

Abstract
Vascular access dysfunction associated with arteriovenous grafts and fistulas contributes to the morbidity and mortality of chronic kidney disease (CKD) patients receiving hemodialysis. We hypothesized that the uremic conditions associated with CKD promote a pathophysiological vascular smooth muscle cell (VSMC) phenotype that contributes to neointimal hyperplasia. We analyzed the effect of culturing human VSMC with uremic serum. Expression of VSMC contractile marker genes was reduced 50-80% in cells exposed to uremic serum and the decreased expression was accompanied by changes in histone marks. There was an increase in proliferation in cells exposed to uremic conditions, with no change in the levels of apoptosis. Interestingly, we found that uremic serum inhibited PDGF-induced migration of VSMC. Histomorphometric analysis revealed venous neointimal hyperplasia in veins from chronic kidney disease (CKD) patients prior to any surgical manipulation as compared to veins from patients with no kidney disease. We conclude that uremia associated with CKD alters VSMC phenotype in vitro and contributes to neointimal hyperplasia formation in vivo contributing to the pathogenesis of vascular access dysfunction in CKD patients.
Keywords: Vascular Smooth Muscle Cells, Chronic Kidney Disease, Uremia, Hyperplasia, Vascular Access Dysfunction, Arteriovenous Fistula, Arteriovenous Grafts
2. INTRODUCTION
There are currently over 400,000 Americans with end-stage renal disease (ESRD) who are dependent on a reliable vascular access for life-preserving hemodialysis (1). Unfortunately, vascular access dysfunction is a primary reason for morbidity in patients requiring hemodialysis (2) with associated annual costs that exceed $1 billion in the US (3). A major cause of vascular access failure in both arteriovenous fistula (AVF) and arteriovenous grafts (AVG) is venous outflow obstruction due to hemodynamically significant peri-anastomotic intimal hyperplasia development (4). Indeed, while AVF is the method of choice for dialysis access in ESRD patients (5), there is still an urgent need to develop better therapeutic strategies to treat vascular access stenosis and subsequent dysfunction as the one-year patency rate of AVF is estimated to be only 63% (6). It appears that the uremic condition present in hemodialysis patients exacerbates endothelial cell dysfunction and may be responsible for the de novo venous intimal hyperplasia and medial hypertrophy that is observed in these patients even before the creation of arteriovenous access (7-9). Several animal studies have shown that chronic kidney disease (CKD) aggravates damage and accelerates neointimal hyperplasia development in AVFs (10, 11). Understanding the cellular and molecular mechanisms that lead to the development of intimal hyperplasia and vascular disease in CKD patients is warranted as cardiovascular disease remains the major cause of death in ESRD patients with a 10-20 fold higher risk compared to the general population (12).
Unlike terminally differentiated cells, vascular smooth muscle cells (VSMC) retain the ability to de-differentiate or drastically modulate their phenotype in response to environmental cues. A group of VSMC specific genes have been identified as useful markers of the relative state of differentiation of VSMCs. The fully differentiated VSMC is associated with high expression of several specific contractile proteins including smooth muscle alpha actin, smooth muscle myosin heavy chain, SM22alpha and calponin (13). In response to vascular injury, VSMCs exhibit a phenotypic change exemplified by loss of contractility and abnormal proliferation, migration and extracellular matrix secretion (13). This “synthetic phenotype” is important for repair of vascular damage but it also plays a role in the development of various vascular pathologies including atherosclerosis, hypertension and post-angioplasty restenosis (14, 15) . A feature of VSMC phenotype plasticity is transcriptional repression of the specific contractile genes (14).
Therefore, we hypothesize that the uremic conditions associated in CKD and ESRD promotes a pathophysiological VSMC phenotype that contributes to vascular access dysfunction in hemodialysis patients. In this study, we examine the effect of culturing VSMC with uremic serum on their phenotype and physiologic function. We show that VSMC cultured with uremic serum indeed exhibit an altered or dedifferentiated phenotype. Furthermore, we show that there is substantial de novo venous intimal hyperplasia in ESRD patients, and the amount of venous intimal hyperplasic lesion correlates significantly with CKD stages prior to any surgical manipulation.
3. MATERIALS AND METHODS
3.1 Subjects
This study was approved by the Temple University School of Medicine Institutional Review Board (IRB). After informed consent, blood samples were obtained from 21 patients with end stage renal disease who were on hemodialysis (HD) and from 20 healthy donors with normal kidney function. In HD, blood was collected prior to routine HD session. Blood samples were centrifuged at 3000 rpm and the serum was aliquoted and stored at −80°C. Anonymous discarded vein tissues were collected with the approval and oversight of the Washington University School of Medicine and the Temple University School of Medicine IRBs.
3.2 Cell Culture
Human aortic smooth muscle cells were obtained from Lonza (CC-2571)and cultured in smooth muscle basal media (SmBM), supplemented with growth factors and 5% FBS (CC-3182), according to the manufacturer’s guidelines. Cells from passage 4-8 were used in the described studies. Throughout the studies, cells from different lots were used. For experimental conditions, growth medium was supplemented with 10% vol/vol normal or uremic serum. We used pooled serum from (3-5) maintenance hemodialysis patients in each experiment. Experiments were repeated using serum from different hemodialysis patients. Normal pooled serum from healthy donors was used for comparison in the different assays.
