

Abstract
The luminal SR protein CSQ2 contains phosphate on roughly half of the serines found in its C-terminus. The sequence around phosphorylation sites in CSQ2 suggest that the in vivo kinase is protein kinase CK2, even though this enzyme is thought to be present only in the cytoplasm and nucleus. To test whether CSQ2 kinase is CK2, we combined approaches that reduced CK2 activity and CSQ2 phosphorylation in intact cells. Tetrabromocinnamic acid, a specific inhibitor of CK2, inhibited both the CSQ2 kinase and CK2 in parallel across a range of concentrations. In intact primary adult rat cardiomyocytes and COS cells, 24 h of drug treatment reduced phosphorylation of overexpressed CSQ2 by 75%. Down-regulation of CK2α subunits in COS cells using siRNA, produced a 90% decrease in CK2α protein levels, and CK2-silenced COS cells exhibited a twofold reduction in CSQ2 kinase activity. Phosphorylation of CSQ2 overexpressed in CK2-silenced cells was also reduced by a factor of two. These data suggested that CSQ2 in intact cells is phosphorylated by CK2, a cytosolic kinase. When phosphorylation site mutants were analyzed in COS cells, the characteristic rough endoplasmic reticulum form of the CSQ2 glycan (GlcNAc2Man9,8) underwent phosphorylation site dependent processing such that CSQ2-nonPP (Ser to Ala mutant) and CSQ2-mimPP (Ser to Glu mutant) produced apparent lower and greater levels of ER retention, respectively. Taken together, these data suggest CK2 can phosphorylate CSQ2 co-translationally at biosynthetic sites in rough ER, a process that may result in changes in its subsequent trafficking through the secretory pathway.
Keywords: Calsequestrin, CK2, Phosphorylation, Sarcoplasmic reticulum, Kinase inhibitors, siRNA, Glycosylation, Rough ER
Introduction
Calsequestrin (CSQ2) is a resident luminal protein of the SR [1–6] where an involvement in junctional Ca2+ release is assumed to be its primary function [7, 8]. The fast-twitch skeletal muscle (CSQ1) and cardiac (CSQ2) isoforms come from independent genes but share a relatively conserved sequence [4, 9]. Mammalian CSQ2 proteins have an 18–25 residue C-terminal extension that contains two or three serines in an otherwise highly acidic sequence, and is not present in CSQ1 [4, 10]. These C-terminal serines are prolific in vitro substrates for an endogenous protein kinase, making CSQ2 the most heavily phosphorylated protein in heart cell homogenates, in the absence of exogenous kinase activators such as cyclic AMP [11]. It is similarly phosphorylated in all mammalian cells following its overexpression [11–13].
CSQ2 phosphorylation state and its Asn316 (N)-linked glycan structure can be simultaneously determined by mass spectrometric analysis of purified
CSQ2 protein [12, 14]. Pathways of CSQ2 trafficking are revealed in the CSQ2 glycan structures, which always comprised only high-mannose structures Man3–9GlcNAc2. The exact distribution of CSQ2 glycoforms depends upon the cell type used as the source, and extent of mannose trimming in that cell type [11, 12, 14]. In cardiomyocytes, CSQ2 traffics directly from juxtanuclear rough ER to successive junctional SR puncta where it is retained by its polymerization [15]. Mannose trimming occurs to a great extent in cardiomyocytes (Man3–5GlcNAc2), which may reflect a relatively slow anterograde trafficking or slow protein breakdown. In nonmuscle cells, the majority of CSQ2 molecules undergo no mannose trimming (Man9GlcNAc2), while a single mannose is trimmed from some CSQ2 molecules (Man9GlcNAc2) [11–13].
Mass spectrometry of intact purified CSQ2 also reveals significant insights into its phosphorylation. In heart tissue, newly synthesized molecules (Man9,8) exist in a completely phosphorylated state [11]. As CSQ2 undergoes mannose trimming during trafficking to junctional SR, the number of phosphorylated molecules dramatically decreases, such that the most highly trimmed glycans contain no phosphate [11]. When analyzed in cultured nonmuscle cells, it is also evident that anterograde trafficking of CSQ2 is accompanied by its dephosphorylation [11–13]. Epitope-tagged forms of CSQ2 that escape ER retention, due to decreased polymerization, are also completely dephosphorylated [13].
The putative identity of CSQ2 kinase derives from the fact that its C-terminal phosphorylation sites (Table 1) match closely a consensus sequence (S/T-X-X-D/E) for the cytosolic serine/threonine protein kinase CK2 (formerly casein kinase 2) [16, 17]. CSQ2 is also a very efficient in vitro substrate for CK2 [11, 18]. Despite considerable knowledge about the CSQ2 phosphorylation of lumenal SR substrates from in vitro studies, the identity of the CSQ2 kinase has not yet been established. Evidence that supports the CK2 present in broken cell preparations as the prominent phosphorylation activity for lumenal SR substrates such as CSQ2 [11, 18], sarcalumenin [19], histidine-rich Ca2+-binding protein (HCP) [19], GRP94 [20], and caln- exin [21] is very strong. In studies of muscle microsomal vesicles [11, 19], some CK2 activity was found to exhibit the kind of detergent-dependent increases predicted for a lumenal kinase. These studies, however, suffered from the fact that kinase activities in vitro cannot be assumed to act on a given substrate unless its co-localization in intact cells can be unequivocally established. Given this experimental limitation of these studies, and the predicted non-entry of CK2 into ER, these data cannot be considered definitive.
Table 1. CSQ2 phosphoform variants used in this study.
| Name | cDNA | Phosphoform |
|---|---|---|
| CSQ-WT | -EDDDDDDGNNSDEESNDDSDDDD391E-COOH | Phosphorylated on Ser 378,382,386 |
| CSQ-nonPP | . . . . . . . . . A . . .A. . . . A . . . . . . | Phosphorylation deficient |
| CSQ-mimPP | . . . . . . . . . E . . .E. . . . .E . . . . . . | Phosphomimetic |
A paradox, however, results if one assumes that CK2 is the CSQ2 kinase, as CK2 has no signal sequence for gaining access to the ER lumen [22], whereas, CSQ2 is thought to be wholly contained within the SR lumen throughout its lifetime [4, 23]. Mammalian protein kinase CK2, in the holoenzyme form, is composed of two separate catalytic subunits and two identical regulatory subunits [24]. The catalytic subunits, CK2α and CK2α′, share approximately 90% homology and are the product of different genes [25–27]. Both catalytic subunits are capable of phosphorylating substrates independent of holoenzyme formation, where binding can occur to different organellar sites within the cytosolic space [24, 28, 29]. CK2 has been shown to promote cell proliferation [30], and has recently become a chemotherapeutic target in order to promote tumor regression [31] leading to the advent of a series of new and highly specific inhibitors [32].
In this study, we have combined cellular and molecular approaches to investigate the actions of protein kinase CK2 on the cardiac-specific C-terminus of CSQ2. Reduced CSQ2 phosphorylation produced by reversible CK2-specific inhibitors, loss of CK2 protein by siRNA, and loss of phosphorylation by sequence mutations support the conclusion that CK2 is the CSQ2 kinase in vivo, and support a hypothesis that phosphorylation of CSQ2 occurs co-translocationally in rough ER to affect its subsequent anterograde trafficking.
