

Abstract
Heart failure is highly influenced by heritability, and nearly 100 genes link to familial cardiomyopathy. Despite the marked genetic diversity that underlies these complex cardiovascular phenotypes, several key genes and pathways have emerged. Hypertrophic cardiomyopathy is characterized by increased contractility and a greater energetic cost of cardiac output. Dilated cardiomyopathy is often triggered by mutations that disrupt the giant protein titin. The energetic consequences of these mutations offer molecular targets and opportunities for new drug development and gene correction therapies.
Keywords: cardiomyopathy, hypertrophic cardiomyopathy, dilated cardiomyopathy, gene mutation, sarcomere
Heart Failure and Cardiomyopathy
The clinical diagnosis of heart failure (HF) arises when cardiac output is insufficient to supply demand. Acute HF can occur from abrupt occlusion of a coronary artery, catastrophic valve dysfunction, malignant hypertension, or other states that provoke an urgent mismatch between supply and demand. Chronic HF is experienced as a slow decline in function, measured over years, as fatigue, breathlessness and often with evidence of end organ vascular insufficiency. Fluid overload and arrhythmias contribute to the HF spectrum. The timeline of chronic HF is punctuated by acute HF exacerbations, and the annual costs associated with HF exceed $30 billion US dollars (Heidenreich et al., 2013). The major costs are calculated in repeated hospitalizations, the need for medical and device intervention, and lost productivity (Dunlay et al., 2011). Because of the chronic and progressive nature of HF, there is opportunity to intervene at early stages.
HF is frequently accompanied by cardiomyopathy, defined as a morphologically abnormal heart. In vivo, echocardiography provides critical information regarding chamber dimensions and function, while magnetic resonance imaging also provides a more in-depth visualization of myocardial tissue composition (Mahrholdt et al., 2005; Rickers et al., 2005). The major forms of cardiomyopathy include hypertrophic, dilated, restrictive, and arrhythmogenic (sometimes referred to as right ventricular) cardiomyopathy (Maron et al., 2006). Each of these forms of cardiomyopathy has a major heritable component and genetic testing is now used in the evaluation of individuals with cardiomyopathy (Arndt and MacRae, 2014; McNally et al., 2013; Teekakirikul et al., 2013). The genes for which there is genetic testing are shown in Figure 1. Overall, there are nearly 100 genes linked to inherited forms of cardiomyopathy. More than 20 genes are implicated in hypertrophic cardiomyopathy (HCM) while fewer genes are linked to arrhythmogenic right ventricular cardiomyopathy (ARVC). Dilated cardiomyopathy (DCM) is the most genetically heterogeneous. The same gene may be implicated in multiple forms of cardiomyopathy, underscoring the importance of genomic context in the pathophysiology of disease-associated variants. In addition to genetic causes, ischemia, toxic insult, and valvular defects contribute to DCM, and more than one etiology may contribute to any form of cardiomyopathy. Despite this heterogeneity, several essential classes of genetic mutations are present around which existing and novel therapies can be applied.
Figure 1.

Shown are the genes that have been linked to human inherited cardiomyopathy. Those genes responsible for HCM (pink) and DCM (blue). There are a number of genes that cause both HCM and DCM (purple). Mutations in genes encoding desmosomal and other proteins cause Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC, green) and there is overlap between mutations in these genes that lead to other forms of cardiomyopathy.
Genetic assessment in Cardiomyopathy
The large number of genes responsible for cardiomyopathy, as well as the myriad of diverse mutations within each of these genes, produces remarkable heterogeneity for this complex disorder. Individual cardiomyopathy-associated genetic variants are infrequent in the general population (< 1 in 500), and individual genetic variants associate with a range of expressivity causing mild and severe forms of disease. For example, deletion of arginine 14 in phospholamban (PLN gene) was described with early onset cardiomyopathy and accompanying lethal arrhythmias (Haghighi et al., 2006). In one population, this same mutation was found in 12% of ARVC and 15% of DCM subjects (van der Zwaag et al., 2012) and a follow up retrospective evaluation of 295 gene mutations carriers confirmed an earlier age of onset of both arrhythmias and cardiomyopathy (van Rijsingen et al., 2014). Curiously, this same mutation was described in individuals with late onset DCM without evidence of ventricular arrhythmias (DeWitt et al., 2006). The range of outcome with the same given variant is consistent with the presence of genetic and environmental factors that modify outcome (Arad et al., 2002; Marian, 2000) (Figure 2). Sex is a modifier of cardiomyopathy expression. Rare truncating mutations in TTN, the gene encoding the giant protein titin, are associated with more severe left ventricular (LV) dysfunction in males compared to females (Herman et al., 2012). Sex differences have also been described in hypertrophic cardiomyopathy (HCM), where males are usually more affected, and this is recapitulated in animal models (Geisterfer-Lowrance et al., 1996; Vikstrom et al., 1996). Sex differences are attributed to a number of factors including hormone levels, gene expression differences, and basic differences in physiology including heart size. Factors other than sex also influence the expression of genetic variants on the pathophysiology of heart disease.
Figure 2.

Familial studies for inherited cardiomyopathy often demonstrate a primary pathogenic variant, and pathogenic variants differ in their effect on phenotypic outcome. Each genome contains many additional variants that serve to modify the expression of the primary pathogenic variant. These secondary modifers may be common or rare in the population. In addition to these genetic modifiers, comorbidities, environmental factors, and sex modulate the expression of cardiomyopathy. The manifestation of cardiomyopathy varies over the lifetime of the individual. Those mutations, or combinations of mutations, with the most potent effect on phenotype manifest earlier in life. Milder mutations may not express until later in life or may remain subclinical throughout the lifetime of the individual. (Dilated, DCM; hypertrophic HCM; arr right ventricular, ARVC).
Secondary or additional genetic variants also contribute to severity or progression of disease. In individuals with more than one mutation, there is often earlier onset and in some cases a more rapid progression of disease (Girolami et al., 2010; Golbus et al., 2014; Ingles et al., 2005; Richard et al., 1999). A recent survey used whole genome sequencing of 11 unrelated individuals to identify the spectrum of cardiomyopathy variants (Golbus et al., 2014). In 9 of 11 individuals, the primary disease-causing variant was identified, usually because it segregated with disease. However, two cases had additional, rare, potentially pathogenic variation. Closer inspection of these families revealed that these secondary variants were segregating with a more severe phenotype, indicating that multiple rare variants may contribute to altered disease course within a family. Common or higher frequency variation also impacts the effects of underlying pathogenic variants. Recent work has revealed an association between severity of cardiomyopathy and mitochondrial DNA haplogroup (Strauss et al., 2013). Human mitochondrial DNA can be divided into groups based on shared genealogy. Strauss and colleagues studied a large Mennonite family with autosomal recessive myopathy and cardiomyopathy caused by a frameshift mutation in the gene coding for adenine nucleotide translocator-1. The authors found considerable variability in the progression and severity of the cardiac phenotype that segregated with the maternal lineage. Sequencing showed segregation of two mitochondrial haplogroups, one of which conferred more severe cardiomyopathy (Strauss et al., 2013).
