

Abstract
Insulin is essential for the regulation of fuel metabolism and triggers the uptake of glucose by skeletal muscle. The imported glucose is either stored or broken down, as insulin stimulates glycogenesis and ATP synthesis. The mechanism by which ATP production is increased is incompletely understood at present and, generally, relatively little functional information is available on the effect of insulin on mitochondrial function. In this paper we have exploited extracellular flux technology to investigate insulin effects on the bioenergetics of rat (L6) and human skeletal muscle myoblasts and myotubes. We demonstrate that a 20-min insulin exposure significantly increases (i) the cell respiratory control ratio, (ii) the coupling efficiency of oxidative phosphorylation, and (iii) the glucose sensitivity of anaerobic glycolysis. The improvement of mitochondrial function is explained by an insulin-induced immediate decrease of mitochondrial proton leak. Palmitate exposure annuls the beneficial mitochondrial effects of insulin. Our data improve the mechanistic understanding of insulin-stimulated ATP synthesis, and reveal a hitherto undisclosed insulin sensitivity of cellular bioenergetics that suggests a novel way of detecting insulin responsiveness of cells.
Abbreviations: 2DG, 2-deoxyglucose; BSA, bovine serum albumin; DMEM, Dulbecco's modified Eagle medium; ECAR, extracellular acidification rate; FCCP, trifluorocarbonylcyanide phenylhydrazone; FCS, fetal calf serum; Hepes, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid; UCP, uncoupling protein
Keywords: Skeletal muscle cells, Insulin sensitivity, Oxidative phosphorylation, Mitochondrial coupling efficiency, Cell respiratory control, Mitochondrial proton leak
Highlights
-
•
Insulin acutely improves mitochondrial function of skeletal muscle cells.
-
•
Mitochondrial proton leak of skeletal muscle cells is attenuated by insulin.
-
•
Insulin sensitises anaerobic glycolysis of skeletal muscle cells to glucose.
-
•
Bioenergetic insulin effects offer novel assays for cellular insulin sensitivity.
1. Introduction
Skeletal muscle plays an important role in the maintenance of blood glucose homeostasis by taking up excess glucose in response to insulin [1]. Defects in insulin responsiveness, as for example manifested in obese subjects, contribute to the development of type 2 diabetes [2]. Obesity-related insulin resistance is at least partly caused by high levels of circulating non-esterified fatty acids [3], and mitochondrial dysfunction has been implicated in this resistance [4]. Insufficient mitochondrial capacity to burn excess fatty acids has been held responsible for obesity-blunted insulin sensitivity [5], but this notion is highly controversial [6]. In healthy muscle, insulin stimulates the storage of imported glucose as glycogen, an anabolic effect that has been recognised for some time [7]. It is becoming increasingly clear, however, that insulin signalling also affects mitochondrial metabolism [8], suggesting a possible additional regulatory role for insulin in oxidative nutrient metabolism. Several independent studies have demonstrated that insulin indeed stimulates ATP synthesis in human muscle, and that this catabolic effect is annulled under pathological conditions relating to type 2 diabetes [9–11]. Insulin-mediated ATP production has been associated with enhanced mitochondrial protein synthesis [9,12] and with increased mRNA levels and activities of mitochondrial enzymes involved in fuel oxidation [9]. Consistently, insulin has been shown to promote glucose oxidation [13]. Insulin is thus thought to stimulate ATP synthesis by increasing the capacity of oxidative phosphorylation [9], but the experimental evidence for this notion is largely circumstantial. Generally, direct functional measurements of the effect of insulin on mitochondrial function in skeletal muscle are relatively scarce.
In this paper, we have exploited recently developed extracellular flux technology [14] to establish insulin effects on the energy metabolism of intact rat and human skeletal muscle cells. Real-time measurements of mitochondrial function reveal that insulin instantly improves the coupling efficiency of oxidative phosphorylation and increases cell respiratory control. We show that these related beneficial insulin effects on mitochondrial function are due to acutely decreased proton leak, and that they are annulled completely in fatty-acid-exposed cells.
2. Materials and methods
2.1. Cell culture
2.1.1. Rat cells
L6 myoblasts were obtained from the European Collection of Cell Culture, and were maintained at 37 °C under a humidified carbogen atmosphere in Dulbecco's Modified Eagle Medium (DMEM) containing 25 mM glucose and 20 mM Hepes, and supplemented with 10% (v/v) fetal calf serum (FCS), 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells between passages 9 and 18 were used for experimentation.
