

Abstract
Background
Cyclophosphamide is an alkylating chemotherapeutic drug administered IV or PO. It is currently assumed that exposure to the active metabolite, 4-hydroxycyclophosphamide (4-OHCP), is the same with either route of administration.
Objectives
To characterize the pharmacokinetics of cyclophosphamide and 4-OHCP in dogs with lymphoma when administered PO or IV.
Animals
Sixteen client-owned dogs with substage A lymphoma were enrolled in the study. Eight dogs received cyclophosphamide IV and 8 received it PO.
Methods
Prospective randomized clinical trial was performed. Blood was collected from each dog at specific time points after administration of cyclophosphamide. The serum was evaluated for the concentration of cyclophosphamide and 4-OHCP with mass spectrometry and liquid chromatography.
Results
Drug exposure to cyclophosphamide measured by area under the curve (AUC)0–inf is significantly higher after intravenous administration (7.14 ± 3.77 μg/h/mL) compared with exposure after oral administration (P-value < .05). No difference in drug exposure to 4-OHCP was detected after IV (1.66 ± 0.36 μg/h/mL) or PO (1.42 ± 0.64 μg/h/mL) administered cyclophosphamide.
Conclusions and Clinical Importance
Drug exposure to the active metabolite 4-OHCP is equivalent after administration of cyclophosphamide either PO or IV.
Keywords: Chemotherapy, Liquid chromatography, Mass spectrometry, Pharmacokinetics
Cyclophosphamide is an alkylating chemotherapeutic drug commonly used in veterinary oncology. It is administered as an intravenous bolus or given PO, often in conjunction with other chemotherapy drugs for use in multidrug protocols. It is used for the treatment of a variety of malignancies. Cyclophosphamide is an oxazaphosphorine prodrug and must undergo activation to form 4-hydroxycyclophosphamide (4-OHCP) before exerting a cytotoxic effect.1–5 Biotransformation of cyclophosphamide occurs in the liver via the cytochrome (CYP) P450 system.1,3 In humans, studies indicate that up to 70% of administered cyclophosphamide is metabolized to 4-OHCP.4 Multiple CYP450 enzymes have been implicated in the mediation of this reaction and include CYP2B6, CYP2C9, CYP2C19, CYP3A4, and CYP3A5.3,6,7
After hydroxylation of cyclophosphamide, 4-OHCP is in equilibrium with its tautomeric form, aldophosphamide. 1,5,8 It is not possible to distinguish 4-OHCP and aldophosphamide with commonly used assays; thus in the literature, 4-OHCP often refers to the combination of both metabolites.1 4-OHCP is able to pass through cellular membranes and thus rapidly enters cells.1,2 Within the cell, 4-OHCP rapidly decomposes to phosphoramide mustard and acrolein.1,2,8 Phosphoramide mustard is the ultimate cytotoxic metabolite of cyclophosphamide, resulting in cross linkage of DNA.1,2 Thus the systemic concentration of 4-OHCP is thought to represent the intracellular cytotoxically active state of cyclophosphamide.1
4-OHCP is unstable and is rapidly metabolized within the cell, which has historically made quantification difficult. 1–5,8 A simple method to quantify 4-OHCP in red blood cells and plasma involves direct stabilization with semicarbazide hydrochloride (SCZ).1 The concentration of 4-OHCP in plasma and red blood cells is the same, indicating measurement of 4-OHCP in the blood can be used as a surrogate marker of intracellular concentration.1
Cyclophosphamide is commercially available in formulations for intravenous and oral administration. Studies in mice and humans indicate that 4-OHCP and phosphoramide mustard are available in equal quantities when administered PO or IV, suggesting that intravenous and oral routes can be used interchangeably in the clinical setting.9
In veterinary medicine, it is assumed that the concentration of the active metabolites is the same with either route of administration although no pharmacokinetic studies have been done in the dog. The decision to administer cyclophosphamide PO or IV is often based on clinician or owner preference. The objective of this prospective study was to characterize the pharmacokinetics of cyclophosphamide and 4-OHCP in the plasma of dogs with lymphoma when administered PO or IV.
