valderrama et al, 2006; Arakawa et al, 2007a,b; Cordeiro et al, 2009; Handa et al, 2013).
It is currently unclear how KSHV-TK auto-phosphorylates itself since it lacks a classical tyrosine kinase signature. Unfortunately, no structure of either the catalytic domain or a full-length gammaherpesvirus TK has been determined. The structure of KSHV-TK or its kinase domain will undoubtedly provide important mechanistic insights into how this viral nucleoside kinase acts as a protein tyrosine kinase. It will also help facilitate the rational design of drugs to inhibit this viral kinase that has generally been overlooked but which should now be added to the list of potent signalling proteins expressed by KSHV. It will now be important to analyse the impact of KSHV-TK in vivo in order to assess its true oncogenic capacity and identify specific inhibitors that block the cellular effects of this viral kinase. In summary, our analysis suggests that KSHV-TK represents a new potential drug target for with which to potentially limit the dissemination of lytically replicating cells infected with KSHV.
Materials and Methods
Expression plasmid
KSHV-TK (ORF21) was amplified and cloned into pEGFPC2 (Clontech), pCherryC2 (a generous gift of Dr. C. Crump, Cambridge, UK) or pcDNA3.1 (−) (Invitrogen). The pEGFPC2 (or pCherryC2)-KSHV-TK-DEAD were generated by PCR-based mutagenesis using the following primers: 5′-gctttacttagaggtggtaatggttgtggtcaaatcaacgctggtcaac-3′ and 5′-gttgaccagcgttgatttgaccacaaccattaccacctctaagtaaagc-3′, which change glycines 260, 263 and 265 to valine residues in the ATP binding site. The KSHV-TK tyrosine to phenylalanine mutants was generated using the following primers using site-directed mutagenesis: Y65F (5′-gtaaaaacctcgtacatatttgacgtggcccaccgtcccga-3′) and (5′-tcgggacggtgggccacgtcaaatatgtacgaggtttttac-3′), Y85F (5′-tgcacgacaactccctctttgcaacgcctaggtttccgcc-3′) and (5′-ggcggaaacctaggcgttgcaaagagggagttgtcgtgca-3′), Y120F (5′-tgacgacgactcgggagactttgcgccaatggatcgcttc-3′) and (5′-gaagcgatccattggcgcaaagtctccgagtcgtcgtca-3′). The pEGFPC2 KSHV-TK N-terminal truncation clones were generated as previously described (Gill et al, 2005).
Full-length FAK was amplified and cloned into pEGFPC2 to generate pEGFPC2-FAK. Paxillin was amplified and cloned into pEGFPN2 to generate pEGFPN2-paxillin. ORFs encoding EBV-TK, MuHV-4-TK and KSHV-TK (and KSHV-TK-DEAD) were cloned into pGEX4T1 (Invitrogen) in frame and downstream of GST. CrkII and CrkL and subdomains (SH2, nSH3, cSH3, plus linker region + cSH3 for CrkL) were amplified and cloned into pEGFPC2.
Cell lines and drugs
HeLa, COS-7, BHK, hTERT-RPE1, FAK+/+ and FAK−/− (Ilic et al, 1995) cells and the cre-transduced derivative 3T3-CRE (Stevenson et al, 2002) were grown in Dulbecco's modified Eagle medium (Invitrogen Corporation) supplemented with 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin and 10% foetal calf serum (PAA Laboratories, Linz, Austria). For FAK+/+ and FAK−/− cells, media were also supplemented with non-essential amino acids (PAA) and 50 μM 2-mercaptoethanol (GIBCO). Cells were treated with 20 μM Y-27652 (Calbiochem), 10 μM blebbistatin (Sigma) or 20 μM Z-VAD-FMK (Promega) 1 h post-transfection and maintained through the transfection in freshly added media.
siRNA treatments
HeLa cells were plated at 5 × 104 cells per well and were transfected with 100 nM total siRNA in OptiMEM (Invitrogen) using Dharmafect 1 (Thermo Scientific) according to manufacturer's instructions. Both a paxillin siRNA pool (catalogue no. 1027416 (Qiagen)) and mock siRNA (sense AUUCUAUCACUAGCGUGACUU/antisense GUCACGCUAGUGAUAGAAUUU) (Thermo Scientific)) were used to transfect HeLa cells. Thirty hours after siRNA addition, the cells were transfected with the pEGFPC2-KSHV-TK using TransIT-LT1 in accordance with the manufacturer's instructions (Mirus). Eighteen hours later, the cells were either harvested to generate cell lysates or fixed using 4% PFA for immunofluorescence. Cell lysates were generated as stated below and analysed by SDS–PAGE and immunoblotting (as described below).