2.1.2. Human cells
Skeletal muscle tissue (~ 350 mg) was removed from vastus lateralis through locally anaesthetised needle biopsy with informed donor consent and approval from the Ethics Committee of the Department of Sport and Health Sciences, College of Life and Environmental Sciences, University of Exeter, UK. Myoblasts were isolated and cultured at the University of Exeter Medical School, St Luke's Campus (Human Tissue Authority licence 12104) according to [15], and cells were passaged at least twice before off-site analysis. Briefly, tissue was collected in cold DMEM containing 5 mM glucose and 20 mM Hepes, minced finely with a sterile scalpel, and cells were dissociated via agitated incubation at 37 °C in digestion medium containing 0.25% (w/v) trypsin, 0.1% (w/v) type IV collagenase, and 0.1% (w/v) bovine serum albumin (BSA). Every 10 min for 30 min in total, dislodged cells were harvested: undigested tissue was spun down gently at 80 g and was then re-incubated with fresh digestion medium; the supernatant was subsequently spun at 150 g to collect dissociated cells. Pooled cells were incubated for 1 h in DMEM (containing 5 mM glucose, 20 mM Hepes, 20% (v/v) FCS, 0.5% (v/v) chick embryo extract, 2 nM insulin, 100 U/mL penicillin, and 100 μg/mL streptomycin) in an uncoated 25 cm2 culture flask to facilitate selective attachment of fibroblasts. Subsequently, the myoblast-enriched supernatant was transferred to a 60 mm collagen-coated culture dish. Attached cells were allowed to reach 80% confluence (5 days' growth approximately), and were then trypsinised for further enrichment: 20-min incubation in a collagen-coated dish to remove relatively rapidly attaching fibroblasts [16], followed by supernatant transfer to another collagen-coated dish. As confirmed by myosin-staining, this method yielded monolayers with more than 95% myoblasts. The initial cell population was allowed to double 4 × before experimentation, and all assays were performed before the population had doubled 8 ×.
2.1.3. Differentiation
Human and L6 myoblasts were seeded on XF24 (Seahorse Bioscience) tissue culture plates at 2 × 104 and 4 × 104 cells/well, respectively, and grown for 48 h in fully supplemented, cell-specific DMEM (specified above). At this point, the FCS level in the respective growth media was lowered to just 2% (v/v) and this ‘light-serum’ medium was refreshed every 2–3 days until myoblasts had turned to myotubes. Visual inspection using a light microscope indicated that complete differentiation took 8–10 days.
2.2. Palmitate
2.2.1. Conjugation
Fatty acid-free BSA (Sigma A7030) was dissolved at 1.6 mM in medium containing 135 mM NaCl, 3.6 mM KCl, 10 mM Hepes (pH 7.4), and 0.5 mM MgCl2. Palmitate (8 mM) was added as powder and stirred continuously for 24–48 h at 35–38 °C until it had dissolved completely. Cooled BSA:palmitate conjugations were filter-sterilised and stored at 4 °C. When added to cultures, the conjugations were diluted 40 × and the total unbound free palmitate level was around 20 nM as estimated from previously published binding parameters [17].
2.2.2. Exposure
Cells were cultured in DMEM containing just 5 mM glucose and 2% (v/v) FCS. This nutrient restriction sensitised the cells to insulin (cf. [18]), and was applied for 10 h in case of glucose uptake assays (Section 2.5) and 24 h in case of the cellular bioenergetics experiments (Section 2.3). At this point, insulin sensitivity was measured either immediately or after a 16-h exposure to palmitate, in which case medium was replaced with FCS-free DMEM containing BSA-conjugated palmitate, or BSA alone.
2.3. Mitochondrial respiration
Mitochondrial bioenergetics were measured in attached cells as described before [19]. Briefly, L6 myoblasts seeded at 4 × 104 cells/well, differentiated, and exposed to palmitate on XF24 plates were washed 4 × with a Krebs Ringer buffer (KRPH) comprising 136 mM NaCl, 3.7 mM KCl, 10 mM Hepes, (pH 7.4), 2 mM NaH2PO4, 1 mM MgCl2, 1.5 mM CaCl2, and 0.1% (w/v) BSA, and were then incubated in this buffer for 1 h at 37 °C under air. Human cells were treated similarly, but were seeded at 2 × 104 cells/well, were not exposed to palmitate, and were not washed into KRPH, but into serum-free DMEM containing 2 mM glucose and 10 mM Hepes. At this point, up to 100 nM insulin was added and cells were incubated for another 20 min. Subsequently, the plates were put in a Seahorse XF24 extracellular flux analyser (controlled at 37 °C) for a 10-min calibration and 3 measurement cycles to record basal cellular respiration. Glucose (2 mM), oligomycin (5 μg/mL), FCCP (2 μM and 20 μM for human and L6 cells, respectively), and a mixture of rotenone (1 μM) plus antimycin A (2 μM) were then added sequentially to, respectively, avoid possible substrate limitation, inhibit the ATP synthase, uncouple oxidative phosphorylation, and gauge non-mitochondrial respiration. Cellular respiratory control was calculated as the ratio of FCCP-stimulated and oligomycin-inhibited mitochondrial respiratory rates [20], and coupling efficiency of oxidative phosphorylation was defined as the oligomycin-sensitive proportion of mitochondrial respiration [20].