3.3 RNA Extraction and Quantitative Real-Time PCR
Cells were subjected to serum starvation for 48-72 h followed by treatment with serum for 24 h. Untreated serum starved cells were used as control. Total RNA from cultured cells was extracted using the RNeasy kit (Qiagen), and cDNA was synthesized with the VILO first-strand synthesis system (Invitrogen). In the real time PCR step, cDNA was amplified with inventoried gene assay products containing two gene specific primers and one FAM dye labeled Taq Man MGB probe using the 7500 Real Time PCR System (Applied Biosystems). Relative gene expression levels were calculated after normalization with internal control eukaryotic 18S gene using the 2-deltadeltaCt method, where Ct is the threshold value.
3.4 Chromatin Immunoprecipitation (ChIP) Assays
ChIP assays were performed, and ChIP-enriched DNA was analyzed by real time qPCR as described (16). Briefly, cells were fixed with 1% formaldehyde at 37C for 10 minutes, fixation was stopped by addition of glycine, and the cells were rinsed twice with cold PBS and harvested into lysis buffer (50mM Hepes-KOH, pH8.0,1mM EDTA, 0.5mM EGTA, 140mM NaCl, 10% glycerol, 0.5%Nonidet P-40, 0.25% Triton X-100 and protease inhibitor cocktail from Roche Applied Science). Resulting nuclei were centrifuged and resuspended in RIPA buffer (10mM Tris–HCl, pH 8.0, 1%Triton-100, 0.1% SDS, 0.1% sodium deoxycholate, 1mM EDTA,0.5mM EGTA, 140 NaCl plus protease inhibitor cocktail from Roche Applied Science). Sonicated samples were centrifuged,precleared with protein G beads/sonicated. Samples were then subjected to immunoprecipitation with specific antibodies with rotation overnight at 4°C.Immunocomplexes were collected by adsorption onto protein G bead slurry, and washed sequentially in buffers from ChIP kit from Upstate–Millipore (Billerica, MA). Antibody-bound chromatin fragments were elutedfrom the beads with 1% SDS in 0.1MNaHCO3. Cross-links were reversed by heating at 65° C overnight. DNA was recovered by phenol-chloroform extraction and precipitation. DNA wasresuspended in 100 ml water and a portion subjected to quantitative Real-Time PCR (7500 Applied Biosystems, FosterCity, CA). Five percent of total soluble chromatin was processed inparallel without immunoprecipitation, and values obtained from this DNA were used to calculate immunoprecipitated DNA as a percentage of input. Triplicate PCRs for each sample were mixedwith 2xSYBR Green PCR master mix (Applied Biosystems). A standard curve of known target DNA was constructed in parallel from which the relative amount of target DNA in the sample was calculated. Control ChIP assays with non-specific IgG were performed in each experiment. Results represent the mean ±SD of at least three independent experiments.
3.5 Proliferation
SMC proliferation was measured using the CyQuant Cell Proliferation Assay Kit (Roche), according to the Prolimanufacturer’s instructions. Cells were plated at a density of 3×103 cells per well in a 96 well plate. Cells were serum starved for 72 hr and then treated with normal human serum or dialysis patient serum for another 72 hr. For the assay, the media was removed from the cells, dye binding solution was added to each well and total cellular nucleic acid was measured using a florescence plate reader (Wallac, Perkin Elmer) using a 485/535nm filter set.
3.6 Assessment of Apoptosis
For detection of apoptosis, quiescent cells were treated with normal human serum or dialysis patient serum. After 24h cells were washed and exposed to 0.5microM staurosporine for 4 h. Detection of Caspase3/7 activity was determined using the Caspase-Glo 3/7 reagent from Promega using their standard assay protocol. Light units were measured using a Veritas microplate reader from Turner Biosystems.
3.7 Cell Migration
Cell migration was evaluated through Boyden chamber method, using triplicate 6.5 mm diameter transwell Boyden chamber plates (Costar) with 8um polycarbonate membrane pore size. VSMC were made quiescent in serum starved media for 48 hours. They were then left untreated, or treated with either normal human serum or dialysis patient serum for 24 hours. VSMC were then seeded (2×104 cells per membrane) in the upper chamber of the transwell. Forty nanograms of PDGF AB were placed in the lower chamber and cells were incubated for 4 hours at 37°C, at which time cells were fixed. The upper layer was scraped free of cells. VSMC that had migrated to the lower surface of the membrane were stained with the nuclear stain DAPI and quantified by counting 4 high powered fields per membrane. Experiments were performed in triplicate from independent groups of VSMC. For directional migration, scratch assay was performed as described (17). VSMC were seeded on 4-chamber glass slides and made quiescent by serum starvation as above, then treated with normal human serum, or dialysis patient sera. A 2mm uniform scratch was made, and the chamber slide was further incubated for 24 hours. The slides were then fixed with paraformaldehyde and stained with the nuclear stain DAPI. Images were captured and the number of migratory cells was evaluated.
