

Abstract
In adaptation to oncogenic signals, pancreatic ductal adenocarcinoma (PDAC) cells undergo epithelial–mesenchymal transition (EMT), a process combining tumor cell dedifferentiation with acquisition of stemness features. However, the mechanisms linking oncogene-induced signaling pathways with EMT and stemness remain largely elusive. Here, we uncover the inflammation-induced transcription factor NFATc1 as a central regulator of pancreatic cancer cell plasticity. In particular, we show that NFATc1 drives EMT reprogramming and maintains pancreatic cancer cells in a stem cell-like state through Sox2-dependent transcription of EMT and stemness factors. Intriguingly, NFATc1–Sox2 complex-mediated PDAC dedifferentiation and progression is opposed by antithetical p53-miR200c signaling, and inactivation of the tumor suppressor pathway is essential for tumor dedifferentiation and dissemination both in genetically engineered mouse models (GEMM) and human PDAC. Based on these findings, we propose the existence of a hierarchical signaling network regulating PDAC cell plasticity and suggest that the molecular decision between epithelial cell preservation and conversion into a dedifferentiated cancer stem cell-like phenotype depends on opposing levels of p53 and NFATc1 signaling activities.
Keywords: cellular plasticity, miRNA, NFATc1, p53, Sox2
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a devastating disease with a survival rate of less than 5% (Hezel et al, 2006; Rustgi, 2006; Maitra & Hruban, 2008; Rhim et al, 2012). A major reason for its aggressive behavior is the remarkable capacity of pancreatic tumor cells to switch phenotypic states in adaptation to microenvironmental cues (Chang et al, 2011b; Rhim et al, 2012; Kapoor et al, 2014). For instance, hypoxic conditions and chronic inflammation can trigger tumor cell dedifferentiation and subsequent conversion into highly invasive cancer stem cell-like phenotypic states. This process is causally linked to activation of an epithelial–mesenchymal transition (EMT) program that induces cytoskeletal reorganization and a progressive loss of cell polarity (Polyak & Weinberg, 2009; Thiery, 2009). A cohort of pleiotropically acting transcription factors, which includes Twist, Snai1, Zeb1, Zeb2, and Slug, drive the EMT program by direct or indirect silencing of epithelial cell adhesion proteins (e.g., E-cadherin) and intermediate filaments (e.g., cytokeratins) (Imamichi et al, 2007; Brabletz et al, 2011). Consequently, EMT-converted tumor cells detach from neighboring cells and migrate away from the epithelial layer in which they originated (Hanahan & Weinberg, 2011). In addition, reprogramming of cancer cells via EMT is often associated with acquisition of stem cell-like features such as self-renewal and the induction of pluripotency factors (e.g., Sox2 and Oct4) (Brabletz et al, 2005). Thus, aberrant activation of EMT endows tumor cells with the necessary traits for dedifferentiation, metastatic seeding and stemness. Uncovering the key molecular pathways that combine oncogenic signals such as inflammation with EMT/stemness is critical to our understanding of tumor progression and hence could reveal new pharmacologic targets and potential therapies to prevent EMT and eliminate PDAC cancer stem cells (Rhim et al, 2012).
The calcium-/calcineurin-responsive nuclear factor of activated T cells (NFATc1) represents a master inflammation-induced transcription factor with essential roles in gene regulation during T-cell activation, differentiation, and apoptosis (Rao et al, 1997; Hogan et al, 2003). In recent years, studies in mice and humans have revealed unexpected functions for NFATc1 beyond the immune system. In fact, NFATc1 has been linked to cell adaptation and differentiation during embryogenesis, where NFATc1 activation promotes EMT during lineage specification (Kao et al, 2009; Li et al, 2011). We have recently demonstrated ectopic expression and activation of nuclear NFATc1 in the majority of advanced human pancreatic cancers and particularly in cancer cells embedded in inflammatory niches (Buchholz et al, 2006). Moreover, using genetically engineered mouse models (GEMM), we confirmed inflammation-induced activation of nuclear NFATc1 in pancreatic epithelial cells and demonstrated profound oncogenic activities of this transcription factor (Baumgart et al, 2014). In fact, NFATc1 activation in pancreatic epithelial cells by either chemically induced inflammation (e.g., caerulein) or transgenic induction [Pdx1-KrasG12D;NFATc1 (KNC)] caused massive acceleration of Kras-driven carcinogenesis, resulting in the formation of precursor lesions and rapid progression toward frank adenocarcinomas (Baumgart et al, 2014).
Here, we explored the role of NFATc1 in pancreatic cancer cell plasticity and demonstrated that EMT and stemness acquisition are tightly regulated by a hierarchical signaling network composed of antagonistic NFATc1–Sox2 and p53–miRNA200c pathways. We show that the molecular decision between epithelial cell preservation and conversion into a dedifferentiated cancer stem cell-like phenotype is made at the level of p53 and NFATc1 signaling activity and that disruption of the tumor suppressor pathway is required for NFATc1–Sox2-driven EMT and stemness. Finally, we provide experimental evidence suggesting that pharmacological inactivation of the novel NFATc1–Sox2 signaling pathway has therapeutic potential in patients with dedifferentiated and metastatic PDAC disease through abolition of a migrating cancer stem-like state, even in a p53-deficient situation.
Results
The significance of p53 deficiency in pancreatic carcinogenesis and PDAC progression has been studied intensively within the last years. Recent studies showed a critical role for p53 in the regulation of differentiation, plasticity, and self-renewal (Pinho et al, 2011b; Spike & Wahl, 2011), suggesting that loss of p53 itself can provoke cancer cell dedifferentiation and progression. Surprisingly, we failed to show a statistical correlation between p53 expression levels and tumor grading in a large series of human PDAC (n = 129, P = 0.1235, Supplementary Table S1). Moreover, PDAC driving GEMMs with combined activation of oncogenic Kras and p53 deficiency based on either mutational disruption or heterozygous deletion [KrasG12D;Trp53R172H (KPC) and KrasG12D;Trp53wt/Δ mice (KPΔC)] preferentially developed well (G1) to moderately (G2) differentiated cancers rather than high-grade PDAC (Hingorani et al, 2005; Morton et al, 2010), indicating that inactivation of p53 alone is not sufficient to drive cancer dedifferentiation in the pancreas. These data allude to additional genetic or signaling-regulated mechanisms that might cooperate with p53 disruption in pancreatic dedifferentiation.
Rising evidence from recent publications suggested multiple implications of the transcription factor NFATc1 in regulation of cellular plasticity and appreciated NFATc1 as an important oncogene in pancreatic carcinogenesis (König et al, 2010). In this context, we aimed to investigate the impact of NFATc1 in pancreatic cell plasticity on the background of p53 inactivation. Strikingly, poorly differentiated (G3) KPC cancers (10/26) displayed exceptionally high levels of nuclear NFATc1 (Fig1A, right panel), whereas the absence of NFATc1 in KPC tumors predicted a good to moderate differentiation state in more than 80% (13/16) of analyzed tumors (Fig1A, left panel). To test whether the suggested correlation of nuclear NFATc1 expression and dedifferentiation in tumors with p53 deficiency also exists in human carcinogenesis, we analyzed a large cohort of human pancreatic adenocarcinomas. Interestingly and in congruence with our results in GEMM, NFATc1-positive human PDAC samples (161 out of 224, 72% of all tested cancer samples) displayed high levels of nuclear p53 staining more frequently in poorly differentiated tumors (59.3% of G3) compared to well- and moderately differentiated tumors (31.4% of G1/G2) (Fig1B and C). Gene sequencing analysis confirmed a predicted correlation between nuclear p53 positivity and mutational gene disruption in G3 cancers, mostly caused by hot spot mutations (Supplementary Fig S1A).