RhoA GTP pull-down assays
1 × 107 HeLa cells were transfected with GFP, GFP-KSHV-TK and GFP-KSHV-TK mutants (DEAD or Y6585F) or mock-challenged using Lipofectamine in accordance with the manufacturer's instructions (Invitrogen). Cell lysates were generated 12 h after the initial transfection in 1 ml using 1× MLB buffer. 500 μl cell lysate was added to 23.1 μl (30 μg) of Rho Assay Reagent (Rhotekin RBD, agarose) (Millipore, catalogue no. 14-383). The mixture was incubated for 45 min at 4°C with gentle agitation. The agarose beads were pelleted (10 s, 14,000 g, 4°C). The beads were washed 3× with MLB and then resuspended in 40 μl of 2× Laemmli's reducing buffer, plus 2 μl of dithiothreitol and boiled for 5 min. The precipitated material was analysed by Western blot analysis for bound RhoA using RhoA using mAb sc-418 (Santa Cruz) (as described below). Input lysates were included as a control.
Electron microscopy
HeLa cells expressing GFP, GFP-KSHV-TK or GFP-KSHV-TK-DEAD were processed for both transmission and scanning electron microscopy as previously described (Bruce et al, 2010). All samples were prepared and processed by the University of Cambridge Multi-Imaging Centre.
Immunofluorescence and antibodies
HeLa, BHK-21 or COS cells grown on glass coverslips were transfected using Lipofectamine™-2000 in accordance with the manufacturer's instructions or infected with 0.5 or 1 p.f.u./cell of MuHV-4 (wild-type), MuHV-4 TK−, MuHV-4 TK Revertant (Coleman et al, 2003) or MuHV-4 [g-KSHV-TK]. 16–18 post-transfection/infection cells were fixed (4% paraformaldehyde, 20 min), permeabilised (0.1% Triton X-100/PBS, 15 min) and blocked (3% bovine serum albumin in PBS/0.1% Tween [T-PBS], 1 h). Primary antibodies were incubated in 3% BSA/T-PBS for 1 h. The following primary antibodies were used: phospho-tyrosine (PY99, sc-7020; Santa Cruz), phospho-FAK Y397 (ab4803; Abcam), phospho-FAK Y861 (ab4804; Abcam), phospho-FAK Y925 (ab38512; Abcam), phospho-paxillin Y31 (ab4832; Abcam), phospho-paxillin Y118 (2541; Cell Signalling), paxillin (610051; BD Transduction laboratories), zyxin (307011; SYSY), FAK (C-20, sc-558; Santa Cruz), vinculin (ab18058; Abcam), VASP (610447; BD Transduction laboratories), parvin (4026; Cell Signalling Technology), CrkII (SAB4500470; Sigma) and CrkL (sc-365092; Santa Cruz) or anti-KSHV-TK (K1-A12) (mAb; Stevenson lab).
Primary antibodies were detected with either Alexa Fluor 568 goat anti-rabbit or Alexa Fluor 568 donkey anti-mouse (Invitrogen). Actin was stained with Alexa568-conjugated phalloidin (Invitrogen). MuHV-4 proteins were stained using mAbs 3F7 (glycoprotein, gN) (May et al, 2005), MG-12B8 (capsid, ORF65) (Gillet et al, 2006), PG-8A7 (Large subunit of RNR, ORF61) (Gill et al, 2010) and BN-6C12 (Tegument component, ORF75c) (Gaspar et al, 2008). Stained cells were mounted in ProLong Gold anti-fade reagent with DAPI (Invitrogen). For time-lapse imaging, live cells were grown on Iwaki glass-based dishes (VWR) housed in a heated incubation chamber with constant CO2. All images and contraction movies were captured using a Leica SP2 Confocal microscope.
Viruses
ORF21 flanking sequences were amplified by PCR to incorporated required restriction sites, LHS flank coordinate positions 31,441–32,879 and RHS flank 34,054–35,931 (based on genome sequence AF105037). The LHS flank was cloned into pSP73 using incorporated BglII and EcoRI restriction sites, generating pSP73-LHSfl. The GFP-KSHV-TK (WT or Y3F mutant) cassette from pEGFPC2-KSHV-TK was digested with Eco47III and SalI and cloned into pSP73-LHSfl. The RHS flank was cloned into the resultant pSP73-LHSfl.-GFP-KSHV-TK plasmid, generating pSP73-LHSfl.-GFP-KSHV-TK-RHSfl. Flanked GFP-KSHV-TK was amplified and sub-cloned blunt into pST76-SR shuttle vector. The GFP-KSHV-TK coding sequence was then recombined into MuHV-4 BAC ORF21 locus by standard protocols (Adler et al, 2000). The MHV-68 BAC (MuHV-4 [g-KSHV-TK]) was reconstituted into infectious virus by transfection into BHK-21 cells using TransIT-LT1 (Mirus). The loxP-flanked BAC cassette was removed by passaging the virus through 3T3-CRE cells. All virus stocks were grown in BHK-21 cells. Cell debris was pelleted by low-speed centrifugation (1,000 g, 3 min), and virions were recovered from the supernatants by high-speed centrifugation (38,000 g, 90 min) and stored at −70°C. Viruses were tittered by plaque assay on BHK-21 cells (de Lima et al, 2004). rKSHV.219/Vero cells (a generous gift from Professor J. Vieira and kindly supplied by Professor D. Blackbourn) (Vieira & O'Hearn, 2004) were lytically activated using 3 mM sodium butyrate. Y-27632 (20 μM) was added 10 h after induction and maintained throughout the experiment. KSHV-lyt virus, a generous gift of Professor W. Brune, was propagated in hTERT-RPE1 cells as previously described (Budt et al, 2011). Filtered virus supernatants were to infect HeLa cells for 30 h in the presence or absence of 20 μM Y-27632.