2.4. Anaerobic glycolysis
Lactate produced during anaerobic glycolysis was approximated by measuring extracellular acidification rates in a Seahorse XF24 extracellular flux analyser [14]. Cells were treated as described in Section 2.2, but were washed, subjected to insulin, and assayed in KRPH (L6 cells) or DMEM (human cells) containing 2 instead of 10 mM Hepes.
2.5. Glucose uptake
2-Deoxyglucose (2DG) uptake was measured according to [21]. In essence, L6 or human myoblasts were seeded at 2 × 104 cells/well and exposed to palmitate on 96-well plates. Following a 10-h nutrient restriction (see Section 2.2) cells were washed 3 × with KRPH (20 mM Hepes, pH 7.4), and incubated in this medium for 20 min at 37 °C with or without 100 nM human insulin. Cells were then incubated for another 20 min in the presence of 2DG, which was added either up to 1 μg/well to establish the dose-dependency of insulin-sensitive glucose uptake (Fig. 1), or at a saturating 30 mg/well in the palmitate exposure experiment (Fig. 6A). Subsequently, cells were washed twice with KRPH, and then lysed via agitated incubation in the presence of 0.1 N NaOH, for 10 min at 65 °C and a further 50–60 min at 85 °C. Lysates were solubilised with 0.1 N HCl, and then incubated, whilst shaking, for 10 min at 37 °C in 200 mM triethanolamine, pH 8. Solubilised lysates were transferred to a 96-well plate containing assay medium with 50 mM triethanolamine (pH 8), 50 mM KCl, 15 U/mL glucose-6-phosphate dehydrogenase, 0.2 U/mL diphorase, 0.1 mM NADP, 0.02% (w/v) BSA, and 2 μM resazurin (Invitrogen). This reaction mixture was incubated at 37 °C for 50 min to allow 2DG-dependent reduction of resazurin [21] to the fluorescent resorufin (λex/em = 540/590 nm), which was detected using a BMG LABTECH PHERAstar FS plate reader.
Fig. 1.
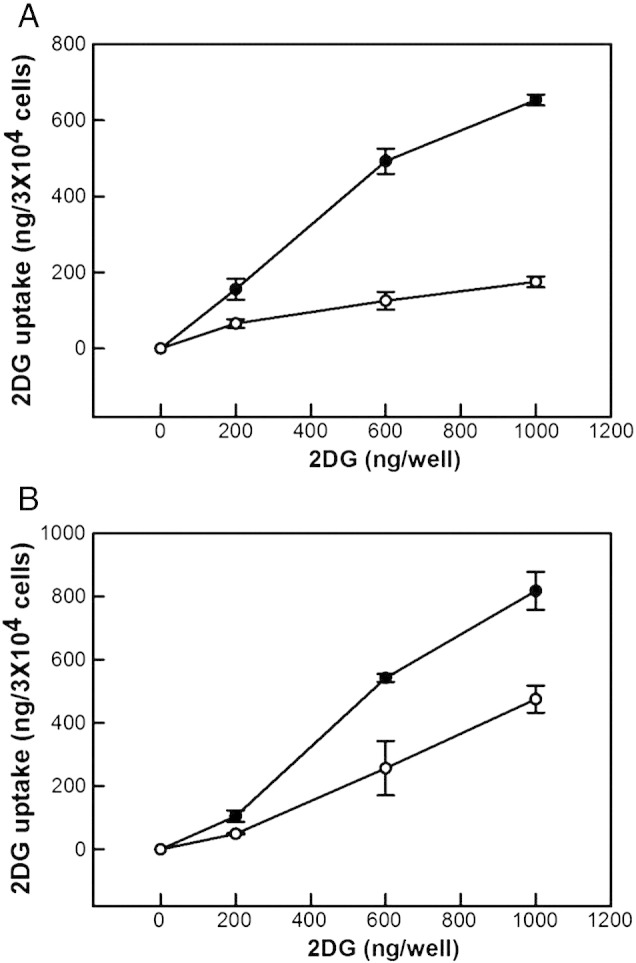
Glucose uptake. 2-Deoxyglucose (2DG) accumulation was measured in L6 and human myoblasts (panels A and B, respectively) without (white symbols) or with 100 nM insulin (black symbols) as fully described in Section 2.5. The data shown are means ± SEM of 3 independent experiments. The differences between insulin-exposed and control cells are statistically significant (P < 0.05) at applied 2DG levels of 600 and 1000 ng/well.