Figure 1.

- Statistical analysis of NFATc1 expression with respect to tumor grading in KPC mice tissues.
- Immunohistochemical detection of NFATc1 and p53, illustrating G1-G2 and G3 in human PDAC tissues. Scale bars, 100 μm.
- Statistical illustration of TMA analysis demonstrating NFATc1 strong nuclear positive tumors (n = 161 patients) with respect to functional status of p53 expression in human PDAC tissues.
- Representative H&E-stained sections as well as immunohistochemical detection of HA-NFATc1 and p53 in well- and poorly differentiated pancreatic tissues of KNC mice. Scale bars, 100 μm.
- Quantification of p53 expression with respect to the tumor grading by evaluating both the intensity of immunostaining and the number of cells with overexpression by means of an immunoreactivity score (IRS) in KNC mice (every dot represents one mouse).
- Incidence of liver metastases in KNC mice (mean values ± SD).
Further evidence for a role of NFATc1 in pancreatic cancer dedifferentiation was gathered by the analysis of tumor grades in KrasG12D;NFATc1 (KNC) mice bearing a wild-type p53 status at birth. One-third (15/41) of the NFATc1-driven (KNC) cancers progressed into highly metastatic, dedifferentiated tumors (classified as tumor grades G3), and this transition was intimately linked to inactivation or silencing of the p53 tumor suppressor pathway, indicated by absent p53 protein expression (Fig1D and E) and the disruption of cellular failsafe mechanisms (Supplementary Fig S1B and C). Moreover, p53 deficiency in KNC mice correlated with a 25-fold induction of liver metastases (Fig1F). Collectively, results from human tissues and GEM models revealed that combined p53 deficiency and NFATc1 activation favors a dedifferentiated and highly aggressive phenotype in pancreatic cancer.
As tumor cell dedifferentiation and metastases require activation of EMT, we questioned whether p53 deficiency is a gate opener for NFATc1 to drive EMT transcription programs in pancreatic cancer. For this purpose, we performed microarray-based expression profiling followed by gene set enrichment analysis (GSEA) in cell lines derived from well-differentiated KNC tumors depleted of p53 (KNC-siRNA control versus KNC-siRNA p53). Loss of p53 caused robust transcriptional activation of gene signatures implicated in EMT, stemness, and metastasis (Fig2A and B, and Supplementary Dataset S1), and these expression changes were accompanied by acquisition of a fast migrating phenotype, evidenced by wound healing experiments and time-lapse microscopy (Fig2C). Differential gene expression analysis further demonstrated that the “EMT master regulators”, Twist, Snai1, and Zeb1, were among the most significantly induced genes in p53-depleted cancer cells (Fig2D and E). Zeb1 and Twist were also highly enriched in dedifferentiated, metastatic KNC tumors with p53 deficiency, while the expression of epithelial markers such as E-cadherin and cytokeratin-19 showed decreased expression levels (Fig2F). Together, these results suggest that loss of p53 permits EMT and the formation of a dedifferentiated phenotype in NFATc1-driven PDAC.
Figure 2.
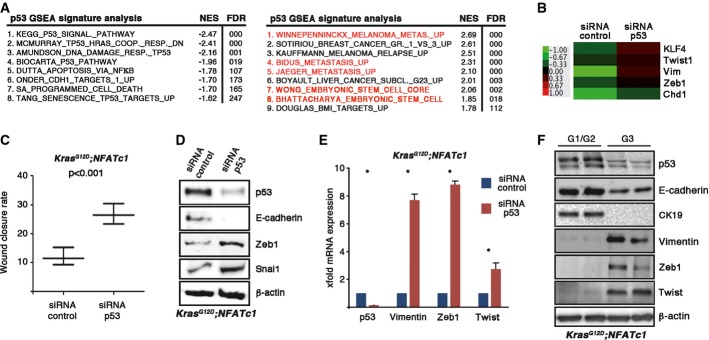
- Genome-wide expression and GSEA analysis show p53-enriched signatures with “EMT” and “stem cell transcript” in KNC tumor cells.
- Heat map showing p53-dependent regulation of “EMT”- and “stemness”-related genes in KNC tumor cells. Fold change relative to control cells is displayed in a green red color scheme for selected genes with FClog2 < 1.0 or FClog2 > −1.0.
- Statistical analysis of wound healing assay performed in primary KNC tumor cells following p53 depletion. Time-lapse analyzer software was used to quantify the wound closure rate. Means ± SD are shown from one out of three independent experiments.
- Western blot analysis revealing protein expression levels upon p53 depletion in KNC tumor cells.
- X-fold mRNA expression of EMT-related marker genes after p53 depletion in KNC tumor cells. Means ± SD from at least three independent experiments are shown. Asterisks display significance (*P < 0.05).
- Representative protein expression of EMT inducers in pancreatic lysates prepared from KNC mice with different tumor gradings.
To verify the significance of this observation for tumor progression in vivo, we combined pancreas-specific NFATc1 expression with mutational or genetic p53 inactivation [KrasG12D;Trp53R172H;NFATc1 (KPNC)] and [KrasG12D;Trp53wt/Δ;NFATc1 (KPΔNC)] (Supplementary Fig S2A). Mice of both genotypes were born at the expected Mendelian ratio with no apparent signs of pancreas abnormalities. However, the majority of mice developed severe cachexia and abdominal distension caused by the accumulation of sanguineous ascites, and died within 6–8 weeks, resulting in a median survival of < 65 days (Fig3A). At necropsy, pancreata were enlarged by solid tumor masses and most tumor-bearing mice also showed liver metastases (Fig3B and Supplementary Fig S2B and C). Virtually, all mice with nuclear NFATc1 activation and p53 deficiency formed dedifferentiated PDAC (Fig3C–E) and there was no phenotypic difference between mice with oncogenic mutations or deletion of p53 (Fig3A–D). Similarly, wound closure assays confirmed accelerated cell migration in primary cell lines derived from KPNC and KNC tumors that was comparable to observations in GEMM cell lines upon siRNA-mediated p53 depletion (Fig2C and Supplementary Fig S2D).
Figure 3.

- Kaplan–Meier curves showing survival of KNC, KPNC, and KPΔNC mice under the control of p48-Cre pancreas-specific promoters (P < 0.0001 for KPNC or KPΔNC cohorts versus KNC, log-rank test, for pairwise combination).
- Pathological features of KPNC and KPΔNC mice before tumor extraction. Pancreatic tissue is encircled.
- H&E and corresponding immunohistochemical stainings of (C) KPNC and (D) KPΔNC mouse tumors. Scale bars, 100 μm.
- Tumor grading of PDAC from KPNC mice (n = 10).
To explore whether the fast migrating phenotype and the enhanced incidence of metastases are functionally correlated with the acceleration of NFATc1-controlled EMT, we compared the expression levels of epithelial marker proteins and EMT key players in pancreatic tissue lysates of KPC and KPNC tumors. Western blot analysis revealed induced expression of Snai1, Twist, and Zeb1 in response to constitutive activation of NFATc1, while the expression of cytokeratin-19 was diminished (Supplementary Fig S2E). Moreover, comparative RNA expression analysis displayed moderate EMT marker expression levels in KPC tumors and well-differentiated KNC pancreatic tissue, while high-grade KNC tumors showed enhanced expression of Zeb1, Twist, Snail, and vimentin. EMT marker expression in G3 KNC tumors was only excelled by lysates derived from KPNC mice (Fig4A). Conclusively, these data illustrate that deletion- or mutation-based inactivation of p53 hastens NFATc1-driven pancreatic carcinogenesis and progression.