Materials and methods
Antibodies and commercial proteins
Monoclonal antibodies specific to CSQ2 were the gift of Dr. Larry Jones, Indiana University School of Medicine. Mouse monoclonal antibody raised to human protein kinase CK2α catalytic subunit (CSNK2A1) and rabbit polyclonal antibody raised to human protein kinase CK2α′ catalytic subunit (CSNK2A2) were purchased from Abcam (ab70774 and ab10474). Human CSNK2A1 (His-tagged CK2α) and CSNK2A2 (GST-tagged CK2α′) recombinant proteins were purchased from Invitrogen (PV3248 and PV3624).
Cell culture
COS-7 African green monkey kidney (COS) and human embryonic kidney 293 cells (HEK-293), purchased from American Type Culture Collection, were cultured in Dulbecco's Modified Eagle's Medium (DMEM) containing 25 mM HEPES, pH 7.4, 10% fetal bovine serum, 100 units/ml penicillin G, 0.1 mg/ml streptomycin and 0.25 μg/ml amphotericin B, at 37°C with 5% CO2.
Cardiomyocyte preparation
Rat ventricular cells were enzymatically dissociated using a Langendorff perfusion apparatus, as previously described [15]. Heart cells were cultured on laminin-coated dishes in modified Medium 199 containing 2% bovine serum.
CSQ2 plasmid and adenoviral constructs
CSQ2 constructs and viral inserts were generated from wild-type canine cardiac CSQ2 cDNA [4]. Creation and purification of adenoviruses for wild-type CSQ2 (CSQ-WT, Ad.CSQ) and the nonphosphorylatable mutant Ad.nonPP (Ser 378,382,386 Ala) were previously described [12]. The phosphorylation mimic CSQ2 mutant Ad.mimPP was produced in an identical manner to that of Ad.nonPP except glutamates were used to replace the C-terminal serines (Ser 378,382,386 Glu). Briefly, the mutating reverse primer (67-mer) contained three single base changes necessary for the Ser to Glu conversion. PCR products were cloned into pBluescript (Stratagene), sequenced by the dideoxy method [33], and then subcloned into the pShuttle plasmid for virus construction (AdEasy System, American Type Culture Collection).
Adenovirus-mediated overexpression
Cells were treated with recombinant adenoviruses at a multiplicity of infection (MOI, pfu/cell) of 50 or 100 for COS and heart cells [12]. Purified viruses were added to cultured cells at the time of plating and left on throughout the overexpression period.
Purification of overexpressed CSQ2
Purification of CSQ2 from COS and cardiomyocyte cultures was carried out 2 days post-infection, as previously described [12] but with minor modifications. Cell pellets were resuspended at roughly 1 mg/ml in extraction buffer containing 10 mM Tris, pH 8.0, 250 mM NaCl, 1% Triton X-100 and 1 mM EGTA. Extracts were centrifuged at 25,000×g for 30 min, bound to 40 μl DEAE-Sephacel (Amersham biosciences), washed, then eluted with 100 μl extraction buffer containing 500 mM NaCl. SDS-PAGE analysis was carried out according to Laemmli [34]. Protein concentrations were determined according to Lowry et al. [35]. Under these conditions, CSQ2 was the major protein in the sample as analyzed by SDS-PAGE and coomassie blue staining.
Preparation of native CSQ2 kinase sources
Freshly isolated adult rat cardiomyocytes and cultured COS cells were pelleted and washed in PBS. The pellets were resuspended in low ionic strength media containing 5 mM MOPS, pH 8.0, 1 mM EGTA and protease inhibition cocktail (Sigma P8340). Suspensions were incubated on ice for 10 min and then passed through a 255/8 ga. needle and glass/teflon homogenizer ten times each. NaCl was added to the resulting lysates (homogenates) to a concentration of 100 mM. Detergent mixtures were created by adding Triton X-100 to the homogenates to a final concentration of 1%. Identical volumes (0.5 ml) of homogenates, detergent-free and detergent extraction mixtures were subjected to high speed centrifugation (30,000×gmax for 15 min) in order to obtain homogenates and extracts.
In vitro phosphorylation of purified CSQ2
The standard CSQ2 phosphorylation reaction mixture for “back phosphorylation” consisted of 20 mM MOPS, pH 8.0, 150 mM NaCl, 0.5 mM EGTA, 10 mM MgCl2, and contained 20 μM [γ-32P]ATP (3000 cpm/pmol), 2 μg purified CSQ2 substrate, and a source of CSQ2 kinase, brought to a total volume of 50 μl with distilled water. As a CSQ2 kinase source, either 2 μg of COS cell or cardiomyocyte homogenate, or its extract, or 0.1–0.4 μg purified human CK2α or CK2α′ kinase, was added as indicated. Quantification of phosphate incorporation was determined by SDS-PAGE, autoradiography, and scintillation counting of radioactive bands excised from dried gels. Measurements of “front phosphorylation”, or endogenous phosphorylation, included a pretreatment with or without 0.01 U acid phosphatase (P0157, Sigma) for 60 min in 10 μl of 30 mM MES buffer, pH 5.8, 0.1 mM EGTA, as previously described [18], then dilution into phosphorylation buffer to a final volume of 50 μl. For comparing effects of inhibitors on cellular CSQ2 kinase and commercial CK2, activities were first normalized based upon activity against 2 μg of CSQ2. CK2α isoforms were normalized based upon manufacturer's stated activities for the peptide substrate RRRDDDSDDD.
Inhibition of CSQ2 kinase using specific inhibitors of protein kinase CK2
The CK2 specific inhibitors tetrabromocinnamic acid (TBCA, Calbiochem), 2-dimethylamino-4,5,6,7-tetra-bromo-1H-benzimidazole (DMAT, Calbiochem), and 4,5,6,7-tetrabromobenzotriazole (TBB, Sigma) were dissolved in DMSO then diluted into phosphorylation reactions to varying micromolar concentrations. To inhibit CSQ2 phosphorylation in intact cells, COS or heart cells were plated in 6-well dishes with Ad.CSQ2 for 24 h. TBCA (100 μM) or DMSO was then added for a total incubation time of 48, then cells were harvested for analysis.
Mass spectrometry
For mass spectrometry of CSQ-WT, CSQ-nonPP, and CSQ-mimPP, recombinant proteins were overexpressed for 48 h in HEK cells following the addition of respective adenoviruses. CSQ2 was purified and prepared for electrospray ionization mass spectrometry as previously described [13].
RNAi
HEK and COS cells were grown in 100 mm diameter culture dishes for 24 h with or without Ad.CSQ2 or Ad.nPP, then transfected using DharmaFECT 1 transfection Reagent (Thermo Scientific T-2001-01) for 48 h with 100 nM Dicer-Substrate RNAi (Integrated DNA Technologies) designed to silence either CSNK2A1 (IDT #HSC.RNAI.N177559.10.3), CSNK2A2 (IDT #HSC.RNAI.N001896.10.2), or a combination of both. Negative controls included no siRNA or DS NC1 universal DsiRNA negative control (Integrated DNA Technologies). Detergent extracts were created for use with standard back and front phosphorylation assays described above, after normalization by protein assay.
mRNA analysis
RNA extraction from COS cells overexpressing Ad.CSQ was performed using RNeasy Fibrous Tissue Mini Kit (Qiagen) according to manufacturer's protocol. RNA concentrations were determined by spectrophotometer at 260 nm. One microgram of DNAase-treated total RNA was reverse transcribed using a high-capacity cDNA reverse transcription kit (Applied Biosystems). Quantitative real-time PCR (qPCR) with fast SYBR green (Applied biosystems step one with version 2.0 software) was performed using CSQ2 primers designed to span one intron. Relative quantitation was then used to evaluate mRNA levels compared to GAPDH internal control. Canine CSQ2 forward primer: 5′-CCA AGC TTG CCA AGA AGC TGG GT-3′, reverse primer: 5′-ACG TCA GCT GCA AAC TCG CCA-3′.