We also speculated that a significant difference would be present for drug exposure of cyclophosphamide as measured by area under the curve (AUC) when administered IV or PO to the same population of dogs. This had also previously been established within the human population by Struck et al.9
Materials and Methods
This was a randomized prospective study. Before starting the study, the protocol was approved by the Institutional Animal Care and Use Committee and written client consent was obtained before dog enrollment. A total of 16 dogs diagnosed with lymphoma that were treatment naïve were enrolled. Criteria for inclusion were dogs with substage A lymphoma, body weight >10 kg, alkaline phosphatase, alanine transferase, and total bilirubin values within the reference range, and no previous history of chemotherapy treatment. Dogs were excluded if they had already been treated with or had received corticosteroids within 30 days of presentation, or were receiving phenobarbitone.
Dogs were randomly assigned to receive cyclophosphamide either POa or IVb at a dose of 250 mg/m2. Intravenous cyclophosphamide was administered as a slow bolus infusion over 10 minutes. The oral dose was rounded to the nearest 25mg tablet and administered at a single time point. All dogs received furosemide (1 mg/kg) subcutaneously immediately before treatment. Whole blood was collected at the following time points, 0, 15, 30, 60 minutes after administration, and then 2, 4, 6, 8, and 24 hours.
After the 24-hour time point collection and study completion, dogs were then treated with a variety of different chemotherapy protocols as selected by the owners. These included 15 or 19-week CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone), single agent doxorubicin, CCNU and L-asparaginase or prednisone alone. As part of the 15-week CHOP protocol, 4 dogs received a dose of vinciristine 24 hours after the initial dose of cyclophosphamide. The remaining 12 dogs did not receive additional chemotherapy, with the exception of prednisone, until 1 week after cyclophosphamide administration. Toxicity and tumor response were assessed at 1-week postcyclophosphamide administration. Hematologic toxicosis was graded according to VCOG-CTCAE.10
Chemicals
Synthetic, “preactivated” (preoxidized) precursor to the cyclophosphamide metabolite 4-OHCP, 4-hydroperoxycyclophosphamide (4-OOHCP; purity 98%) was purchased from IIT GmbH/ NIOMECH.c Cyclophosphamide monohydrate, hexamethylphosphoramide (internal standard [IS]), SCZ, and all other reagents (methanol, acetonitrile, and ammonium hydroxide) used were analytical grade purchased from Sigma-Aldrich.d
Sample Handling
The method for sample handling used was described by Huitema et al, 1 and is briefly explained here. Whole blood was collected from dogs and placed into sodium heparin tubes at the assigned time points. The sample was immediately placed on ice and centrifuged at 2,500 × g for 10 minutes at −4°C. One milliliter of plasma was placed into a 10mL polypropylene tube with 100 μL of 2M SCZ in order to trap the 4-OHCP in the stable form. The sample was then vortexed for 30 seconds and stored at −80°C until analysis. Samples were stored for between 2 weeks and 8 months before analysis. Huitema et al1 determined that the samples were stable when stored for up to 12 months.
Standard dilutions of free cyclophosphamide and 4-OOHCP were prepared in water at 10 and 1 mg/mL, respectively, and each were added immediately to naïve dog plasma to result in a standard curve ranging from 12.5 to 5,000 ng/mL. 4-OOHCP spontaneously converts to 4-OHCP in aqueous solutions which was trapped upon the addition of 2M SCZ to produce semicarbizide derivative of 4-OHCP (4-OHCP-SCZ) and vortexed for 30 seconds.10 Samples were aliquoted and stored at −80°C until analysis.
On the day of sample analysis, dog and standard curve samples were thawed, 100 μL of each was added to a microcentrifuge tube containing 25 ng of IS (10 μL or 2.5 μg/mL solution) and vortexed briefly. Proteins were then precipitated with the addition of 300 μL of 1 : 1 (v : v) methanol : acetonitrile and brief mixing. Samples were then centrifuged at approximately 21,000 × g for 15 minutes, 50 μL of supernatant was then added to 400 μL of 1mM ammonium hydroxide (pH 10.1), mixed and transferred to HPLC vials.