Fig. 6.

Palmitate annuls insulin sensitivity of mitochondrial function. L6 myoblasts were exposed to BSA-conjugated palmitate (PA) or BSA as described in Section 2.2. 2-Deoxyglucose (2DG) uptake (panel A), cell respiratory control (panel B), and coupling efficiency (panel C) were measured following incubation without (white bars) or with 100 nM insulin (black bars). The data shown are means ± SEM of 5 independent experiments. The asterisks indicate statistical significance between insulin-exposed and control cells (P < 0.05).
2.6. Statistics
The significance of mean differences was tested by ANOVA – applying Fisher's LSD multiple comparison post-hoc analysis – using SPSS v17 (IBM) or Stat Graphics Plus v5.1 (Statistical Graphics Corporation) software.
3. Results
3.1. Insulin improves mitochondrial function
Wishing to establish the effect of insulin on mitochondrial function of skeletal muscle cells, we first checked the insulin sensitivity of our myoblasts. The glucose uptake data shown in Fig. 1 confirm that cultured rat myoblasts (L6) and primary human myoblasts both respond well to insulin: following a 20-min exposure, dose-dependent accumulation of 2DG is more than tripled by 100 nM insulin in L6 cells and almost doubled in human cells. The comparatively modest insulin response of human myoblasts is due to a relatively high basal glucose uptake (Fig. 1B). Next, we measured mitochondrial oxygen uptake in real-time using non-invasive extracellular flux technology [14]. The typical respiratory traces presented in Fig. 2 demonstrate that basal cellular respiration by nutrient-restricted (cf. Section 2.2) L6 myoblasts and L6 myotubes is not limited by glucose (Fig. 2A and B, respectively), as its addition during the assay does not notably change respiration rates. As expected, both myoblast and myotube respiration – in rat and human systems alike – decreases significantly after inhibition of the ATP synthase with oligomycin (Fig. 2). Also conforming to textbook knowledge [22], oligomycin-inhibited respiration in all our muscle models is stimulated substantially by FCCP, and the resultant maximum respiratory activity, which is uncoupled from ATP synthesis, is annulled largely by the electron transfer inhibitors rotenone and antimycin A.
Fig. 2.

Real-time non-invasive measurement of cell respiration. L6 myoblasts (panel A), L6 myotubes (panel B), human myoblasts (panel C) and human myotubes (panel D) were incubated in the absence (white symbols) or the presence of 10 nM (grey symbols) or 100 nM (black symbols) insulin for 20 min, and then transferred to a Seahorse analyser for respiratory analysis (see section 2.3 for detail). Glucose (GLU), oligomycin (OLI), FCCP and a mixture of rotenone and antimycin A (R/A) were added at times indicated by the dotted lines. The shown traces are representative for the respective skeletal muscle cells, and each data point represents an average respiratory rate ± SEM of 7 wells. All respiratory rates (Jo) were normalised to the final measurement in the presence of oligomycin.
As illustrated by the normalised respiratory traces in Fig. 2, the extent to which FCCP stimulates oligomycin-inhibited respiration is increased markedly in skeletal muscle cells that had been exposed to 10 or 100 nM insulin for 20 min prior to the experiment. This interesting immediate effect of insulin on the cell respiratory control ratio [20] is quantified in Fig. 3A, revealing that 100 nM insulin increases this control ratio in L6 cells from 10 or 9 (myoblasts and myotubes, respectively) to approximately 12. Cell respiratory control in human cells is lower than in their rat counterparts, but mirroring the observations in L6 cells, the control ratios in both human myoblasts and human myotubes (just below 4 and 6, respectively) are increased significantly by insulin (Fig. 3A). Similarly, the coupling efficiency of oxidative phosphorylation, which is another internally normalised bioenergetic parameter [20] that may be derived from data as shown in Fig. 2, is increased by insulin (Fig. 3B). Coupling efficiency, i.e., the proportion of respiratory activity that is used to make ATP, is roughly 80% in L6 myoblasts and myotubes. Exposure to insulin increases the coupling efficiency moderately, but significantly and instantly, to approximately 85% (Fig. 3B). Coupling efficiency in human myoblasts and human myotubes (70% and 67% on average, respectively) is lower than in their rat equivalents, but the stimulating effect of insulin in human systems is comparatively large (Fig. 3B). Together, the data in Fig. 3 demonstrate that insulin improves mitochondrial function in muscle cells by acutely increasing cellular respiratory control and the proportion of basal respiration that is used to make ATP.