Figure 4.

- Quantitative RT-PCR analysis of EMT-related markers obtained from tumors derived from mice of the indicated genotypes (n = 3, means ± SD).
- Relative mRNA expression of NFATc1- and EMT-related marker genes on administration of CsA (1 μM) in KPNC cells. Data represents means ± SD from at least three independent experiments. Asterisks show significance (*P < 0.05).
- Western blot analysis revealing protein expression levels in KPNC cells from the indicated proteins following transfection of siRNA control or siRNA toward NFATc1, respectively.
Source data are available online for this figure.
Next, we sought to analyze the impact of NFATc1 on EMT regulators and marker proteins in p53-deficient KPNC cells and performed differential expression analyses following pharmacological NFATc1 inhibition by application of the calcineurin inhibitor cyclosporin A (CsA) or upon siRNA-mediated depletion of the transcription factor. NFATc1 depletion decreased expression of the EMT regulators Twist, Zeb1, and Snai1 and, presumably as a consequence of this, lowered the levels of mesenchymal markers, for example, vimentin and N-cadherin (Fig4B and C). These findings suggested that NFATc1 expression is required for appropriate expression and function of EMT regulators in pancreatic cancer.
To dissect the mechanism by which NFATc1 controls EMT regulators and governs cell plasticity in favor of a dedifferentiated phenotype, we focused on the stemness marker Sox2 (sex-determining region Y (SRY)-Box2), the expression of which was strongly indu-ced in all NFATc1-driven PDAC models and primary cell lines with p53 inactivation (Fig5A–D and Supplementary Fig S3A). Depletion of NFATc1 or its pharmacological inhibition decreased Sox2 expression in human and mouse PDAC cells (Fig5E–G), indicating that Sox2 is a downstream target of NFATc1. Accordingly, chromatin immunoprecipitation experiments (ChIP) revealed recruitment of NFATc1 to binding elements within the Sox2 gene enhancers, which are highly conserved among vertebrates (Fig5H and I, and Supplementary Fig S3B). Most importantly, NFATc1 inhibition led to reduced recruitment of RNA polymerase II to the Sox2 promoter (Fig5J), indicating that NFATc1 transcriptionally activates Sox2.
Figure 5.

- Sox2 mRNA expression of pancreatic lysates from KNC, KPNC, and KPC mice, analyzed by real-time PCR in three independent experiments (means ± SD).
- Western blot analysis shows Sox2 expression in pancreatic lysates of (B) KNC mice with indicated tumor grading and in (C) KPNC and KPC mice.
- Protein expression levels after siRNA-mediated p53 depletion in KNC tumor cells.
- NFATc1 and Sox2 protein expression after (E) NFATc1 depletion or (F) upon CsA (1 μM, 24 h) treatment in Panc1 cells.
- Sox2 and E-cadherin mRNA expression after NFATc1 depletion in L3.6 cells.
- Sequence alignment of 5′ and 3′ enhancer regions show highly conserved NFATc1 binding in different species; putative NFAT binding sides within the enhancer regions of the Sox2 promoter are highlighted in red.
- ChIP assays show NFATc1 binding to Sox2 enhancer and promoter in KNC tumor cells (I) and RNA polymerase II binding to Sox2 promoter upon CsA (1 μM, 24 h) treatment in L3.6 cells (J).
- Co-immunoprecipitation of endogenous HA-NFATc1 and Sox2 was performed in KPNC cells.
- ChIP experiments show NFATc1 binding along with H3K27 acetylation (H3K27ac) mark at the selected Snai1 enhancer region and RNA polymerase II binding at Snai1 promoter in the presence or absence of Sox2 in KPNC tumor cells. Representative results from at least three independent experiments are shown (means ± SD;P < 0.05).
- Relative mRNA expression levels of EMT-related marker genes upon Sox2 depletion in KPNC tumor cells (P < 0.05).
- Zeb1 and Snail mRNA levels in L3.6 cells after shRNA-mediated depletion of Sox2 and transient overexpression of NFATc1 shows that loss of Sox2 significantly rescues NFATc1-mediated induction of EMT genes (*P < 0.05).
Source data are available online for this figure.
A unique feature of NFATc1-driven gene regulation is the necessity to partner with other DNA binding proteins, mostly transcription factors, which help to contact DNA. Considering the critical roles for both NFATc1 and Sox2 in PDAC, we questioned whether NFATc1 not only induces gene expression but also directly interacts with Sox2 to form transcription complexes at genes encoding for “EMT master transducers”. To address this question, we carried out co-immunoprecipitation experiments in p53-deficient PDAC cells and identified robust NFATc1–Sox2 complex formation (Fig5K). Notably, the NFATc1–Sox2 pathway is also operative in human PDAC as we found concomitant expression of NFATc1 and Sox2 in a significant portion of freshly extracted primary human PDAC cells (Supplementary Fig S3C) and in tumor cell nuclei of 79% of human pancreatic cancers (Supplementary Fig S3D and E).
To explore the impact of Sox2 activation on EMT marker genes, we conducted ChIP analysis in KPNC cells following depletion of Sox2 expression. Sox2 deficiency abolished NFATc1 recruitment to the Snail enhancer, and this was accompanied by reduced enhancer and promoter activity and visualized by diminished H3K27 acetylation and declined RNA polymerase II binding, respectively (Fig5L). In line with our chromatin studies, loss of Sox2 displayed a significant reduction in EMT marker expression (Fig5M) and rescued NFATc1-mediated up-regulation of EMT genes in human pancreatic cancer cell lines (Fig5N and Supplementary Fig S3F and G). Collectively, NFATc1-mediated Sox2 induction and complex formation enable NFATc1 to drive transcription of “EMT master transducers” in p53-deficient PDAC.
Since NFATc1 and Sox2 are enriched in dedifferentiated cancer, we next investigated whether NFATc1 and Sox2 were increased under conditions driving the enrichment of cancer stem-like cells. Consistent with this notion, we found that both NFATc1 and Sox2 were highly induced in both human and mouse pancreatic cancer cell lines under conditions selecting for cancer stem-like cells (Fig6A and B). In fact, we identified a fourfold to eightfold increase in NFATc1 and Sox2 expression in spheroid forming cells and particularly in those cells with high levels of the stem cell marker CD44 (Fig6C). Moreover, CsA treatment and RNAi toward NFATc1 inhibited the formation of CD44-positive cancer stem-like cells (Supplementary Fig S4A and B) and impaired sphere formation (Fig6D and E, and Supplementary Fig S4C–E). Together, these experiments suggest that NFATc1–Sox2 signaling promotes EMT and associated stem cell-like characteristics in PDAC.
Figure 6.
