

Abstract
Background
Defects of cytoarchitectural proteins can cause left ventricular noncompaction (LVNC), which is often associated with conduction system diseases. We have previously identified a p.D117N mutation in the LDB3-encoding Z-band Alternatively Spliced PDZ motif gene (ZASP) in a patient with LVNC and conduction disturbances. We sought to investigate a role of p.D117N mutation in the LBD3 NM_001080114.1 isoform (ZASP1-D117N) in the regulation of cardiac sodium channel (Nav1.5) that plays an important role in the cardiac conduction system.
Methods and Results
Effects of ZASP1-wt and ZASP1-D117N on Nav1.5 were studied in HEK-293 cells and neonatal rat cardiomyocytes (NRCMs). Patch-clamp study demonstrated that ZASP1-D117N significantly attenuated INa by 27% in HEK-293 cells and by 32% in NRCMs. In addition, ZASP1-D117N rightward shifted the voltage-dependent activation and inactivation in both systems. In silico simulation using Luo-Rudy phase 1 model demonstrated that altered Nav1.5 function can reduce cardiac conduction velocity by 28% compared to the control. Pull-down assays showed that both wt and ZASP1-D117N can complex with Nav1.5 and telethonin/T-Cap, which required intact PDZ domains. Immunohistochemical staining in NRCMs demonstrates that ZASP1-D117N did not significantly disturb the Z-line structure. Disruption of cytoskeletal networks with ML-7 and cytochalasin D abolished the effects of ZASP1-D117N on the Nav1.5.
Conclusions
ZASP1 can form protein complex with telethonin/T-Cap and Nav1.5. The LVNC-specific ZASP1 mutation can cause loss-of-function of Nav1.5 without significant alteration of the cytoskeletal protein complex. Our study suggests that electrical remodeling can occur in LVNC subject due to a direct effect of mutant ZASP on Nav1.5.
Keywords: ZASP, sodium channel, cardiac conduction disturbance, left ventricular noncompaction
Introduction
Left ventricular noncompaction (LVNC) is associated with heart failure and arrhythmias such as atrio-ventricular (AV) block and intraventricular conduction disturbance (IVCD). Recently, we have demonstrated that genetic defects of the LIM domain-binding protein 3 (LDB3)-encoding Z-band alternatively spliced PDZ-motif protein (ZASP) are associated with LVNC.1 In the study, we identified ZASP1-D117N mutation in two Caucasian males who suffer from systolic dysfunction, dilated left ventricle with hypertrabeculated myocardium, and IVCD. Both patients showed sporadic heterozygote mutation. One of the patients had a family history of sudden cardiac death.
ZASP is one of the major components of the Z-disk proteins in cardiac muscle,2 which plays an important role in stabilizing the Z-disk structure through its PDZ-mediated interaction with α-actinin-2 (ACTN2), the main component of the Z-disk actin cross-linker, and F-actin, the main cytoarchitectural protein of cardiomyocytes.3 Global ablation of the murine ZASP homolog cypher can disorganize both sarcomere and cytoskeleton, leading to severe cardiomyopathy and skeletal myopathy in mice and humans, termed “zaspopathy”,4 while the cardiac-specific ablation of cypher led to dilated cardiomyopathy (DCM) and sudden cardiac death.5 However, the detail underlying mechanisms of arrhythmias in zaspopathy remains unclear.
On the other hand, loss-of-function of sodium channel (Nav1.5) has been recognized as a key pathophysiological mechanism of inherited conduction diseases.6 In addition, recent studies proposed a novel concept that the defects of non-ion channel proteins or channel interacting proteins (ChIPs) can affect the function of various ion channels, leading to secondary channelopathies.7-10
Along these lines of evidence, we hypothesized that mutant ZASP in LVNC patients might affect Nav1.5. The effects of ZASP1-D117N on the function of Nav1.5 and the anatomical remodeling of cytoarchitecture were studied in mammalian expression systems and neonatal rat cardiomyocytes (NRCMs). The amplitude and voltage-dependency of activation and inactivation of Nav1.5 were affected by ZASP1-D117N, which can lead to conduction disturbances confirmed by a computer simulation. These findings uncover some key elements of the mechanisms leading to conduction disturbances in “Zaspopathy.”
Materials and Methods
HEK-293 cell preparation and transient expression of wt and mutant ZASP1
The detail methods were described in Expanded Methods. Briefly, HEK-293 cells stably expressing hNav1.5 were transfected with the plasmid pcDNA3.1-CT-GFP-TOPO containing wt or mutant ZASP1 cDNA.
Neonatal rat cardiomyocyte (NRCM) isolation and transient expression of wt and mutant ZASP1
This research protocol was approved by the Institutional Animal Care and Use Committee. The neonatal rat cardiomyocytes were isolated according to the procedure described as previously (see Expanded Methods).10
Patch-clamp
Whole cell configuration of the voltage-clamp technique was used to record INa and IK as previously described (see Expanded Methods).10
Computer simulation
Computer simulations of conduction were done in a 3 cm-long cable of coupled myocytes described the Luo-Rudy phase 1 model.11 The voltage is governed by the following partial differential equation:
where Cm = 1 μF/cm2 and D = 0.001 cm2/ms is the diffusion constant. This value of the diffusion constant corresponds to a lumped myoplasmic and gap junctional intercellular conductance of 0.92 μS.12 The differential equations were integrated with a finite difference method with a spatial step of 100 μm and a time step of 0.01 ms. The maximum sodium channel conductance was set as GNa =16 mS/cm2 which gives rise to a conduction velocity (CV) of 57.4 cm/s.
Immunohistochemistry
Immunohistochemical (IHC) staining was performed by the standard technique as described in Expanded Methods.
In vitro interaction pull-down assay
Interactions between ZASP1 and Nav1.5 were studied with His-tagged pull-down assays (ProFound pull-down PolyHis Protein: Protein Interaction Kit; Pierce, IL: see details in Expanded Methods).
Statistics
Comparison of the continuous variables between two groups (ZASP1-wt vs. ZASP1-D117N) was performed using Mann-Whitney-Wilcoxon test. Kruskal-Wallis test was conducted to compare continuous variables among 3 groups, with post-hoc Mann-Whitney-Wilcoxon test to compare differences between any 2 groups. All comparisons were performed to test 2-tailed methods and p<0.05 was considered statistically significant. Statistical analyses were performed using SPSS PASW Statistics 17 software (IBM, Chicago, IL). Data in the text and figures are presented as median [25th percentile; 75th percentile] or mean ± SD unless otherwise stated.