Fig. 3.
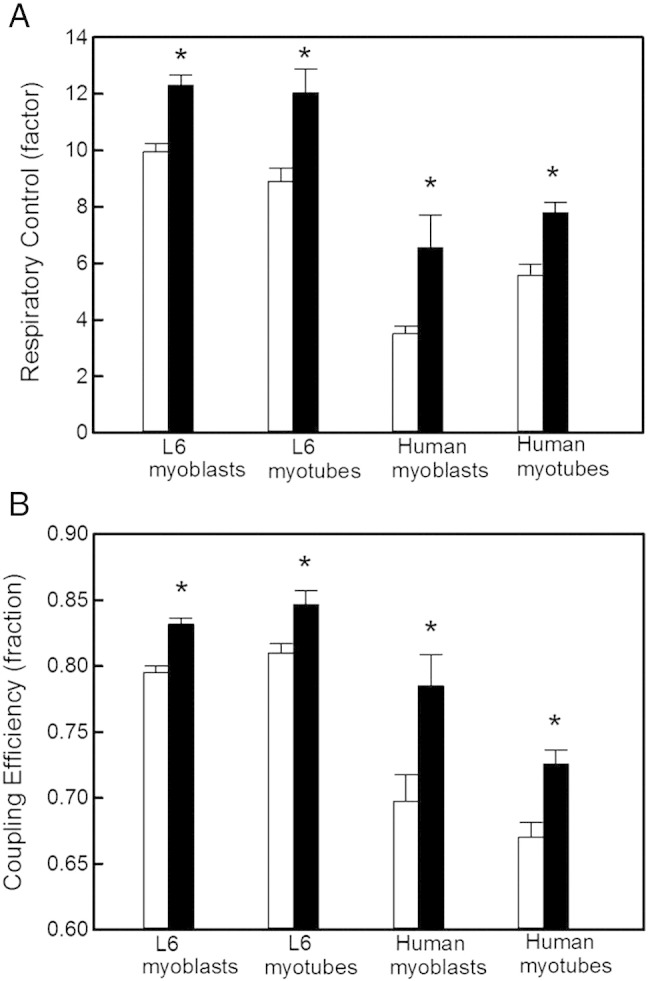
Insulin improves mitochondrial function. The cell respiratory control ratio (panel A) and coupling efficiency of oxidative phosphorylation (panel B) were calculated from respiratory data (exemplified in Fig. 2) as described fully in Section 2.3. As indicated by the asterisks, these internally normalised parameters were higher, in all muscle systems, in the presence of 100 nM insulin (black bars) than in its absence (white bars): P < 0.05 and P < 0.01 for differences between respiratory control ratios and coupling efficiencies, respectively. The data shown are means ± SEM of 3–6 independent experiments.
3.2. Insulin lowers mitochondrial proton leak
Both the cell respiratory control ratio and mitochondrial coupling efficiency are bioenergetic parameters that reflect multiple processes underlying oxidative phosphorylation, including substrate oxidation, proton leak across the mitochondrial inner membrane, and, in case of coupling efficiency, ATP turnover [19,20]. These processes can be quantified individually by inspecting absolute oxygen uptake activities, as is illustrated by Fig. 4, in which respiratory rates exhibited by human muscle cells incubated under different energetic conditions are reported as oxygen consumption per assayed well. Given the relatively small variation in cell number between wells (data not shown), and assuming that insulin does not instantly affect cell density, this normalisation allows insulin effects to be estimated within an experimental system. Because of density differences, absolute myoblast and myotube activities can not be compared. Notwithstanding this caveat, the effects of insulin on absolute basal respiration (Fig. 4A), respiration coupled to ATP synthesis (Fig. 4B), respiration associated with proton leak (Fig. 4C), and maximum respiration (Fig. 4D), shed light on the mechanism by which insulin increases the cell respiratory control ratio and mitochondrial coupling efficiency in human muscle cells. Insulin attenuates both basal respiratory activity (Fig. 4A) and, more pronouncedly, mitochondrial proton leak (Fig. 4C) in myotubes – similar, but statistically insignificant, effects are apparent in myoblasts. Insulin does not significantly affect the FCCP-uncoupled (Fig. 4D) or oligomycin-sensitive respiration (Fig. 4B). As proton leak is a major determinant of both cell respiratory control and coupling efficiency [20], its decreased activity upon acute insulin exposure most likely accounts for the insulin effects reported in Fig. 3.
Fig. 4.