3.8 Protein Extraction and Western Blot Analysis
VSMC were washed with cold PBS and resuspended in RIPA buffer (20mM Tris-HCl, ph7.5, 150mM NaCl, 1mM Na2EDTA,1mM EGTA, 1% NP40 1% sodium deoxycholate, 2.5mM sodium pyrophosphate, 1mM b-glycerophosphate 1mM Na3VO4 1ug/ml leupeptin plus protease inhibitor mixture (SIGMA) and incubated on ice for 15 minutes. The lysate was centrifuged at 13,000 x g for 10 min at 4°C. Equal whole cell lysates were analyzed by SDS-PAGE on 4-15% gradient gels followed by transfer to nitrocellulose membranes. After blocking, the membranes were incubated with cleaved caspase-3 antibody (Cell Signaling #9664), and subsequently with anti-beta actin antibody (Abcam). The blots were then incubated with horseradish peroxidase-conjugated anti-rabbit antibodies (Roche Applied Science, Indianapolis, IN) and developed with SuperSignal West Dura chemiluminescence substrate (ThermoFisher Scientific, Chicago, IL).
3.9 Quantitative Histomorphometry
Patient vein samples were harvested processed and embedded in paraffin using standard methods. Serial sections of 5-μm thickness every 100 μm of the vein segment were obtained. Sections were stained for hematoxylin and eosin as well as for elastic-Van Gieson (Vvg) to visualize morphology. The areas within the lumen and the areas circumscribed by the internal elastic lamina (IEL) and external elastic lamina (EEL) were determined by tracing along the respective vessel regions. The media was defined as the region between the EEL and the IEL, and the intima was measured as the region between the lumen and the IEL. The intimal/ medial areas and lumen sizes in Vvg-stained sections were measured using the NIH Image J program, and the intimal/media ratios were calculated.
3.10 Data Analysis
All experiments were performed at least three times, and results were expressed as themean ± standard deviation (S.D.). Statistical comparison of single parameters between 2 groups was performed by paired Student t test. The Kruskal-Wallis 1-way ANOVA was used to comparethe means of multiple groups and were followed by Dunn’s test. Data were considered statistically significant if p was <0.05.
4. RESULTS
4.1 Exposure of VSMC to uremic serum attenuates expression of contractile marker genes
VSMC retain remarkable phenotypic plasticity in response to a large variety of environmental cues. It is generally accepted that VSMC trans-differentiation is implicated in the pathophysiology of vascular remodeling following vascular injury. To investigate the possible role of CKD in altering VSMC phenotype, we evaluated cultured primary human VSMC. Quiescent cells which exhibit a contractile phenotype, were exposed to healthy human serum or pooled uremic serum from hemodialysis patients for 24 hours. Quantitative RT-PCR analysis was used to determine the expression of genes encoding contractile markers, calponin, SM22alpha SMalpha-actin (ACTA) and smooth muscle myosin heavy chain (MHC). In each case expression was compared to the level of gene expressed by quiescent untreated cells. The results showed that relative mRNA expression of these smooth muscle specific genes was attenuated by 40 to 85% in the cells exposed to uremic serum (Figure 1A). In contrast, the expression of myocardin, the cofactor that regulates their expression was not affected by uremic serum. In addition, VSMC exposed to dialysis patient serum had (40%) increased expression of the extracellular matrix gene Col1a1 compared to cells exposed to normal human serum or quiescent cells. Furthermore, exposure to uremic serum also decreased SM-22alpha and SM-alpha actin protein expression compared to cells exposed to normal human serum (Figure 1B).
Figure 1.

Exposure to uremic serum affects VSMC gene expression. (A) VSMC were serum starved for 48 h and cells were either left untreated (ctrl) or subsequently exposed to normal human serum (NS) or dialysis patient serum (DS) for 24 h, followed by qRT-PCR analysis. Results were normalized to eukaryotic 18S expression and the control cells, the relative mRNA levels are presented with each experiment conducted in triplicate. Results shown are mean ± SD of 3-5 independent experiments. p values between normal serum NS and DS are shown. (B) Uremic serum decreases expression of SM-22alpha and SM-alpha actin. Cultured vascular smooth cells were serum starved for 48 hours and then either left untreated (ctrl) or treated with normal (NS) or dialysis patient serum (DS) for 24 hours. Total protein lysates were subjected to Western blot analysis of SM-22alpha and SMalpha actin. tubulin was used as a loading control.