Results
ZASP1-D117N causes loss-of-function of hNav1.5 in HEK-293 cells
To study the effects of wt and mutant ZASP1 on hNav1.5 function, HEK-293 cells stably expressing hNav1.5 were transiently transfected with GFP-tagged ZASP1-wt or ZASP1-D117N plasmids (5 μg). Figure 1A represents superimposed macroscopic hNav1.5 current (INa) traces at various depolarization levels obtained from the cells expressing ZASP1-wt, or ZASP1-D117N. The peak INa densities were significantly reduced in the cells transfected with ZASP1-D117N compared to the ZASP1-wt. Compared to the ZASP1-wt, ZASP1-D117N shifted the onset and the peak voltage of the I-V curve toward positive by 10 mV and 15 mV, respectively (Figure 1B).
Figure 1.
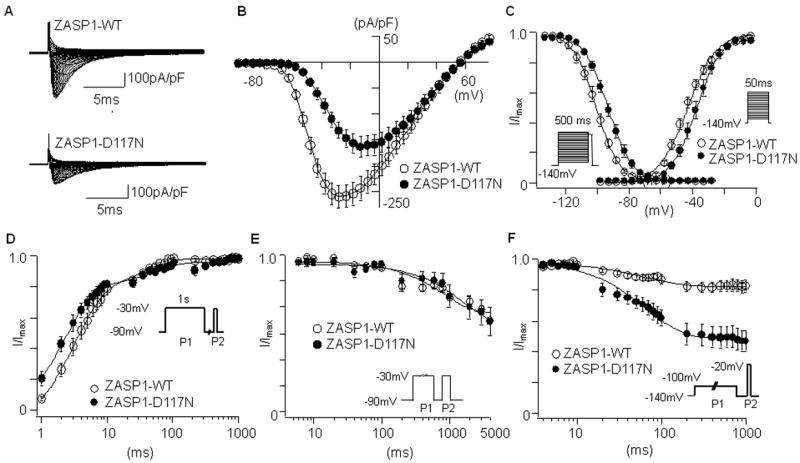
The effects of ZASP1-wt and ZASP1-D117N on hNav1.5 in HEK-293 cells. (A) Superimposed whole-cell current traces induced by a step-pulse protocol (20 ms between -120 mV and 80 mV) from a holding potential of -140 mV. (B) I-V relationships. (C) Voltage-dependence of peak conductance and steady-state fast inactivation. Conductance G (V) was calculated by the equation: G (V) = I / (Vm - Erev), where I is the peak currents, Erev is the measured reversal potential, Vm is the membrane potential. The normalized peak conductance was plotted as a function of membrane potentials. Steady-state inactivation was estimated by pre-pulse protocols (500 ms) from a holding potential of −140 mV. The normalized peak currents were plotted as a function of membrane potentials. Steady state activation and inactivation were fitted with the Boltzmann equation: y = [1 + exp ((Vh - Vm)/k)]-1, where y represents variables; Vh, midpoint; k, slope factor; Vm, membrane potential. Data were represented as mean ± SD. (D) Recovery from the fast-inactivation estimated by a double pulse protocol shown in inset. The recovery time course was fitted with a double exponential function: I (t)/Imax= C - Af × exp (-t/τf) - As × exp (-t/τs), where t is the recovery time; Af and As are the fraction of fast and slow components; τf and τs are the time constants of fast and slow components of recovery. (E) Time-course for the development of slow inactivation. The pulse protocol is shown as inset. Voltage was stepped from a holding potential of -90 mV to -30 mV for various times, stepped to -90 mV for 20 ms to allow recovery from fast inactivation, and then stepped to -30 mV (20 ms). To generate the curves, the current amplitude during the test pulse (second pulse) was normalized as a fraction of the current amplitude during the first pulse, and plotted against the duration of control pulse. The curve was fit with a double exponential equation.
(F) Closed-state inactivation. The membrane potential was held at -100 mV for various durations (Δt) from a holding potential of -140 mV before the test pulse (20 ms at -20 mV). The normalized currents by the maximal currents (t = 1 ms) were plotted against Δt. Data are presented as mean ± SD.
To study voltage-dependency of steady-state activation and inactivation, the peak conductance and peak currents induced by a pre-pulse protocol were plotted against the membrane potentials, and fitted with the Boltzman equation. ZASP1-D117N was found to right-ward shift the activation curve by 7 mV, and rightward shift the inactivation curve by 5 mV compared to the ZASP1-wt (Figure 1C). The slope factors (k) in the steady-state activation were also affected by ZASP1-D117N. Despite the smaller shift of inactivation curve, the larger shift of activation curve might increase the voltage threshold required to activate INa, contributing to the decrease of conduction velocity.13
Recovery time course from fast inactivation was examined by a double-pulse protocol. ZASP1-D117N was found to accelerate the fast-component of recovery time compared to the ZASP1-wt (Figure 1D). Figure 1E represents the development of slow inactivation. The time course was comparable between the two groups. In addition, direct transition from closed-state to inactivation-state without channel opening (closed-state inactivation) was studied. Interestingly, the time course of closed-state inactivation was accelerated by ZASP1-D117N compared to the ZASP1-wt (Figure 1F). Table 1 and 2 summarizes the parameters of INa studied in HEK-293 cells.
Table 1.
Nav1.5 parameters
| HEK-293 cells | NRCMs | |||
|---|---|---|---|---|
|
| ||||
| WT (n) | D117N (n) | WT (n) | D117N (n) | |
|
| ||||
| Peak current density (pA/pF) | (20) | (19) | (16) | (13) |
| -265.1 [-306.4; -181.8] | -142. 2[-207.0; -82.7]* | -756.8 [-1079.6; -545.5] | -650.0 [-701.8; -576.0] | |
|
| ||||
| Activation | (23) | (18) | (19) | (11) |
|
| ||||
| Vh (mV) | -45.4[-52.2; -41.3] | -36.7[-43.8; -30.3] † | -46.8 [-51.6; -38.8] | -32.1 [-39.4; -30.8]† |
|
| ||||
| k | 7.26[5.9; 8.4] | 6.55[5.8; 8.1]* | 7.3 [6.4; 7.7] | 7.4 [7.1; 7.9] |
|
| ||||
| Fast-inactivation | (16) | (19) | (16) | (8) |
|
| ||||
| Vh (mV) | -97.0[-103.0; -95.4] | -92.8[-98.1; -90.9] † | -95.7 [-101.9; -92.1] | -85.7 [-99.9; -77.6] † |
|
| ||||
| k | 6.1[5.8; 6.5] | 7.2[6.4; 7.8] | 6.9 [6.1; 7.4] | 7.5 [6.7; 7.8] |
Vh, represents midpoint voltage of maximal activation/inactivation; k, slope factor; n, the number of patches.
p<0.05;
p<0.01 vs. ZASP1-wt.