Immunoprecipitation and immunoblotting
Transfected or infected cells were lysed (20 min, 4°C) in 10 mM Tris, pH 7.6, 150 mM NaCl, 5 mM EDTA, 10% glycerol, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, 1 mM Na3V04, 1 mM NaF plus complete protease inhibitors (Roche Diagnostics). Insoluble debris was removed by centrifugation (13,000 g, 15 min, 4°C). For immunoprecipitation studies, 1 μg of antibody specific to FAK (C-20) (sc-558; Santa Cruz), CrkII (610035; BD Transduction laboratories), CrkL (B-1, sc-365092; Santa Cruz), the p85 subunit of PI3-Kinase (06-195; Millipore), GFP (ab290; Abcam) or mCherry (M11217; Invitrogen) was incubated with pre-cleared cell lysates o/n at 4°C with constant rotation, followed by incubation with Protein A/G UltraLink Resin (Thermo Scientific) for 2 h at 4°C with rotation. The resin was pelleted and washed 5× in cell lysis buffer minus SDS and sodium deoxycholate and further washed using 50 mM Hepes, pH 7.4, 150 mM NaCl, 1% Triton X-100, 10% glycerol and precipitated proteins eluted by heating (95°C, 5 min) in Laemmli's buffer, resolved by SDS–PAGE and transferred to PVDF membranes. The membranes were then probed for either FAK (C20 antibody), CrkII, CrkIIY221 (34915S; Cell Signalling), CrkL, CrkLY207 (3181S; Cell Signalling), PI3K-p85, mCherry or GFP (mAb JL-8; Clontech), Src (mAb GD11; Millipore) or paxillin (610051; BD Transduction laboratories). For siRNA depletion studies, the actin mAb AC-40 (Sigma) was also used. Primary antibodies were detected using either polyclonal rabbit anti-mouse (Dako) or horseradish peroxidase-conjugated anti-rabbit Ig (GE Healthcare, UK), followed by enhanced chemiluminescence substrate development (AP Biotech). Phosphotyrosine was detected using PY99 and MuHV-4 TK with mAb CS1-4A5 (May et al, 2008).
Expression and purification of GST fusion proteins, and in vitro kinase assays
BL21 (DE3) Rosetta cells containing pGEX4T1-EBV-TK, MuHV-4-TK, KSHV-TK, KSHV-TK-DEAD or pOPHT-CrkII (His tagged) were grown overnight at 37°C. The next day the bacterial culture was split 1:10, grown initially for 90 min. Expression was induced by the addition of 0.4 mM IPTG for 3 h at 37°C. The bacteria were resuspended in lysis buffer (150 mM NaCl, 50 mM Tris, pH 7.4, 1% Triton X-100, plus PMSF and protease inhibitors), incubated on ice for 2 h and sonicated. The lysates were clarified by centrifugation and incubated with either glutathione sepharose (for GST-TK preparations) or Ni-NTA agarose (for His-CrkII) for 1 h at 4°C with rotation. The beads were washed four times in lysis buffer and fusion proteins eluted using 100 mM glutathione (for GST fusions) or 250 imidazole (for His-CrkII) in 50 mM Tris, pH 8.8, 200 mM NaCl. The eluted fusion protein was dialysed overnight against 1× PBS and stored at −70°C until required. Dephosphorylation assay: GST fusion proteins were incubated with reaction buffer (25 mM HEPES, pH 7, 50 mM Nacl, 2.5 mM EDTA, 2.5 mM DTT) plus PTP1B (539735; Calbiochem) and incubated at 30°C for 30 min. Samples were extensively washed post-incubation if used in subsequent kinase reactions. Kinase assay: GST fusion proteins were incubated with kinase reaction buffer (10 mM HEPES, pH 7.7, 75 mM NaCl, 5 mM MgCl2, 0.5 mM DTT) and incubated at 30°C for 30 m. The reactions were stopped by the addition of 2× Laemmli's sample buffer and analysed by Western blotting.