These data underscore how the expressivity and penetrance of specific cardiomyopathy gene variants varies widely (Hershberger et al., 2013). In silico algorithms score pathogenicity on numeric scales, relying on conservation data and less so on structural information (Ritchie and Flicek, 2014). Based on segregation with clinical phenotype, more highly penetrant mutations have been described, but even highly penetrant mutations may require the context of specific genetic backgrounds or ethnicities to fully manifest, and this “background effect” has been modeled in mice (Semsarian et al., 2001; Suzuki et al., 2002; Wheeler et al., 2009). With the emergence of sequence data from large numbers of ethnically diverse humans, it has become clear that “pathogenic” variation is found at a higher than expected rate. Specifically, previously described pathogenic mutations are present at a frequency higher than the prevalence of cardiomyopathy (Andreasen et al., 2013; Golbus et al., 2012; Pan et al., 2012). Not all genetic variants induce the same degree of cardiac dysfunction and, for primary mutation and secondary modifiers there are “mild” and “severe” mutations. Determining the expressivity of given mutations is challenging and computational algorithms, however imperfect, are emerging now serve as an adjunct to interpreting the pathogenicity of cardiomyopathy mutations. Genetic variation remains a strong predictor of risk for developing cardiomyopathy, particularly within families where a primary gene mutation has been identified.
Recent work has focused on reclassifying the potential pathogenicity of variants based on frequency in the population at large, with higher frequency variants considered less pathogenic (MacArthur et al., 2014). This methodology assumes that pathogenic alleles will be found in a frequency in the population less than or equal to the disease prevalence and assumes that individual variants are sufficient to cause disease. Studies of penetrance and expressivity indicate that the entire genomic context as well as the environment, dictate the role of particular variants (Hershberger et al., 2013). Pathogenicity of particular variants must be considered within the phenotype context, as many cardiomyopathic genetic variants are necessary but not sufficient to cause disease. Most large human genetic datasets include individuals who have not been specifically evaluated for subtle signs of cardiomyopathy and/or individuals who are too young to have yet developed disease. Similarly, it is expected that additional genetic and environmental stimuli are necessary to express the full phenotype of cardiomyopathy and heart failure. In addition to these secondary genetic and environmental modifiers, epigenetic influences may markedly alter the expression of mutant alleles or alternative genetic pathways that diminish or enhance pathogenicity. As sparks do not cause fire in the absence of oxygen, cardiomyopathy mutations require context to fully manifest. Variants are only pathogenic in a larger context that includes both the susceptible genetic and environmental conditions (Figure 2). Exploring and defining the genetic and environment stimuli necessary for cardiomyopathy expression is critical, as these modifiers influence outcome and are targets for intervention.
Hypertrophic Cardiomyopathy and thick filament gene mutations
Hypertrophic cardiomyopathy (HCM) is estimated at 1:500 in younger individuals and is enriched in families. This estimate derives from a population-based survey of individuals 23-35 years of age (Maron et al., 1995). Given the broad age range of HCM and the appreciation that some genetic mutations have later onset, the overall population prevalence is higher. An Olmstead County study conducted in 1985 identified a similar prevalence for HCM (19.7 per 100,000) and a higher prevalence for DCM (36.5 per 100,000). This study and the previous rely on older methods of detection, and as such likely underestimate the prevalence of HCM. Hypertrophy of the ventricular myocardium arises in response to physiological stimuli, such as exercise, and pathological stimuli such as hypertension or aortic stenosis. In genetic HCM, autosomal dominant mutations in the MYH7 and MYBPC3 genes account for nearly 80% of inherited HCM (Kensler et al., 2011). These genes encode the sarcomere thick filament proteins-myosin heavy chain (MYH7) and cardiac myosin binding protein-C (cMyBP-C). Although they are both highly associated with HCM, the mechanisms of the HCM-causing mutations in these two genes differ. The majority of pathogenic variants in MYH7 that cause HCM result in amino acid substitutions in critical residues and domains that adversely affect function. In contrast, the majority of pathogenic HCM-causing MYBPC3 variants are premature stop codons or frame shifting mutations, frequently resulting in absence of protein. MYBPC3 are thought to have a milder disease course with later onset then mutations in MYH7 (Charron et al., 1998; Maron et al., 2001), which may be attributed to the difference in pathogenic mechanism between mutations in these genes.
MYH7 encodes myosin heavy chain ( MHC), the thick filament protein responsible for hydrolyzing ATP to produce force. Myosin can be divided into the globular head domain and its coiled-coil rod domain. The myosin head is attached to an arm that articulates away from the rod region on a flexible hinge to extend into the interfilament space (Figure 3). The rod domain mediates the formation of the thick filament with its characteristic periodicity (Moore et al., 2012). Mutations in MYH7 have been identified in all regions of the protein, with more mutations concentrating in the ATPase domain, actin binding domain, and domains responsible for force transmission (Walsh et al., 2010). Although occurring at lower frequency, mutations in the rod domain have also been linked to HCM (Blair et al., 2002). Modeling MYH7 mutations has been achieved using materials from human tissues, in vitro or cell-based expression, or genetic engineering in mice. Each of these methods has limitations and the results from distinct approaches have not always produced consistent findings. Mice, like other small mammals, express α-MHC as the major cardiac myosin (encoded by MYH6), rather than MHC like the larger human heart. MHC, while similar in overall structure to MHC, has an intrinsically faster rate of ATP hydrolysis and contractile kinetics (Korte et al., 2005). The intrinsic capabilities of α-MHC versus β-MHC can lead to the same mutation demonstrating different biophysical characteristics (Lowey et al., 2008).
Figure 3.
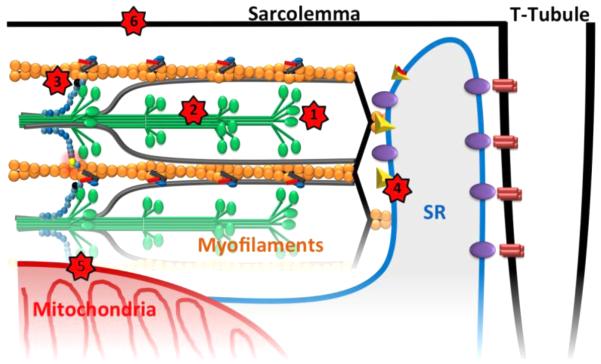
Myosin heads protrude from the thick filaments (green) to interact with actin containing thin filaments (yellow). Multiple sites throughout the sarcomere and cardiomyocyte are now the targets for new drug development for heart failure (Red stars). 1. Small molecules like omecamtiv are aimed at myosin ATPase activity to increase or decrease contractility. 2. Antisense or RNAi approaches are being tested to silence mutant alleles but not normal alleles. 3. cMyBP-C phosphorylation can be modified through kinase/phosphatases to modulate its “brake effect” on cross-bridge cycling. 4. Calcium handing in the sarcoplasmic reticulum is a target in development. 5. Palmitoyltransferase-1 can be altered using perhexiline to shift metabolic substrate usage from fatty acid oxidation to glycolysis. 6. The regulation of nitric oxide synthase can be used to change cellular redox state and prevent glutathionylation and dysregulation of myofilament proteins.
There are hundreds of distinct missense MYH7 mutations responsible for HCM, and a clear unifying hypothesis has been elusive. Clinically, HCM is often characterized by a hyperdynamic state in which there is an increase in left ventricular ejection fraction from 60% to 70% or more. For example, the R453C MYH7 HCM mutation displays an impaired catalytic cycle of ATP hydrolysis despite few biochemical alterations in the ATPase domain, and this mutation counter-intuitively results in increased contractility (Bloemink et al., 2014; Sommese et al., 2013). Increasing evidence suggests that HCM mutations in MYH7 cause increased energy usage due to a less efficient myosin motor and that this energetic mismatch results in perturbed metabolic state (Crilley et al., 2003). As one indication of this energetic mismatch, reduced phospho-creatine levels (PCr) have been observed using 31P NMR spectroscopy of animal models of HCM and materials from human HCM patients (Ingwall, 2014; Witjas-Paalberends et al., 2014a; Witjas-Paalberends et al., 2014b). Human hearts expressing the MYH7 R403Q mutation generate increased tension and faster actin sliding velocities, but at a higher energetic cost (Alpert et al., 2005). Mice engineered with the R403Q HCM-associated mutation had reduced rate of relaxation and increased end diastolic pressure after inotropic stimulation (Tyska et al., 2000). A similar decrease in PCr and increased ADP was seen in these hearts, consistent with a higher energetic cost of contraction (Spindler et al., 1998).