Table 2.
Time-dependent parameters of Nav1.5
| WT (n) | D117N (n) | |
|---|---|---|
| Recovery from fast-inactivation | (10) | (8) |
| τf (ms) | 4.2 [3.2; 5.2] | 1.9 [1.8; 2.2] |
| τs (ms) | 24.6 [22.2; 27.0] | 54.0 [42.1; 61.1]* |
| Af (%) | 0.3 [0.2; 0.3] | 0.1 [0.1; 0.2]* |
| As (%) | 0.9 [0.8; 0.9] | 0.9 [0.9; 1.1] |
| Closed-state inactivation | (14) | (19) |
| τ (ms) | 68.2 [50.4; 80.9] | 81.8 [72.0; 113.4]* |
| Slow inactivation | (8) | (8) |
| τ (ms) | 800.1 [761.4; 838.8] | 1202.6 [1145.5; 1347.0] |
τf and τs the time constants of fast and slow components of recovery; Af and As, the fraction of fast and slow components; n, the number of patches.
p<0.05, vs. ZASP1-wt.
The effects of ZASP1-D117N on the late INa were also studied in the HEK-293 cells. ZASP1-D117N did not significantly affect the 30 μM tetrodotoxin-sensitive late INa induced by a long depolarization pulse (Supplemental Figure S-I).
Since our patients were heterozygote,1 we also studied the effects of different ratios of ZASP1-wt and ZASP1-D117N plasmids on INa. While the transfection with 5 μg of ZASP1-wt and 1 μg of ZASP1-D117N plasmids did not significantly reduce the INa compared to 5 μg of ZASP1-wt, the transfection with 3 μg of ZASP1-wt and 3 μg of ZASP1-D117N, which might mimic the heterozygotic situation, significantly decreased the INa and rightward shifted the peak voltage (Supplemental Figure S-II).
Effects of ZASP1-D117N on delayed rectifier potassium channels
We also studied whether ZASP1-D117N can affect two major components of delayed rectifier potassium channels (slow component, IKs and rapid component, IKr). ZASP1-D117N did not significantly affect IKs and IKr in mammalian cell lines (Supplemental Figure S-III). These results indicate that ZASP1-D117N specifically alters the Nav1.5 functions.
The expression of ZASP1 and cytoarchitectural proteins in HEK-293 cells
The expression of mRNAs from genes encoding various cytoarchitectural proteins in HEK-293 cells suggests that this cell line may express cardiomyocyte-like proteins.14 The expression of various cytoskeletal proteins were studied using proteins extracted from HEK-293 cells stably expressing Flag-tagged hNav1.5 and transiently expressing V5-tagged ZASP1-wt or ZASP1-D117N. Figure 2 demonstrates that exogenous ZASP1-wt and ZASP1-D117N (V5-tagged) as well as endogenous ZASP, ACTN2, DMD, SNTA1, T-Cap, and β-actin were expressed in the HEK-293 cells.
Figure 2.
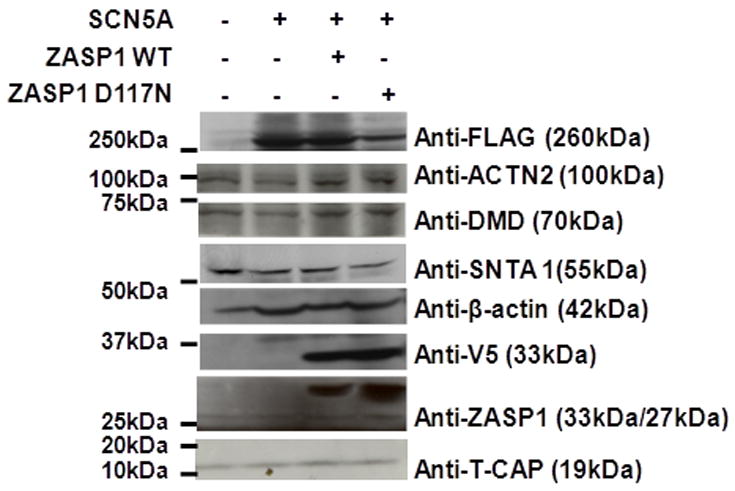
Western blot detects various cytoskeletal proteins in HEK-293 cells. Lane 1: samples from non-transfected HEK-293 cells; Lane 2: stable HEK-293 cells expressing Flag-tagged Nav1.5; Lane 3: stable HEK-293 cells transfected with V5-tagged ZASP1-wt; Lane 4: stable HEK-293 cells transfected with V5-tagged ZASP1-D117N. Note that the staining demonstrates the expression of ACTN2, DMD, SNTA1, and ZASP previously unknown to be produced in HEK-293 cells.
Protein interaction between Nav1.5 and ZASP1-telethonin/T-Cap complex
To study the protein-protein interaction between hNav1.5 and ZASP1-wt or ZASP1-D117N, pull-down assays were performed (Figure 3). First, three batches of E. coli were transformed with the plasmids containing SCN5A, V5/His-tagged ZASP1-wt, or V5/His-tagged ZASP1-D117N. The protein expression of Nav1.5 and ZASP was confirmed with Western blot using anti-pan sodium channel and anti-V5 antibodies (Panel A left blot). Second, the proteins extracted from the HEK-293 cells stably expressing hNav1.5 were incubated with V5/His-tagged ZASP1-wt or ZASP1-D117N produced in the E. coli transformed with the corresponding plasmids. The protein mixture was immunoprecipitated with the beads coated with anti-His antibody. The precipitants were analyzed with Western blot using anti-pan sodium channel antibody, anti T-Cap antibody, and anti V5 antibody. The results showed that both ZASP1-wt and ZASP1-D117N can form a protein complex with hNav1.5 and T-Cap (Panel A mid blot). Third, the same protocol was performed using the NRCM lysates. Panel A right blot shows that both ZASP1-wt and ZASP1-D117N can form a protein complex with rat Nav1.5 and T-Cap.
Figure 3.