Mass Spectrometry and Liquid Chromatography Conditions
Positive ion electrospray ionization mass spectra were obtained with a MDS Sciex 3,200 Q-TRAP triple quadrupole mass spectrometer e with a turbo ionspray source interfaced to a Shimadzu Prominence HPLC systemf and a CTC Analytics HTC PAL System autosampler.g Samples were chromatographed with an Alltech Hypersil ODS (C18) 3U; 150 × 4.6mmh protected by a Security-Guard C18 cartridge (4 × 2.0mm I.D)i maintained at room temperature. An isocratic mobile phase consisting of 55% 10mM ammonium hydroxide (pH 10.1) and 45% acetonitrile was used at a flow rate of 800 μL/min. Sample injection volume was 25 μL, and the analysis run time was 10 minutes. Mass spectrometer compound specific settings were optimized to monitor the transitions.1
Analytes were quantified by IS reference monitoring of the ion transitions m/z 261.2 → 140.2 for cyclophosphamide, m/z 334.3 → 221.2 for the semicarbizide derivative of 4-OHCP (4-OHCP-SCZ), and m/z 180.0 → 135.0 for the IS. The mass spectrometer settings were optimized as follows: turbo ionspray temperature, 550°C; ion spray voltage, 3,500 V; declustering potential, 21V (CP), 46V (4-OHCP-SCZ) and 21V (IS); entrance potential, 4V for all 3 analytes; collision energy (CE), 17V (CP), 28V (4-OHCP-SCZ) and 10V (IS); collision cell entrance potential, 16V (CP and 4-OHCP-SCZ) and 15.2V (IS); collision cell exit potential, 4.5V (CP), 2.3V (4-OHCP-SCZ) and 3.0V (IS); curtain gas, N2 (CUR), 45 units; collision gas, N2 (CAD), 3; nebulizer as, N2, 75 units; and auxiliary gas, N2, 60 units.
Statistical Analysis
Pharmacokinetic parameters for both cyclophosphamide and 4-OHCP were examined and compared between the groups. These parameters were calculated by noncompartmental analysis and included maximum concentration (Cmax), clearance, AUC0–inf, half life (t1/2λ), time to maximum concentration (Tmax). AUC0–inf and Cmax corrected for dose were also included in the analysis. All parameters are accompanied by standard deviation. The 2 groups were compared using Mann-Whitney analysis. Values were considered significantly different if the P-value was <.05.
Results
Sixteen dogs were enrolled in the study. Eight dogs received cyclophosphamide PO, and 8 IV. For those that received the drug PO the median dose was 272 mg/m2 with a range of 250–325 mg/m2.
The AUC0–inf is significantly higher when cyclophosphamide was administered IV, 7.14 ± 3.77 μg/h/ mL, versus 1.85 ± 2.58 μg/h/mL (Table 1, Fig 1A). As expected the AUC0–inf corrected for dose also demonstrated a statistically significant difference between the intravenous (29.30 ± 15.37 ng/h/mL/mg) and oral (7.18 ± 10.43 ng/h/mL/mg). The Cmax of intravenous cyclophosphamide was reached immediately after administration, and then decreased steadily over time. After oral administration, Cmax was reached at approximately 0.78 hours (± 0.394), and then decreased more slowly in comparison with intravenous administration. The Cmax of cyclophosphamide after intravenous administration was significantly higher, 20.49 ± 12.95 versus 1.16 ± 1.08 μg/mL, than the oral group. The Cmax corrected for dose was also significantly higher within the intravenous group (82.61 ± 42.53 ng/mL/mg) compared with the oral group (4.54 ± 7.23 ng/mL/mg). No difference was observed in the terminal half life with either route of administration.
Table 1.
Pharmacokinetic parameters of cyclophosphamide following IV or oral dosing in dogs.
| PK Parametera | Units | IV | Oral |
|---|---|---|---|
| AUC0→inf | μg/h/mL | 7.14 ± 3.77 | 1.85 ± 2.58* |
| Cmax | μg/mL | 20.49 ± 12.95 | 1.16 ± 1.80* |
| t1/2λ | hour | 0.53 ± 0.12 | 0.78 ± 0.39 |
| CL (CLs or CLoral) | L/h | 42.54 ± 22.38 | 327.66 ± 206.94 |
| Tmax | hour | — | 1.81 ± 1.07 |
| AUC/dose | ng/h/mL/mg | 29.30 ± 15.37 | 7.18 ± 10.43* |
| Cmax/dose | ng/mL/mg | 82.61 ± 42.53 | 4.54 ± 7.23* |
Pharmacokinetic parameters accompanied by standard deviation.
PK parameters were calculated by noncompartmental analysis.
Significantly different (P < .05) from IV dosing parameter as determined by Mann-Whitney analysis.
Fig 1.