Results
Characterization of CSQ2 phosphorylation in cellular extracts
Native and overexpressed canine CSQ2 is phosphorylated in heart or nonmuscle cells on the same sites within the C-terminus, and the reactions are thought to occur by a similar or identical CSQ2 kinase [11, 12]. We analyzed CSQ2 kinase activity from both primary rat cardiomyocytes and COS cells. Both cell types were homogenized in the presence of Triton X-100 detergent to allow us to assay CSQ2 kinase using purified canine CSQ2 in vitro. Determinations of reaction linearity as a function of protein (kinase source) were carried out with varying amounts of homogenate protein. In contrast to an expected linear relationship between protein and kinase activity, increasing protein concentration actually exhibited an inverse relationship with CSQ2 phosphorylation (Fig. 1a, b); that is, increasing homogenate protein produced decreasing levels of [32P]-phosphate incorporation. To address this extreme nonlinearity, we tested whether linearity could be better achieved using either a mild detergent extract or nondetergent extraction to separate the CSQ2 kinase from possible interfering phosphatases or ATPases that might account for this effect. We found that linearity was partially restored with extractions carried out in detergent, and more fully restored if detergents were removed (Fig. 1c). Whether partial restoration of linearity was due to removal of CSQ2 phosphatase or cellular ATPases was not further investigated. From these determinations, we selected 2 μg of detergent extract to measure CSQ2 kinase activities. The efficiency of CSQ2 kinase extraction based upon measures of its activity could not be determined in these experiments because of this nonlinearity. The efficiency of protein kinase CK2 extraction, however, could be determined by immunoblot, and was found to be about 75 and 50% for detergent and detergent-free extracts, respectively (Fig. 1d).
Fig. 1.
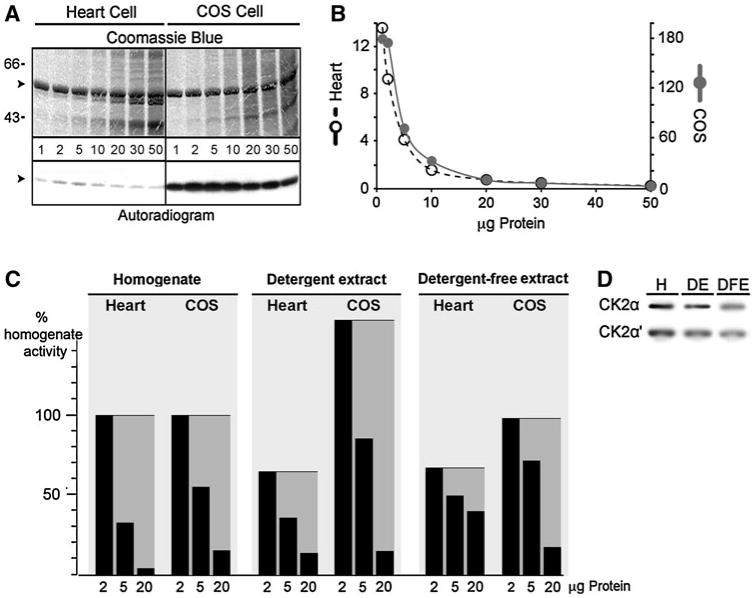
Effects of cellular protein on CSQ2 kinase activity. a Increasing concentrations, 1, 2, 5, 10, 20, 30 or 50 μg of rat heart cell detergent extract or COS cell homogenate was used to phosphorylate 2 μg of purified native canine CSQ2 (arrowheads). Reaction mixtures were analyzed by SDS-PAGE and [32P]autoradiography. b Radioactive incorporation was determined, and kinase activity is shown on the y-axis. CSQ2 kinase activities are shown in units of mmol Pi incorporation/mol CSQ2/μg protein. c CSQ2 kinase activity was measured in 2, 5 or 20 μg of homogenate, from a detergent extraction of 2, 5 or 20 μg of homogenate, or from a detergent-free extraction of 2, 5 or 20 μg of homogenate derived from cardiomyocytes or COS cells. Gray shading highlights the levels for each preparation that would constitute linearity of CSQ2 kinase activity. d Immunoblot of CK2α or CK2α′ subunits from 50 μg of COS cell homogenate (H), detergent extract (DE), or detergent-free extract (DFE)
Pharmacological inhibition of CSQ2 phosphorylation in vitro
Owing to our previous in vitro evidence implicating CK2 as CSQ2 kinase [18], we tested the effects of new competitive inhibitors of CK2 [36] on CSQ2 kinase activity in cell extracts. To allow us to compare effects of CK2-specific inhibitors, we first normalized endogenous CSQ2 kinase activities in myocytes, COS cells, and purified CK2 by comparing their relative ability to phosphorylate 2 μg pure CSQ2. Based upon these determinations alone, one would calculate that about 0.07% of cardiomyocyte protein and 1.7% of COS cell protein is CK2, if CK2 and CSQ2 kinase are the same protein and purified CK2α maintains its native activity. A value based upon immunoblotting of pure CK2 and COS cell extracts was closer to 1% of total COS protein, and consistent with a 25-fold lower level in cardiomyocytes (data not shown).
Equal CSQ2 kinase activities from all three sources were then used to test inhibitors in vitro, using 2, 5, 10, 20, 50, or 100 μM of CK2-specific inhibitors TBB [37], DMAT [38] and TBCA [39] (Fig. 2a–c). All three inhibitors were capable of inhibiting endogenous CSQ2 kinase activity in both cardiomyocyte and COS cell extracts, and over the same range concentrations as those required to inhibit commercial CK2 phosphorylation of CSQ2. The most potent of the three inhibitors was TBCA, which inhibited both endogenous CSQ2 kinase as well as commercial CK2 phosphorylation of CSQ2 at an IC50 of about 10 μM. TBCA produced a near identical inhibition curve for all three kinase sources (Fig. 2c). TBB and DMAT were also capable of inhibiting CSQ2 kinase and commercial CK2, but at higher micromolar concentrations (IC50 approximately 50 and 60 μM, respectively). TBCA was chosen for subsequent experiments in cultured cells because it is reportedly the most specific and potent inhibitor of the three [39], and because we found TBCA to be the most soluble in culture media (data not shown).
Fig. 2.