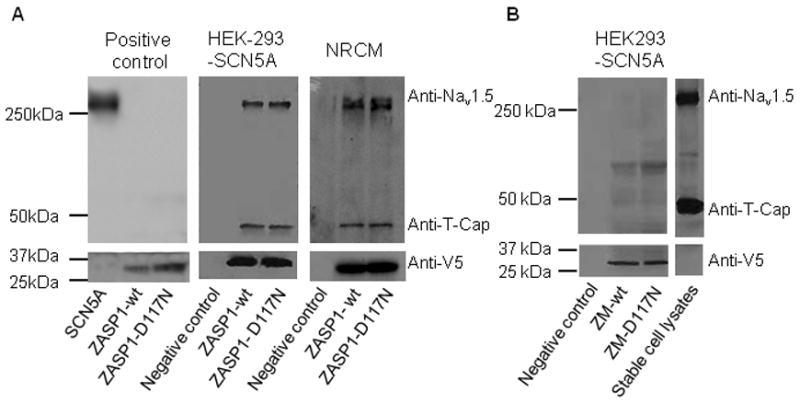
ZASP1, telethonin/T-Cap and Nav1.5 form a complex. (A) Left gel: Positive controls using Nav1.5, ZASP1-wt, and ZASP1-D117N produced in E.coli. Mid gel: Pull down assay using HEK-293 stable cells. Right panel: Pull down assay using NRCMs. (B) Pull down assay using ZASP fragments. ZM-wt: ZASP-wt lacking the PDZ domain; ZM-D117N: ZASP-D117N lacking the PDZ domain.
Since ZASP1 possesses N-terminal PDZ domain that is important in protein-protein interaction, we examined whether the PDZ domain is essential for the formation of protein complex. A pull-down assay using a ZASP1 segment (ZM) that lacks the PDZ domain was performed. The proteins extracted from the stable HEK-293 cells were incubated with V5/His-tagged ZM-wt or ZM-D117N produced in the E. coli. The protein mixture was immunoprecipitated with anti-His antibody, and the precipitants were analyzed with Western blot. Figure 3B shows that neither Nav1.5 nor T-Cap was detected in the immunoprecipitants, indicating that the PDZ domain is critically involved in the formation of protein complex between ZASP1, Nav1.5, and T-Cap.
ZASP1-D117N alters the Nav1.5 functions in NRCMs
The interaction and functional modification of INa by ZASP in HEK-293 cells may substantially differ from those in cardiomyocytes. Therefore, we studied the effects of ZASP1-wt and ZASP1-D117N on INa in NRCMs. The isolated NRCMs were transfected with GFP-tagged ZASP1-wt or ZASP1-D117N using electroporation. Figure 4A represents superimposed macroscopic INa traces obtained from the NRCMs transfected with ZASP1-wt or ZASP1-D117N. Similar to the observation in HEK-293 cells, ZASP1-D117N decreased the INa densities, and rightward shifted the onset and the peak voltage of I-V relationships by 15 mV compared to the ZASP1-wt (Figure 4B). ZASP1-D117N was found to rightward shift the voltage-dependency of steady-state activation and inactivation by 11.8 mV and 8.4 mV, respectively (Figure 4C). The slope factors in the steady-state activation and inactivation were not significantly affected by ZASP1-D117N (Figure 4C). These data indicate that ZASP1-D117N can cause loss-of-function of cardiac sodium channel in NRCMs similar to the effects on human Nav1.5 in mammalian cell lines. Table 1 summarizes the INa parameters in NRCMs.
Figure 4.
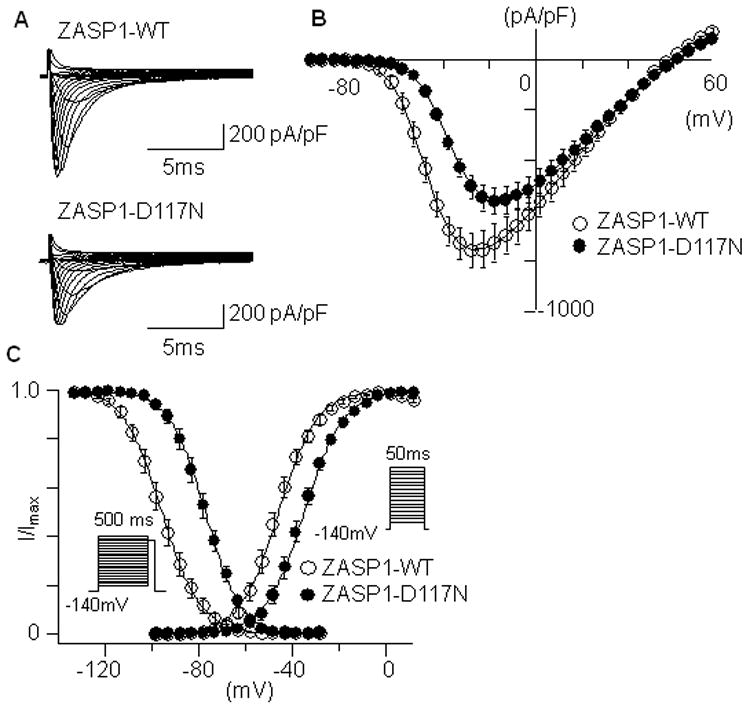
The effects of ZASP1-wt and ZASP1-D117N on Nav1.5 in NRCM. (A) Superimposed whole-cell current traces induced by a step-pulse protocol from a holding potential of −140 mV obtained from the NRCM transfected with ZASP1-wt or ZASP1-D117N. (B) I-V relationships. (C) Voltage-dependency of peak conductance and steady-state fast inactivation. The data were obtained and analyzed using same protocols and equations used in Figure 1. Data are presented as mean ± SD.
The localization of ZASP1 and cytoarchitectural proteins in NRCMs
Next, we studied the localization of ZASP-wt and ZASP-D117N in NRCMs. NRCMs were transfected with GFP-tagged ZASP1-wt or ZASP1-D117N. The cells were cultured for 24-48 hours, then fixed and stained with antibodies against various cytoarchitectural proteins. The fluorescence confocal images demonstrated that both ZASP1-wt and ZASP1-D117N exists along the Z-lines and were clearly overlapped with the Z-disk proteins (i.e., ACTN2, T-Cap, and actin; top and bottom panels in Figure 5). On the contrary, DMD and SNTA1 showed less clear overlap with ZASP compared to the Z-disk proteins (2nd and 3rd panels in Figure 5). These data suggest that ZASP1-D117N can affect INa without significant disruption of the Z-disk protein complex in NRCMs.
Figure 5.
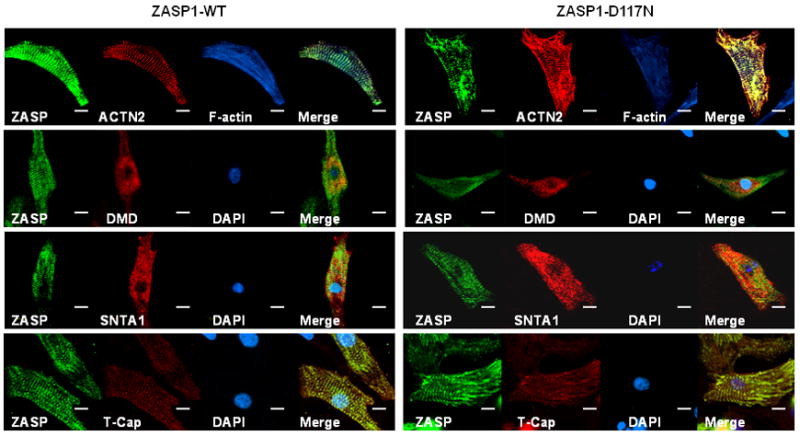
Immunohistochemical analysis identifies the expression of critical cytoskeletal proteins in NRCM. Confocal fluorescence images of NRCM were captured after transient transfection with GFP-tagged ZASP1-wt and ZASP1-D117N. The NRCM were fixed and stained with antibodies against ACTN2, phalloidin (F-actin), DMD, SNTA1, and telethonin/T-Cap, respectively. The scale bars indicate 10 μm.