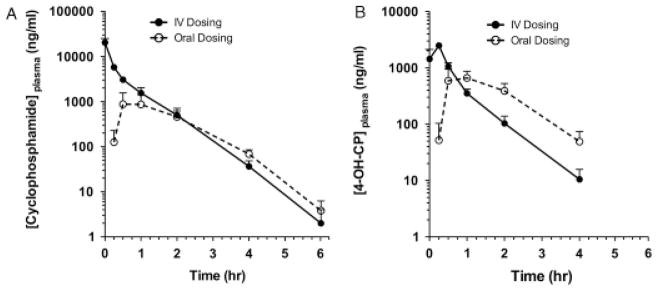
Plasma concentration versus time curves for cyclophosphamide and 4-OHCP. (A) Plasma concentration of cyclophosphamide when administered PO or IV to dogs with substage A lymphoma. (B) Plasma concentration for 4-OHCP when administered PO or IV to dogs with substage A lymphoma.
As expected, drug exposure as measured by AUC0–inf for 4-OHCP was not significantly different when administered IV (1.66 ± 0.36 μg/h/mL) or PO (1.42 ± 0.64 μg/h/ mL). Analysis of AUC0–inf corrected for dose was also performed, and no difference was appreciated between the intravenous (7.60 ± 4.18 ng/h/mL/mg) or oral (5.38 ± 2.63 ng/h/mL/mg) group. This indicates that regardless of the route of administration, exposure to the active metabolite of cyclophosphamide is equivalent. The pharmacokinetic values measured for 4-OHCP are presented in Table 2 and Figure 1B.
Table 2.
Pharmacokinetic parameters of 4-OH-cyclophosphamide following IV or oral dosing in dogs.
| PK Parametera | Units | IV | Oral |
|---|---|---|---|
| AUC0→inf | μg/h/mL | 1.66 ± 0.36 | 1.42 ± 0.64 |
| Cmax | μg/mL | 2.79 ± 1.34 | 1.21 ± 0.80* |
| t1/2λ | hour | 0.67 ± 0.45 | 0.59 ± 0.29 |
| Tmax | hour | 0.22 ± 0.09 | 1.25 ± 0.65* |
| AUC/dose | ng/h/mL/mg | 7.60 ± 4.18 | 5.38 ± 2.63 |
| Cmax/dose | ng/mL/mg | 10.3 ± 5.1 | 4.6 ± 3.3* |
Pharmacokinetic parameters accompanied by standard deviation.
PK parameters were calculated by noncompartmental analysis.
Significantly different (P < .05) from IV dosing parameter as determined by Mann-Whitney analysis.
As with cyclophosphamide, the Cmax of 4-OHCP was significantly higher, 2.79 ± 1.34 μg/mL, and the time to achieve this concentration (Tmax), 0.22 ± 0.09 hour, was significantly shorter for the intravenous group. The Cmax for the oral group was reached approximately at 1.25 ± 0.65 hours after administration. However, as opposed to the intravenous group, the concentration of 4-OHCP was sustained for a longer period of time (Fig 1B). The concentration of 4-OHCP in the intravenous group declined steadily after Cmax was also corrected for dose (Cmax/dose), and a statistically significant difference was detected between the intravenous (10.3 ± 5.1 ng/mL/mg) and oral (4.6 ± 3.3 ng/mL/mg) groups. Despite the significant difference between AUC0–inf of cyclophosphamide in the different groups, drug exposure to the active metabolite was ultimately the same. Drug exposure, as measured by AUC0–inf, to 4-OHCP (1.66 ± 0.36 μg/h/ mL) in the intravenous group was approximately 5-fold less than exposure to cyclophosphamide (7.14 ± 3.77 μg/ h/mL). Within the oral group, the exposure to 4-OHCP and cyclophosphamide was similar.