Epigenetic mechanisms which include histone post-translational modifications are crucial regulators of cellular homeostasis and control gene expression (18). The uremic environment present in CKD patients may alter chromatin accessibility and compromise the regulation of contractile marker genes. We wanted to determine if the dialysis induced changes in gene expression in VSMC were associated with changes in the active or repressive epigenetic marks at the promoters of contractile marker genes. We performed chromatin immunoprecipitation assays (ChIP) assays with acetylated histone 4 (AcH4) and lysine 9 trimethylated histone H3 (H3k9me3) -specific antibodies, markers of transcriptional activation and repression respectively. ChIP-enriched DNA samples were analyzed by quantitative PCR using primers spanning a CArG binding site at the SMalpha actin promoter and at a non CArG site for the calponin promoter. Levels of AcH4 were significantly reduced at both the SMalphasm actin and calponin promoters in cells exposed to dialysis patient sera. Levels of the repressive H3K9me3 were increased at these promoters (Figure2). These results confirmed that CKD can downregulate contractile phenotype gene expression and it may be caused at least in part through epigenetic histone modifications at their promoters.
Figure 2.

Histone modifications at contractile marker gene promoters. Levels of acetylated H4 and H3K9me3 at the SM alpha-actin and calponin promoters were examined by chromatin immunoprecipitation assays. Anti-histone immunoprecipitated DNA and input DNA were subjected to qPCR using primers specific for the indicated gene promoters to measure enrichment levels. Results shown are mean ± SD of three independent experiments.
4.2 Uremic conditions increase VSMC proliferation and do not affect staurosporine induced apoptosis
It is known that increased proliferation of VSMC is associated with intimal hyperplasia formation and vascular remodeling (15). In order to determine whether CKD had an effect on proliferative capacity of VSMC, we next examined proliferation of cultured cells in the presence of normal human serum or uremic serum for three days. As expected, cell proliferation was stimulated in the presence of growth medium (GM) compared with cells cultured in the presence of 0.1% FBS, control basal medium (Ctrl). Cells exposed to 10% uremic serum exhibited significantly greater proliferation compared to cells grown in 10% normal human serum (Figure 3A).
Figure 3.
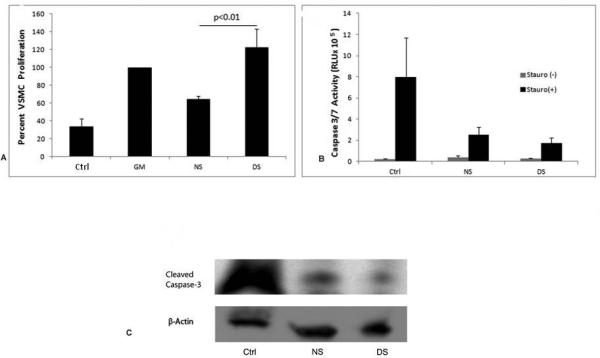
Uremic serum increases proliferation of vascular smooth muscle cells (VSMC) but has not effect on apoptosis. (A) VSMC were plated on 96 well plates (3×1033 cells/well) in growth media. Cells were subsequently serum starved for 72 hours and then treated with basal media (Ctrl 0.1%FBS), growth media GM (5%FBS), normal serum NS (10%) or dialysis patient serum DS (10%) for another 72 hours. Proliferation was measured using the Cyquant reagent. For the assay, the media was removed from the cells, dye binding solution was added to each well and nucleic acid was measured using a fluorescence plate reader using a 485/535nm filter set. (B) VSMC were plated on 96 well plates 5×103 (cells/well) after adhesion cells were serum starved for 48 hours and then treated for 24 hours in either, basal media (ctrl), normal human serum (NS) or dialysis patient serum (DS). Cells were then washed in HBSS and exposed to 0.5M staurosporine for 4 hours at which time supernatant was removed and 100ul of Caspase3/7 substrate was added and further incubated for 1 hour at room temperature. Enzymatic activity was detected using a luminometer. Results shown are mean ± SD of 3-5 independent experiments. (C) Expression of Cleaved Caspase 3 was measured by Western blot analysis in VSMC lysates of cells that were serum starved (ctrl), treated with 10% normal healthy serum (NS), or treated with dialysis patient serum (DS) and then treated with staurosporine for 6 hours.
Apoptosis of VSMC is an important process in both normal and pathological remodeling of vessel walls (19). The uremic conditions present in CKD may alter the regulation of apoptosis in VSMC. In order to determine if CKD affects VSMC apoptosis we next examined if uremic serum could change levels of apoptosis. Staurosporine is a broad range protein kinase inhibitor that triggers apoptosis. Quiescent cells and cells cultured with normal human serum or uremic serum for 24 hr were washed and treated with 0.5microM staurosporine. Levels of apoptosis were determined using a Caspase 3/7 luminescent assay. As shown in Figure 3B, quiescent cells contained high levels of caspase 3/7 activity, while cells exposed to both normal and uremic serum had low levels of active enzyme activity. Similarly, there was very little if any expression of cleaved caspase 3 observed in lysates from cells exposed to either normal or uremic serum as measured by western blot analysis, Figure 3C.