Disruption of actin abolished the alteration of Nav1.5 function by ZASP1-D117N
Next, we employed ML-7 (5-iodonaphthalene-1-sulfonyl homopiperazine), a highly specific inhibitor of myosin light chain kinase, and Cytochalasin D, a potent inhibitor of actin polymerization (Cyto-D). Both reagents are known to disrupt cytoskeletal structures mainly by disassembling actin fibers that are important members of Z-line structure.15,16
The NRCMs transiently transfected with GFP-tagged ZASP1-wt or ZASP1-D117N were incubated with ML-7 (30 μM) or Cyto-D (2 μM) for 30 minutes before patch-clamp experiments. Very interestingly, the reduction of INa and the shift of I-V curves by ZASP1-D117N were abolished with these treatments (Figure 6A and 6B). Figure 6C demonstrates the voltage-dependency of the steady-state activation and inactivation were not affected by ZASP1-D117N after ML-7 treatment. Similar results were obtained in the cells treated with Cyto-D (Figure 6D to 6F). Table 3 summarizes the parameters with ML-7 and Cyto-D treatments.
Figure 6.
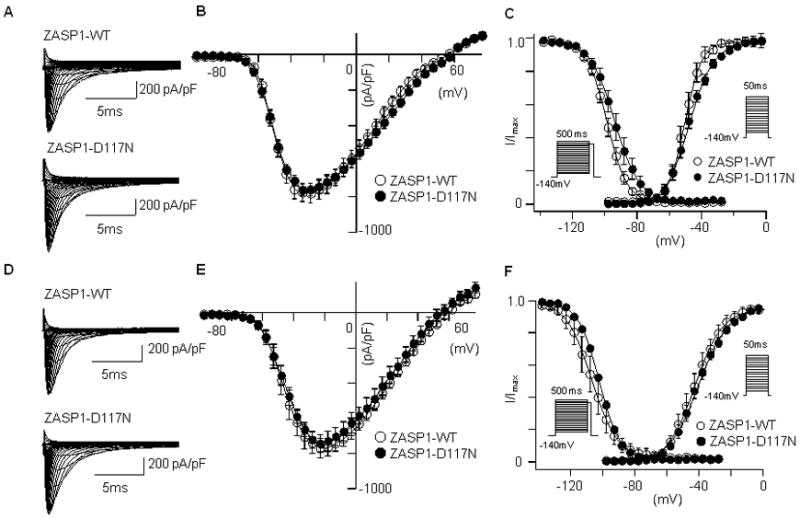
The effects of ML-7 and cytochalasin D on the hNav1.5 in NRCM. (A) Superimposed whole-cell current traces induced by a step-pulse protocol from a holding potential of -140 mV in the cells incubated with ML-7. (B) I-V relationships with ML-7 treatments. (C) Voltage-dependency of the peak conductance and steady-state fast inactivation. The data were obtained and analyzed using same protocols and equations described in Figure 1. (D-F) Same protocols were performed in the cells treated with Cyto-D. Data are presented as mean ± SD.
Table 3.
Effect of ML-7 and Cytochalasin D on Nav1.5 parameters
| ML-7 | Cytochalasin D | |||
|---|---|---|---|---|
| WT (n=15) | D117N (n=10) | WT (n=10) | D117N (n=11) | |
| Peak current density (pA/pF) | -231.6 [-288.4; -193.9] | -230.8 [-282.3; -159.1] | -143.8 [-219.6; -120.2] | -203.0 [-234.0; -177.3] |
| Activation | ||||
| Vh (mV) | -36.2 [-38.7; -35.2] | -42.2 [-47.2; -39.4] | -34.8 [-35.3; -26.9] | -45.7 [-50.2; -36.2] |
| k | 7.5 [5.7; 8.3] | 7.4 [6.8; 7.8] | 8.3 [7.1; 10.4] | 6.7 [6.5; 7.4] |
| Fast-inactivation | ||||
| Vh (mV) | -99.9 [-104.6; -96.4] | -95.0 [-101.7; -91.2] | -93.9 [-103.0; -88.8] | -97.9 [-106.0; -95.1] |
| k | 6.8 [6.3; 7.2] | 6.2 [5.6; 7.1] | 7.1 [5.4; 7.7] | 5.8 [5.6; 6.6] |
Vh, represents midpoint voltage of maximal activation/inactivation; k, slope factor; n, the number of patches.
Decline of Na+ conductance by ZASP1-D117N modulates CV and conduction failure
An important question is whether these modifications of Nav1.5 by ZASP-D117N can cause conduction disturbances in actual human hearts. It has been believed that cardiac conduction in ventricles depends on the magnitude of the available Na+ conductance, which in turn depends on the voltage dependence of channel activation and inactivation. Computer simulations were performed using the Luo-Rudy phase 1 model using the gating kinetic parameters of the sodium channel provided in Table 1 (HEK-293 cells). The results demonstrated that the alteration of Na+ channels mediated by ZASP1-D117N would have a distinct effect in depolarized tissue compared with that by ZASP1-wt. Figure 7 shows the influence of the sodium channel conductance and kinetics on CV. At control, CV was 57.4 cm/s. A 30% reduction of GNa to 11.2 mS/cm2, CV was reduced by about 12% to 50.5 cm/s. A right shift of 10 mV in both the activation and inactivation curves resulted in a 16% reduction to 48.1 cm/s. The combined effect of both changes applied at the same time result in a reduction of 27% in CV to 41.8 cm/s.
Figure 7.