In addition to MYH7 missense mutations in HCM, MYH7 missense variants are also found in DCM. DCM-associated MYH7 missense mutations have been modeled in mice, albeit in the context of Myh6. However, these mutations show an increased tension cost, with more ATP required for a given amount of shortening, depressed actin sliding velocities and gross dilation (Schmitt et al., 2006). In contrast to the hypercontractile HCM mutations, DCM mutations when modeled in expressed myosin, cause a hypocontractile state and quickly lead to HF (Bloemink et al., 2014; Sommese et al., 2013).
The second major thick filament protein implicated in HCM is MYBPC3, which encodes cardiac myosin binding protein C (cMyBP-C). MYPBC3 and MYH7 mutations are at nearly equal frequency in HCM cohorts, each representing approximately 40% of identified mutations. In contrast to MYH7 HCM mutations, which are mainly nonsynonymous single nucleotide polymorphisms (nsSNPs), MYBPC3 mutations more commonly disrupt the reading frame. By disrupting the carboxyl-terminus of cMybp-C, these mutations alter the myosin and titin binding sites and disallow mutant cMyBP-C incorporation in the sarcomere. cMyBP-C is incorporated into the thick filament through direct binding of its carboxy-terminus to the myosin rod and also to the giant protein titin (Freiburg and Gautel, 1996; Gilbert et al., 1996). cMyBP-C includes eight immunoglobulin (Ig) domains along its length with three fibronectin (Fn) domains towards its carboxy-terminus. cMyBP-C extends transversely into the interfilament space where its amino-terminal region interacts with the myosin S2 region and actin in a phosphorylation-dependent manner (Barefield and Sadayappan, 2010; Bezold et al., 2013; Kunst et al., 2000). The phosphorylation state of three serine residues mediate cMyBP-C’s ability to regulate cross-bridge cycling, and dysregulation of these residues results in cardiomyopathy (Sadayappan et al., 2005; Sadayappan et al., 2006). Recent work has clearly defined the role of cMyBP-C as a “molecular brake” on myosin cross-bridge cycling, resulting in slowing of actin sliding velocities while in a dephosphorylated state (Previs et al., 2012). However, phosphorylation of cMyBP-C by various upstream pathways, notably PKA, reduces the inhibitory effect of cMyBP-C in a graded manner, with phosphorylation of additional sites providing additional relief of myosin inhibition, allowing throttling of this effect (Weith et al., 2012a; Weith et al., 2012b). In addition, it has been recently shown that cMyBP-C’s interaction with actin displaces α-tropomyosin, thus modifying thin-filament activation (Mun et al., 2014)
Oxidative modifications, often accompanying pathological remodeling of the heart, have been reported having detrimental effects on cMybp-C and sarcomere function. Using a hypertensive mouse model demonstrating diastolic dysfunction prior to hypertrophic remodeling, cMyBP-C was determined to be glutathionylated at three cysteine residues (Patel et al., 2013). This oxidative modification resulted in increased myofilament calcium sensitivity in isolated myofilaments. Treatment with tetrahydrobiopterin prevented cMy-BP-C glutathionylation and improved relaxation kinetics and diastolic dysfunction (Jeong et al., 2013). While additional work is required to determine whether these channels are wholly mediated by cMyBP-C oxidation, the possibility of treatment prior to the onset of hypertrophy with anti-oxidant agents warrants further investigation.
With these data, it is now clear that cMyBP-C regulates contractility and alters sarcomere energetics. HCM-associated MYBPC3 mutations, especially those that reduce the amount of cMyBP-C, promote a loss of cross-bridge cycling inhibition. The loss of cMyBP-C regulation has been shown to decrease maximal force development in samples of human tissue from mutation carriers (van Dijk et al., 2014) and in mouse models of disease (Barefield et al., 2014a; Harris et al., 2002). However, these MYBPC3 mutations have been shown to increase the energetic cost of contraction similar to MYH7 mutations (Witjas-Paalberends et al., 2014b). Whether MYBPC3 mutations act by haploinsufficiency or dominant negative activity has been examined, and evidence for truncated cMyBP-C protein has been lacking (Marston et al., 2009). Thus, MYBPC3 mutations appear to act mainly by reducing protein content (Barefield et al., 2014b; van Dijk et al., 2009). Treatment has therefore focused on restoring protein levels through gene therapy, with some notable recent success in a mouse model (Mearini et al., 2014).
In South Asian populations, it is estimated that 4% of the population carries a 25 bp deletion in intron 32 of MYBPC3 (Dhandapany et al., 2009). This variant increases the risk for heart failure and cardiomyopathy. This deletion induces skipping of the downstream exon near the 3’ end of MYBPC3. The true prevalence throughout South Asia has been estimated to be lower, but larger population samples are likely needed (Simonson et al., 2010). This deletion has also been linked to increased left ventricular dysfunction after myocardial infarction (Srivastava et al., 2011) consistent with this variant increasing susceptibility to heart failure, especially in combination with other cardiac insults. Rare instances of individuals with two mutant alleles of MYBPC3 and early onset lethal disease have been described, and often associate with the feature of noncompaction (Dellefave et al., 2009; Lekanne Deprez et al., 2006; Schaefer et al., 2014; Wessels et al., 2014). There is an emerging view that HCM can be subdivided into “sarcomere” vs “nonsarcomere”, and that pathophysiology and outcome are different between these groups (Olivotto et al., 2008; Olivotto et al., 2011)
The thin filament
Cross-bridge formation between myosin heads and actin filaments is largely regulated by the proteins of the thin filament. The thin filament is composed of actin, tropomyosin, the troponin complex including troponin T (the tropomyosin binding subunit), troponin I (the inhibitory subunit) and troponin C (the Ca2+ binding subunit). Titin and other Z-disk related proteins also contribute to this regulation. Regulation of cross-bridge formation depends on the Ca2+ and ATP availability and the conformation of the troponin-tropomyosin complex on the actin filament (Lehman et al., 2000). The complex exists in three states each of which determines the extent of actin and myosin interaction (McKillop and Geeves, 1993). HCM-causing mutations have been identified in each of these components and whether these proteins exert their effect through changing sarcomere energetics and the cost of contraction has been suggested (Tardiff, 2011).
Recently, Moore and colleagues examined the tropomyosin-binding region of cardiac troponin T, a region that harbors severe and phenotypically diverse HCM mutations (Moore et al., 2014). In vitro motility assays showed that specific mutations in cardiac troponin T disrupted weak electrostatic interactions between the thin filament and myosin. Complementary in vivo data indicates that these same mutations cause cardiac remodeling and disarray of the myofiber, suggesting that the weak cross-bridge formation causes destabilization of the myofilament structure ultimately resulting in disease (Moore et al., 2014).