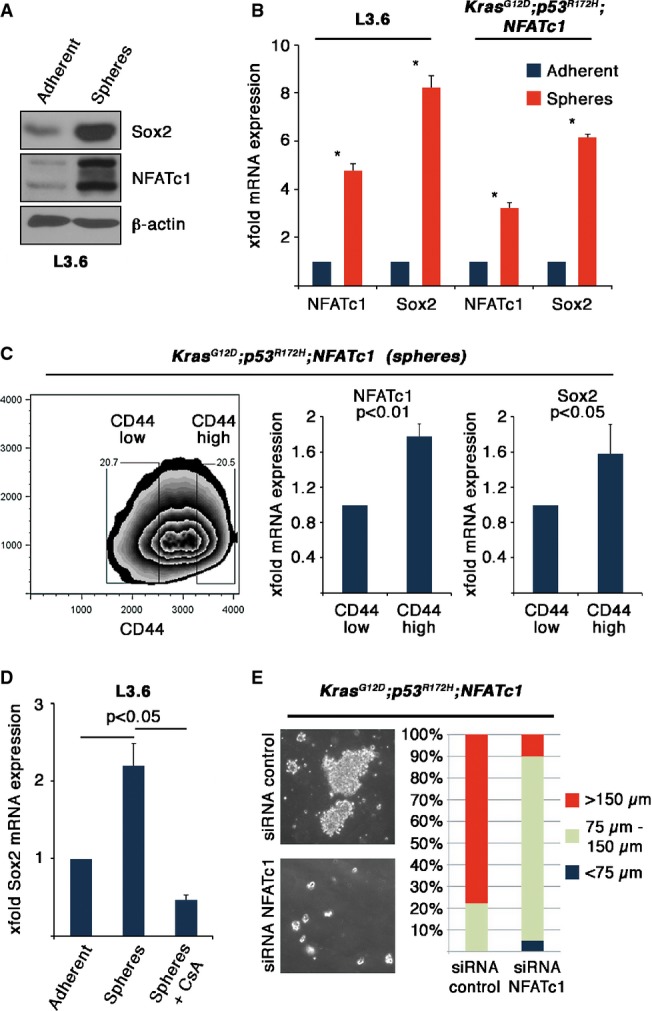
- NFATc1 and Sox2 protein expression in L3.6 cells (adherent versus spheres).
- Relative mRNA expression of NFATc1 and Sox2 in L3.6 cells as well as in KPNC mice tumor cells (adherent versus spheres). Graph represents data from three independent experiments (means ± SD). Asterisks display significance (*P < 0.05).
- Cell sorting was performed to collect CD44 high and low populations in KPNC spheres (left panel), which were analyzed for NFATc1 and Sox2 mRNA expression (right panel).
- Sox2 mRNA expression upon CsA (1 μM) treatment in L3.6 cells (adherent versus spheres) (*P < 0.05).
- Bright-field microscopy images as well as quantification of KPNC spheres after transient NFATc1 depletion.
Given that NFATc1–Sox2 pathway activation and EMT programming requires p53 deficiency, we next hypothesized that p53 is dominant over NFATc1–Sox2 signaling in governing cell fate decisions. p53 counteracts EMT in many cell types through activation of specific subsets of microRNA (miR), most notably members of the miR-200 and miR-34 families (Kim et al, 2011a,b). Accordingly, loss of miR-200c expression was recently reported in dedifferentiated human PDAC with poor survival outcome (Kent et al, 2009). Moreover, Sox2 recently has been shown to represent a direct target of miR-200c in colon cancer, in which the micro-RNA controls Sox2 expression and attenuates stemness features (Lu et al, 2014). We therefore tested the existence of a p53-miRNA pathway in the regulation of pancreatic cancer cell plasticity. As expected, we found a substantial loss of expression of miR-200c and miR-34a in poorly differentiated tumors, in primary cells derived from KPNC mice and following transient depletion of p53 in KNC cells (Supplementary Fig S5A–C), while re-expression of p53 in KPNC cells caused induction of miR-200c and miR-34a expression (Fig7A). Importantly, reactivation of the pathway by either viral-mediated transduction of p53 (Fig7B and Supplementary Fig S6A) or following re-expression of miR-200c, but not miR-34a (Fig7C and D, and Supplementary Fig S5D) decreased Sox2 expression and consequently caused transcriptional silencing of EMT regulators in KPΔNC and KPNC cells through the disruption of NFATc1/Sox2 chromatin association (Fig7F and G). In contrast to that, depletion of miR-200c resulted in a significant induction of EMT markers (Fig7E and Supplementary Fig S5E). Finally, consistent with the loss of this pathway driving EMT and dedifferentiation, restoration of the p53-miR200c signaling axis prevented cell migration (Supplementary Fig S5F) and the acquisition of cancer stem cell-like properties, as indicated by reduced expression of stem cell antigens CD44 and ALDH1 (Fig S6B), EMT markers (Supplementary Figs S5G and S6C–E), and diminished sphere-forming capacity (Fig7H). Thus, loss of p53 function correlates with NFATc1 activation in higher grade PDAC, supporting the notion of p53 as a gatekeeper for NFATc1-induced cancer cell plasticity.
Figure 7.

- Expression analysis of indicated miRNAs upon adenoviral re-expression of p53 in KPΔNC tumor cells, detected by real-time PCR (P < 0.05).
- Protein expression of EMT marker genes after adenoviral re-expression of GFP-tagged p53 in KPΔNC tumor cells.
- Protein (C) and mRNA (D) expression levels of EMT and stemness marker genes upon miRNA-34a and miRNA-200c overexpression using specific mimics in KPNC tumor cells. Representative results from at least three independent experiments are shown (means ± SD;P < 0.05).
- Western blot analysis of EMT marker expression in KNC cells following depletion of miR-200c.
- ChIP experiments showing NFATc1 binding along with H3K27 acetylation mark at the selected Snai1 enhancer region as well as Sox2 and RNA polymerase II binding at Snai1 promoter after miR-200c overexpression in KPNC tumor cells. Means ± SD are shown from one out of three independent experiments.
- ChIP analysis in KPΔNC tumor cells after adenoviral GFP-tagged p53 overexpression. NFATc1 and H3K27ac binding are illustrated on Snai1 enhancer, and Sox2 and RNA polymerase II binding are demonstrated on Snai1 promoter. Means ± SD are shown from one out of three independent experiments.
- Quantification of KPΔNC spheres after adenoviral GFP-tagged p53 overexpression.
Discussion
More than any other solid carcinoma, pancreatic cancer is characterized by persistent inflammation, highly invasive growth, and early seeding of tumor cells to liver and lung. Cellular plasticity is a requirement for dedifferentiation, EMT, stemness, and ultimately dissemination of transformed cells, thus significantly contributing the extremely poor clinical prognosis of pancreatic cancer patients. Major tumor suppressor genes such as p53 tightly control cellular plasticity, and recent studies showed a critical role for p53 in the regulation of differentiation, plasticity, and self-renewal (Pinho et al, 2011a; Spike & Wahl, 2011; Szychot et al, 2013). However, frequent deficiency in p53 function, by either deletion or dominant-negative mutation of the p53 gene in well-differentiated tumors, argues against this concept and suggests that the loss of p53 function on its own may not be sufficient to drive EMT and stemness (Brabletz, 2012). Given the prominent oncogenic role of inflammation-induced NFATc1 during pancreatic tumorigenesis and progression (Jauliac et al, 2002; Lagunas & Clipstone, 2009; Koenig et al, 2010; Baumgart et al, 2014), and recent reports implicating NFAT in neural differentiation and early lineage specification of mouse embryonic stem cells (Nagamoto-Combs & Combs, 2010; Li et al, 2011), we hypothesized that NFATc1 could be a central regulator of p53-mediated stemness and EMT. In support of our hypothesis, GEMMs harboring an oncogenic mutation of p53 preferentially develop well- to moderately differentiated pancreatic tumors. Moreover, a large set of clinical specimen showed no significant correlation between p53 expression/mutation and tumor grading or metastasis. However, poor differentiation was strongly associated with the activation of the transcription factor NFATc1. Combined with extensive biochemical and functional in vitro and in vivo investigations using genetic knockin and knockout strategies, as well as pharmacological assays, our data suggest that NFAT overexpression is an independent hallmark feature during pancreatic tumorigenesis driving progression via dedifferentiation, stemness, and EMT that requires p53 inactivation.