4.3 Decreased Migration Exhibited by VSMC exposed to uremic serum
In addition to proliferation, cell migration plays an essential role in the development of intimal lesion (20). We evaluated the migratory properties of VSMC exposed to uremic serum compared to cells exposed to normal human serum. Surprisingly, we found that cells pretreated with dialysis serum displayed significantly less PDGF induced migration than cells pretreated with normal human serum in a transwell assay (Figure 4A). We also performed a scratch wound assay in which directional migration of a VSMC monolayer could be assessed. Equal numbers of VSMC were incubated in healthy or dialysis patient serum for 24 hours at which time dishes were scraped to create a 2-mm wound track devoid of cells. Normal or dialysis sera was added again and cells were fixed and stained 24 hours later to avoid potential effects on proliferation. Data presented on Figure 4B demonstrate that cells exposed to uremic serum migrate into the wound more slowly than cells incubated with normal serum. Taken together these findings suggest that exposure to uremic serum has an inhibitory effect on VSMC migration.
Figure 4.

Effects of uremic serum on VSMC migration. (A) Boyden Chamber Assay. One hundred microliters of VSMC suspension (2×104) that was pre-treated with normal human serum (NS), dialysis patient serum (DS), or without serum (Ctrl) was placed in the upper chemotaxis chamber, and 600microl of basal media containing 40ng PDGF-beta was placed in the lower chamber. The chamber was incubated at 37°C and 5% CO2 for 6 hr. The transmigrated cells on the filter membrane were fixed and stained and subsequently quantified. Values are means ± S.D. from three independent experiments(P<0.05). (B) Quantitative analysis of scratch wound area from three different groups of VSMC. Quiescent cells were plated in basal media a scratch wound was made and cells were either left untreated (Ctrl) or exposed to 10% normal human serum (NS) or dialysis patient serum (DS) for 24 hours. Cells were stained with DAPI. Significant difference from 3 different slides of DS treated cells versus NS (P<0.0001).
4.4 Significant de novo venous intimal hyperplasia in chronic kidney disease (CKD) patients prior to arteriovenous (AV) access surgery
It is known that CKD patients have arterial pathologies such as atherosclerosis and calcification (21, 22). In addition, some studies suggest that there is pre-existing venous intimal hyperplasia in CKD patients prior to dialysis access surgery (9). Therefore, we examined the histopathology of the superficial arm (cephalic) veins of patients with normal kidney function and CKD patients from different stages, and compared the amount of de novo venous intimal hyperplasia in these populations. For analysis, we divided the CKD patients into three groups. Control group consisted of patients with normal kidney function and CKD patients with stages 1 and 2. Group 2 consisted of CKD patients with stages 3 and 4, and group 3 were end stage renal disease (ESRD) patients consisted of CKD patients with stage 5. Indeed, we found that de novo venous intimal hyperplasia is associated with increasing CKD stage, Figure 5A. Many patients with ESRD encounter multiple hospitalizations requiring upper extremity intravenous catheter placement, phlebotomy, and blood pressure cuff measurement. In order to demonstrate that the venous intimal hyperplasia is not secondary to these iatrogenic maneuvers, we collected lower extremity superficial (great saphenous) veins from discarded tissue following lower extremity and coronary bypass grafting and major leg amputations. Intravenous catheter placement, phlebotomy, and blood pressure cuff measurement are not routinely performed in the lower extremities at the participating medical centers. Indeed, we also found that de novo superficial vein intimal hyperplasia development correlated significantly with patient CKD stages in lower extremity veins (Figure 5B). Quantitation of the histomorphometry was performed as described in the materials and methods, and summarized in Figure 5 C-F. The data analyzed for upper extremity included samples from 8 patients for group control group and 14 patients for ESRD. For lower extremity data analyzed included samples from 32 patients from control group, 25 patients for group 2 and 7 patients for group ESRD. For both upper and lower extremities there were significantly greater intimal lesion volume in patients with higher stage of CKD disease and corresponding intima to media ratio.
Figure 5.

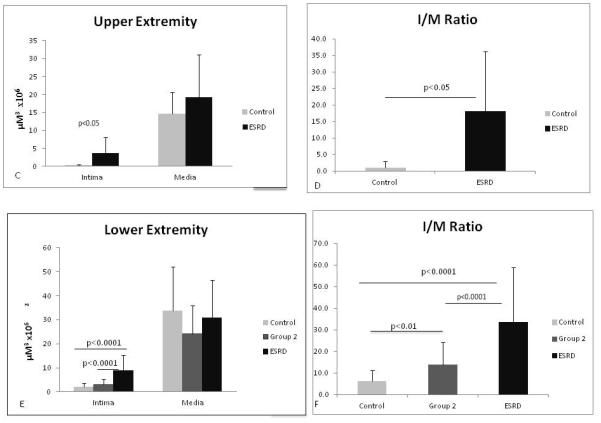
Correlation of venous intimal hyperplasia lesion with chronic kidney disease (CKD) staging. Representative micrographs are shown from vein sections after Vvg staining of cephalic veins (A) control samples include patients with normal kidney function and CKD stages 1 and 2, (ESRD) end stage renal disease patients or chronic kidney disease stage 5. (B) saphenous veins sections from CKD patients. Control group includes patients with normal kidney function and CKD stages 1 and 2. Group 2 includes patients with CKD stages 3 and 4, and ESRD CKD stage 5. All sections are shown at a magnification of 4X. Intimal hyperplasia lesions were evaluated and quantified as described in methods. (C) Bar graphs representing lesion area and corresponding I/M ratio (D) for upper extremity veins are shown. (E) Quantified lesion area and I/M ratio (F) for lower extremity veins. Values represent mean ± SD.