Titin, the third filament, a gene for DCM
The giant protein titin is necessary for the passive forces that maintain sarcomere integrity, and these passive forces play a critical role in left ventricular mechanics, especially filling during diastole. Titin is the largest known protein and spans half the sarcomere from Z-disk to M-line (Furst et al., 1988). Adjacent to the M-line titin contains a titin kinase domain (TK) (Gautel, 2011). The A-band portion of titin is composed of both Ig and fibronectin domains (Labeit et al., 1992). The I-band portion is composed of two “spring-like” domains in cardiac muscle; the N2B, and PEVK along with tandem immunoglobulin (Ig) segment. These “spring-like” domains are responsible for passive force during sarcomere stretch (reviewed in (Anderson and Granzier, 2012)).
Several mouse models have been created to dissect titin’s role in the sarcomere. Lee and colleagues deleted the region of TTN encoding its N2B region, and found that passive tension was elevated triggering an increase in calcium sensitivity at long sarcomere length (Lee et al., 2010). Increases in calcium sensitivity result in increased length-dependent activation, indicating that passive-tension induced by titin is a factor in the Frank-Starling mechanism of the heart (Katz, 2002). In further support of the idea that increased titin-induced passive tension results in increased length-dependent activation, N2B deleted mice have higher LV diastolic stiffness and diastolic dysfunction (Lee et al., 2010). Truncation of the PEVK region, another spring-like element, also exhibited increased passive tension along with diastolic dysfunction (Granzier et al., 2009). A mouse lacking the tandem Ig segment showed similar physiology (Chung et al., 2013). Disrupting the I-band/A-band junction of titin also increased strain of the spring regions of the I-band and caused diastolic dysfunction in the mice (Granzier et al., 2014). Currently, mouse models specifically lacking the A-band region of titin are lacking. However, a recent study has shown that the A-band of titin is not required for thick filament assembly in zebrafish (Myhre et al., 2014). These zebrafish have a truncated TTN, lacking the C-terminal, A-band associated rod domain. The zebrafish form normal muscle with grossly normal thick and thin filament assembly, and only after embryonic development does the sarcomere break down, consistent with TTN having a role in sarcomere maintenance (Myhre et al., 2014). The zebrafish also display reduced heartbeat and cardiac edema, consistent with cardiac dysfunction.
TTN has recently been identified as a major cardiomyopathy gene in humans (Herman et al., 2012). Herman et al. captured and sequenced the 360 exons of TTN in large cohorts of more than 300 DCM and 200 HCM subjects as well as a control group with normal heart function. They identified a large number of missense mutations in TTN, even in the control population, making these variants difficult to interpret. Instead, they focused on truncating variants that created frameshifts, stops and splice site alterations. Approximately 25% of DCM patients had a truncating variant in TTN, while only 1% of HCM and 3% of the control population had truncating variants, making TTN mutations the most common genetic source of DCM to date. Truncating variants identified in the DCM cohort disproportionately occurred in the A-band region of TTN (Herman et al., 2012). A recent study supports that DCM-associated TTN truncating variants fall into the A band of titin (Roberts et al., 2015). Furthermore, the truncating variants in the general population fall into TTN isoforms expressed at much lower levels in the heart.
Implications for gene-based therapy and drug development
Gene-based correction is now possible by targeting RNA using reduction strategies and other methods that “bypass” mutations, creating internally truncated proteins. For dominant diseases, especially in MYH7, targeting the mutant allele with RNAi is possible in a mutation specific manner (Jiang et al., 2013). Developing individualized treatment plans with mutation-specific sequences may be cumbersome and require unique validation methods. As an alternative, it is possible to target more common variation, present on the mutant but not normal allele. In this manner, sequences could be developed to treat larger numbers of patients and providing a more feasible regulatory approval pathway. The degree to which the mutant allele must be reduced can be guided by data from human hearts, where the expression of mutant proteins is often less than 50% (Helms et al., 2014). Genetic “bypass” methods are also being developed, and these methods manipulate RNA using anti-sense sequences to induce alternative splicing to avoid the mutation. This approach, referred to as exon skipping, takes advantage of naturally occurring splice forms or creates newly engineered, internally truncated proteins (Veltrop and Aartsma-Rus, 2014). As long as there is physiological evidence that such internally truncated proteins can compensate, these approaches may be useful for some genes linked to cardiomyopathy. Complete functional restoration may not be needed, as even partial improvement may be sufficient to improve phenotype. Given the structural data available on β-MHC, it is unlikely that internal truncation methods are suitable for MYH7 gene mutations. However, for proteins such as titin that are composed of repetitive domains, internal truncations may be a viable alternative (Freiburg et al., 2000). Newer DNA-based gene editing methods are an active area of research, currently being tested in cell-based models (Li et al., 2014; Xie et al., 2014). These methods have been employed to correct muscle disease in the mdx mouse model (Long et al., 2014), and are currently being tested in cardiomyopathy models.
The energetic and metabolic deficits in heart failure offer opportunities for small molecule based therapy. Modulating contractility can occur by either blunting hypercontractile states, or by using positive inotropic agents to improve deficits in contractility. A recent potential therapeutic for reduced cardiac performance is omecamtiv mecarbil, which directly activates MHC through enhancement of contractility, and with a corresponding improvement in cardiac output (Cleland et al., 2011; Malik et al., 2011). Identifying targets that regulate the cardiomyocyte metabolic state has also shown some promising preliminary results. Modest improvements in the HCM phenotype were observed in a mouse model carrying a MYBPC3 mutation following the application of perhexiline, a molecule that targets mitochondrial palmitoyltransferase-1, altering metabolic substrate usage from fatty acid oxidation to glycolysis (Gehmlich et al., 2014). However, more work remains to be done to evaluate the efficacy of altering metabolic substrate usage. The differences between the mouse and human hearts, reflected by their difference MHC usage, will likely mandate that these approaches be validated in larger mammalian hearts.
Conclusions
The genetic complexity underlying cardiomyopathy is challenging the concept of “single gene disorders”. The number of genes and individual mutations is greater than had been expected, and it has only been through the availability of next generation sequencing that such genetic diversity has been appreciated. While this genetic landscape is daunting in scope and breadth, key themes have emerged. HCM, with its distinct phenotype, is largely linked to two key thick filament proteins. A wealth of data supports an energetically inefficient myocardium in HCM. DCM, while more genetically heterogeneous than HCM, has one major gene, TTN, which now begins to focus the etiology of a sizable subset of disease. With improved DNA sequencing, it is now possible to identify combinations of genetic mutations that contribute to cardiomyopathy. HCM can now be subdivided into sarcomere and non-sarcomere HCM with clinically meaningful differences in physiology, and the same classification will develop for DCM. A better understanding of the molecular subtypes of cardiomyopathy will help more precisely apply existing and evolving therapies.