Mass spectrometry
Partially purified GST-KSHV-TK was run on a Coomassie gel and tryptic digested, and the fractionated peptides were analysed by LC-MS/MS to map phosphorylated peptides. MS/MS analysis was completed by Cambridge Centre for Proteomics Core Services (http://www.bioc.cam.ac.uk/uto/deery.html). The identity of each peptide and phosphorylation sites was determined using Mascot as well as manual interpretation of the spectra.
Statistical analysis
Data are presented as mean ± standard error of the mean and were analysed by Student's t-test using Prism 6.0 (GraphPad Software, CA). A P-value of < 0.05 was considered statistically significant.
Acknowledgments
This work was supported by Medical Research Council Grant G0701185 to PGS and MBG. MW is supported by Cancer Research UK. We would like to thank Erik Sahai (London Research Institute, Cancer Research UK) and Margaret Frame (Institute of Genetics and Molecular Medicine University of Edinburgh) for insights and constructive comments. We would also like to thank Susanna Colaco and Janet May for excellent technical support.
Author contributions
MBG designed and performed most of the experiments and contributed to manuscript writing. RT assisted in experiments. PGS provided funding and resources. MW assisted with experiment design and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Movie S1
Supplementary Movie S2
Supplementary Movie S3
Legends for Supplementary Figures and Movies
Review Process File
Source Data for Figure 6C
Source Data for Figure 7
Source Data for Figure 8
References
- Adler H, Messerle M, Wagner M, Koszinowski UH. Cloning and mutagenesis of the murine gammaherpesvirus 68 genome as an infectious bacterial artificial chromosome. J Virol. 2000;74:6964–6974. doi: 10.1128/jvi.74.15.6964-6974.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakawa Y, Cordeiro JV, Schleich S, Newsome TP, Way M. The release of vaccinia virus from infected cells requires RhoA-mDia modulation of cortical actin. Cell Host Microbe. 2007a;1:227–240. doi: 10.1016/j.chom.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Arakawa Y, Cordeiro JV, Way M. F11L-mediated inhibition of RhoA-mDia signaling stimulates microtubule dynamics during vaccinia virus infection. Cell Host Microbe. 2007b;1:213–226. doi: 10.1016/j.chom.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Birge RB, Kalodimos C, Inagaki F, Tanaka S. Crk and CrkL adaptor proteins: networks for physiological and pathological signaling. Cell Commun Signal. 2009;7:13. doi: 10.1186/1478-811X-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussiotis VA, Freeman GJ, Berezovskaya A, Barber DL, Nadler LM. Maintenance of human T cell anergy: blocking of IL-2 gene transcription by activated Rap1. Science. 1997;278:124–128. doi: 10.1126/science.278.5335.124. [DOI] [PubMed] [Google Scholar]
- Bruce EA, Digard P, Stuart AD. The Rab11 pathway is required for influenza A virus budding and filament formation. J Virol. 2010;84:5848–5859. doi: 10.1128/JVI.00307-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budt M, Hristozova T, Hille G, Berger K, Brune W. Construction of a lytically replicating Kaposi's sarcoma-associated herpesvirus. J Virol. 2011;85:10415–10420. doi: 10.1128/JVI.05071-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge K, Turner CE, Romer LH. Tyrosine phosphorylation of paxillin and pp125FAK accompanies cell adhesion to extracellular matrix: a role in cytoskeletal assembly. J Cell Biol. 1992;119:893–903. doi: 10.1083/jcb.119.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med. 1995;332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science. 1994;266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- Charras G, Paluch E. Blebs lead the way: how to migrate without lamellipodia. Nat Rev Mol Cell Biol. 2008;9:730–736. doi: 10.1038/nrm2453. [DOI] [PubMed] [Google Scholar]