Metastasis as the final result of cellular motility and plasticity is suppressed by p53 through miRNA induction. Recent work unveiled a p53-dependent suppressor network which includes the miRNA-200 and miRNA-34 family (Chang et al, 2011a; Choi et al, 2011; Kim et al, 2011a). Members of both families can target zinc finger transcription factors such as Zeb1, Zeb2, or Slug and suppress migration and invasion (Kim et al, 2011a; Liu et al, 2014). The importance of these transcription factors as well as the impact of partner factors such as Snail and Twist for cellular motility and the induction of EMT have been widely shown in numerous studies (Lamouille et al, 2014). In this context, the crucial role of Zeb1 expression for tumor cell dissemination and resettlement has been recently demonstrated in mouse pancreatic cancer cells (Rhim et al, 2012) and Zeb1 expression is linked to tumorigenicity, invasion, and metastasis (Wellner et al, 2009). The fact that Zeb1 is also expressed in floating tumor cells with induced stem cell markers (Rhim et al, 2012, 2014) suggests an important role of this transcription factor for anchorage-independent survival and points toward a role of these proteins as mediators between EMT and stem-like properties (Brabletz, 2012; Sánchez-Tilló et al, 2012). Notably, our study provides first mechanistic evidence that NFATc1 controls expression of Snail and Zeb1. Moreover, the activity of this oncogenic axis is dominantly counteracted by the p53-miRNA-200c tumor suppressor pathway, which efficiently suppresses cellular dedifferentiation, EMT, and stem-like properties in PDAC.
To regulate target gene expression, NFATc1 relies on partner proteins which determine target gene specificity and the mode of transcriptional action (Rao et al, 1997). In the context of cellular plasticity, we provide first evidence that Sox2 acts as strong NFATc1 partner in the regulation of Snai1 and Zeb1. Sox2 is a transcription factor with pivotal roles in pluripotency during mammalian development and at different stages of carcinogenesis (Cox et al, 2012; Leis et al, 2012). Specifically, Sox2 maintains self-renewal of undifferentiated embryonic stem cells and promotes tumorigenesis in various epithelial tumors including pancreatic cancer through its ability to regulate gene regulation (Herreros-Villanueva et al, 2013; Sarkar & Hochedlinger, 2013). Our biochemical approaches and chromatin studies suggest that Sox2 not only functions as coactivator and interaction partner of NFATc1 but also represents a target of the calcineurin-dependent transcription factor. Thus, our data demonstrate that key features of cell plasticity are tightly regulated in PDAC by a hierarchical signaling network composed of opposed NFATc1–Sox2 and p53–miRNA-200c pathways and highlight the importance of EMT to promote the generation of highly tumorigenic cancer stem-like cells with increased motility and self-renewing capacities.
Collectively, our experimental findings suggest a hierarchical signaling and transcription network driving pancreatic cancer cell plasticity. Importantly, we show that the molecular decision between epithelial cell preservation and conversion into a dedifferentiated cancer stem cell-like phenotype is made at the level of p53 and NFATc1 signaling activity. Based on these findings, we propose the existence of a powerful antithetical NFATc1 and p53 pathway in cell plasticity and suggest a model in which the p53–miR-200c tumor suppressor pathway is dominant over NFATc1–Sox2 signaling in transcriptional regulation of EMT and stemness. Importantly, disruption of the p53 tumor suppressor pathway is required for NFATc1-driven dedifferentiation in PDAC (Fig8). Finally, our results highlight that pharmacological inactivation of NFATc1–Sox2 signaling might hold great therapeutic potential in patients with dedifferentiated and metastatic PDAC disease through abolition of a migrating cancer stem-like state, even in a p53-deficient situation.
Figure 8.
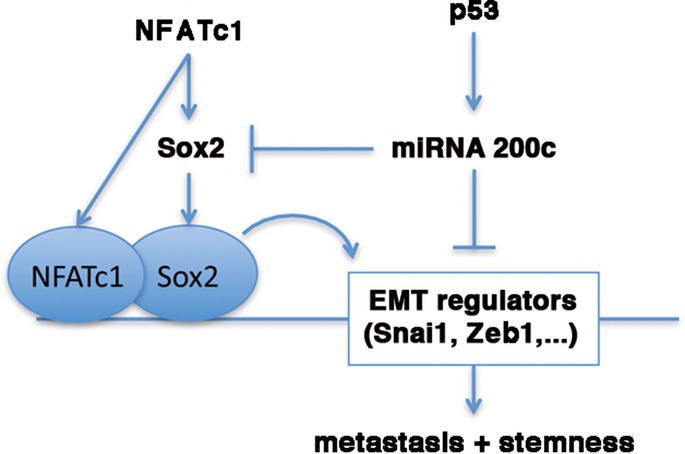
Proposed model of antithetical NFATc1–Sox2 and p53–miR-200c network in pancreatic cancer plasticity
A proposed model shows an antidromic NFATc1–Sox2 and p53–miR200c transcriptional network controlling EMT and stemness in pancreatic cancer.
Materials and Methods
Generation and characterization of mice
Generation and characterization of p48-Cre,LSL-KrasG12D, p53R172H, p53wt/Δ (Jackson et al, 2001; Meuwissen et al, 2003; Gu et al, 2004; Hingorani et al, 2005; Nakhai et al, 2008), and c.n.NFATc1 (Baumgart et al, 2014) mice have been described before. These mice strains were interbred to obtain KrasG12D;NFATc1;p48Cre (KNC) mice, KrasG12D;p53R172H;NFATc1;p48Cre (KPNC), and KrasG12D;p53wt/Δ;NFATc1;p48Cre (KPΔNC) tetra mutant animals. These mouse strains were genotyped by PCR as previously described (Baumgart et al, 2014), and only mice with correct genotype have been used for the study. All mouse strains are of Black6 background, and gender was irrelevant for tumor development. Animals were sacrificed/analyzed between 6 weeks and 12 months after birth as specified in legends. All animal experiments were carried out using protocols approved by the Institutional Animal Care and Use Committee at the Philipps University of Marburg.
Cell culture
Human cell line Panc-1 (CRL-1469) was obtained from ATCC. L3.6 cells were obtained from Daniel D Billadeau and described previously (Herreros-Villanueva et al, 2013). Isolation and culture of mouse primary tumor cells were performed as described previously (Baumgart et al, 2014). Mouse pancreatic tumor cells were generated from primary pancreatic tumors isolated from KPNC or KPΔNC, respectively. Mouse pancreatic tumor cells were cultured in DMEM containing 10% fetal bovine serum with 2 mM L-glutamine, 1% nonessential amino acids, penicillin (50 U ml−1), and streptomycin (50 U ml−1). Human pancreatic cancer L3.6 cells were cultured in 10% fetal bovine serum containing MEM medium (Life Technologies), and Panc-1 cells were cultured in DMEM containing 10% fetal bovine serum with 2 mM L-glutamine medium.