References
- Alpert NR, Mohiddin SA, Tripodi D, Jacobson-Hatzell J, Vaughn-Whitley K, Brosseau C, Warshaw DM, Fananapazir L. Molecular and phenotypic effects of heterozygous, homozygous, and compound heterozygote myosin heavy-chain mutations. American journal of physiology. Heart and circulatory physiology. 2005;288:H1097–1102. doi: 10.1152/ajpheart.00650.2004. [DOI] [PubMed] [Google Scholar]
- Anderson BR, Granzier HL. Titin-based tension in the cardiac sarcomere: molecular origin and physiological adaptations. Progress in biophysics and molecular biology. 2012;110:204–217. doi: 10.1016/j.pbiomolbio.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen C, Nielsen JB, Refsgaard L, Holst AG, Christensen AH, Andreasen L, Sajadieh A, Haunso S, Svendsen JH, Olesen MS. New population-based exome data are questioning the pathogenicity of previously cardiomyopathy-associated genetic variants. Eur J Hum Genet. 2013;21:918–928. doi: 10.1038/ejhg.2012.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arad M, Seidman JG, Seidman CE. Phenotypic diversity in hypertrophic cardiomyopathy. Hum Mol Genet. 2002;11:2499–2506. doi: 10.1093/hmg/11.20.2499. [DOI] [PubMed] [Google Scholar]
- Arndt AK, MacRae CA. Genetic testing in cardiovascular diseases. Curr Opin Cardiol. 2014;29:235–240. doi: 10.1097/HCO.0000000000000055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barefield D, Kumar M, de Tombe PP, Sadayappan S. Contractile dysfunction in a mouse model expressing a heterozygous MYBPC3 mutation associated with hypertrophic cardiomyopathy. American journal of physiology. Heart and circulatory physiology. 2014a;306:H807–815. doi: 10.1152/ajpheart.00913.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barefield D, Kumar M, Gorham J, Seidman JG, Seidman CE, de Tombe PP, Sadayappan S. Haploinsufficiency of MYBPC3 exacerbates the development of hypertrophic cardiomyopathy in heterozygous mice. Journal of molecular and cellular cardiology. 2014b doi: 10.1016/j.yjmcc.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barefield D, Sadayappan S. Phosphorylation and function of cardiac myosin binding protein-C in health and disease. Journal of molecular and cellular cardiology. 2010;48:866–875. doi: 10.1016/j.yjmcc.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezold KL, Shaffer JF, Khosa JK, Hoye ER, Harris SP. A gain-of-function mutation in the M-domain of cardiac myosin-binding protein-C increases binding to actin. The Journal of biological chemistry. 2013;288:21496–21505. doi: 10.1074/jbc.M113.474346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair E, Redwood C, de Jesus Oliveira M, Moolman-Smook JC, Brink P, Corfield VA, Ostman-Smith I, Watkins H. Mutations of the light meromyosin domain of the beta-myosin heavy chain rod in hypertrophic cardiomyopathy. Circulation research. 2002;90:263–269. doi: 10.1161/hh0302.104532. [DOI] [PubMed] [Google Scholar]
- Bloemink M, Deacon J, Langer S, Vera C, Combs A, Leinwand L, Geeves MA. The hypertrophic cardiomyopathy myosin mutation R453C alters ATP binding and hydrolysis of human cardiac beta-myosin. The Journal of biological chemistry. 2014;289:5158–5167. doi: 10.1074/jbc.M113.511204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charron P, Dubourg O, Desnos M, Isnard R, Hagege A, Bonne G, Carrier L, Tesson F, Bouhour JB, Buzzi JC, et al. Genotype-phenotype correlations in familial hypertrophic cardiomyopathy. A comparison between mutations in the cardiac protein-C and the beta-myosin heavy chain genes. Eur Heart J. 1998;19:139–145. doi: 10.1053/euhj.1997.0575. [DOI] [PubMed] [Google Scholar]
- Chung CS, Hutchinson KR, Methawasin M, Saripalli C, Smith JE, 3rd, Hidalgo CG, Luo X, Labeit S, Guo C, Granzier HL. Shortening of the elastic tandem immunoglobulin segment of titin leads to diastolic dysfunction. Circulation. 2013;128:19–28. doi: 10.1161/CIRCULATIONAHA.112.001268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleland JG, Teerlink JR, Senior R, Nifontov EM, Mc Murray JJ, Lang CC, Tsyrlin VA, Greenberg BH, Mayet J, Francis DP, et al. The effects of the cardiac myosin activator, omecamtiv mecarbil, on cardiac function in systolic heart failure: a double-blind, placebo-controlled, crossover, dose-ranging phase 2 trial. Lancet. 2011;378:676–683. doi: 10.1016/S0140-6736(11)61126-4. [DOI] [PubMed] [Google Scholar]
- Crilley JG, Boehm EA, Blair E, Rajagopalan B, Blamire AM, Styles P, McKenna WJ, Ostman-Smith I, Clarke K, Watkins H. Hypertrophic cardiomyopathy due to sarcomeric gene mutations is characterized by impaired energy metabolism irrespective of the degree of hypertrophy. Journal of the American College of Cardiology. 2003;41:1776–1782. doi: 10.1016/s0735-1097(02)03009-7. [DOI] [PubMed] [Google Scholar]
- Dellefave LM, Pytel P, Mewborn S, Mora B, Guris DL, Fedson S, Waggoner D, Moskowitz I, McNally EM. Sarcomere mutations in cardiomyopathy with left ventricular hypertrabeculation. Circ Cardiovasc Genet. 2009;2:442–449. doi: 10.1161/CIRCGENETICS.109.861955. [DOI] [PubMed] [Google Scholar]
- DeWitt MM, MacLeod HM, Soliven B, McNally EM. Phospholamban R14 deletion results in late-onset, mild, hereditary dilated cardiomyopathy. Journal of the American College of Cardiology. 2006;48:1396–1398. doi: 10.1016/j.jacc.2006.07.016. [DOI] [PubMed] [Google Scholar]
- Dhandapany PS, Sadayappan S, Xue Y, Powell GT, Rani DS, Nallari P, Rai TS, Khullar M, Soares P, Bahl A, et al. A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nat Genet. 2009;41:187–191. doi: 10.1038/ng.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlay SM, Shah ND, Shi Q, Morlan B, VanHouten H, Long KH, Roger VL. Lifetime costs of medical care after heart failure diagnosis. Circ Cardiovasc Qual Outcomes. 2011;4:68–75. doi: 10.1161/CIRCOUTCOMES.110.957225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freiburg A, Gautel M. A molecular map of the interactions between titin and myosin-binding protein C. Implications for sarcomeric assembly in familial hypertrophic cardiomyopathy. European journal of biochemistry / FEBS. 1996;235:317–323. doi: 10.1111/j.1432-1033.1996.00317.x. [DOI] [PubMed] [Google Scholar]
- Freiburg A, Trombitas K, Hell W, Cazorla O, Fougerousse F, Centner T, Kolmerer B, Witt C, Beckmann JS, Gregorio CC, et al. Series of exon-skipping events in the elastic spring region of titin as the structural basis for myofibrillar elastic diversity. Circulation research. 2000;86:1114–1121. doi: 10.1161/01.res.86.11.1114. [DOI] [PubMed] [Google Scholar]
- Furst DO, Osborn M, Nave R, Weber K. The organization of titin filaments in the half-sarcomere revealed by monoclonal antibodies in immunoelectron microscopy: a map of ten nonrepetitive epitopes starting at the Z line extends close to the M line. J Cell Biol. 1988;106:1563–1572. doi: 10.1083/jcb.106.5.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautel M. Cytoskeletal protein kinases: titin and its relations in mechanosensing. Pflugers Archiv : European journal of physiology. 2011;462:119–134. doi: 10.1007/s00424-011-0946-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehmlich K, Dodd MS, William Allwood J, Kelly M, Bellahcene M, Lad HV, Stockenhuber A, Hooper C, Ashrafian H, Redwood CS, et al. Changes in the cardiac metabolome caused by perhexiline treatment in a mouse model of hypertrophic cardiomyopathy. Molecular bioSystems. 2014 doi: 10.1039/c4mb00594e. [DOI] [PubMed] [Google Scholar]