5. DISCUSSION
The present study was designed to elucidate the impact of uremia on VSMC phenotype. Our findings demonstrate that uremic culture conditions have the ability to modify VSMC phenotype directly, which may contribute to intimal hyperplasia and vascular dysfunction. Normal VSMC cultured with dialysis patient sera for only 24 hours led to loss of VSMC phenotypic markers representative of the contractile phenotype including SM alpha actin, SM22alpha, calponin and myosin heavy chain. However, the expression of the coactivator myocardin was not affected by uremic conditions. This is not surprising since it has been shown that in addition to myocardin expression itself, myocardin can modulate VSMC phenotype through protein-protein interactions (23). Interestingly, there was an increase in expression of the extracellular matrix gene collagen1a1 in the cells grown in uremic media, suggesting a synthetic phenotype (15).
The long term exposure to the uremic environment in CKD patients may affect epigenetic programs regulating chromatin accessibility and gene expression. Indeed, we found evidence of changes in chromatin structure at the promoters of VSMC specific genes in cells exposed to uremic sera. There was a decrease in histone H4 acetylation, which is considered to be a marker of active chromatin, at both the SMA promoter and the calponin promoter. At the same time, there was an increase at these promoters in H3K9 trimethylation, which generally correlates with gene silencing and transcriptional repression (24). Therefore, our data is in agreement with the notion that epigenetic mechanisms play a role in the control of VSMC phenotypic switching (25) and may contribute to detrimental vascular remodeling.
We observed significantly more proliferation of VSMC in cells cultured with dialysis patient sera than cells exposed to normal human serum. In the context of atherosclerosis, it has been demonstrated that increased proliferation, migration and production of extracellular matrix components by phenotypically modulated VSMC plays a critical role in lesion development (26). We believe a similar mechanism is in play in the development of anastomotic intimal hyperplasia in ESRD patients. The pathological factors in CKD that contribute to vascular dysfunction may include chronic inflammation and increased levels of oxidative stress. Studies have shown that CKD aggravates damage and accelerates development of neointimal hyperplasia in AVFs (10,11). A better understanding of the cellular and molecular mechanisms that lead to the development of neointimal hyperplasia in CKD is warranted.
We did not find increased apoptosis in VSMC cultured with 10% uremic serum for 24 hours or 48 hours (data not shown). This is in contrast with some studies that suggest that uremia increases apoptosis of VSMC. This may be accounted by differences in experimental conditions. In one study the effect of individual toxins, urea and inorganic phosphate was examined, and not whole uremic serum (27). Furthermore, our findings are in agreement with studies which demonstrated that uremic serum promoted proliferation, but not apoptosis of vascular endothelial cells (28).
Surprisingly, VSMC cultured in uremic serum exhibited less migration than cells cultured in normal serum. Although PDGF-B is a known mediator of VSMC modulation, inducing both proliferation and migration (15), exposure of VSMC to uremic serum inhibited PDGF B-induced migration. These cells also displayed impaired migration in the wound assay (Figure 4) suggesting that the uremic serum may contain anti-migratory molecules. This observation is in contrast with was reported in vivo where VSMC derived from uremic mice exhibited increased migration in an ex vivo assay (11). Therefore, it is likely that additional signals are provided in vivo, for example from the uremic endothelium that may result in increased VSMC migration in vivo. However, it appears that in vitro, the uremic milieu activates VSMC to a dedifferentiated proliferative state with compromised migration. This confirms the idea that dedifferentiation of VSMC involves different subtypes of phenotypically modulated VSMC (25). We believe that this dedifferentiated VSMC contributes to NH development and vascular access dysfunction in CKD patients undergoing hemodialysis.
Vascular access failure is characterized by vein stenosis leading to thrombosis in AVF and grafts (29), therefore, we also examined the veins of CKD patients. We performed immuno-histochemical analysis and observed that patients with ESRD had significant pre-existing venous neointimal hyperplasia lesions compared to patients with normal renal function. These findings are in accordance with previously reported studies (9). Importantly, we also found that the NH lesion correlated with increase in CKD stage, which has not been previously documented. This further supports the idea that the uremic environment contributes to the development of NH.
In conclusion, we have demonstrated that both arterial VSMC exposed in vitro to uremic conditions and veins from CKD patients exhibit phenotypic changes associated with vascular remodeling and vascular access dysfunction. Therefore, it is likely that we need to initiate targeted therapies aimed at preventing VSMC dedifferentiation and dysfunction prior to dialysis access surgery.
Table 1.
Demographics of Dialysis Patients (In vitro studies serum)
| Age (mean +/− SD) | 58.4+/− 16.97 |
|---|---|
| Gender, men (%) | 11 (52%) |
| Coronary disease | 10 (48%) |
| Diabetes | 9 (43%) |
6. ACKNOWLEDGEMENT
This study was supported by Temple University funds.