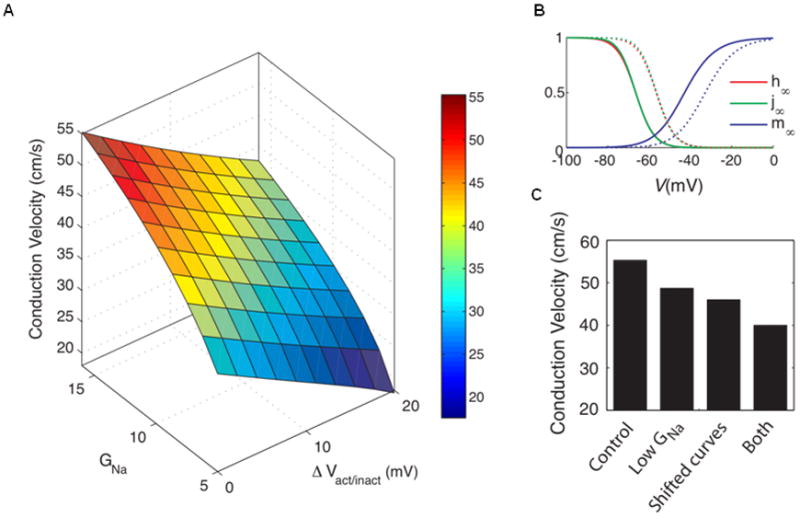
Changes in CV in a cable of Luo-Rudy 1 model cells due to changes in the sodium properties. (A) Measured CV as a function of GNa and of DVact/inact (the shift in activation and inactivation curves). (B) Standard steady-state activation and inactivation curves (solid lines) and the activation and inactivation curves shifted by +10 mV (dashed lines). (C) Change in CV from the situation with standard parameter values (‘Control’) due to a reduction in the maximum sodium channel conductance to 11.2 mS/cm2 (‘Low GNa’), a shift by 10 mV in the positive direction of the sodium channel activation and inactivation curves (‘Shifted curves’) and both of these changes (‘Both’).
Discussion
In this study, we demonstrated several underlying mechanisms by which ZASP1-D117N, a ZASP mutation identified in the patients suffering from DCM/LVNC, may cause IVCD: (1) ZASP1-D117N can cause loss-of-function of Nav1.5 in human cell lines as well as in neonatal cardiomyocytes; (2) in silico simulation using the Luo-Rudy model shows that the extent of functional disturbances of Nav1.5 caused by ZASP-D117N is sufficient enough to delay cardiac conduction in human hearts; (3) the interaction between ZASP1 and Nav1.5 requires a preservation of Z-disk protein complex; and (4) the modification of Nav1.5 by ZASP1-D117N occurs without significant disruption of Z-line structures in cardiomyocytes.
How does ZASP1 interact with Nav1.5?
In cardiomyocytes, various cytoarchitectural proteins interact with each other to form structural networks: (1) ZASP1 binds to ACNT2; (2) F-actin fibers are anchored to the Z-line via ACNT2; (3) DMD binds to F-actin,17 and (4) SNTA1 forms a complex with DMD (DAPC: dystrophin-associated protein complex).18 It has been reported that these proteins can alter the function and localization of Nav1.5. For example, DAPC can directly interact with the three last residues of Nav1.5 (Ser-Ile-Val: SIV) that constitutes a PDZ-binding motif, and plays a role in anchoring Nav1.5 to the lateral membranes. Ablation of dystrophin can attenuate cardiac conduction due to a reduction of INa.18 Telethonin/T-Cap, a member of Z-disk protein complex, can interact with Nav1.5, and a mutation of T-Cap can alter the function of Nav1.5.19 Very recently, SAP97 was found to interact with SIV motif of Nav1.5, and contribute to the localization of Nav1.5 in intercalated disks.20 However, no data have been previously reported with regard to an interaction between ZASP1, telethonin/T-Cap, and Nav1.5.
Our study demonstrates that: (1) both wt and mutant ZASP1 can form a protein complex with Nav1.5 and telethonin/T-Cap (Figure 3A); (2) ZASP1-D117N does not alter the association with Nav1.5 and telethonin/T-Cap (Figure 3A); (3) the PDZ domain is necessary to form the protein complex between ZASP1, Nav1.5, and telethonin/T-Cap (Figure 3B); and (4) Z-line structures were not significantly disrupted by ZASP1-D117N in cardiomyocytes (Figure 5). Therefore, we speculated that stability of Z-line structure network is important for interaction between ZASP1 and Nav1.5, and that ZASP1-D117N might alter the function of Nav1.5 by affecting local protein conformational change. In fact, when we disbanded the linkage between ZASP and the cytoskeleton with ML-7 and Cyto-D, the modulation of Nav1.5 by ZASP1-D117N was abolished. However, our study also showed that the treatment of NRCMs with ML-7 and Cyto-D did not alter the INa in the cells transfected with ZASP1-wt. This suggests that ML-7 and Cyto-D may affect unknown signal transduction pathways by which ZASP1-D117N can cause the modification of Nav1.5.
Another speculative underlying mechanism is that a local conformational change of Z-line complex by ZASP1-D117N may alter the mechanical stress between F-actin and DMD/SNTA1 complex, which may ultimately lead to modification of Nav1.5 function. Although we do not have a direct evidence of this mechanism, a similar concept has been proposed by a recent study using atomic force microscope that directly demonstrated a critical role of telethonin/T-Cap molecule in regulation of mechanical tension of cytoskeletal networks.21
Localization of Nav1.5 via ZASP/telethonin complex
While Nav1.5 preferentially localizes at the intercalated disc via SAP97 and lateral membranes via DAPC (2 pools), localization at the T-tubular system has also been recognized.22-24 It appears that Nav1.5 upon post-translational modification remains attached to the cytoskeleton probably linked to multiprotein complexes, and stored in subcellular compartments. Recent study showed that Nav1.5 localizes at the cardiomyocyte membrane along the sarcomeric Z-lines via α-actinin-2, thus connecting Nav1.5 to actin filaments.25 ZASP/telethonin may contribute to localizing Nav1.5 to the T-tubule membrane at the Z-line. Therefore, it is unlikely that the ZASP1-Nav1.5-telethonin/T-Cap protein complex represents a new pool, but rather it brings further evidence of multiprotein complex associated with α-actinin-2.
Electrical remodeling may precede anatomical remodeling
Our study suggests that electrical remodeling may precede anatomical remodeling in LVNC associated with ZASP: the loss-of-function of Nav1.5 by the mutated ZASP1 can occur without significant disruption of cytoarchitectural networks. This is particularly important in a clinical situation since patients who carry ZASP1-D117N may develop arrhythmias even before manifesting heart failure symptoms. Although the conduction disturbances observed in our patients might be caused by degeneration or fibrosis of conduction system as a consequence of anatomical remodeling, it is reasonable to speculate that the loss-of-function of Nav1.5 by ZASP1-D117N might exaggerate the conduction delay even if it is caused by anatomical remodeling.