- Geisterfer-Lowrance AA, Christe M, Conner DA, Ingwall JS, Schoen FJ, Seidman CE, Seidman JG. A mouse model of familial hypertrophic cardiomyopathy. Science. 1996;272:731–734. doi: 10.1126/science.272.5262.731. [DOI] [PubMed] [Google Scholar]
- Gilbert R, Kelly MG, Mikawa T, Fischman DA. The carboxyl terminus of myosin binding protein C (MyBP-C, C-protein) specifies incorporation into the A-band of striated muscle. Journal of cell science. 1996;109:101–111. doi: 10.1242/jcs.109.1.101. Pt 1. [DOI] [PubMed] [Google Scholar]
- Girolami F, Ho CY, Semsarian C, Baldi M, Will ML, Baldini K, Torricelli F, Yeates L, Cecchi F, Ackerman MJ, et al. Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. Journal of the American College of Cardiology. 2010;55:1444–1453. doi: 10.1016/j.jacc.2009.11.062. [DOI] [PubMed] [Google Scholar]
- Golbus JR, Puckelwartz MJ, Dellefave-Castillo L, Fahrenbach JP, Nelakuditi V, Pesce LL, Pytel P, McNally EM. Targeted Analysis of Whole Genome Sequence Data to Diagnose Genetic Cardiomyopathy. Circ Cardiovasc Genet. 2014 doi: 10.1161/CIRCGENETICS.113.000578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golbus JR, Puckelwartz MJ, Fahrenbach JP, Dellefave-Castillo LM, Wolfgeher D, McNally EM. Population-based variation in cardiomyopathy genes. Circ Cardiovasc Genet. 2012;5:391–399. doi: 10.1161/CIRCGENETICS.112.962928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzier HL, Hutchinson KR, Tonino P, Methawasin M, Li FW, Slater RE, Bull MM, Saripalli C, Pappas CT, Gregorio CC, et al. Deleting titin's I-band/A-band junction reveals critical roles for titin in biomechanical sensing and cardiac function. Proc Natl Acad Sci U S A. 2014;111:14589–14594. doi: 10.1073/pnas.1411493111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzier HL, Radke MH, Peng J, Westermann D, Nelson OL, Rost K, King NM, Yu Q, Tschope C, McNabb M, et al. Truncation of titin's elastic PEVK region leads to cardiomyopathy with diastolic dysfunction. Circulation research. 2009;105:557–564. doi: 10.1161/CIRCRESAHA.109.200964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighi K, Kolokathis F, Gramolini AO, Waggoner JR, Pater L, Lynch RA, Fan GC, Tsiapras D, Parekh RR, Dorn GW, 2nd, et al. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc Natl Acad Sci U S A. 2006;103:1388–1393. doi: 10.1073/pnas.0510519103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SP, Bartley CR, Hacker TA, McDonald KS, Douglas PS, Greaser ML, Powers PA, Moss RL. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circulation research. 2002;90:594–601. doi: 10.1161/01.res.0000012222.70819.64. [DOI] [PubMed] [Google Scholar]
- Heidenreich PA, Albert NM, Allen LA, Bluemke DA, Butler J, Fonarow GC, Ikonomidis JS, Khavjou O, Konstam MA, Maddox TM, et al. Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ Heart Fail. 2013;6:606–619. doi: 10.1161/HHF.0b013e318291329a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms AS, Davis FM, Coleman D, Bartolone SN, Glazier AA, Pagani F, Yob JM, Sadayappan S, Pedersen E, Lyons R, et al. Sarcomere mutation-specific expression patterns in human hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2014;7:434–443. doi: 10.1161/CIRCGENETICS.113.000448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–628. doi: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10:531–547. doi: 10.1038/nrcardio.2013.105. [DOI] [PubMed] [Google Scholar]
- Ingles J, Doolan A, Chiu C, Seidman J, Seidman C, Semsarian C. Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling. J Med Genet. 2005;42:e59. doi: 10.1136/jmg.2005.033886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingwall JS. The energetic cost of contraction is higher in the myocardium of patients with hypertrophic cardiomyopathy. Cardiovascular research. 2014;103:192–193. doi: 10.1093/cvr/cvu145. [DOI] [PubMed] [Google Scholar]
- Jeong EM, Monasky MM, Gu L, Taglieri DM, Patel BG, Liu H, Wang Q, Greener I, Dudley SC, Jr., Solaro RJ. Tetrahydrobiopterin improves diastolic dysfunction by reversing changes in myofilament properties. Journal of molecular and cellular cardiology. 2013;56:44–54. doi: 10.1016/j.yjmcc.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Wakimoto H, Seidman JG, Seidman CE. Allele-specific silencing of mutant Myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science. 2013;342:111–114. doi: 10.1126/science.1236921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz AM. Ernest Henry Starling, his predecessors, and the "Law of the Heart". Circulation. 2002;106:2986–2992. doi: 10.1161/01.cir.0000040594.96123.55. [DOI] [PubMed] [Google Scholar]
- Kensler RW, Shaffer JF, Harris SP. Binding of the N-terminal fragment C0-C2 of cardiac MyBP-C to cardiac F-actin. Journal of structural biology. 2011;174:44–51. doi: 10.1016/j.jsb.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte FS, Herron TJ, Rovetto MJ, McDonald KS. Power output is linearly related to MyHC content in rat skinned myocytes and isolated working hearts. American journal of physiology. Heart and circulatory physiology. 2005;289:H801–812. doi: 10.1152/ajpheart.01227.2004. [DOI] [PubMed] [Google Scholar]
- Kunst G, Kress KR, Gruen M, Uttenweiler D, Gautel M, Fink RH. Myosin binding protein C, a phosphorylation-dependent force regulator in muscle that controls the attachment of myosin heads by its interaction with myosin S2. Circulation research. 2000;86:51–58. doi: 10.1161/01.res.86.1.51. [DOI] [PubMed] [Google Scholar]
- Labeit S, Gautel M, Lakey A, Trinick J. Towards a molecular understanding of titin. The EMBO journal. 1992;11:1711–1716. doi: 10.1002/j.1460-2075.1992.tb05222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EJ, Peng J, Radke M, Gotthardt M, Granzier HL. Calcium sensitivity and the Frank-Starling mechanism of the heart are increased in titin N2B region-deficient mice. Journal of molecular and cellular cardiology. 2010;49:449–458. doi: 10.1016/j.yjmcc.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman W, Hatch V, Korman V, Rosol M, Thomas L, Maytum R, Geeves MA, Van Eyk JE, Tobacman LS, Craig R. Tropomyosin and actin isoforms modulate the localization of tropomyosin strands on actin filaments. Journal of molecular biology. 2000;302:593–606. doi: 10.1006/jmbi.2000.4080. [DOI] [PubMed] [Google Scholar]