Interanimal variability was observed with regards to cyclophosphamide within the 2 groups. Greater variability in pharmacokinetic parameters was seen within the oral group. Individual differences to cyclophosphamide exposure, as measured by AUC0–inf, within the intravenous groups was approximately 5.6-fold (range 151–842 μg/h/mL), compared with 20-fold (range 23.9– 487 μg/h/mL) within the oral group. Substantial variation was also observed with regards to Cmax. Variability within the intravenous group was approximately 5.6-fold (range 8,020–45,000 ng/mL), compared with 46-fold difference for the oral group (range 119–5,470 ng/mL). No variability with regards to Tmax was observed within the intravenous group; however, an 8-fold difference was observed for the oral group (range 30–240 minutes). Variability within the intravenous and oral groups was also observed for the pharmacokinetic parameters of 4-OHCP. However, unlike cyclophosphamide, the degree of interanimal variability between the groups was similar. Variability for drug exposure was approximately 1.8-fold for the intravenous (range 67.7–125.6 μg/h/mL), and 3.3-fold for the oral group (range 41.6–138.3 μg/h/mL). Interanimal variability for Cmax was 8.3-fold in the intravenous group (range 619–5,180 ng/mL) and 7.2-fold for the oral (range 340–2,450 ng/mL). Variability for Tmax was 2.5-fold for the intravenous group (range 10–25 minutes) and 4-fold for the oral group (range 30–120 minutes).
One week after drug administration, the dogs were reexamined and a CBC was performed. Seven of the 8 dogs in the intravenous group were re-evaluated at this time. Five dogs were in a partial remission (72%), 1 was in a complete remission (14%), and 1 dog had progressive disease (14%). The overall response rate for dogs in the intravenous group was 85%. All 8 dogs enrolled in the oral group were available for re-evaluation. Six dogs were in a partial remission (75%), and 2 dogs had a complete remission (25%). The overall response rate for the oral groups was 100%.
No episodes of sterile hemorrhagic cystitis (SHC) were reported after administration of cyclophosphamide at any time during treatment.
After administration of cyclophosphamide, dogs received a variety of chemotherapy protocols. These included; CCNU and L-asparaginase, 19-week multidrug CHOP protocol, 15-week multidrug CHOP protocol, COP, or single agent doxorubicin. Three dogs who received the 15-week CHOP protocol were administered vincristine (0.7 mg/m2, IV) 24 hours after cyclophosphamide at the completion of the study.
Four dogs developed hematological toxicoses 7 days after cyclophosphamide administration. Two dogs associated with the oral and 2 with the intravenous administration. Two episodes of grade 3 neutropenia were reported, 1 in each group. Both of these dogs had been administered vincristine 24 hours after cyclophosphamide, as part of the 15-week CHOP protocol. A single episode of grade 2 and grade 1 neutropenia were reported among the oral and intravenous group, respectively. No episodes of gastrointestinal toxicoses were reported.
Discussion
We did not detect a significant difference in the availability of the active metabolite, 4-OHCP when cyclophosphamide is administered IV or PO to dogs; however, a significant decrease in drug exposure as measured by the AUC0–inf for cyclophosphamide occurs with the oral administration. This finding supports the human literature, and it is likely that oral and intravenous cyclophosphamide can be used interchangeably with the same exposure of active metabolite being achieved in dogs with lymphoma.9
When administered PO the bioavailability of cyclophosphamide is estimated at ~25% from the results of this study. This is likely because of differences in absorption through the gastrointestinal tract, and first-pass elimination through the liver.1,2,11,12 As cyclophosphamide is metabolized to 4-OHCP in the liver, only a small percentage will reach the systemic circulation for sampling. Thus exposure to cyclophosphamide when administered PO reflects the portion of the prodrug not converted to the active metabolite by the liver.
In contrast, the exposure to cyclophosphamide when administered IV is significantly higher (P < .05) as there is no first-pass elimination through the liver, and sampling reflects exposure before metabolism in the liver.1,13 However, despite the higher exposure to the prodrug, exposure to the active metabolite is not statistically different from that achieved with oral dosing. This difference in exposure to the prodrug is attributed to a combination of excretion of prodrug unchanged in the urine, and absorption or distribution to organs other than the liver not allowing for metabolic activation. 1,3,4,14 It has been established that within the human population approximately 25% of cyclophosphamide after intravenous administration is excreted unchanged in urine.4 Unfortunately, this information is not currently available for dogs and was not examined in this study; however, it is assumed to be similar.