Effect of insulin on absolute oxygen consumption. The basal mitochondrial respiratory rate (panel A), the oligomycin-sensitive rate (ATP turnover, panel B), the oligomycin-resistant rate (proton leak, panel C), and the FCCP-stimulated rate (maximum respiration, panel D) were expressed as total oxygen consumed per minute per XF24 well, and were calculated for human muscle cells incubated without (white bars) or with 100 nM insulin (black bars). The data shown are means ± SEM of 3–6 independent experiments. Because of density variation between myoblasts and myotubes, only internal comparisons ± insulin were tested statistically – the asterisks indicate significant differences (P < 0.05).
3.3. Insulin sensitises anaerobic glycolysis to glucose
In addition to oxidative phosphorylation, we measured anaerobic glycolysis by monitoring the extracellular acidification rate (ECAR), which is proportional to glycolytic lactate production. The typical ECAR traces presented in Fig. 5 (panels A–C) reveal distinct, albeit somewhat variable, effects of insulin on the time-resolved response of anaerobic glycolysis to glucose. When normalised to basal ECAR, the immediate response of both L6 and human myoblasts, and of human myotubes, to glucose is a 1.5 to 2-fold rate increase – this glucose response is amplified significantly (achieving 2.5 to 4-fold rate increases) in cells exposed to insulin (Fig. 5D).
Fig. 5.

Insulin enhances glucose sensitivity of anaerobic glycolysis. Typical XF24 extra-cellular acidification (ECAR) traces reflecting the glycolytic behaviour of L6 myoblasts (panel A), human myoblasts (panel B), and human myotubes (panel C). Glucose (GLU) was added at times indicated by the dotted lines to cells incubated without (white symbols) and with 100 nM insulin (black symbols). The shown traces are representative for the respective skeletal muscle cells, and each data point represents an average ECAR ± SEM of 4–5 wells. All rates were normalised to the final basal ECAR measurement. The ECAR response was quantified by normalising the average value of the (first) 4 readings after glucose addition to the final reading before glucose addition (panel D). The responses shown are means ± SEM of 3 independent experiments. As indicated by the asterisks, all differences between cells without (white bars) and with 100 nM insulin (black bars) were statistically significant (P < 0.05).
3.4. Palmitate annuls mitochondrial insulin effects
The results presented in Figs. 2–5 demonstrate that insulin provokes significant and relatively acute bioenergetic responses in muscle cells. To confirm that insulin-resistant cells do not exhibit such responses, we exposed L6 myoblasts to palmitate (see Section 2.2). The glucose uptake experiments presented in Fig. 6A confirm that serum-deprived control cells, which were exposed to BSA only, still accumulate 2DG in an insulin-sensitive manner, and that the insulin-mediated 2DG uptake is fully blunted in palmitate-exposed cells. Similar bioenergetic experiments reveal that such palmitate-induced insulin resistance is indeed reflected by mitochondrial dysfunction: the insulin stimulation of both the cell respiratory control ratio (Fig. 6B) and the mitochondrial coupling efficiency (Fig. 6C) observed in BSA control cells, are fully annulled, perhaps even reversed, in palmitate-exposed cells.
4. Discussion
The results reported in this paper reveal that insulin improves mitochondrial function of both rat and human skeletal muscle cells. By decreasing the proton leak across the mitochondrial inner membrane (Fig. 4), a short insulin exposure leads to immediate increases in coupling efficiency of oxidative phosphorylation and cell respiratory control (Fig. 3). These findings highlight the relative strength of real-time bioenergetic measurements in cells as they shed novel mechanistic light on the emerging role of insulin in oxidative nutrient catabolism. Moreover, the instant insulin effects on both mitochondrial function (Figs. 2–4) and anaerobic glycolysis (Fig. 5) suggest a new way of detecting cellular insulin sensitivity. The mechanism by which insulin decreases mitochondrial proton leak is unclear at present.
4.1. Mechanistic implications
In addition to stimulating glycogenesis and glucose storage [7], there is growing awareness that insulin enhances oxidative glucose catabolism. Increased capacity of oxidative phosphorylation [9] has been suggested to explain insulin-stimulated ATP synthesis in human muscle [9–11]. Indeed, this notion is supported indirectly by insulin-induced increases in the activities of mitochondrial enzymes such as pyruvate dehydrogenase [23], citrate synthase [9], and cytochrome oxidase [9], and by insulin-stimulated expression of genes encoding for mitochondrial respiratory complex components [9]. Moreover, insulin has been reported to stimulate both mitochondrial protein synthesis [9,12] and glucose oxidation [13]. Our results do not provide evidence for an insulin-mediated increase in oxidative capacity as maximum respiration is not acutely affected by insulin (Fig. 4D). Similarly, basal respiration is not increased by insulin either, but is in fact decreased (Fig. 4A). A pronounced decrease of proton leak (Fig. 4C) is largely responsible for this lowered basal respiration. The discrepancy between our data and published findings likely relates to different exposure times: evidence for increased oxidative capacity has been gathered after 4–8 h persistent insulin exposure, whereas our improved coupling efficiency manifests itself after a mere 20 min. Increased mitochondrial coupling efficiency is clearly consistent with insulin-stimulated ATP synthesis [9–11], and it may well be that such stimulation results from an immediate improvement of the efficiency of ATP production and a somewhat delayed increase in the production capacity. Insulin-stimulated ATP synthesis is decreased in muscle from type 2 diabetic patients [9], their insulin-resistant offspring [10], or in muscle exposed to lipid excess [11]. Consistently, palmitate exposure fully annuls the mitochondrial responses to insulin in L6 myoblasts (Fig. 6B and C).