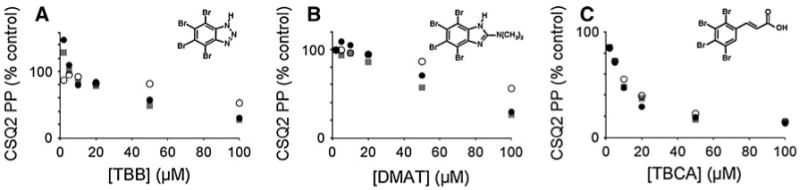
Effects of CK2 inhibition on CSQ2 kinase activity. Measurements of CSQ2 kinase activities were carried out as described in Experimental procedures using equivalent amounts of activity from cardiomyocytes (closed circles), COS cells (closed squares), or pure CK2α (open circles). Reactions included 0, 2, 5, 10, 20, 50 and 100 μM concentrations of the CK2 inhibitors TBB (a), DMAT (b) or TBCA (c) (structures shown in respective insets). Activities are shown (y-axis) as percent of control (untreated)
Pharmacological inhibition of CSQ2 phosphorylation in cultured cells
Our ability to inhibit cardiomyocyte and COS cell CSQ2 kinase in vitro, led us to determine whether TBCA would inhibit phosphorylation of CSQ2 during its overexpression in COS cells. For both cell types, CSQ2 was overexpressed using an adenovirus to achieve high rates of biosynthesis which should, as previously hypothesized, support a higher rate of CSQ2 phosphorylation [11]. We previously showed using 32Pi-labeling in rat cardiomyocytes that endogenous CSQ2 phosphorylation is very low compared to similar amounts of virally overexpressed CSQ2 [11]. In experiments here, cardiomyocytes and COS cells were treated with Ad.CSQ for 24 h before an additional treatment with either 100 μM TBCA or vehicle (DMSO). Levels of endogenous phosphate on CSQ2 were determined from the difference between CSQ2 phosphorylation with and without phosphatase pretreatment. We found that TBCA produced a potent inhibition of phosphate incorporation into CSQ2 in cultured cardiomyocytes and COS cells of 82 and 66%, respectively, compared to vehicle controls (Fig. 3). The reduced effect of TBCA on CSQ2 phosphorylation in COS cells compared to heart cells might reflect the high levels of kinase in COS cells (Fig. 1b).
Fig. 3.
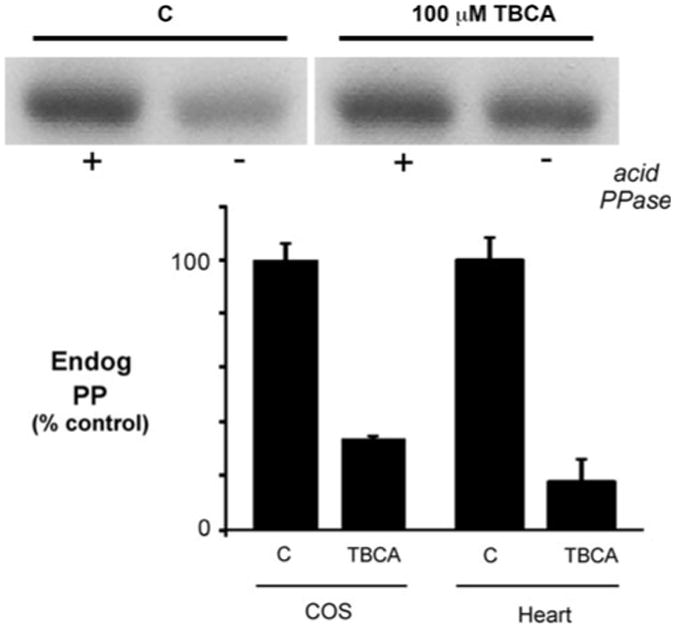
Reduction in endogenous CSQ2 phosphorylation in cells treated with TBCA. COS cells or cardiomyocytes (Heart) were treated with Ad.CSQ for 24 h in culture, then DMSO (C) or 100 μM TBCA was added for 24 h. Cells were extracted in detergent, and levels of phosphate on the overexpressed CSQ2 (Endog PP) were determined for equal volumes of C or TBCA-treated cell extracts using the front phosphorylation assay. Endogenous phosphate on CSQ2 corresponds to the difference in radioactivity between 32P-incorporation after (+) and before (−) acid phosphatase (PPase) treatment. In this assay, levels of unoccupied phosphorylation sites were measured using [32P]ATP and purified CK2 (see materials and methods). A sample autoradiogram is shown for a measurement of endogenous CSQ2-phosphate content following overexpression in COS cells (upper panel), along with summary data for COS cells and cardiomyocytes for C or TBCA-treated cells (lower panel, means ± S.E.M., n = 3)
siRNA knockdown of CK2 catalytic subunits
To corroborate the pharmacological data that supported identification of CSQ2 kinase as CK2, RNAi experiments were performed. Three separate siRNA mixtures that were tested were successful in silencing CK2α and CK2α′ catalytic subunits at 100 nM in either HEK or COS cells, as determined by immunoblotting of cell homogenates (data not shown). CK2α and CK2α′ siRNA with the greatest knockdown efficiencies were used for subsequent experiments in cultured COS cells. Transfection of COS cells with siRNA directed at both CK2α and CK2α′ led to roughly 95% reduction in CK2α and CK2α′ protein levels, as determined by immunoblotting (Fig. 4a, b). Extracts prepared from these siRNA-cells exhibited an approximate twofold reduction in CSQ2 kinase activity (Fig. 4c). This decrease in CSQ2 kinase activity is notable given the very high levels of CK2 present in COS cells (Fig. 1b) and the potential that siRNA does not silence all cellular pools of CK2 equally, such as those with lower rates of turnover. Silencing of individual CK2 isoforms led to apparent compensatory increases in the other CK2α isoform after 48 h, preventing determination of isoform-specific CSQ2 kinase activities (data not shown).
Fig. 4.
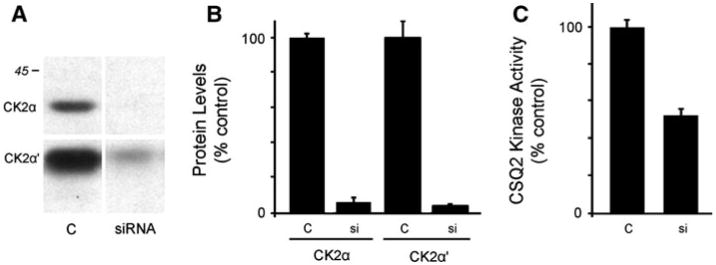
Knockdown of CK2α and CK2α′ using siRNA. COS cells were transfected for 48 h with siRNA against either CK2α or CK2α′, then extracted in detergent to measure CSQ2 kinase activities from detergent. a Untreated cells (C) or siRNA-treated cell samples (50 μg) were analyzed by immunoblotting using specific antibodies to detect CK2α (∼40 kDa) or CK2α′ (∼35 kDa); compare to 45 kDa MW standard. b Shows the average levels of CK2α and CK2α′ knockdown (si) as a percent of untreated (C) cells. c Shows the effects of CK2α plus CK2α′ knockdown on CSQ2 kinase activity levels measured in detergent extracts of treated (si) and untreated (C) COS cells. Plots show means ± S.E.M., n = 3
Measuring effects of CK2-siRNA on endogenous CSQ2 phosphate in COS cells was complicated by increases in cellular protein apparent by SDS-PAGE and protein staining, following 48 h of CK2-siRNA (Fig. 5b). Significant increases in CSQ2 protein levels also occurred with CK2-siRNA treatment (4.6 ± 0.1-fold, mean ± S.E.M., average of three separate experiments). Analysis by qPCR of CSQ2 mRNA showed a roughly twofold increase in response to CK2 siRNA (Fig. 5c). Increased rates of protein synthesis during a 2 h [35S]methionine labeling of cells further supported the overall picture of transcriptional or translational changes in cellular protein, although the mechanism was not further investigated. As an alternative to the use of cell extracts to measure CSQ2 front phosphorylation, therefore, measurements of CSQ2 phosphorylation were carried out on CSQ2 purified from cellular extracts following normalization of amounts by antibody binding (immunoblotting). In accordance with the roughly twofold decrease in CSQ2 kinase activity caused by CK2 siRNA transfection (Fig. 4c), we measured a twofold decrease in the levels of phosphate in purified CSQ2 in COS cells (Fig. 5a). This loss of endogenous phosphate occurred despite a 24 h period of overexpression prior to siRNA treatment. Moreover, even with CK2 protein diminishing over the course of the treatment period, an ongoing phosphorylation of CSQ2 would have occurred due to the exquisite affinity of CSQ2 for CK2 [18].