Immunohistochemistry and immunoblotting
H&E staining, immunohistochemistry, and Western blotting were performed as described previously (Baumgart et al, 2012) with antibodies directed against HA (6E2, #2367, Cell Signalling), p53 (1C12, #2524, Cell Signalling; CM5, NCL-p53-CM5p, Leica), Sox2 (ab97959, ab59776, Abcam), ZEB1 (E-20, sc-10572, Santa Cruz), Snai1 (L70G2, #3895, Cell Signalling), Twist1 (ab49254, Abcam), p21 (F-5, sc-6246, Santa Cruz), caspase 3 (#9661, Cell Signalling; ab1476-1 Epitomics), vimentin (RV202, 550513, BD Bioscience), CK19 (ab15463, Abcam), E-cadherin (36/e-cad, 610181, BD Bisoscience), NFATc1 (7A6, #sc7294, Santa Cruz), and beta-actin (AC-15, A3854, Sigma). For protein analysis of animal tissues, each lane represents an independent animal and experiments are repeated twice. For all other Western blot experiments, every image is a representative out of at least three independent experiments. Immunohistochemistry or pictures from animal tumors are representative out of four independent animals. Immunohistochemistry from human tissue are representative out of at least 19 samples.
Transfection and viral transduction
Micro-RNA was isolated using RNeasy Plus Mini Kit (Qiagen) followed by reverse transcription using the TaqMan miRNA RT Kit (Life Technologies). For miRNAs overexpression, cells were transfected with miR-34a and miR-200c or miR-control (Life Technologies) using Lipofectamine 2000 (Life Technologies). Gene expression was quantified by real-time PCR experiment using TaqMan MicroRNA Assays (U6, miR-34a, miR 200c, Life Technologies). The NFATc1 construct was gifted by Neil Clipstone and was transfected using Lipofectamine 2000 reagent. SiRNAs were purchased from Ambion or Dharmacon and transfected with siLentFect lipid reagent (Bio-Rad) according to the manufacturer's protocol. A set of two to three siRNAs was used per target. Generation and use of recombinant adenoviruses for wild-type p53 have been previously described (Schlereth et al, 2010). Cells were infected with adenovirus encoding GFP (as a control) or GFP together with wild-type p53. Production of retrovirus was performed using standard calcium phosphate method. Phoenix cells were transfected with 4-hydroxytamoxifen-activated pBABE-Puro-p53ERT or pBABE-Puro as a control. 48 h after transfection, the supernatant containing retroviral particles were filtered through a 0.45-μm filter and supplemented with 8 μg ml−1 Polybrene to infect the KPΔNC cells. Transduced cells were selected using 3 μg ml−1 puromycin (Life Technologies). Stable depletion of Sox2 was performed by using lentivirus-mediated shRNA expression with target sequence of 5-cagctcgcagacctacatgaa-3′, as previously described (Herreros-Villanueva et al, 2013).
Real-time PCR, microarray, and bioinformatics analyses
Total RNAs were extracted from cells by using RNeasy kit (Qiagen) according to the manufacturer's instructions. RNAs were reverse-transcribed by using Superscript II kit (Life Technologies), and experiments were performed in triplicate. Real-time PCR analysis was performed using an ABI-7500 instrument (Applied Biosystem/Life Technologies), and the quantification of RNA levels was normalized to RPLP0. Primer sequences used in this study are listed in the Supplementary Information. All results for qRT-PCR experiments are tested for statistical significance, which was always P < 0.05. Microarray and bioinformatic analyses were performed as described previously (Baumgart et al, 2014). All data obtained from microarray analysis were deposited (MIAME, accession number: E-MTAB-2324).
Sphere assay
For sphere-formation assay, adherent pancreatic tumor cells were dissociated to single cells by 0.05% trypsin–EDTA solution (Life Technologies) and plated at 100,000 cells per ml in serum-free DMEM containing insulin (Life Technologies), Albumin Bovine Fraction V (Sigma), N-2 Plus media, B-27 (Life Technologies), EGF (Sigma), and HB-EGF (Peprotech) into low-attachment dishes (Corning, Corning, NY, USA), as described previously (Herreros-Villanueva et al, 2013). Cells were incubated for 5 days before sphere formation was assayed or gene expression was analyzed.
Flow cytometry, co-immunoprecipitation, and ChIP analysis
For flow cytometry analysis, PE (phycoerythrin)-conjugated anti-CD44 (BD Biosciences) antibody was used. ALDH1 activity was detected using the ALDEFLUOR assay kit (Stem Cell Technologies) as described by the manufacture instructions. Samples were measured with a FACS Calibur Flow Cytometer (BD Biosciences) and analyzed with FlowJo software (TreeStar, Stanford, CA). Co-immunoprecipitation and ChIP experiments were performed as described previously (Koenig et al, 2010). Following ChIP antibodies were used: IgG, NFATc1 (7A6, #sc7294, Santa Cruz), H3K27ac (ab4729, Abcam), RNA polymerase II (Millipore), and Sox2 (ab97959, ab59776, Abcam).
Wound healing and time-lapse experiments
For wound healing experiments, cells were seeded at 5 × 104 cells per well and cultured in 6-well plates and grown to a 80% confluent monolayer. Subsequently, cells were transfected with siRNAs against p53 or control siRNA for 24 h. The confluent cell layers were scratched with a p200 pipette tip. Wound width and relative wound density were measured by time-lapse analyzer software as described previously (Huth et al, 2010, 2011).
TMA staining and analysis
All stainings carried out on human specimens were approved by the Mayo Clinic Institutional Review Board (Rochester, MN). Written confirmation was obtained from each patient. TMAs containing samples from PDAC patients were stained as indicated and analyzed for NFATc1 and p53 expression in the Pathology Research Core. TMA slides were placed in the BOND III (Leica Biosystems) stainer for online processing. They were treated with Epitope Retrieval 2 solution for 20 min and stained with NFATc1 (Abcam Cat #25916, 1:100) or p53 (Santa Cruz Biotechnology Cat #sc-126, 1:100) for 15 min, and detection was achieved using the Polymer Refine Detection kit per manufactures instructions (Leica Biosystems). Counter staining was performed for 5 min with hematoxylin. Slides were dehydrated through increasing concentrations of alcohol, cleared in xylene, and cover-slipped in xylene-based mounting media.
Data analysis
The TMAs were evaluated for NFATc1 and p53 expression (both nuclear and cytosolic) by a trained pancreatic pathologist and were scored as positive (score 2 or 3) or negative (score 0 or 1). Stain positivity across the multiple evaluable cores per patient was reduced to 1 observation per unique subject by using the core, which stained with the maximum intensity for each stain. A per-patient score (intensity of stain × extent of stain) was calculated and averaged across all patients for the purpose of exploring associations with clinical variables. Clinical variables are presented as mean (standard deviation) for continuous variables and frequency for categorical variables. Wilcoxon/Kruskal–Wallis rank sum tests were used to explore the association between categorical clinical variables and continuous average score for each stain. Log-rank tests were used to examine the associations of survival and age of surgery with each stain categorized into quartiles. A subsequent analysis which dichotomized the nuclear p53 stain at the median was also considered for age at surgery.
Statistical analyses
Data are presented as mean ± standard deviations (SD) or ± standard errors (SE). Significance was tested by Student's t-test using GraphPad Prism software or one-way ANOVA for multiple comparisons. For mice survivals analysis, Kaplan–Meier curves were determined by log-rank test using GraphPad Prism software. P < 0.05 is considered to be statistically significant.