4.2. Novel insulin sensitivity assay
The cell respiratory control ratio (Fig. 3A), coupling efficiency of oxidative phosphorylation (Fig. 3B), and the glucose sensitivity of anaerobic glycolysis (Fig. 5D) are parameters that may be measured simultaneously with a Seahorse XF extracellular flux analyser, either on 24-well or 96-well platforms. The hitherto unknown insulin sensitivity of these bioenergetic parameters suggests a novel, rapid and sensitive way of detecting insulin responsiveness of cells. Indeed, we show that the mitochondrial responses to insulin are annulled (Fig. 6B and C) in cells exposed to lipotoxic conditions that lead to insulin resistance (Fig. 6A). The assay reported here complements traditional glucose uptake assays [24] and offers some specific advantages. For example, the cellular bioenergetic method does not rely on the use of radioisotopes and comprises many fewer experimental steps, and is thus likely subject to less variation, than the non-radioactive glucose uptake procedure [21] summarised in Section 2.5. Moreover, the method is non-invasive and allows measurements in intact, attached cells. Importantly, the obtained bioenergetic parameters (Figs. 3 and 5D) are internally normalised, which renders the assay free from possible well-to-well variation in cell number, and thus greatly improves its sensitivity. Finally, potential application of our assay in screens for insulin-sensitising compounds bears the additional advantage of detecting possible mitochondrial side effects of such drugs early. This information would be invaluable to the pharmaceutical industry given the growing appreciation that it pays to test drug candidates for adverse effects on mitochondria at an early stage of discovery [25].
Acknowledgments
We thank Prof. Andrew Jones and Mr Lee Wiley (Sport & Health Sciences, College of Life & Environmental Sciences, University of Exeter) for the human skeletal muscle biopsy, Prof. Paul Winyard (Exeter University Medical School, UK) for providing licenced facilities to work with human tissue, and Dr Jane Carré (Exeter University Medical School, UK) for help with the human muscle cell isolation. Work in our lab is supported by the Medical Research Council [New Investigator Research Grant G1100165 to CA] and Plymouth University [salary support for RBN].
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Contributor Information
Raid B. Nisr, Email: raid.nisr@plymouth.ac.uk.
Charles Affourtit, Email: charles.affourtit@plymouth.ac.uk.
References
- 1.DeFronzo R.A., Jacot E., Jequier E., Maeder E., Wahren J., Felber J.P. The effect of insulin on the disposal of intravenous glucose: results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes. 1981;30:1000–1007. doi: 10.2337/diab.30.12.1000. [DOI] [PubMed] [Google Scholar]
- 2.Alonso-Magdalena P., Quesada I., Nadal A. Endocrine disruptors in the etiology of type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2011;7:346–353. doi: 10.1038/nrendo.2011.56. [DOI] [PubMed] [Google Scholar]
- 3.Muoio D.M., Newgard C.B. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and β-cell failure in type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008;9:193–205. doi: 10.1038/nrm2327. [DOI] [PubMed] [Google Scholar]
- 4.Lowell B.B., Shulman G.I. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- 5.Goodpaster B.H. Mitochondrial deficiency is associated with insulin resistance. Diabetes. 2013;62:1032–1035. doi: 10.2337/db12-1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holloszy J.O. “Deficiency” of mitochondria in muscle does not cause insulin resistance. Diabetes. 2013;62:1036–1040. doi: 10.2337/db12-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen P., Nimmo H.G., Proud C.G. How does insulin stimulate glycogen synthesis? Biochem. Soc. Symp. 1978:69–95. [PubMed] [Google Scholar]