Interestingly, we have previously reported a similar observation in a cardiac-specific transgenic mouse model of DCM caused by p.S196L mutation in ZASP4, another ZASP isoform highly expressed in human hearts. In mice expressing ZASP4-S196L, cardiac conduction defects and atrioventricular block were observed at 3-months of age before myocardial structural remodeling and ventricular dysfunction occurred at 10 months of age.26 In this model, we observed attenuation of L-type Ca2+ currents and rightward shift of voltage dependency of Nav1.5, which was different from the electrical remodeling by ZASP1-D117N.
Since we do not have multiple biopsy samples of patients’ cardiac muscles at different time points due to ethical reasons, and a transgenic mouse model of ZASP1-D117N was embryonic lethal (data not shown), we cannot infer whether ZASP1-D117N, also, follows the same pattern of ZASP4-S196L.
Can the biophysical modification of Nav1.5 by ZASP1-D117N cause conduction blocks?
Our computer simulation demonstrated that the biophysical modification of INa by ZASP-D117N can reduce the CV by 27%. However, since the actual CV in our patients was unknown, whether such decrease in CV can cause intraventricular conduction delay remains undetermined in this study. Recently, Smits et al. reported that a SCN5A mutation E161K identified in a patient suffered from overlap disease of Brugada syndrome and conduction block can decrease the peak INa and rightward shift the voltage-dependency of steady-state activation, resulting in the reduction of the CV in ventricle from 65 cm/s to 50 cm/s.27 This suggests that the 27% reduction in CV may be sufficient to cause conduction disturbances although further clinical electrophysiological studies are warranted.
Conclusion
We conclude that ZASP1-D117N, a mutation previously identified in patients with cardiomyopathy, might contribute to conduction-system diseases associated with the structural cardiac disease. These data confirm our previous hypothesis and provide novel insights into the relationship between cytoskeletal proteins and ion channels. Although further studies elucidating the detailed dynamics of wt and mutant ZASP in human arrhythmias and heart failure are warranted, we believe that our data contribute to understanding the mechanisms of arrhythmias in this LVNC subject, and provide a new framework for therapeutic interventions in patients suffering from arrhythmias associated with cardiomyopathy.
Study limitation
Our system employs overexpression of ZASP1-D117N in isolated cells rather than using a genetically engineered mouse model. However, our rationale originated from the observation that the mRNA level of ZASP is increased in human idiopathic dilated cardiomyopathy,28 thus we tested whether the overexpression of ZASP in NRCMs might recapitulate the cytoarchitectural environment in cardiomyocytes. In addition, although we have identified some components interacting with Nav1.5 in a complex, other sodium channel components such as SCN1B, SCN2B, SCN3B, and SCN4B and/or currently unknown proteins could also be involved in the localization and regulation of Nav1.5. Therefore, it is likely that Nav1.5 complex is much more multifaceted and further investigation to elucidate these intricate connections is warranted.
Supplementary Material
Acknowledgments
Authors are grateful to Shahrzad Abbasi for her technical assistance.
Funding Sources: Dr. Vatta was funded by grants from the National Institutes of Health, National Heart, Lung, & Blood Institute (HL07887). Dr. Ai was supported by Methodist Research Institute Showalter Cardiovascular Research Awards. The financial support of the Foundation Cariparo, Italy (Progetto Eccellenza 2010 CHROMUS) to Dr. Georgine Faukner is gratefully acknowledged. Financial support to Dr. Brunelli was provided by a grant from the American Heart Association.
Footnotes
Conflicts of Interest Disclosures: None.
References
- 1.Vatta M, Mohapatra B, Jimenez S, Sanchez X, Faulkner G, Perles Z, Sinagra G, Lin JH, Vu TM, Zhou Q, Bowles KR, Di Lenarda A, Schimmenti L, Fox M, Chrisco MA, Murphy RT, McKenna W, Elliott P, Bowles NE, Chen J, Valle G, Towbin JA. Mutations in cypher/zasp in patients with dilated cardiomyopathy and left ventricular non-compaction. J Am Coll Cardiol. 2003;42:2014–2027. doi: 10.1016/j.jacc.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 2.Faulkner G, Pallavicini A, Formentin E, Comelli A, Ievolella C, Trevisan S, Bortoletto G, Scannapieco P, Salamon M, Mouly V, Valle G, Lanfranchi G. Zasp: A new z-band alternatively spliced pdz-motif protein. J Cell Biol. 1999;146:465–475. doi: 10.1083/jcb.146.2.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klaavuniemi T, Ylanne J. Zasp/cypher internal zm-motif containing fragments are sufficient to co-localize with alpha-actinin--analysis of patient mutations. Exp Cell Res. 2006;312:1299–1311. doi: 10.1016/j.yexcr.2005.12.036. [DOI] [PubMed] [Google Scholar]
- 4.Zhou Q, Chu PH, Huang C, Cheng CF, Martone ME, Knoll G, Shelton GD, Evans S, Chen J. Ablation of cypher, a pdz-lim domain z-line protein, causes a severe form of congenital myopathy. J Cell Biol. 2001;155:605–612. doi: 10.1083/jcb.200107092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zheng M, Cheng H, Li X, Zhang J, Cui L, Ouyang K, Han L, Zhao T, Gu Y, Dalton ND, Bang ML, Peterson KL, Chen J. Cardiac-specific ablation of cypher leads to a severe form of dilated cardiomyopathy with premature death. Hum Mol Genet. 2009;18:701–713. doi: 10.1093/hmg/ddn400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abriel H. Roles and regulation of the cardiac sodium channel na v 1.5: Recent insights from experimental studies. Cardiovasc Res. 2007;76:381–389. doi: 10.1016/j.cardiores.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 7.London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, Viswanathan PC, Pfahnl AE, Shang LL, Madhusudanan M, Baty CJ, Lagana S, Aleong R, Gutmann R, Ackerman MJ, McNamara DM, Weiss R, Dudley SC., Jr Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (gpd1-l) decreases cardiac na+ current and causes inherited arrhythmias. Circulation. 2007;116:2260–2268. doi: 10.1161/CIRCULATIONAHA.107.703330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ueda K, Valdivia C, Medeiros-Domingo A, Tester DJ, Vatta M, Farrugia G, Ackerman MJ, Makielski JC. Syntrophin mutation associated with long qt syndrome through activation of the nnos-scn5a macromolecular complex. Proc Natl Acad Sci U S A. 2008;105:9355–9360. doi: 10.1073/pnas.0801294105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, Tester DJ, Balijepalli RC, Foell JD, Li Z, Kamp TJ, Towbin JA. Mutant caveolin-3 induces persistent late sodium current and is associated with long-qt syndrome. Circulation. 2006;114:2104–2112. doi: 10.1161/CIRCULATIONAHA.106.635268. [DOI] [PubMed] [Google Scholar]