Acknowledgments
We are grateful to Kristina Reutlinger, Sarah Hanheide (both University Medical center Göttingen), Bettina Geisel, and Gevin Giel (Philipps University of Marburg) for technical support. This work was generously supported by the Deutsche Forschungsgemeinschaft (V.E.: KFO210, SFB-TR17), the German Cancer Research Foundation (to V.E.: N°-109423 “Inflammation and Cancer” and A.K.: “Mildred Scheel” Fellowship), Mayo Foundation for Medical Research, NCI Pancreas SPORE Grant P50 CA102701 to D.D.B. and H.C.C., and NIH CA172045 to M.H.
Author contributions
SKS, N-MC, EH, GS, ML, NV, SB, SV, AS, JG, MM, J-SZ, X-KL, AK, and VE designed and performed experiments and analyzed results. SKS, ML, and TS generated and performed viral p53 depletion. SKS and CB performed FACS analysis. IE performed and analyzed sequencing of p53. HCC, WRB, TCS and DDB planned, performed, and analyzed the TMA analysis. JS generated tumor cell lines. SKS, EH, DDB, MH, AN, AK, and VE assembled the results and wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Dataset S1
Source Data for Supplementary Figure S2
Source Data for Supplementary Figure S3
Source Data for Supplementary Figure S4
Source Data for Supplementary Figure S6
Review Process File
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
References
- Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat Rev Cancer. 2005;5:744–749. doi: 10.1038/nrc1694. [DOI] [PubMed] [Google Scholar]
- Brabletz S, Bajdak K, Meidhof S, Burk U, Niedermann G, Firat E, Wellner U, Dimmler A, Faller G, Schubert J, Brabletz T. The ZEB1/miR-200 feedback loop controls Notch signalling in cancer cells. EMBO J. 2011;30:770–782. doi: 10.1038/emboj.2010.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart S, Glesel E, Singh G, Chen N-M, Reutlinger K, Zhang J, Billadeau DD, Fernandez-Zapico ME, Gress TM, Singh SK, Ellenrieder V. Restricted heterochromatin formation links NFATc2 repressor activity with growth promotion in pancreatic cancer. Gastroenterology. 2012;142:388–398. doi: 10.1053/j.gastro.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabletz T. EMT and MET in metastasis: where are the cancer stem cells? Cancer Cell. 2012;22:699–701. doi: 10.1016/j.ccr.2012.11.009. [DOI] [PubMed] [Google Scholar]
- Baumgart S, Chen N-M, Siveke JT, König A, Zhang J-S, Singh SK, Wolf E, Bartkuhn M, Esposito I, Heßmann E, Reinecke J, Nikorowitsch J, Brunner M, Singh G, Fernandez-Zapico ME, Smyrk T, Bamlet WR, Eilers M, Neesse A, Gress TM, et al. Inflammation-induced NFATc1-STAT3 transcription complex promotes pancreatic cancer initiation by KrasG12D. Cancer Discov. 2014;4:688–701. doi: 10.1158/2159-8290.CD-13-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchholz M, Schatz A, Wagner M, Michl P, Linhart T, Adler G, Gress TM, Ellenrieder V. Overexpression of c-myc in pancreatic cancer caused by ectopic activation of NFATc1 and the Ca2+/calcineurin signaling pathway. EMBO J. 2006;25:3714–3724. doi: 10.1038/sj.emboj.7601246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C-J, Chao C-H, Xia W, Yang J-Y, Xiong Y, Li C-W, Yu W-H, Rehman SK, Hsu JL, Lee H-H, Liu M, Chen C-T, Yu D, Hung M-C. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011a;13:317–323. doi: 10.1038/ncb2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S-C, Mulloy B, Magee AI, Couchman JR. Two distinct sites in sonic Hedgehog combine for heparan sulfate interactions and cell signaling functions. J Biol Chem. 2011b;286:44391–44402. doi: 10.1074/jbc.M111.285361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YJ, Lin C-P, Ho JJ, He X, Okada N, Bu P, Zhong Y, Kim SY, Bennett MJ, Chen C, Ozturk A, Hicks GG, Hannon GJ, He L. miR-34 miRNAs provide a barrier for somatic cell reprogramming. Nat Cell Biol. 2011;13:1353–1360. doi: 10.1038/ncb2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox JL, Wilder PJ, Desler M, Rizzino A. Elevating SOX2 levels deleteriously affects the growth of medulloblastoma and glioblastoma cells. PLoS ONE. 2012;7:e44087. doi: 10.1371/journal.pone.0044087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu G, Wells JM, Dombkowski D, Preffer F, Aronow B, Melton DA. Global expression analysis of gene regulatory pathways during endocrine pancreatic development. Development. 2004;131:165–179. doi: 10.1242/dev.00921. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Herreros-Villanueva M, Zhang J-S, Koenig A, Abel EV, Smyrk TC, Bamlet WR, de Narvajas AA-M, Gomez TS, Simeone DM, Bujanda L, Billadeau DD. SOX2 promotes dedifferentiation and imparts stem cell-like features to pancreatic cancer cells. Oncogenesis. 2013;2:e61. doi: 10.1038/oncsis.2013.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, DePinho RA. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006;20:1218–1249. doi: 10.1101/gad.1415606. [DOI] [PubMed] [Google Scholar]
- Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–483. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- Huth J, Buchholz M, Kraus JM, Schmucker M, von Wichert G, Krndija D, Seufferlein T, Gress TM, Kestler HA. Significantly improved precision of cell migration analysis in time-lapse video microscopy through use of a fully automated tracking system. BMC Cell Biol. 2010;11:24. doi: 10.1186/1471-2121-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huth J, Buchholz M, Kraus JM, Mølhave K, Gradinaru C, von Wichert G, Gress TM, Neumann H, Kestler HA. TimeLapseAnalyzer: multi-target analysis for live-cell imaging and time-lapse microscopy. Comput Methods Programs Biomed. 2011;104:227–234. doi: 10.1016/j.cmpb.2011.06.002. [DOI] [PubMed] [Google Scholar]
- Imamichi Y, König A, Gress T, Menke A. Collagen type I-induced Smad-interacting protein 1 expression downregulates E-cadherin in pancreatic cancer. Oncogene. 2007;26:2381–2385. doi: 10.1038/sj.onc.1210012. [DOI] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jauliac S, López-Rodriguez C, Shaw LM, Brown LF, Rao A, Toker A. The role of NFAT transcription factors in integrin-mediated carcinoma invasion. Nat Cell Biol. 2002;4:540–544. doi: 10.1038/ncb816. [DOI] [PubMed] [Google Scholar]