- Chen MS, Summers WP, Walker J, Summers WC, Prusoff WH. Characterization of pyrimidine deoxyribonucleoside kinase (thymidine kinase) and thymidylate kinase as a multifunctional enzyme in cells transformed by herpes simplex virus type 1 and in cells infected with mutant strains of herpes simplex virus. J Virol. 1979;30:942–945. doi: 10.1128/jvi.30.3.942-945.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coen DM, Kosz-Vnenchak M, Jacobson JG, Leib DA, Bogard CL, Schaffer PA, Tyler KL, Knipe DM. Thymidine kinase-negative herpes simplex virus mutants establish latency in mouse trigeminal ganglia but do not reactivate. Proc Natl Acad Sci USA. 1989;86:4736–4740. doi: 10.1073/pnas.86.12.4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman HM, de Lima B, Morton V, Stevenson PG. Murine gammaherpesvirus 68 lacking thymidine kinase shows severe attenuation of lytic cycle replication in vivo but still establishes latency. J Virol. 2003;77:2410–2417. doi: 10.1128/JVI.77.4.2410-2417.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro JV, Guerra S, Arakawa Y, Dodding MP, Esteban M, Way M. F11-mediated inhibition of RhoA signalling enhances the spread of vaccinia virus in vitro and in vivo in an intranasal mouse model of infection. PLoS One. 2009;4:e8506. doi: 10.1371/journal.pone.0008506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Canto AJ, Virgin HW, 4th, Speck SH. Ongoing viral replication is required for gammaherpesvirus 68-induced vascular damage. J Virol. 2000;74:11304–11310. doi: 10.1128/jvi.74.23.11304-11310.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison AJ, Stow ND. New genes from old: redeployment of dUTPase by herpesviruses. J Virol. 2005;79:12880–12892. doi: 10.1128/JVI.79.20.12880-12892.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degreve B, Johansson M, De Clercq E, Karlsson A, Balzarini J. Differential intracellular compartmentalization of herpetic thymidine kinases (TKs) in TK gene-transfected tumor cells: molecular characterization of the nuclear localization signal of herpes simplex virus type 1 TK. J Virol. 1998;72:9535–9543. doi: 10.1128/jvi.72.12.9535-9543.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efstathiou S, Kemp S, Darby G, Minson AC. The role of herpes simplex virus type 1 thymidine kinase in pathogenesis. J Gen Virol. 1989;70(Pt 4):869–879. doi: 10.1099/0022-1317-70-4-869. [DOI] [PubMed] [Google Scholar]
- Elion GB. Mechanism of action and selectivity of acyclovir. Am J Med. 1982;73:7–13. doi: 10.1016/0002-9343(82)90055-9. [DOI] [PubMed] [Google Scholar]
- Elion GB. Acyclovir: discovery, mechanism of action, and selectivity. J Med Virol. 1993;41(Suppl 1):2–6. doi: 10.1002/jmv.1890410503. [DOI] [PubMed] [Google Scholar]
- Fackler OT, Grosse R. Cell motility through plasma membrane blebbing. J Cell Biol. 2008;181:879–884. doi: 10.1083/jcb.200802081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field AK, Biron KK. “The end of innocence” revisited: resistance of herpesviruses to antiviral drugs. Clin Microbiol Rev. 1994;7:1–13. doi: 10.1128/cmr.7.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame MC. Newest findings on the oldest oncogene; how activated src does it. J Cell Sci. 2004;117:989–998. doi: 10.1242/jcs.01111. [DOI] [PubMed] [Google Scholar]
- Galarraga MC, Gomez E, de Ona M, Rodriguez A, Laures A, Boga JA, Melon S. Influence of ganciclovir prophylaxis on citomegalovirus, human herpesvirus 6, and human herpesvirus 7 viremia in renal transplant recipients. Transplant Proc. 2005;37:2124–2126. doi: 10.1016/j.transproceed.2005.03.123. [DOI] [PubMed] [Google Scholar]
- Gangappa S, Kapadia SB, Speck SH, Virgin HW., 4th Antibody to a lytic cycle viral protein decreases gammaherpesvirus latency in B-cell-deficient mice. J Virol. 2002;76:11460–11468. doi: 10.1128/JVI.76.22.11460-11468.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar G, De Clercq E, Neyts J. Human herpesvirus 8 gene encodes a functional thymidylate synthase. J Virol. 2002;76:10530–10532. doi: 10.1128/JVI.76.20.10530-10532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar M, Gill MB, Losing JB, May JS, Stevenson PG. Multiple functions for ORF75c in murid herpesvirus-4 infection. PLoS One. 2008;3:e2781. doi: 10.1371/journal.pone.0002781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelkop S, Babichev Y, Isakov N. T cell activation induces direct binding of the Crk adapter protein to the regulatory subunit of phosphatidylinositol 3-kinase (p85) via a complex mechanism involving the Cbl protein. J Biol Chem. 2001;276:36174–36182. doi: 10.1074/jbc.M100731200. [DOI] [PubMed] [Google Scholar]