Fig. 5.
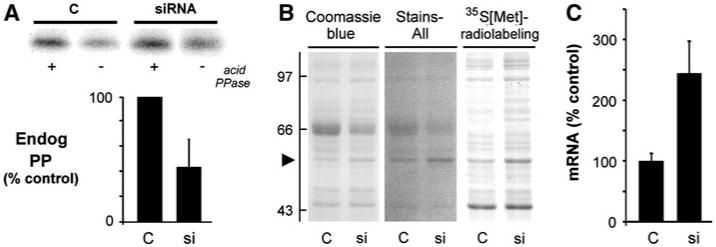
Reduction in endogenous CSQ2 phosphorylation in cells treated with CK2 siRNA. COS cells were treated with Ad.CSQ for 24 h in culture, then transfected with siRNA against CK2α plus CK2α′ for an additional 48 h. a Cells were extracted in detergent, and levels of phosphate on partially purified CSQ2 (Endog PP) were determined. Individual samples of CSQ2 were normalized by immunoblotting. a Sample autoradiogram is shown for purified CSQ2 samples for C and siRNA-treated COS cells (upper panel), along with summary results from analyses of endogenous CSQ2-phosphate content for C and siRNA-treated cells (lower panel, means ± S.E.M., n = 4). b Similar Ad.CSQ-treated COS cells (C or siRNA-treated) were metabolically labeled with 35S[Met] for 2 h, and identical sample volumes were analyzed by SDS-PAGE. The gel was stained with stains-all to highlight CSQ2 as a dark-blue protein band on a pink background (arrowhead), then destained and re-stained with coomassie blue, then dried and placed against film for autoradiography. MW standards (left) are in kDa. The 66 kDa standard is albumin, which also contaminates the sample cultures from the serum in the medium. c Effects of CK2 siRNA on CSQ2 mRNA levels determined by triplicate qPCR measurements (means ± S.E.M., n = 3)
Effects of CSQ2 phosphorylation site mutation on protein trafficking in nonmuscle cells
To determine whether CSQ2 phosphorylation could affect its trafficking, we examined purified wild-type (CSQ-WT) and phosphorylation site mutants (Table 1) by electrospray mass spectrometry to determine the details of its polymorphic structure [12]. CSQ-WT was composed primarily of molecules having glycans with structures of Man9Glc- NAc2 and Man8GlcNAc2 (Man9,8) with only minor amounts or no Man7 (Fig. 6a). In addition, these CSQ-WT molecules contained peaks of phosphorylated protein with a spacing of 81 Da (Fig. 6a, shaded) as previously described [12, 13]. Replacement of phosphorylatable serines with alanines (CSQ-nonPP) led to loss of all CSQ2 phosphorylation along with the appearance of lower-mass forms of the protein that correspond to increased levels of mannose trimming (Fig. 6b). We previously showed in COS cells that mannose trimming beyond Man9,8 occurs for CSQ2 molecules that are not efficiently retained in ER [13].
Fig. 6.
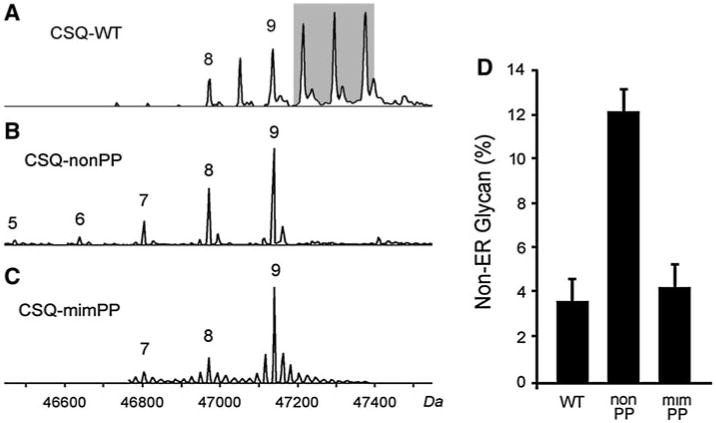
Effects of CSQ2 phosphorylation site sequence on glycan structure in nonmuscle cells. Cultured nonmuscle cells were treated with recombinant adenoviruses encoding either the wild-type CSQ2 sequence (WT), the Ser(378,382,386)Ala triple point mutant (nonPP), or the Ser(378,382,386)Glu mutant (mimPP) for 48 h, then CSQ2 was purified and analyzed by mass spectrometry as described in Experimental Procedures. Sample mass spectra are shown for each purified sequence of CSQ2 (a–c). Numbers 5–9 refer to the number of mannoses in the glycan that correspond to the mass values shown along the bottom axis. Minor levels of peaks that differ in mass by 20–40 Da, are likely to correspond to cation-bound molecules, and varied among spectra. Mannose content for the three CSQ2 structures are shown as aligned, although there were minor differences in mass due to the sequence mutations. Molecular weights (bottom) are those corresponding to the WT sequence. d The percentages of total CSQ2 molecules that exhibited mannosidase trimming to below eight mannoses (non-ER glycan) are shown from multiple (n = 3) experiments (means ± S.E.M.)
To determine whether this effect on CSQ2 trafficking was purely due to the loss of phosphorylatable serines, we carried out the same analysis following replacement of serines with glutamates (CSQ-mimPP). In contrast to CSQ-nonPP, CSQ-mimPP molecules gave structures similar to that of CSQ-WT (Fig. 6c). Three separate overexpression experiments and mass spectrometric analyses were carried out for CSQ-WT, CSQ-nonPP, and CSQ-mimPP samples. CSQ2 glycans containing less than eight mannoses is used as a parameter representing reduced ER retention (Fig. 6d). These data support the view of a CSQ2 phosphorylation occurring co-translationally, and inhibiting CSQ2 from further anterograde movement.