CYP P450 is predominantly responsible for phase 1 metabolism in the liver, as is the case with cyclophosphamide. 6,7,13–16 The genetic polymorphisms in certain CYP P450 isozymes, including CYP2B6, CYP2C9, CYP2C19, CYP3A4, and CYP3A5, can contribute to the interanimal variability commonly seen among dogs receiving cyclophosphamide.6,7,14–16 In some cases, these polymorphisms were found to be predictors for intoxication and clinical response.6,7,14,17
Given the importance of CYP P450 isozymes in hepatic drug metabolism, concurrent liver disease and the effect, if any, it has on cyclophosphamide warrants further investigation. To assist in controlling for some interanimal variability, we only enrolled dogs with liver parameters, including total bilirubin, within the normal reference range. These inclusion criteria have been used in studies of cyclophosphamide pharmacokinetics in humans.4,9
Interpatient variability among the human population receiving cyclophosphamide has also been associated with sex, age, ethnicity, and the administration of concurrent medications.4,7,14 Environmental factors such as the influence of diets, are yet to be evaluated. Recognition of interanimal variability has resulted in individual dose adjustments in the aim of tailoring therapy to maximize efficacy and minimize severity of toxicoses.2,4,6,7,12,14,18 In addition to interanimal variability, intra-animal variability occurs over time within some individuals, indicating that dose adaptations made during the treatment course might be warranted to reduce toxicity and maximize efficacy.2,12
In our study, the focus was not on establishing the causes of interanimal variability within the canine population. In an attempt to minimize interanimal variability, we only enrolled dogs greater than 10 kg, dogs were not permitted to have received prednisone or dexamethasone 30 days before administration of cyclophosphamide, and dogs receiving drugs known to be inducers of hepatic microsomal mixed function oxygenase activity were excluded.19 Additional chemotherapeutic drugs were not administered until the study was completed to prevent unknown pharmacokinetic interactions.12
We observed variability between dogs within both groups in relation to cyclophosphamide and 4-OHCP. Greater variability was observed for cyclophosphamide within the oral group. The increased variability within the oral group is likely associated with environmental factors such as diet, as well as individual differences in gastrointestinal absorption of the prodrug, and individual differences in metabolism.13 Variability between the groups for the measured pharmacokinetic values of 4-OHCP was similar. The reason for interanimal variability within the canine population remains unknown.
A significantly shorter time to Cmax of 4-OHCP was seen in the intravenous group. This is expected as absorption of the prodrug after intravenous administration is not a factor compared with the oral route.13 A significantly increased Cmax of 4-OHCP was also achieved in the intravenous group. However, after Cmax the concentration of 4-OHCP in the plasma decreased more rapidly in the IV than the oral group. Again this is likely attributed to the slower rate of absorption from the gastrointestinal tract, and subsequently a greater amount of cyclophosphamide was available for conversion to 4-OHCP over time. It is unknown if the more sustained higher concentration of 4-OHCP with oral administration is beneficial over that observed in the intravenous group.
We did not observe any substantial difference in overall response rate or episodes of hematological toxicity between the 2 groups. However, our study was not adequately powered to make any direct conclusions regarding the risk of toxicoses associated with the different routes of administration, and not all dogs received the same chemotherapy protocol before assessment of toxicity. No episodes of SHC were reported among the study population after the administration of cyclophosphamide. To help prevent uroepithelial toxicity associated with cyclophosphamide all dogs were treated with furosemide administered before cyclophosphamide, received the medication in the morning, were encouraged to drink fresh water, and were taken outside frequently to encourage urination.12,20 Dogs receiving cyclophosphamide IV have an increased risk for developing SHC, which is reduced with the administration of furosemide.3,12,21,22 The frequency of SHC after oral administration is reported less in the literature, and usually associated with long-term treatment.12 It has been suggested that the occurrence of SHC associated with oral cyclophosphamide can be further reduced by dividing the dose over a 3–4 day period.20
In this study, we focused on determining if the concentration of active metabolite, 4-OHCP, and thus the cytotoxic capability of the parent drug cyclophosphamide were the same if administered PO or IV. To accomplish this, we administered the total oral cyclophosphamide dose at a single time point. At this time, it is unknown what effect of dividing the oral dose over a 3–4 day period has on pharmacokinetic or pharmacodynamic parameters when compared with bolus administration via the intravenous route.
In conclusion, dog exposure to the active metabolite of cyclophosphamide, 4-OHCP, is the same in dogs with lymphoma when administered PO or IV. A significant difference in exposure to the prodrug is observed. This data suggests that oral and intravenous administration of cyclophosphamide can be used interchangeably with the same exposure of active metabolite being achieved in dogs with lymphoma.