- Gill MB, Murphy JE, Fingeroth JD. Functional divergence of Kaposi's sarcoma-associated herpesvirus and related gamma-2 herpesvirus thymidine kinases: novel cytoplasmic phosphoproteins that alter cellular morphology and disrupt adhesion. J Virol. 2005;79:14647–14659. doi: 10.1128/JVI.79.23.14647-14659.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill MB, Kutok JL, Fingeroth JD. Epstein-Barr virus thymidine kinase is a centrosomal resident precisely localized to the periphery of centrioles. J Virol. 2007;81:6523–6535. doi: 10.1128/JVI.00147-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill MB, Wright DE, Smith CM, May JS, Stevenson PG. Murid herpesvirus-4 lacking thymidine kinase reveals route-dependent requirements for host colonization. J Gen Virol. 2009;90:1461–1470. doi: 10.1099/vir.0.010603-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill MB, May JS, Colaco S, Stevenson PG. Important role for the murid herpesvirus 4 ribonucleotide reductase large subunit in host colonization via the respiratory tract. J Virol. 2010;84:10937–10942. doi: 10.1128/JVI.00828-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillet L, Gill MB, Colaco S, Smith CM, Stevenson PG. Murine gammaherpesvirus-68 glycoprotein B presents a difficult neutralization target to monoclonal antibodies derived from infected mice. J Gen Virol. 2006;87:3515–3527. doi: 10.1099/vir.0.82313-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenney JR, Jr, Zokas L. Novel tyrosine kinase substrates from Rous sarcoma virus-transformed cells are present in the membrane skeleton. J Cell Biol. 1989;108:2401–2408. doi: 10.1083/jcb.108.6.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson EA, Chillemi AC, Sage DR, Fingeroth JD. The Epstein-Barr virus thymidine kinase does not phosphorylate ganciclovir or acyclovir and demonstrates a narrow substrate specificity compared to the herpes simplex virus type 1 thymidine kinase. Antimicrob Agents Chemother. 1998;42:2923–2931. doi: 10.1128/aac.42.11.2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson EA, Schinazi RF, Fingeroth JD. Human herpesvirus 8 open reading frame 21 is a thymidine and thymidylate kinase of narrow substrate specificity that efficiently phosphorylates zidovudine but not ganciclovir. J Virol. 2000;74:684–692. doi: 10.1128/jvi.74.2.684-692.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa Y, Durkin CH, Dodding MP, Way M. Vaccinia virus F11 promotes viral spread by acting as a PDZ-containing scaffolding protein to bind myosin-9A and inhibit RhoA signaling. Cell Host Microbe. 2013;14:51–62. doi: 10.1016/j.chom.2013.06.006. [DOI] [PubMed] [Google Scholar]
- Holton RH, Gentry GA. The Epstein-Barr virus genome encodes deoxythymidine kinase activity in a nested internal open reading frame. Intervirology. 1996;39:270–274. doi: 10.1159/000150528. [DOI] [PubMed] [Google Scholar]
- Hong YK, Foreman K, Shin JW, Hirakawa S, Curry CL, Sage DR, Libermann T, Dezube BJ, Fingeroth JD, Detmar M. Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma-associated herpesvirus. Nat Genet. 2004;36:683–685. doi: 10.1038/ng1383. [DOI] [PubMed] [Google Scholar]
- Huang X, Wu D, Jin H, Stupack D, Wang JY. Induction of cell retraction by the combined actions of Abl-CrkII and Rho-ROCK1 signaling. J Cell Biol. 2008;183:711–723. doi: 10.1083/jcb.200801192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichiba T, Hashimoto Y, Nakaya M, Kuraishi Y, Tanaka S, Kurata T, Mochizuki N, Matsuda M. Activation of C3G guanine nucleotide exchange factor for Rap1 by phosphorylation of tyrosine 504. J Biol Chem. 1999;274:14376–14381. doi: 10.1074/jbc.274.20.14376. [DOI] [PubMed] [Google Scholar]
- Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–544. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- Iwasaki T, Nakata A, Mukai M, Shinkai K, Yano H, Sabe H, Schaefer E, Tatsuta M, Tsujimura T, Terada N, Kakishita E, Akedo H. Involvement of phosphorylation of Tyr-31 and Tyr-118 of paxillin in MM1 cancer cell migration. Int J Cancer. 2002;97:330–335. doi: 10.1002/ijc.1609. [DOI] [PubMed] [Google Scholar]
- Lammermann T, Sixt M. Mechanical modes of ‘amoeboid’ cell migration. Curr Opin Cell Biol. 2009;21:636–644. doi: 10.1016/j.ceb.2009.05.003. [DOI] [PubMed] [Google Scholar]
- Lamorte L, Rodrigues S, Sangwan V, Turner CE, Park M. Crk associates with a multimolecular Paxillin/GIT2/beta-PIX complex and promotes Rac-dependent relocalization of Paxillin to focal contacts. Mol Biol Cell. 2003;14:2818–2831. doi: 10.1091/mbc.E02-08-0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laukaitis CM, Webb DJ, Donais K, Horwitz AF. Differential dynamics of alpha 5 integrin, paxillin, and alpha-actinin during formation and disassembly of adhesions in migrating cells. J Cell Biol. 2001;153:1427–1440. doi: 10.1083/jcb.153.7.