- Lekanne Deprez RH, Muurling-Vlietman JJ, Hruda J, Baars MJ, Wijnaendts LC, Stolte-Dijkstra I, Alders M, van Hagen JM. Two cases of severe neonatal hypertrophic cardiomyopathy caused by compound heterozygous mutations in the MYBPC3 gene. J Med Genet. 2006;43:829–832. doi: 10.1136/jmg.2005.040329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HL, Fujimoto N, Sasakawa N, Shirai S, Ohkame T, Sakuma T, Tanaka M, Amano N, Watanabe A, Sakurai H, et al. Precise Correction of the Dystrophin Gene in Duchenne Muscular Dystrophy Patient Induced Pluripotent Stem Cells by TALEN and CRISPR-Cas9. Stem Cell Reports. 2014 doi: 10.1016/j.stemcr.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long C, McAnally JR, Shelton JM, Mireault AA, Bassel-Duby R, Olson EN. Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA. Science. 2014;345:1184–1188. doi: 10.1126/science.1254445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowey S, Lesko LM, Rovner AS, Hodges AR, White SL, Low RB, Rincon M, Gulick J, Robbins J. Functional effects of the hypertrophic cardiomyopathy R403Q mutation are different in an alpha- or beta-myosin heavy chain backbone. The Journal of biological chemistry. 2008;283:20579–20589. doi: 10.1074/jbc.M800554200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR, Adams DR, Altman RB, Antonarakis SE, Ashley EA, et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508:469–476. doi: 10.1038/nature13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahrholdt H, Wagner A, Judd RM, Sechtem U, Kim RJ. Delayed enhancement cardiovascular magnetic resonance assessment of non-ischaemic cardiomyopathies. Eur Heart J. 2005;26:1461–1474. doi: 10.1093/eurheartj/ehi258. [DOI] [PubMed] [Google Scholar]
- Malik FI, Hartman JJ, Elias KA, Morgan BP, Rodriguez H, Brejc K, Anderson RL, Sueoka SH, Lee KH, Finer JT, et al. Cardiac myosin activation: a potential therapeutic approach for systolic heart failure. Science. 2011;331:1439–1443. doi: 10.1126/science.1200113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marian AJ. Pathogenesis of diverse clinical and pathological phenotypes in hypertrophic cardiomyopathy. Lancet. 2000;355:58–60. doi: 10.1016/s0140-6736(99)06187-5. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 1995;92:785–789. doi: 10.1161/01.cir.92.4.785. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Niimura H, Casey SA, Soper MK, Wright GB, Seidman JG, Seidman CE. Development of left ventricular hypertrophy in adults in hypertrophic cardiomyopathy caused by cardiac myosin-binding protein C gene mutations. Journal of the American College of Cardiology. 2001;38:315–321. doi: 10.1016/s0735-1097(01)01386-9. [DOI] [PubMed] [Google Scholar]
- Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, Jalilzadeh S, Carballo S, Redwood C, Watkins H. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circulation research. 2009;105:219–222. doi: 10.1161/CIRCRESAHA.109.202440. [DOI] [PubMed] [Google Scholar]
- McKillop DF, Geeves MA. Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophysical journal. 1993;65:693–701. doi: 10.1016/S0006-3495(93)81110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally EM, Golbus JR, Puckelwartz MJ. Genetic mutations and mechanisms in dilated cardiomyopathy. The Journal of clinical investigation. 2013;123:19–26. doi: 10.1172/JCI62862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mearini G, Stimpel D, Geertz B, Weinberger F, Kramer E, Schlossarek S, Mourot-Filiatre J, Stoehr A, Dutsch A, Wijnker PJ, et al. Mybpc3 gene therapy for neonatal cardiomyopathy enables long-term disease prevention in mice. Nat Commun. 2014;5:5515. doi: 10.1038/ncomms6515. [DOI] [PubMed] [Google Scholar]
- Moore JR, Leinwand L, Warshaw DM. Understanding cardiomyopathy phenotypes based on the functional impact of mutations in the myosin motor. Circulation research. 2012;111:375–385. doi: 10.1161/CIRCRESAHA.110.223842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RK, Abdullah S, Tardiff JC. Allosteric effects of cardiac troponin TNT1 mutations on actomyosin binding: a novel pathogenic mechanism for hypertrophic cardiomyopathy. Archives of biochemistry and biophysics. 2014;552553:21–28. doi: 10.1016/j.abb.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mun JY, Previs MJ, Yu HY, Gulick J, Tobacman LS, Beck Previs S, Robbins J, Warshaw DM, Craig R. Myosin-binding protein C displaces tropomyosin to activate cardiac thin filaments and governs their speed by an independent mechanism. Proc Natl Acad Sci U S A. 2014;111:2170–2175. doi: 10.1073/pnas.1316001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myhre JL, Hills JA, Prill K, Wohlgemuth SL, Pilgrim DB. The titin A-band rod domain is dispensable for initial thick filament assembly in zebrafish. Dev Biol. 2014;387:93–108. doi: 10.1016/j.ydbio.2013.12.020. [DOI] [PubMed] [Google Scholar]
- Olivotto I, Girolami F, Ackerman MJ, Nistri S, Bos JM, Zachara E, Ommen SR, Theis JL, Vaubel RA, Re F, et al. Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin Proc. 2008;83:630–638. doi: 10.4065/83.6.630. [DOI] [PubMed] [Google Scholar]
- Olivotto I, Girolami F, Sciagra R, Ackerman MJ, Sotgia B, Bos JM, Nistri S, Sgalambro A, Grifoni C, Torricelli F, et al. Microvascular function is selectively impaired in patients with hypertrophic cardiomyopathy and sarcomere myofilament gene mutations. Journal of the American College of Cardiology. 2011;58:839–848. doi: 10.1016/j.jacc.2011.05.018. [DOI] [PubMed] [Google Scholar]
- Pan S, Caleshu CA, Dunn KE, Foti MJ, Moran MK, Soyinka O, Ashley EA. Cardiac structural and sarcomere genes associated with cardiomyopathy exhibit marked intolerance of genetic variation. Circ Cardiovasc Genet. 2012;5:602–610. doi: 10.1161/CIRCGENETICS.112.963421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel BG, Wilder T, Solaro RJ. Novel control of cardiac myofilament response to calcium by S-glutathionylation at specific sites of myosin binding protein C. Front Physiol. 2013;4:336. doi: 10.3389/fphys.2013.00336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Previs MJ, Beck Previs S, Gulick J, Robbins J, Warshaw DM. Molecular mechanics of cardiac myosin-binding protein C in native thick filaments. Science. 2012;337:1215–1218. doi: 10.1126/science.1223602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard P, Isnard R, Carrier L, Dubourg O, Donatien Y, Mathieu B, Bonne G, Gary F, Charron P, Hagege M, et al. Double heterozygosity for mutations in the beta-myosin heavy chain and in the cardiac myosin binding protein C genes in a family with hypertrophic cardiomyopathy. J Med Genet. 1999;36:542–545. [PMC free article] [PubMed] [Google Scholar]
- Rickers C, Wilke NM, Jerosch-Herold M, Casey SA, Panse P, Panse N, Weil J, Zenovich AG, Maron BJ. Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation. 2005;112:855–861. doi: 10.1161/CIRCULATIONAHA.104.507723. [DOI] [PubMed] [Google Scholar]
- Ritchie GR, Flicek P. Computational approaches to interpreting genomic sequence variation. Genome Med. 2014;6:87. doi: 10.1186/s13073-014-0087-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, Buchan RJ, Walsh R, John S, Wilkinson S, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. 2015;7 doi: 10.1126/scitranslmed.3010134. 270ra276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadayappan S, Gulick J, Osinska H, Martin LA, Hahn HS, Dorn GW, 2nd, Klevitsky R, Seidman CE, Seidman JG, Robbins J. Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circulation research. 2005;97:1156–1163. doi: 10.1161/01.RES.0000190605.79013.4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadayappan S, Osinska H, Klevitsky R, Lorenz JN, Sargent M, Molkentin JD, Seidman CE, Seidman JG, Robbins J. Cardiac myosin binding protein C phosphorylation is cardioprotective. Proc Natl Acad Sci U S A. 2006;103:16918–16923. doi: 10.1073/pnas.0607069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer E, Helms P, Marcellin L, Desprez P, Billaud P, Chanavat V, Rousson R, Millat G. Next-generation sequencing (NGS) as a fast molecular diagnosis tool for left ventricular noncompaction in an infant with compound mutations in the MYBPC3 gene. Eur J Med Genet. 2014;57:129–132. doi: 10.1016/j.ejmg.2014.02.015. [DOI] [PubMed] [Google Scholar]