Abbreviations
- AUC
area under the curve
- Cmax
maximum concentration
- CHOP
cyclophosphamide, doxorubicin, vincristine, prednisone
- COP
cyclophosphamide, vincristine, prednisone
- CYP
cytochrome
- 4-OHCP
Q4-hydroxycyclophosphamide
- SCZ
semicarbazide hydrochloride
- SHC
sterile hemorrhagic cystitis
- Tmax
time to maximum concentration
- VCOG-CTCAE
Veterinary Co-operative Oncology Group-common terminology criteria for adverse events
Footnotes
Cytoxan, Bristol-Myers Squibb Company, Princeton, NJ
Cytoxan, Baxter Healthcare Corporation, Deerfield, IL
Bielefeld, Germany
St Louis, MO
Applied Biosystems Inc, Foster City, CA
Shimadzu Prominence HPLC system, Columbia, MD
Leap Technologies, Carrboro, NC
Grace Davison Discovery Sciences, Deerfield, IL
Phenomenex, Torrance, CA
This work was done at the Animal Cancer Center, Colorado State University—Veterinary Teaching Hospital, Fort Collins, CO.
This work was presented by E. Warry, S. Lana, R. Hansen, D. Gustafson: Pharmacokinetics of cyclophosphamide administered PO or IV to dogs with lymphoma. Ninth Annual Colorado State University, College of Veterinary Medicine and Biomedical Research, Annual Scientific Research Day, January, 2010.
References
- 1.Huitema A, Tibben M, Kerbusch T, et al. High performance liquid chromatographic determination of the stabilized cyclophosphamide metabolite 4-hydroxycyclophosphamide in plasma and red blood cells. J Liq Chrom Rel Technol. 2000;23:1725–1744. [Google Scholar]
- 2.Ren S, Kalhorn T, McDonald F, et al. Pharmacokinetics of cyclophosphamide and its metabolites in bone marrow transplantation patients. Clin Pharmacol Ther. 1998;64:289–301. doi: 10.1016/S0009-9236(98)90178-3. [DOI] [PubMed] [Google Scholar]
- 3.Xie H, Griskevicius L, Stahle L, et al. Pharmacogenetics of cyclophosphamide in patients with hematological malignancies. Eur J Pharmaceut Sci. 2006;27:54–61. doi: 10.1016/j.ejps.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 4.McCune J, Salinger D, Vicini P, et al. Population pharmacokinetics of cyclophosphamide and metabolites in children with Pharmacokinetics of Cyclophosphamide in the Dog 907 neuroblastoma: A report from the children’s oncology group. J Clin Pharmacol. 2009;49:88–102. doi: 10.1177/0091270008325928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Jonge M, van Dam S, Hillebrand M, et al. Simultaneous quantification of cyclophosphamide, 4-hydroxycyclophosphamide, N,N′,N″-triethylenethiophophoramide (thiotepa) and N,N′,N″-triethylenephosphoramide (tepa) in human plasma by high performance liquid chromatography couples with electrospray ionization tandem mass spectrometry. J Mass Spectrom. 2004;39:262–271. doi: 10.1002/jms.570. [DOI] [PubMed] [Google Scholar]
- 6.Holford H. Pharmacokinetics and pharmacodynamics: Rational dosing and time course of drug action. In: Katzung B, Masters S, Trevor A, editors. Basic and Clinical Pharmacology. 11. New York: McGraw-Hill Companies; 2009. pp. 37–66. [Google Scholar]
- 7.Wang H, Tompkins L. CYP2B6: New insights into a historically overlooked cytochrome P450 isozyme. Curr Drug Metab. 2008;9:598–610. doi: 10.2174/138920008785821710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takada K, Arefayene M, Desta Z, et al. Cytochrome P450 pharmacogenetics as a predictor of toxicity and clinical response to pulse cyclophosphamide in lupus nephritis. Arthritis Rheum. 2004;50:2202–2210. doi: 10.1002/art.20338. [DOI] [PubMed] [Google Scholar]
- 9.Nakajima M, Komagata S, Fujiki Y, et al. Genetic polymorphisms of CYP2B6 affects the pharmacokinetics/pharmacodynamics of cyclophosphamide in Japanese cancer patients. Pharmacogenet Genomics. 2007;17:431–445. doi: 10.1097/FPC.0b013e328045c4fb. [DOI] [PubMed] [Google Scholar]