- Kao S-C, Wu H, Xie J, Chang C-P, Ranish JA, Graef IA, Crabtree GR. Calcineurin/NFAT signaling is required for neuregulin-regulated Schwann cell differentiation. Science. 2009;323:651–654. doi: 10.1126/science.1166562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, Zhong Y, Wu C-J, Sadanandam A, Hu B, Chang Q, Chu GC, Al-Khalil R, Jiang S, Xia H, Fletcher-Sananikone E, Lim C, Horwitz GI, Viale A, Pettazzoni P, et al. Yap1 activation enables bypass of oncogenic kras addiction in pancreatic cancer. Cell. 2014;158:185–197. doi: 10.1016/j.cell.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent OA, Mullendore M, Wentzel EA, López-Romero P, Tan AC, Alvarez H, West K, Ochs MF, Hidalgo M, Arking DE, Maitra A, Mendell JT. A resource for analysis of microRNA expression and function in pancreatic ductal adenocarcinoma cells. Cancer Biol Ther. 2009;8:2013–2024. doi: 10.4161/cbt.8.21.9685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NH, Kim HS, Li X-Y, Lee I, Choi H-S, Kang SE, Cha SY, Ryu JK, Yoon D, Fearon ER, Rowe RG, Lee S, Maher CA, Weiss SJ, Yook JI. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J Cell Biol. 2011a;195:417–433. doi: 10.1083/jcb.201103097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Veronese A, Pichiorri F, Lee TJ, Jeon Y-J, Volinia S, Pineau P, Marchio A, Palatini J, Suh S-S, Alder H, Liu C-G, Dejean A, Croce CM. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med. 2011b;208:875–883. doi: 10.1084/jem.20110235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig A, Linhart T, Schlengemann K, Reutlinger K, Wegele J, Adler G, Singh G, Hofmann L, Kunsch S, Büch T, Schäfer E, Gress TM, Fernandez-Zapico ME, Ellenrieder V. NFAT-induced histone acetylation relay switch promotes c-Myc-dependent growth in pancreatic cancer cells. Gastroenterology. 2010;138:1189–1199. doi: 10.1053/j.gastro.2009.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König A, Fernandez-Zapico ME, Ellenrieder V. Primers on molecular pathways–the NFAT transcription pathway in pancreatic cancer. Pancreatology. 2010;10:416–422. doi: 10.1159/000315035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagunas L, Clipstone NA. Deregulated NFATc1 activity transforms murine fibroblasts via an autocrine growth factor-mediated Stat3-dependent pathway. J Cell Biochem. 2009;108:237–248. doi: 10.1002/jcb.22245. [DOI] [PubMed] [Google Scholar]
- Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leis O, Eguiara A, Lopez-Arribillaga E, Alberdi MJ, Hernandez-Garcia S, Elorriaga K, Pandiella A, Rezola R, Martin AG. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene. 2012;31:1354–1365. doi: 10.1038/onc.2011.338. [DOI] [PubMed] [Google Scholar]
- Li X, Zhu L, Yang A, Lin J, Tang F, Jin S, Wei Z, Li J, Jin Y. Calcineurin-NFAT signaling critically regulates early lineage specification in mouse embryonic stem cells and embryos. Cell Stem Cell. 2011;8:46–58. doi: 10.1016/j.stem.2010.11.027. [DOI] [PubMed] [Google Scholar]
- Liu Y, Sánchez-Tilló E, Lu X, Huang L, Clem B, Telang S, Jenson AB, Cuatrecasas M, Chesney J, Postigo A, Dean DC. The ZEB1 transcription factor acts in a negative feedback loop with miR200 downstream of Ras and Rb1 to regulate Bmi1 expression. J Biol Chem. 2014;289:4116–4125. doi: 10.1074/jbc.M113.533505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y-X, Yuan L, Xue X-L, Zhou M, Liu Y, Zhang C, Li J-P, Zheng L, Hong M, Li X-N. Regulation of colorectal carcinoma stemness, growth, and metastasis by an miR-200c-Sox2-negative feedback loop mechanism. Clin Cancer Res. 2014;20:2631–2642. doi: 10.1158/1078-0432.CCR-13-2348. [DOI] [PubMed] [Google Scholar]
- Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. 2008;3:157–188. doi: 10.1146/annurev.pathmechdis.3.121806.154305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, Berns A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell. 2003;4:181–189. doi: 10.1016/s1535-6108(03)00220-4. [DOI] [PubMed] [Google Scholar]
- Morton JP, Karim SA, Graham K, Timpson P, Jamieson N, Athineos D, Doyle B, McKay C, Heung M-Y, Oien KA, Frame MC, Evans TRJ, Sansom OJ, Brunton VG. Dasatinib inhibits the development of metastases in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology. 2010;139:292–303. doi: 10.1053/j.gastro.2010.03.034. [DOI] [PubMed] [Google Scholar]
- Nagamoto-Combs K, Combs CK. Microglial phenotype is regulated by activity of the transcription factor, NFAT (nuclear factor of activated T cells) J Neurosci. 2010;30:9641–9646. doi: 10.1523/JNEUROSCI.0828-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakhai H, Siveke JT, Mendoza-Torres L, Schmid RM. Conditional inactivation of Myc impairs development of the exocrine pancreas. Development. 2008;135:3191–3196. doi: 10.1242/dev.017137. [DOI] [PubMed] [Google Scholar]
- Pinho AV, Rooman I, Real FX. p53-dependent regulation of growth, epithelial-mesenchymal transition and stemness in normal pancreatic epithelial cells. Cell Cycle. 2011a;10:1312–1321. doi: 10.4161/cc.10.8.15363. [DOI] [PubMed] [Google Scholar]
- Pinho AV, Rooman I, Reichert M, De Medts N, Bouwens L, Rustgi AK, Real FX. Adult pancreatic acinar cells dedifferentiate to an embryonic progenitor phenotype with concomitant activation of a senescence programme that is present in chronic pancreatitis. Gut. 2011b;60:958–966. doi: 10.1136/gut.2010.225920. [DOI] [PubMed] [Google Scholar]
- Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, Leach SD, Stanger BZ. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–361. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhim AD, Thege FI, Santana SM, Lannin TB, Saha TN, Tsai S, Maggs LR, Kochman ML, Ginsberg GG, Lieb JG, Chandrasekhara V, Drebin JA, Ahmad N, Yang Y-X, Kirby BJ, Stanger BZ. Detection of circulating pancreas epithelial cells in patients with pancreatic cystic lesions. Gastroenterology. 2014;146:647–651. doi: 10.1053/j.gastro.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustgi AK. The molecular pathogenesis of pancreatic cancer: clarifying a complex circuitry. Genes Dev. 2006;20:3049–3053. doi: 10.1101/gad.1501106. [DOI] [PubMed] [Google Scholar]
- Sánchez-Tilló E, Liu Y, de Barrios O, Siles L, Fanlo L, Cuatrecasas M, Darling DS, Dean DC, Castells A, Postigo A. EMT-activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cell Mol Life Sci. 2012;69:3429–3456. doi: 10.1007/s00018-012-1122-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar A, Hochedlinger K. The sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell. 2013;12:15–30. doi: 10.1016/j.stem.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlereth K, Beinoraviciute-Kellner R, Zeitlinger MK, Bretz AC, Sauer M, Charles JP, Vogiatzi F, Leich E, Samans B, Eilers M, Kisker C, Rosenwald A, Stiewe T. DNA binding cooperativity of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2010;38:356–368. doi: 10.1016/j.molcel.2010.02.037. [DOI] [PubMed] [Google Scholar]
- Spike BT, Wahl GM. p53, stem cells, and reprogramming: tumor suppression beyond guarding the genome. Genes Cancer. 2011;2:404–419. doi: 10.1177/1947601911410224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szychot E, Brodkiewicz A, Peregud-Pogorzelski J. Will therapies that target tumour suppressor genes be useful in cancer treatment? Adv Clin Exp Med. 2013;22:861–864. [PubMed] [Google Scholar]
- Thiery J-P. [Epithelial-mesenchymal transitions in cancer onset and progression] Bull Acad Natl Med. 2009;193:1969–1978. discussion 1978–1979. [PubMed] [Google Scholar]
- Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D, Hausen zur A, Brunton VG, Morton J, Sansom O, Schüler J, Stemmler MP, Herzberger C, Hopt U, Keck T, Brabletz S, Brabletz T. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487–1495. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Dataset S1
Source Data for Supplementary Figure S2
Source Data for Supplementary Figure S3
Source Data for Supplementary Figure S4
Source Data for Supplementary Figure S6
Review Process File
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7