Discussion
CSQ2 kinase as protein kinase CK2
In mammalian heart tissue, CSQ2 exists as a highly phosphorylated protein [11, 12, 18]. Endogenous phosphate is found only on a cluster of two or three serine residues (depending upon the species) that exist in a cardiac-specific C-terminal extension. CSQ2 phosphorylation appears to be very similar in all mammalian cells examined to date, whether one examines native or overexpressed CSQ2 [12, 13]. In intact heart tissue, CSQ2 appears to be phosphorylated early in its biosynthesis in rough ER based upon the fact that newly synthesized molecules (Man 9,8,7) only exist in the fully phosphorylated state, whereas the most highly trimmed glycans (Man 2,3,4) are mostly devoid of phosphate [11]. A critical question that has remained unresolved in this process, however, is whether the CSQ2 kinase is protein kinase CK2. We previously showed that CSQ2 phosphorylation sites adhere closely to the consensus sequence for protein kinase CK2, and CSQ2 is exquisitely sensitive to CK2 in vitro [23]. However, if CK2 is the CSQ2 kinase, it raises the important question of how CSQ2, a luminal ER protein, can be phosphorylated by what is predicted to be a cytosolic enzyme. CK2 has no known mechanism for localizing to the ER/SR lumen where CSQ2 is thought to be wholly contained throughout its lifetime [40]. Only when the CSQ2 C-terminus is in the process of traversing the translocon pore complex could such an association theoretically occur [41]. Such a process suggests that CSQ2 phosphorylation is a co-translational event, and CK2 could be associated with the translocon, similar to the association of the oligosaccharide transferase complex (OST) that brings about co-translational N-linked glycosylation. CK2 has previously been shown to play a role in assembly of the translocon pore complex and signal sequence through phosphorylation of sec63 [42]. Calnexin, another resident ER protein, is a transmembrane protein that undergoes interaction with the ribosome in response to phosphorylation of its cytosolically exposed CK2 site [43]. We have previously reported that a number of resident ER/SR proteins are substrates for CK2 in vitro [40], perhaps suggesting that the reaction mechanism described in this article may play a wider role in ER/SR biology.
CSQ2 kinase activity and its inhibition
Whether CK2 activity was competitively inhibited using TBCA, a highly specific inhibitor of CK2 [39], or bio-specifically down-regulated using RNAi technology, CSQ2 kinase activity was diminished, suggesting that CK2 is responsible for the high phosphate levels found on newly synthesized CSQ2 from heart tissue.
The advent of highly specific pharmacological inhibitors of CK2 has also been driven by the fact that these compounds exhibit antigrowth and anticancer activities [44, 45], reflecting an important role of CK2 in cell growth. The fact that CSQ2 is the major substrate for CK2 in heart homogenates is consistent with a possible role of CSQ2 phosphorylation in cardiac hypertrophy. We have previously shown that CSQ2 phosphorylation is doubled in canine tachycardia-induced heart failure [14].
In contrast to the relatively immediate inhibition of all available CK2 catalytic subunits using TBCA, siRNA inhibition was expected to be more gradual requiring turnover of the existing kinase pool, as suggested by Zhu et al. [45]. Furthermore, we cannot be certain that turnover of all CK2 pools within the cell are the same, whereas ATP binding sites are likely to be identical. As both subunits are capable of efficiently phosphorylating CSQ2 in vitro (data not shown), simultaneous CK2α/α′ knockdown was needed to verify the identity of CK2 as CSQ2 kinase. Analysis by immunoblot usually showed a putative compensatory increase in its subunit partner in single subunit down-regulation experiments that was not further investigated. In spite of these differences in mechanisms, both CK2 inhibition techniques produced qualitative decreases in CSQ2 kinase activity, and each was able to lower phosphate incorporation in overexpressed CSQ2. The finding that 95% inhibition of protein kinase CK2 produced only a 50% decrease in the ability of cell extracts to phosphorylate-purified CSQ2 (Fig. 5a) may reflect both the exquisite sensitivity of CSQ2 for CK2 [18], and the relative high levels of CK2 in COS cells (Fig. 1).
Experimental determinations of CSQ2 kinase and phosphorylation
Measurements of in vitro CSQ2 phosphorylation were surprisingly “nonlinear”; and in fact produced lower levels of CSQ2 phosphorylation using higher levels of protein in the phosphorylation assay. Endogenous ATPases or phosphatases represent potential sources of these countervailing activities. Support for the involvement of transmembrane enzyme activities was the fact that CSQ2 kinase activity was closest to linearity when cell homogenates were extracted without use of detergents (Fig. 1c, d). Understanding the limitations of the assay for CSQ2 kinase activity allowed us to use CK2 inhibition as cellular probes.
Studies aimed at determining effects of CK2 reduction on phosphate levels in CSQ2 were carried out in CSQ2-adenovirus treated cells. As previously observed from 32P-labeling reactions in cultured rat cardiomyocytes [11], the higher rate of CSQ2 biosynthesis obtained through Ad.CSQ2 treatment was needed to observe phosphorylation, presumably because of its elevated rate of biosynthesis and co-translational phosphorylation. CK2 silencing itself, however, produced increases in cellular and CSQ2 protein synthesis that affected steady state protein levels after 48 h in culture, as well as [35S] met incorporation over a 2 h period (Fig. 5b). CSQ2 purification by DEAE chromatography permitted measurements of CSQ2 phosphorylation state using equal amounts of CSQ2 in our standard assay. This approach, while correcting for differences in CSQ2 levels that resulted from CK2-siRNA treatments, assumes that effects of CK2 silencing on levels of phosphate in endogenous CSQ2 were the same proportion of molecules in the two samples.
Protein kinase CK2 regulation of CSQ2 trafficking
Based upon our identification of CSQ2 kinase in intact cells as the cytosolic protein kinase CK2, our previous hypothesis that CSQ2 phosphorylation is a co-translational event [11] can be expanded. In our new model (Fig. 7), we hypothesize that CSQ2 phosphorylation occurs before the C-terminal tail has entered the lumen of the protein pore complex (sec61) or ER lumen [46]. One possible mechanism might be a translocation pause wherein the ribosome separates from the translocon permitting a transient co-localization of CK2 and CSQ2 [46, 47]. If such a process, so proximal in the secretory pathway, were to affect the subsequent trafficking of CSQ2 through the secretory pathway, affects on its mannose trimming would not be unexpected. Intracellular localization of CSQ2 can consistently be predicted from its N-linked glycan structure in a number of mammalian cell types including cardiomyocytes [13, 41]. In this study, comparisons of glycan structures for CSQ-WT, CSQ-nonPP, and CSQ-mimPP showed that only the CSQ-nonPP form exhibits excessive mannose trimming, indicative of increased trafficking away from rough ER. Glutamate, a commonly used phosphomimetic substitution, showed a marked reversal of this effect on glycan structure. These results support the idea that CSQ2 phosphorylation is associated with its biosynthesis and affects retention of CSQ2 in ER.
Fig. 7.
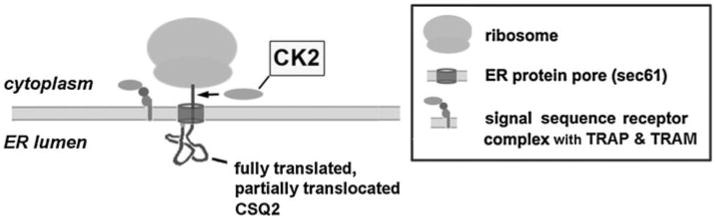
Hypothetical mechanism of cardiac CSQ2 phosphorylation by CK2 in rough ER during translocation across the ER membrane. A mechanism whereby an ER protein such as CSQ2 becomes exposed to cytoplasmic CK2 would require that the ER-bound ribosome permits access to the nascent CSQ2 polypeptide after completion of translation but during its translocation through the translocon. Other known translocon accessory proteins, such as TRAP and TRAM [15] are present in cardiomyocyte rough ER and could function in such translocational regulation [47]
In heart failure, the detailed structure of CSQ2 is dramatically shifted. A peak of newly synthesized CSQ2 (with Man9,8 glycans) accumulates instead of continuing through its normal trafficking cycle [14], indicating that this newly synthesized protein is being retained within rough ER structures. Rough ER, as we have recently demonstrated, is localized to juxtanuclear cisternae in cardiomyocytes [15]. Together with findings reported here, we hypothesize that changes in CK2 activity and/or localization in heart failure increase CSQ2 phosphorylation with consequent ER retention, to favor a perinuclear Ca2+ microdomain shift to facilitate increased transcription or translation in hypertrophy.