Abbreviations
- VSMC
Vascular smooth muscle cell
- CKD
Chronic kidney disease
- ESRD
End stage renal disease
7. REFERENCES
- 1.Hsu C.-y., Vittinghoff E, Lin F, Shlipak MG. The Incidence of End-Stage Renal Disease Is Increasing Faster than the Prevalence of Chronic Renal Insufficiency. Annals of Internal Medicine. 2004;141(2):95–101. doi: 10.7326/0003-4819-141-2-200407200-00007. [DOI] [PubMed] [Google Scholar]
- 2.Feldman H, Kobrin S, Wasserstein A. Hemodialysis vascular access morbidity. Journal of the American Society of Nephrology. 1996;7(4):523–535. doi: 10.1681/ASN.V74523. [DOI] [PubMed] [Google Scholar]
- 3.Lee H, Manns B, Taub K, Ghali WA, Dean S, Johnson D, Donaldson C. Cost analysis of ongoing care of patients with end-stage renal disease: The impact of dialysis modality and dialysis access. American Journal of Kidney Diseases. 2002;40(3):611–622. doi: 10.1053/ajkd.2002.34924. doi:10.1053/ajkd.2002.34924. [DOI] [PubMed] [Google Scholar]
- 4.Lee T, Roy-Chaudhury P. Advances and New Frontiers in the Pathophysiology of Venous Neointimal Hyperplasia and Dialysis Access Stenosis. Advances in Chronic Kidney Disease. 2009;16(5):329–338. doi: 10.1053/j.ackd.2009.06.009. doi:10.1053/j.ackd.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tordoir J, Canaud B, Haage P, Konner K, Basci A, Fouque D, Kooman J, Martin-Malo A, Pedrini L, Pizzarelli F, Tattersall J, Vennegoor M, Wanner C, ter Wee P, Vanholder R. EBPG on Vascular Access. Nephrology Dialysis Transplantation. 2007;22(suppl 2):ii88–ii117. doi: 10.1093/ndt/gfm021. doi:10.1093/ndt/gfm021. [DOI] [PubMed] [Google Scholar]
- 6.Biuckians A, Scott EC, Meier GH, Panneton JM, Glickman MH. The natural history of autologous fistulas as first-time dialysis access in the KDOQI era. Journal of Vascular Surgery. 2008;47(2):415–421. doi: 10.1016/j.jvs.2007.10.041. doi:10.1016/j.jvs.2007.10.041. [DOI] [PubMed] [Google Scholar]
- 7.Wali MA, Eid RA, Dewan M, Al-Homrany MA. Intimal Changes in the Cephalic Vein of Renal Failure Patients before Arterio-Venous Fistula (AVF) Construction. Jornal of Smooth Muscle Research. 2003;39(4):95–105. doi: 10.1540/jsmr.39.95. [DOI] [PubMed] [Google Scholar]
- 8.Ku YM, Kim YO, Kim JI, Choi YJ, Yoon SA, Kim YS, Song SW, Yang CW, Kim YS, Chang YS, Bang BK. Ultrasonographic measurement of intima-media thickness of radial artery in pre-dialysis uraemic patients: comparison with histological examination. Nephrology Dialysis Transplantation. 2006;21(3):715–720. doi: 10.1093/ndt/gfi214. doi:10.1093/ndt/gfi214. [DOI] [PubMed] [Google Scholar]
- 9.Lee T, Chauhan V, Krishnamoorthy M, Wang Y, Arend L, Mistry MJ, El-Khatib M, Banerjee R, Munda R, Roy-Chaudhury P. Severe venous neointimal hyperplasia prior to dialysis access surgery. Nephrology Dialysis Transplantation. 2011;26:2264–2270. doi: 10.1093/ndt/gfq733. doi:10.1093/ndt/gfq733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langer S, Kokozidou M, Heiss C, Kranz J, Kessler T, Paulus N, Kruger T, Jacobs MJ, Lente C, Koeppel TA. Chronic kidney disease aggravates arteriovenous fistula damage in rats. Kidney Int. 2010;78(12):1312–1321. doi: 10.1038/ki.2010.353. [DOI] [PubMed] [Google Scholar]
- 11.Kokubo T, Ishikawa N, Uchida H, Chasnoff SE, Xie X, Mathew S, Hruska KA, Choi ET. CKD Accelerates Development of Neointimal Hyperplasia in Arteriovenous Fistulas. Journal of the American Society of Nephrology. 2009;20(6):1236–1245. doi: 10.1681/ASN.2007121312. doi:10.1681/asn.2007121312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarnak MJ, Foley RN. Cardiovascular Mortality in the General Population Versus Dialysis: A Glass Half Full or Empty? American Journal of Kidney Diseases. 2011;58(1):4–6. doi: 10.1053/j.ajkd.2011.04.004. doi:10.1053/j.ajkd.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 13.Owens GK. Regulation of differentiation of vascular smooth muscle cells. Physiological Reviews. 1995;75(3):487–517. doi: 10.1152/physrev.1995.75.3.487. [DOI] [PubMed] [Google Scholar]