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lembo D, Donalisio M, Hofer A, Cornaglia M, Brune W, Koszinowski U, Thelander L, Landolfo S. The ribonucleotide reductase R1 homolog of murine cytomegalovirus is not a functional enzyme subunit but is required for pathogenesis. J Virol. 2004;78:4278–4288. doi: 10.1128/JVI.78.8.4278-4288.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lima BD, May JS, Stevenson PG. Murine gammaherpesvirus 68 lacking gp150 shows defective virion release but establishes normal latency in vivo. J Virol. 2004;78:5103–5112. doi: 10.1128/JVI.78.10.5103-5112.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littler E, Zeuthen J, McBride AA, Trost Sorensen E, Powell KL, Walsh-Arrand JE, Arrand JR. Identification of an Epstein-Barr virus-coded thymidine kinase. EMBO J. 1986;5:1959–1966. doi: 10.1002/j.1460-2075.1986.tb04450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littler E, Arrand JR. Characterization of the Epstein-Barr virus-encoded thymidine kinase expressed in heterologous eucaryotic and procaryotic systems. J Virol. 1988;62:3892–3895. doi: 10.1128/jvi.62.10.3892-3895.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JN, Ganem DE, Osmond DH, Page-Shafer KA, Macrae D, Kedes DH. Sexual transmission and the natural history of human herpesvirus 8 infection. N Engl J Med. 1998;338:948–954. doi: 10.1056/NEJM199804023381403. [DOI] [PubMed] [Google Scholar]
- May JS, Colaco S, Stevenson PG. Glycoprotein M is an essential lytic replication protein of the murine gammaherpesvirus 68. J Virol. 2005;79:3459–3467. doi: 10.1128/JVI.79.6.3459-3467.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May JS, Smith CM, Gill MB, Stevenson PG. An essential role for the proximal but not the distal cytoplasmic tail of glycoprotein M in murid herpesvirus 4 infection. PLoS One. 2008;3:e2131. doi: 10.1371/journal.pone.0002131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moodie SA, Willumsen BM, Weber MJ, Wolfman A. Complexes of Ras.GTP with Raf-1 and mitogen-activated protein kinase kinase. Science. 1993;260:1658–1661. doi: 10.1126/science.8503013. [DOI] [PubMed] [Google Scholar]
- Moore PS, Gao SJ, Dominguez G, Cesarman E, Lungu O, Knowles DM, Garber R, Pellett PE, McGeoch DJ, Chang Y. Primary characterization of a herpesvirus agent associated with Kaposi's sarcomae. J Virol. 1996;70:549–558. doi: 10.1128/jvi.70.1.549-558.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Yano H, Uchida H, Hashimoto S, Schaefer E, Sabe H. Tyrosine phosphorylation of paxillin alpha is involved in temporospatial regulation of paxillin-containing focal adhesion formation and F-actin organization in motile cells. J Biol Chem. 2000;275:27155–27164. doi: 10.1074/jbc.M000679200. [DOI] [PubMed] [Google Scholar]
- Neyts J, De Clercq E. In vitro and in vivo inhibition of murine gamma herpesvirus 68 replication by selected antiviral agents. Antimicrob Agents Chemother. 1998;42:170–172. doi: 10.1128/aac.42.1.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolopoulos SN, Turner CE. Actopaxin, a new focal adhesion protein that binds paxillin LD motifs and actin and regulates cell adhesion. J Cell Biol. 2000;151:1435–1448. doi: 10.1083/jcb.151.7.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman L, Sengupta K, Aranda-Espinoza H. Blebbing dynamics during endothelial cell spreading. Eur J Cell Biol. 2010;90:37–48. doi: 10.1016/j.ejcb.2010.09.013. [DOI] [PubMed] [Google Scholar]
- Paluch EK, Raz E. The role and regulation of blebs in cell migration. Curr Opin Cell Biol. 2013;25:582–590. doi: 10.1016/j.ceb.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrin S, Mellor H. Actin stress fibres. J Cell Sci. 2007;120:3491–3499. doi: 10.1242/jcs.018473. [DOI] [PubMed] [Google Scholar]
- Pinner S, Sahai E. Imaging amoeboid cancer cell motility in vivo. J Microsc. 2008;231:441–445. doi: 10.1111/j.1365-2818.2008.02056.x. [DOI] [PubMed] [Google Scholar]
- Reardon JE, Spector T. Herpes simplex virus type 1 DNA polymerase. Mechanism of inhibition by acyclovir triphosphate. J Biol Chem. 1989;264:7405–7411. [PubMed] [Google Scholar]
- Sahai E, Marshall CJ. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol. 2003;5:711–719. doi: 10.1038/ncb1019. [DOI] [PubMed] [Google Scholar]
- Sakakibara A, Ohba Y, Kurokawa K, Matsuda M, Hattori S. Novel function of Chat in controlling cell adhesion via Cas-Crk-C3G-pathway-mediated Rap1 activation. J Cell Sci. 2002;115:4915–4924. doi: 10.1242/jcs.00207. [DOI] [PubMed] [Google Scholar]
- Saraste M, Sibbald PR, Wittinghofer A. The P-loop–a common motif in ATP- and GTP-binding proteins. Trends Biochem Sci. 1990;15:430–434. doi: 10.1016/0968-0004(90)90281-f. [DOI] [PubMed] [Google Scholar]