In conclusion, findings generated from multiple experimental approaches in this study support an emerging picture of CSQ2 phosphorylation as a co-translational process, and we now hypothesize that it is a co-translocational event. Verification that CK2 is the in vivo CSQ2 kinase implicates the involvement of the rough ER translocon in this biological reaction. The dependence of CSQ2 glycan structure on its state of phosphorylation has consistently revealed important features of this reaction, both here using CSQ2 phosphorylation site mutants as well as for CSQ2 in failing hearts. These studies should guide future investigations by establishing a basic biochemical paradigm for CSQ2 phosphorylation in the mammalian cell.
Acknowledgments
We wish to thank Lauren Dovantzis for expert technical assistance. This study was supported by grant HL62586 from the NIH/NHLBI.
Abbreviations
- CSQ2
Cardiac calsequestrin
- CSQ1
Fast-twitch skeletal muscle calsequestrin
- CSQ-WT
Wild-type cardiac calsequestrin sequence
- SR
Sarcoplasmic reticulum
- ER
Endoplasmic reticulum
- Ad.CSQ
CSQ-WT adenovirus
- Ad.nonPP
Adenovirus for CSQ2 Ser → Ala mutant
- Ad.mimPP
adenovirus for CSQ2 Ser →Glu mutant
- MOI
Multiplicity of infection
- TBCA
Tetrabromocinnamic acid
- DMAT
2-Dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole
- TBB
4,5,6,7-Tetrabromobenzotriazole
References
- 1.MacLennan DH, Wong PT. Isolation of a calcium-sequestering protein from sarcoplasmic reticulum. Proc Natl Acad Sci. 1971;68:1231–1235. doi: 10.1073/pnas.68.6.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ikemoto N, Bhatnager GM, Gergely J. Fractionation of solubilized sarcoplasmic reticulum. Biochem Biophys Res Commun. 1971;44:1510–1517. doi: 10.1016/s0006-291x(71)80257-7. [DOI] [PubMed] [Google Scholar]
- 3.Campbell KP, MacLennan DH, Jorgensen AO, Mintzer MC. Purification and characterization of calsequestrin from canine cardiac sarcoplasmic reticulum and identification of the 53, 000 D glycoprotein. J Biol Chem. 1983;258:1197–1204. [PubMed] [Google Scholar]
- 4.Scott BT, Simmerman HK, Collins JH, Nadal-Ginard B, Jones LR. Complete amino acid sequence of canine cardiac calsequestrin deduced by cDNA cloning. J Biol Chem. 1988;263:8958–8964. [PubMed] [Google Scholar]
- 5.Jones LR, Cala SE. Biochemical evidence for functional heterogeneity of cardiac sarcoplasmic reticulum vesicles. J Biol Chem. 1981;256:11809–11818. [PubMed] [Google Scholar]
- 6.Jorgensen AO, McLeod AG, Campbell KP, Denney GH. Evidence for the presence of calsequestrin in both peripheral and interior regions of sheep purkinje fibers. Circ Res. 1984;55:267–270. doi: 10.1161/01.res.55.2.267. [DOI] [PubMed] [Google Scholar]
- 7.Terentyev D, Viatchenko-Karpinski S, Gyorke I, Volpe P, Williams SC, Gyorke S. Calsequestrin determines the functional size and stability of cardiac intracellular calcium stores: mechanism for hereditary arrhythmia. Proc Natl Acad Sci. 2003;100:11759–11764. doi: 10.1073/pnas.1932318100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knollmann BC. New roles of calsequestrin and triadin in cardiac muscle. J Physiol. 2009;587:3081–3087. doi: 10.1113/jphysiol.2009.172098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fliegel L, Ohnishi M, Carpenter MR, Khanna VK, Reithmeier RA, MacLennan DH. Amino acid sequence of rabbit fast-twitch skeletal muscle calsequestrin deduced from cDNA and peptide sequencing. Proc Natl Acad Sci. 1987;84:1167–1171. doi: 10.1073/pnas.84.5.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yano K, Zarain-Herzberg A. Sarcoplasmic reticulum calsequestrins: structural and functional properties. Mol Cell Biochem. 1994;135:61–70. doi: 10.1007/BF00925961. [DOI] [PubMed] [Google Scholar]
- 11.Ram ML, Kiarash A, Marsh JD, Cala SE. Phosphorylation and dephosphorylation of calsequestrin on CK2-sensitive sites in heart. Mol Cell Biochem. 2004;266:209–217. doi: 10.1023/b:mcbi.0000049164.28580.56. [DOI] [PubMed] [Google Scholar]
- 12.O'Brian JJ, Ram ML, Kiarash A, Cala SE. Mass spectrometry of cardiac calsequestrin characterizes microheterogeneity unique to heart and indicative of complex intracellular transit. J Biol Chem. 2002;277:37154–37160. doi: 10.1074/jbc.M204370200. [DOI] [PubMed] [Google Scholar]
- 13.Houle TD, Ram ML, McMurray WJ, Cala SE. Different endoplasmic reticulum trafficking and processing pathways for calsequestrin (CSQ) and epitope-tagged CSQ. Exp Cell Res. 2006;312:4150–4161. doi: 10.1016/j.yexcr.2006.09.010. Epub 2006 Sep 4120. [DOI] [PubMed] [Google Scholar]
- 14.Kiarash A, Kelly C, Phinney B, Valdivia H, Abrams J, Cala S. Defective glycosylation of calsequestrin in heart failure. Cardiovasc Res. 2004;63:264–272. doi: 10.1016/j.cardiores.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 15.McFarland TP, Milstein ML, Cala SE. Rough endoplasmic reticulum to junctional sarcoplasmic reticulum trafficking of calsequestrin in adult cardiomyocytes. J Mol Cell Cardiol. 2010;49:556–564. doi: 10.1016/j.yjmcc.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuenzel EA, Mulligan JA, Sommercorn J, Krebs EG. Substrate specificity determinants for casein kinase II as deduced from studies with synthetic peptides. J Biol Chem. 1987;262:9136–9140. [PubMed] [Google Scholar]
- 17.Marin O, Meggio F, Marchiori F, Borin G, Pinna LA. Site specificity of casein kinase-2 (TS) from rat liver cytosol. A study with model peptide substrates. Eur J Biochem. 1986;160:239–244. doi: 10.1111/j.1432-1033.1986.tb09962.x. [DOI] [PubMed] [Google Scholar]
- 18.Cala SE, Jones LR. Phosphorylation of cardiac and skeletal muscle calsequestrin isoforms by casein kinase II. Demonstration of a cluster of unique rapidly phosphorylated sites in cardiac calsequestrin. J Biol Chem. 1991;266:391–398. [PubMed] [Google Scholar]
- 19.Shoshan-Barmatz V, Orr I, Weil S, Meyer H, Varsanyi M, Heilmeyer LM. The identification of the phosphorylated 150/160 kDa proteins of sarcoplasmic reticulum, their kinase and their association with the ryanodine receptor. Biochim Biophys Acta. 1996;1283:89–100. doi: 10.1016/0005-2736(96)00079-x. [DOI] [PubMed] [Google Scholar]