- 10.Wu G, Ai T, Kim JJ, Mohapatra B, Xi Y, Li Z, Abbasi S, Purevjav E, Samani K, Ackerman MJ, Qi M, Moss AJ, Shimizu W, Towbin JA, Cheng J, Vatta M. Alpha-1-syntrophin mutation and the long-qt syndrome: A disease of sodium channel disruption. Circ Arrhythm Electrophysiol. 2008;1:193–201. doi: 10.1161/CIRCEP.108.769224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luo CH, Rudy Y. A model of the ventricular cardiac action potential. Depolarization, repolarization, and their interaction. Circ Res. 1991;68:1501–1526. doi: 10.1161/01.res.68.6.1501. [DOI] [PubMed] [Google Scholar]
- 12.Shaw RM, Rudy Y. Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and l-type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res. 1997;81:727–741. doi: 10.1161/01.res.81.5.727. [DOI] [PubMed] [Google Scholar]
- 13.Tan HL, Bink-Boelkens MT, Bezzina CR, Viswanathan PC, Beaufort-Krol GC, van Tintelen PJ, van den Berg MP, Wilde AA, Balser JR. A sodium-channel mutation causes isolated cardiac conduction disease. Nature. 2001;409:1043–1047. doi: 10.1038/35059090. [DOI] [PubMed] [Google Scholar]
- 14.Shaw G, Morse S, Ararat M, Graham FL. Preferential transformation of human neuronal cells by human adenoviruses and the origin of hek 293 cells. Faseb J. 2002;16:869–871. doi: 10.1096/fj.01-0995fje. [DOI] [PubMed] [Google Scholar]
- 15.Seguchi O, Takashima S, Yamazaki S, Asakura M, Asano Y, Shintani Y, Wakeno M, Minamino T, Kondo H, Furukawa H, Nakamaru K, Naito A, Takahashi T, Ohtsuka T, Kawakami K, Isomura T, Kitamura S, Tomoike H, Mochizuki N, Kitakaze M. A cardiac myosin light chain kinase regulates sarcomere assembly in the vertebrate heart. J Clin Invest. 2007;117:2812–2824. doi: 10.1172/JCI30804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Undrovinas AI, Shander GS, Makielski JC. Cytoskeleton modulates gating of voltage-dependent sodium channel in heart. Am J Physiol. 1995;269:H203–214. doi: 10.1152/ajpheart.1995.269.1.H203. [DOI] [PubMed] [Google Scholar]
- 17.Fatkin D, Graham RM. Molecular mechanisms of inherited cardiomyopathies. Physiol Rev. 2002;82:945–980. doi: 10.1152/physrev.00012.2002. [DOI] [PubMed] [Google Scholar]
- 18.Gavillet B, Rougier JS, Domenighetti AA, Behar R, Boixel C, Ruchat P, Lehr HA, Pedrazzini T, Abriel H. Cardiac sodium channel nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ Res. 2006;99:407–414. doi: 10.1161/01.RES.0000237466.13252.5e. [DOI] [PubMed] [Google Scholar]
- 19.Mazzone A, Strege PR, Tester DJ, Bernard CE, Faulkner G, De Giorgio R, Makielski JC, Stanghellini V, Gibbons SJ, Ackerman MJ, Farrugia G. A mutation in telethonin alters nav1.5 function. J Biol Chem. 2008;283:16537–16544. doi: 10.1074/jbc.M801744200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petitprez S, Zmoos AF, Ogrodnik J, Balse E, Raad N, El-Haou S, Albesa M, Bittihn P, Luther S, Lehnart SE, Hatem SN, Coulombe A, Abriel H. Sap97 and dystrophin macromolecular complexes determine two pools of cardiac sodium channels nav1.5 in cardiomyocytes. Circ Res. 2011;108:294–304. doi: 10.1161/CIRCRESAHA.110.228312. [DOI] [PubMed] [Google Scholar]
- 21.Bertz M, Wilmanns M, Rief M. The titin-telethonin complex is a directed, superstable molecular bond in the muscle z-disk. Proc Natl Acad Sci U S A. 2009;106:13307–133310. doi: 10.1073/pnas.0902312106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scriven DR, Dan P, Moore ED. Distribution of proteins implicated in excitation-contraction coupling in rat ventricular myocytes. Biophys J. 2000;79:2682–2691. doi: 10.1016/S0006-3495(00)76506-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brette F, Orchard CH. Density and sub-cellular distribution of cardiac and neuronal sodium channel isoforms in rat ventricular myocytes. Biochem Biophys Res Commun. 2006;348:1163–1166. doi: 10.1016/j.bbrc.2006.07.189. [DOI] [PubMed] [Google Scholar]
- 24.Dominguez JN, de la Rosa A, Navarro F, Franco D, Aranega AE. Tissue distribution and subcellular localization of the cardiac sodium channel during mouse heart development. Cardiovasc Res. 2008;78:45–52. doi: 10.1093/cvr/cvm118. [DOI] [PubMed] [Google Scholar]
- 25.Ziane R, Huang H, Moghadaszadeh B, Beggs AH, Levesque G, Chahine M. Cell membrane expression of cardiac sodium channel na(v)1.5 is modulated by alpha-actinin-2 interaction. Biochemistry. 2010;49:166–178. doi: 10.1021/bi901086v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Z, Ai T, Samani K, Xi Y, Tzeng HP, Xie M, Wu S, Ge S, Taylor MD, Dong JW, Cheng J, Ackerman MJ, Kimura A, Sinagra G, Brunelli L, Faulkner G, Vatta M. A zasp missense mutation, s196l, leads to cytoskeletal and electrical abnormalities in a mouse model of cardiomyopathy. Circ Arrhythm Electrophysiol. 2010;3:646–656. doi: 10.1161/CIRCEP.109.929240. [DOI] [PubMed] [Google Scholar]
- 27.Smits JP, Koopmann TT, Wilders R, Veldkamp MW, Opthof T, Bhuiyan ZA, Mannens MM, Balser JR, Tan HL, Bezzina CR, Wilde AA. A mutation in the human cardiac sodium channel (e161k) contributes to sick sinus syndrome, conduction disease and brugada syndrome in two families. J Mol Cell Cardiol. 2005;38:969–981. doi: 10.1016/j.yjmcc.2005.02.024. [DOI] [PubMed] [Google Scholar]
- 28.Grzeskowiak R, Witt H, Drungowski M, Thermann R, Hennig S, Perrot A, Osterziel KJ, Klingbiel D, Scheid S, Spang R, Lehrach H, Ruiz P. Expression profiling of human idiopathic dilated cardiomyopathy. Cardiovasc Res. 2003;59:400–411. doi: 10.1016/s0008-6363(03)00426-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.