- Schaller MD, Parsons JT. pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high-affinity binding site for Crk. Mol Cell Biol. 1995;15:2635–2645. doi: 10.1128/mcb.15.5.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer DD, Mitra SK. Multiple connections link FAK to cell motility and invasion. Curr Opin Genet Dev. 2004;14:92–101. doi: 10.1016/j.gde.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Sia IG, Patel R. New strategies for prevention and therapy of cytomegalovirus infection and disease in solid-organ transplant recipients. Clin Microbiol Rev. 2000;13:83–121. doi: 10.1128/cmr.13.1.83-121.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smee DF, Martin JC, Verheyden JP, Matthews TR. Anti-herpesvirus activity of the acyclic nucleoside 9-(1,3-dihydroxy-2-propoxymethyl)guanine. Antimicrob Agents Chemother. 1983;23:676–682. doi: 10.1128/aac.23.5.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smee DF, Boehme R, Chernow M, Binko BP, Matthews TR. Intracellular metabolism and enzymatic phosphorylation of 9-(1,3-dihydroxy-2-propoxymethyl)guanine and acyclovir in herpes simplex virus-infected and uninfected cells. Biochem Pharmacol. 1985;34:1049–1056. doi: 10.1016/0006-2952(85)90608-2. [DOI] [PubMed] [Google Scholar]
- Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, King F, Roberts T, Ratnofsky S, Lechleider RJ, Neel BG, Birge RB, Fajardo JE, Chou MM, Hanafusa H, Schaffhausen B, Cantley LC. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d'Agay MF, Clauvel JP, Raphael M, Degos L, et al. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood. 1995;86:1276–1280. [PubMed] [Google Scholar]
- Staskus KA, Zhong W, Gebhard K, Herndier B, Wang H, Renne R, Beneke J, Pudney J, Anderson DJ, Ganem D, Haase AT. Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J Virol. 1997;71:715–719. doi: 10.1128/jvi.71.1.715-719.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson PG, May JS, Smith XG, Marques S, Adler H, Koszinowski UH, Simas JP, Efstathiou S. K3-mediated evasion of CD8(+) T cells aids amplification of a latent gamma-herpesvirus. Nat Immunol. 2002;3:733–740. doi: 10.1038/ni818. [DOI] [PubMed] [Google Scholar]
- Takino T, Tamura M, Miyamori H, Araki M, Matsumoto K, Sato H, Yamada KM. Tyrosine phosphorylation of the CrkII adaptor protein modulates cell migration. J Cell Sci. 2003;116:3145–3155. doi: 10.1242/jcs.00632. [DOI] [PubMed] [Google Scholar]
- Tozluoglu M, Tournier AL, Jenkins RP, Hooper S, Bates PA, Sahai E. Matrix geometry determines optimal cancer cell migration strategy and modulates response to interventions. Nat Cell Biol. 2013;15:751–762. doi: 10.1038/ncb2775. [DOI] [PubMed] [Google Scholar]
- Tsubouchi A, Sakakura J, Yagi R, Mazaki Y, Schaefer E, Yano H, Sabe H. Localized suppression of RhoA activity by Tyr31/118-phosphorylated paxillin in cell adhesion and migration. J Cell Biol. 2002;159:673–683. doi: 10.1083/jcb.200202117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner CE. Paxillin and focal adhesion signalling. Nat Cell Biol. 2000;2:E231–E236. doi: 10.1038/35046659. [DOI] [PubMed] [Google Scholar]
- Valderrama F, Cordeiro JV, Schleich S, Frischknecht F, Way M. Vaccinia virus-induced cell motility requires F11L-mediated inhibition of RhoA signaling. Science. 2006;311:377–381. doi: 10.1126/science.1122411. [DOI] [PubMed] [Google Scholar]
- Valles AM, Beuvin M, Boyer B. Activation of Rac1 by paxillin-Crk-DOCK180 signaling complex is antagonized by Rap1 in migrating NBT-II cells. J Biol Chem. 2004;279:44490–44496. doi: 10.1074/jbc.M405144200. [DOI] [PubMed] [Google Scholar]
- Valyi-Nagy T, Gesser RM, Raengsakulrach B, Deshmane SL, Randazzo BP, Dillner AJ, Fraser NW. A thymidine kinase-negative HSV-1 strain establishes a persistent infection in SCID mice that features uncontrolled peripheral replication but only marginal nervous system involvement. Virology. 1994;199:484–490. doi: 10.1006/viro.1994.1150. [DOI] [PubMed] [Google Scholar]
- Vieira J, O'Hearn PM. Use of the red fluorescent protein as a marker of Kaposi's sarcoma-associated herpesvirus lytic gene expression. Virology. 2004;325:225–240. doi: 10.1016/j.virol.2004.03.049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Movie S1
Supplementary Movie S2
Supplementary Movie S3
Legends for Supplementary Figures and Movies
Review Process File
Source Data for Figure 6C
Source Data for Figure 7
Source Data for Figure 8