- 14.Regan CP, Adam PJ, Madsen CS, Owens GK. Molecular mechanisms of decreased smooth muscle differentiation marker expression after vascular injury. The Journal of Clinical Investigation. 2000;106(9):1139–1147. doi: 10.1172/JCI10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Owens GK, Kumar MS, Wamhoff BR. Molecular Regulation of Vascular Smooth Muscle Cell Differentiation in Development and Disease. Physiological Reviews. 2004;84(3):767–801. doi: 10.1152/physrev.00041.2003. doi:10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 16.Slupianek A, Yerrum S, Safadi FF, Monroy MA. The chromatin remodeling factor SRCAP modulates expression of prostate specific antigen and cellular proliferation in prostate cancer cells. Journal of Cellular Physiology. 2010;224(2):369–375. doi: 10.1002/jcp.22132. doi:10.1002/jcp.22132. [DOI] [PubMed] [Google Scholar]
- 17.Gabunia K, Jain S, England RN, Autieri MV. Anti-inflammatory cytokine interleukin-19 inhibits smooth muscle cell migration and activation of cytoskeletal regulators of VSMC motility. American Journal of Physiology - Cell Physiology. 2011;300(4):C896–C906. doi: 10.1152/ajpcell.00439.2010. doi:10.1152/ajpcell.00439.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 19.Hotamisligil GS. Endoplasmic reticulum stress and atherosclerosis. Nat Med. 2010;16(4):396–399. doi: 10.1038/nm0410-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schwartz SM. Smooth muscle migration in vascular development and pathogenesis. Transplant Immunology. 1997;5(4):255–260. doi: 10.1016/s0966-3274(97)80005-6. doi:http://dx.doi.org/10.1016/S0966-3274(97)80005-6. [DOI] [PubMed] [Google Scholar]
- 21.Go AS, Chertow GM, Fan D, McCulloch CE, Hsu C.-y. Chronic Kidney Disease and the Risks of Death, Cardiovascular Events, and Hospitalization. New England Journal of Medicine. 2004;351(13):1296–1305. doi: 10.1056/NEJMoa041031. doi:doi:10.1056/NEJMoa041031. [DOI] [PubMed] [Google Scholar]
- 22.Brunet P, Gondouin B, Duval-Sabatier A, Dou L, Cerini C, Dignat-George F, Jourde-Chiche N, Argiles A, Burtey S. Does Uremia Cause Vascular Dysfunction. Kidney and Blood Pressure Research. 2011;34(4):284–290. doi: 10.1159/000327131. [DOI] [PubMed] [Google Scholar]
- 23.Liu Z-P, Wang Z, Yanagisawa H, Olson EN. Phenotypic Modulation of Smooth Muscle Cells through Interaction of Foxo4 and Myocardin. Developmental Cell. 2005;9(2):261–270. doi: 10.1016/j.devcel.2005.05.017. doi:http://dx.doi.org/10.1016/j.devcel.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 24.Jenuwein T, Allis CD. Translating the Histone Code. Science. 2001;293(5532):1074–1080. doi: 10.1126/science.1063127. doi:10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 25.Alexander MR, Murgai M, Moehle CW, Owens GK. Interleukin-1β modulates smooth muscle cell phenotype to a distinct inflammatory state relative to PDGF-DD via NF-κB-dependent mechanisms. Physiological Genomics. 2012;44(7):417–429. doi: 10.1152/physiolgenomics.00160.2011. doi:10.1152/physiolgenomics.00160.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davies MG, P.O H. Pathobiology of intimal hyperplasia. Br. J Surg. 1994;81:1254–1269. doi: 10.1002/bjs.1800810904. [DOI] [PubMed] [Google Scholar]
- 27.Trécherel E, Godin C, Louandre C, Benchitrit J, Poirot S, Mazière J-C, Massy ZA, Galmiche A. Upregulation of BAD, a pro-apoptotic protein of the BCL2 family, in vascular smooth muscle cells exposed to uremic conditions. Biochemical and Biophysical Research Communications. 2012;417(1):479–483. doi: 10.1016/j.bbrc.2011.11.144. doi:http://dx.doi.org/10.1016/j.bbrc.2011.11.144. [DOI] [PubMed] [Google Scholar]
- 28.Serradell M, Díaz-Ricart M, Cases A, Petriz J, Ordinas A, Escolar G. Uraemic medium accelerates proliferation but does not induce apoptosis of endothelial cells in culture. Nephrology Dialysis Transplantation. 2003;18(6):1079–1085. doi: 10.1093/ndt/gfg161. doi:10.1093/ndt/gfg161. [DOI] [PubMed] [Google Scholar]
- 29.Roy-Chaudhury M. C. R. a. P. Vascular access in haemodialysis: strengthening the Achilles’ heel. Nat. Rev. Nephrol. 2013;9:348–357. doi: 10.1038/nrneph.2013.76. [DOI] [PubMed] [Google Scholar]