- 20.Cala SE, Jones LR. GRP94 resides within cardiac sarcoplasmic reticulum vesicles and is phosphorylated by casein kinase II. J Biol Chem. 1994;269:5926–5931. [PubMed] [Google Scholar]
- 21.Cala SE, Ulbright C, Kelley JS, Jones LR. Purification of a 90 kDa protein (Band VII) from cardiac sarcoplasmic reticulum. Identification as calnexin and localization of casein kinase II phosphorylation sites. J Biol Chem. 1993;268:2969–2975. [PubMed] [Google Scholar]
- 22.Lozeman FJ, Litchfield DW, Piening C, Takio K, Walsh KA, Krebs EG. Isolation and characterization of human cDNA clones encoding the alpha and the alpha' subunits of casein kinase II. Biochemistry. 1990;29:8436–8447. doi: 10.1021/bi00488a034. [DOI] [PubMed] [Google Scholar]
- 23.Cala SE, Jones LR. Rapid purification of calsequestrin from cardiac and skeletal muscle sarcoplasmic reticulum vesicles by Ca2--dependent elution from phenyl-sepharose. J Biol Chem. 1983;258:11932–11936. [PubMed] [Google Scholar]
- 24.Olsten ME, Litchfield DW. Order or chaos? An evaluation of the regulation of protein kinase CK2. Biochem Cell Biol. 2004;82:681–693. doi: 10.1139/o04-116. [DOI] [PubMed] [Google Scholar]
- 25.Wirkner U, Voss H, Lichter P, Ansorge W, Pyerin W. The human gene (CSNK2A1) coding for the casein kinase II subunit alpha is located on chromosome 20 and contains tandemly arranged Alu repeats. Genomics. 1994;19:257–265. doi: 10.1006/geno.1994.1056. [DOI] [PubMed] [Google Scholar]
- 26.Litchfield DW. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J. 2003;369:1–15. doi: 10.1042/BJ20021469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang-Feng TL, Naiman T, Kopatz I, Eli D, Dafni N, Canaani D. Assignment of the human casein kinase II alpha' subunit gene (CSNK2A1) to chromosome 16p13.2–p13.3. Genomics. 1994;19:173. doi: 10.1006/geno.1994.1032. [DOI] [PubMed] [Google Scholar]
- 28.Alvarado-Diaz CP, Tapia JC, Antonelli M, Moreno RD. Differential localization of alpha' and beta subunits of protein kinase CK2 during rat spermatogenesis. Cell Tissue Res. 2009;338:139–149. doi: 10.1007/s00441-009-0847-1. [DOI] [PubMed] [Google Scholar]
- 29.Yu IJ, Spector DL, Bae YS, Marshak DR. Immunocyto-chemical localization of casein kinase II during interphase and mitosis. J Cell Biol. 1991;114:1217–1232. doi: 10.1083/jcb.114.6.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guerra B, Issinger OG. Protein kinase CK2 and its role in cellular proliferation, development and pathology. Electrophoresis. 1999;20:391–408. doi: 10.1002/(SICI)1522-2683(19990201)20:2<391::AID-ELPS391>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 31.Tawfic S, Yu S, Wang H, Faust R, Davis A, Ahmed K. Protein kinase CK2 signal in neoplasia. Histol Histopathol. 2001;16:573–582. doi: 10.14670/HH-16.573. [DOI] [PubMed] [Google Scholar]
- 32.Sarno S, Pinna LA. Protein kinase CK2 as a druggable target. Mol Biosyst. 2008;4:889–894. doi: 10.1039/b805534c. [DOI] [PubMed] [Google Scholar]
- 33.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 35.Lowry HO, Rosebrough NJ, Farr AL, Randall RJ. Protein measurements with folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 36.Duncan JS, Gyenis L, Lenehan J, Bretner M, Graves LM, Haystead TA, Litchfield DW. An unbiased evaluation of CK2 inhibitors by chemoproteomics: characterization of inhibitor effects on CK2 and identification of novel inhibitor targets. Mol Cell Proteomics. 2008;7:1077–1088. doi: 10.1074/mcp.M700559-MCP200. [DOI] [PubMed] [Google Scholar]
- 37.Ruzzene M, Penzo D, Pinna LA. Protein kinase CK2 inhibitor 4,5,6,7-tetrabromobenzotriazole (TBB) induces apoptosis and caspase-dependent degradation of haematopoietic lineage cell-specific protein 1 (HS1) in jurkat cells. Biochem J. 2002;364:41–47. doi: 10.1042/bj3640041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schneider CC, Hessenauer A, Gotz C, Montenarh M. DMAT, an inhibitor of protein kinase CK2 induces reactive oxygen species and DNA double strand breaks. Oncol Rep. 2009;21:1593–1597. doi: 10.3892/or_00000392. [DOI] [PubMed] [Google Scholar]
- 39.Pagano MA, Poletto G, Di Maira G, Cozza G, Ruzzene M, Sarno S, Bain J, Elliott M, Moro S, Zagotto G, Meggio F, Pinna LA. Tetrabromocinnamic acid (TBCA) and related compounds represent a new class of specific protein kinase CK2 inhibitors. Chembiochem. 2007;8:129–139. doi: 10.1002/cbic.200600293. [DOI] [PubMed] [Google Scholar]
- 40.Cala SE, Scott BT, Jones LR. Intralumenal sarcoplasmic reticulum Ca(2+)-binding proteins. Semin Cell Biol. 1990;1:265–275. [PubMed] [Google Scholar]
- 41.Helenius A, Aebi M. Intracellular functions of N-linked glycans. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 42.Wang X, Johnsson N. Protein kinase CK2 phosphorylates Sec63p to stimulate the assembly of the endoplasmic reticulum protein translocation apparatus. J Cell Sci. 2005;118:723–732. doi: 10.1242/jcs.01671. [DOI] [PubMed] [Google Scholar]
- 43.Chevet E, Wong HN, Gerber D, Cochet C, Fazel A, Cameron PH, Gushue JN, Thomas DY, Bergeron JJ. Phosphorylation by CK2 and MAPK enhances calnexin association with ribosomes. EMBO J. 1999;18:3655–3666. doi: 10.1093/emboj/18.13.3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaminska B, Ellert-Miklaszewska A, Oberbek A, Wisniewski P, Kaza B, Makowska M, Bretner M, Kazimierczuk Z. Efficacy and mechanism of anti-tumor action of new potential CK2 inhibitors toward glioblastoma cells. Int J Oncol. 2009;35:1091–1100. doi: 10.3892/ijo_00000424. [DOI] [PubMed] [Google Scholar]
- 45.Zhu D, Hensel J, Hilgraf R, Abbasian M, Pornillos O, Deyanat-Yazdi G, Hua XH, Cox S. Inhibition of protein kinase CK2 expression and activity blocks tumor cell growth. Mol Cell Biochem. 2010;333:159–167. doi: 10.1007/s11010-009-0216-0. [DOI] [PubMed] [Google Scholar]
- 46.Hegde RS, Lingappa VR. Regulation of protein biogenesis at the endoplasmic reticulum membrane. Trends Cell Biol. 1999;9:132–137. doi: 10.1016/s0962-8924(99)01504-4. [DOI] [PubMed] [Google Scholar]
- 47.Hegde RS, Kang SW. The concept of translocational regulation. J Cell Biol. 2008;182:225–232. doi: 10.1083/jcb.200804157. [DOI] [PMC free article] [PubMed] [Google Scholar]