- 8.Cheng Z., Tseng Y., White M.F. Insulin signaling meets mitochondriain metabolism. Trends Endocrinol. Metab. 2010;21:589–598. doi: 10.1016/j.tem.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stump C.S., Short K.R., Bigelow M.L., Schimke J.M., Nair K.S. Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc. Natl. Acad. Sci. U. S. A. 2003;100:7996–8001. doi: 10.1073/pnas.1332551100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petersen K.F., Dufour S., Shulman G.I. Decreased insulin-stimulated ATP synthesis and phosphate transport in muscle of insulin-resistant offspring of type 2 diabetic parents. PLoS Med. 2005;2:e233. doi: 10.1371/journal.pmed.0020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brehm A., Krssak M., Schmid A.I., Nowotny P., Waldhäusl W., Roden M. Increased lipid availability impairs insulin-stimulated ATP synthesis in human skeletal muscle. Diabetes. 2006;55:136–140. [PubMed] [Google Scholar]
- 12.Boirie Y., Short K.R., Ahlman B., Charlton M., Nair K.S. Tissue-specific regulation of mitochondrial and cytoplasmic protein synthesis rates by insulin. Diabetes. 2001;50:2652–2658. doi: 10.2337/diabetes.50.12.2652. [DOI] [PubMed] [Google Scholar]
- 13.Gaster M., Beck-Nielsen H. The reduced insulin-mediated glucose oxidation in skeletal muscle from type 2 diabetic subjects may be of genetic origin—evidence from cultured myotubes. Biochim. Biophys. Acta. 2004;1690:85–91. doi: 10.1016/j.bbadis.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 14.Wu M., Neilson A., Swift A.L., Moran R., Tamagnine J., Parslow D., Armistead S., Lemire K., Orrell J., Teich J., Chomicz S., Ferrick D.A. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am. J. Physiol. 2006;292:C125–C136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
- 15.Thompson D.B., Pratley R., Ossowski V. Human primary myoblast cell cultures from non-diabetic insulin resistant subjects retain defects in insulin action. J. Clin. Invest. 1996;98:2346–2350. doi: 10.1172/JCI119046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Springer M.L., Rando T.A., Blau H.M. Isolation and growth of human myoblasts, Adapted from: Gene delivery to muscle, current protocols in human genetics. 2002. http://onlinelibrary.wiley.com/doi/10.1002/0471142905.hg1304s31/full [DOI] [PubMed]
- 17.Huber A.H., Kampf J.P., Kwan T., Zhu B., Kleinfeld A.M. Fatty acid-specific fluorescent probes and their use in resolving mixtures of unbound free fatty acids in equilibrium with albumin. Biochemistry. 2006;45:14263–14274. doi: 10.1021/bi060703e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ching J.K., Rajguru P., Marupudi N., Banerjee S., Fisher J.S. A role for AMPK in increased insulin action after serum starvation. Am. J. Physiol. 2010;299:C1171–C1179. doi: 10.1152/ajpcell.00514.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Affourtit C., Brand M.D. Measuring mitochondrial bioenergetics in INS-1E insulinoma cells. Methods Enzymol. 2009;457:405–424. doi: 10.1016/S0076-6879(09)05023-X. [DOI] [PubMed] [Google Scholar]
- 20.Brand M.D., Nicholls D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamamoto N., Kawasaki K., Kawabata K., Ashida H. An enzymatic fluorimetric assay to quantitate 2-deoxyglucose and 2-deoxyglucose-6-phosphate for in vitro and in vivo use. Anal. Biochem. 2010;404:238–240. doi: 10.1016/j.ab.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 22.Nicholls D.G., Ferguson S. Academic Press; London: 2013. Bioenergetics 4. [Google Scholar]
- 23.Henry R.R., Abrams L., Nikoulina S., Ciaraldi T.P. Insulin action and glucose metabolism in nondiabetic control and NIDDM subjects: comparison using human skeletal muscle cell cultures. Diabetes. 1995;44:936–946. doi: 10.2337/diab.44.8.936. [DOI] [PubMed] [Google Scholar]
- 24.Sasson S., Oron R., Cerasi E. Enzymatic assay of 2-deoxyglucose 6-phosphate for assessing hexose uptake rates in cultured cells. Anal. Biochem. 1993;215:309–311. doi: 10.1006/abio.1993.1594. [DOI] [PubMed] [Google Scholar]
- 25.Will Y., Dykens J.A., Nadanaciva S., Hirakawa B., Jamieson J., Marroquin L.D., Hynes J., Patyna S., Jessen B.A. Effect of the multitargeted tyrosine kinase inhibitors imatinib, dasatinib, sunitinib, and sorafenib on mitochondrial function in isolated rat heart mitochondria and H9c2 cells. Toxicol. Sci. 2008;106:153–161. doi: 10.1093/toxsci/kfn157. [DOI] [PubMed] [Google Scholar]