- 10.Vail D. Veterinary co-operative oncology group – common terminology criteria for adverse events (VCOG-CTCAE) following chemotherapy or biological antineoplastic therapy in dogs and cats v1. 0. Vet Comp Oncol. 2004;2:194–213. doi: 10.1111/j.1476-5810.2004.0053b.x. [DOI] [PubMed] [Google Scholar]
- 11.Cros S, Theilen G, Madewell B, et al. Cyclophosphamide induced cystitis in the dog and cat. J Am Vet Med Assoc. 1977;171:259–262. [PubMed] [Google Scholar]
- 12.Petros W, Broadwater G, Berry D, et al. Association of high dose cyclophosphamide, cisplatin and carmustine pharmacokinetics with survival, toxicity and dosing weight in patients with primary breast cancer. Clin Cancer Res. 2002;8:698–705. [PubMed] [Google Scholar]
- 13.Frye R, Zgheib N, Matzke G, et al. Liver disease selectively modulates cytochrome P450 mediated metabolism. Clin Pharmacol Ther. 2006;80:235–245. doi: 10.1016/j.clpt.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 14.Struck R, Alberts D, Horne K, et al. Plasma pharmacokinetics of cyclophosphamide and its cytotoxic metabolites after intravenous versus oral administration in a randomized, crossover trial. Cancer Res. 1987;47:2723–2726. [PubMed] [Google Scholar]
- 15.Ludeman S, Shulman-Roskes E, Wong K, et al. Oxime derivatives of the intermediary oncostatic metabolites of cyclophosphamide and ifosfamide: Synthesis and deuterium labeling for applications to metabolite quantification. J Pharm Sci. 1995;84:393–398. doi: 10.1002/jps.2600840403. [DOI] [PubMed] [Google Scholar]
- 16.Charney S, Bergman P, Hohenhaus A, et al. Risk factors for sterile hemorrhagic cystitis in dogs with lymphoma receiving cyclophosphamide with or without concurrent administration of furosemide: 216 cases (1990–1996) J Am Vet Med Assoc. 2003;222:1388–1393. doi: 10.2460/javma.2003.222.1388. [DOI] [PubMed] [Google Scholar]
- 17.Sladek N, Doeden D, Powers J, et al. Plasma concentrations of 4-hyroxycyclophosphamide and phosphoramide mustard in patients repeatedly given high doses of cyclophosphamide in preparation for bone marrow transplantation. Cancer Treat Rep. 1984;68:1247–1254. [PubMed] [Google Scholar]
- 18.Petros W, Hopkins P, Spruill S, et al. Associations between drug metabolism genotype, chemotherapy pharmacokinetics and overall survival in patients with breast cancer. J Clin Oncol. 2005;23:6117–6125. doi: 10.1200/JCO.2005.06.075. [DOI] [PubMed] [Google Scholar]
- 19.Timm R, Kaiser R, Lotsch J, et al. Association of cyclophosphamide pharmacokinetics to polymorphic cytochrome P450 2C19. Pharmacogenom J. 2005;5:365–373. doi: 10.1038/sj.tpj.6500330. [DOI] [PubMed] [Google Scholar]
- 20.Salinger D, McCune J, Ren A, et al. Real time dose adjustment of cyclophosphamide in a preparative regimen for hematopoietic cell transplant: A Bayesian pharmacokinetic approach. Clin Cancer Res. 2006;12:4888–4898. doi: 10.1158/1078-0432.CCR-05-2079. [DOI] [PubMed] [Google Scholar]
- 21.Peterson J, Couto C, Hammer A, et al. Acute sterile hemorrhagic cystitis after a single administration of cyclophosphamide in 3 dogs. J Am Vet Med Assoc. 1992;201:1572–1574. [PubMed] [Google Scholar]
- 22.Pinto N, Ludeman S, Dolan M. Pharmacogenetic studies related to cyclophosphamide based therapy. Pharmacogenomics. 2009;10:1897–1903. doi: 10.2217/pgs.09.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hong P, Srigritsanapol A, Chan K. Pharmacokinetics of 4-hydroxcyclophosphamide and metabolites in the rat. Drug Metab Dispos. 1989;19:1–7. [PubMed] [Google Scholar]
- 24.Chun R, Garrett L, Vail D. Cancer chemotherapy. In: Withrow S, Vail D, editors. Withrow and MacEwen’s Small Animal Clinical Oncology. 4. London: Saunders; 2007. pp. 163–193. [Google Scholar]