- Schmitt JP, Debold EP, Ahmad F, Armstrong A, Frederico A, Conner DA, Mende U, Lohse MJ, Warshaw D, Seidman CE, et al. Cardiac myosin missense mutations cause dilated cardiomyopathy in mouse models and depress molecular motor function. Proc Natl Acad Sci U S A. 2006;103:14525–14530. doi: 10.1073/pnas.0606383103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semsarian C, Healey MJ, Fatkin D, Giewat M, Duffy C, Seidman CE, Seidman JG. A polymorphic modifier gene alters the hypertrophic response in a murine model of familial hypertrophic cardiomyopathy. Journal of molecular and cellular cardiology. 2001;33:2055–2060. doi: 10.1006/jmcc.2001.1466. [DOI] [PubMed] [Google Scholar]
- Simonson TS, Zhang Y, Huff CD, Xing J, Watkins WS, Witherspoon DJ, Woodward SR, Jorde LB. Limited distribution of a cardiomyopathy-associated variant in India. Ann Hum Genet. 2010;74:184–188. doi: 10.1111/j.1469-1809.2010.00561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommese RF, Sung J, Nag S, Sutton S, Deacon JC, Choe E, Leinwand LA, Ruppel K, Spudich JA. Molecular consequences of the R453C hypertrophic cardiomyopathy mutation on human beta-cardiac myosin motor function. Proc Natl Acad Sci U S A. 2013;110:12607–12612. doi: 10.1073/pnas.1309493110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spindler M, Saupe KW, Christe ME, Sweeney HL, Seidman CE, Seidman JG, Ingwall JS. Diastolic dysfunction and altered energetics in the alphaMHC403/+ mouse model of familial hypertrophic cardiomyopathy. The Journal of clinical investigation. 1998;101:1775–1783. doi: 10.1172/JCI1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A, Garg N, Mittal T, Khanna R, Gupta S, Seth PK, Mittal B. Association of 25 bp deletion in MYBPC3 gene with left ventricle dysfunction in coronary artery disease patients. PLoS One. 2011;6:e24123. doi: 10.1371/journal.pone.0024123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss KA, DuBiner L, Simon M, Zaragoza M, Sengupta PP, Li P, Narula N, Dreike S, Platt J, Procaccio V, et al. Severity of cardiomyopathy associated with adenine nucleotide translocator-1 deficiency correlates with mtDNA haplogroup. Proc Natl Acad Sci U S A. 2013;110:3453–3458. doi: 10.1073/pnas.1300690110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Carlson KM, Marchuk DA, Rockman HA. Genetic modifier loci affecting survival and cardiac function in murine dilated cardiomyopathy. Circulation. 2002;105:1824–1829. doi: 10.1161/01.cir.0000014926.32463.89. [DOI] [PubMed] [Google Scholar]
- Tardiff JC. Thin filament mutations: developing an integrative approach to a complex disorder. Circulation research. 2011;108:765–782. doi: 10.1161/CIRCRESAHA.110.224170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teekakirikul P, Kelly MA, Rehm HL, Lakdawala NK, Funke BH. Inherited cardiomyopathies: molecular genetics and clinical genetic testing in the postgenomic era. J Mol Diagn. 2013;15:158–170. doi: 10.1016/j.jmoldx.2012.09.002. [DOI] [PubMed] [Google Scholar]
- Tyska MJ, Hayes E, Giewat M, Seidman CE, Seidman JG, Warshaw DM. Single-molecule mechanics of R403Q cardiac myosin isolated from the mouse model of familial hypertrophic cardiomyopathy. Circulation research. 2000;86:737–744. doi: 10.1161/01.res.86.7.737. [DOI] [PubMed] [Google Scholar]
- van der Zwaag PA, van Rijsingen IA, Asimaki A, Jongbloed JD, van Veldhuisen DJ, Wiesfeld AC, Cox MG, van Lochem LT, de Boer RA, Hofstra RM, et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: evidence supporting the concept of arrhythmogenic cardiomyopathy. European journal of heart failure. 2012;14:1199–1207. doi: 10.1093/eurjhf/hfs119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk SJ, Boontje NM, Heymans MW, Ten Cate FJ, Michels M, Dos Remedios C, Dooijes D, van Slegtenhorst MA, van der Velden J, Stienen GJ. Preserved cross-bridge kinetics in human hypertrophic cardiomyopathy patients with MYBPC3 mutations. Pflugers Archiv : European journal of physiology. 2014;466:1619–1633. doi: 10.1007/s00424-013-1391-0. [DOI] [PubMed] [Google Scholar]
- van Dijk SJ, Dooijes D, dos Remedios C, Michels M, Lamers JM, Winegrad S, Schlossarek S, Carrier L, ten Cate FJ, Stienen GJ, et al. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation. 2009;119:1473–1483. doi: 10.1161/CIRCULATIONAHA.108.838672. [DOI] [PubMed] [Google Scholar]
- van Rijsingen IA, van der Zwaag PA, Groeneweg JA, Nannenberg EA, Jongbloed JD, Zwinderman AH, Pinto YM, Dit Deprez RH, Post JG, Tan HL, et al. Outcome in phospholamban R14del carriers: results of a large multicentre cohort study. Circ Cardiovasc Genet. 2014;7:455–465. doi: 10.1161/CIRCGENETICS.113.000374. [DOI] [PubMed] [Google Scholar]
- Veltrop M, Aartsma-Rus A. Antisense-mediated exon skipping: taking advantage of a trick from Mother Nature to treat rare genetic diseases. Exp Cell Res. 2014;325:50–55. doi: 10.1016/j.yexcr.2014.01.026. [DOI] [PubMed] [Google Scholar]
- Vikstrom KL, Factor SM, Leinwand LA. Mice expressing mutant myosin heavy chains are a model for familial hypertrophic cardiomyopathy. Mol Med. 1996;2:556–567. [PMC free article] [PubMed] [Google Scholar]
- Walsh R, Rutland C, Thomas R, Loughna S. Cardiomyopathy: a systematic review of disease-causing mutations in myosin heavy chain 7 and their phenotypic manifestations. Cardiology. 2010;115:49–60. doi: 10.1159/000252808. [DOI] [PubMed] [Google Scholar]
- Weith A, Sadayappan S, Gulick J, Previs MJ, Vanburen P, Robbins J, Warshaw DM. Unique single molecule binding of cardiac myosin binding protein-C to actin and phosphorylation-dependent inhibition of actomyosin motility requires 17 amino acids of the motif domain. Journal of molecular and cellular cardiology. 2012a;52:219–227. doi: 10.1016/j.yjmcc.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weith AE, Previs MJ, Hoeprich GJ, Previs SB, Gulick J, Robbins J, Warshaw DM. The extent of cardiac myosin binding protein-C phosphorylation modulates actomyosin function in a graded manner. Journal of muscle research and cell motility. 2012b;33:449–459. doi: 10.1007/s10974-012-9312-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessels MW, Herkert JC, Frohn-Mulder IM, Dalinghaus M, van den Wijngaard A, de Krijger RR, Michels M, de Coo IF, Hoedemaekers YM, Dooijes D. Compound heterozygous or homozygous truncating MYBPC3 mutations cause lethal cardiomyopathy with features of noncompaction and septal defects. Eur J Hum Genet. 2014 doi: 10.1038/ejhg.2014.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler FC, Tang H, Marks OA, Hadnott TN, Chu PL, Mao L, Rockman HA, Marchuk DA. Tnni3k modifies disease progression in murine models of cardiomyopathy. PLoS Genet. 2009;5:e1000647. doi: 10.1371/journal.pgen.1000647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witjas-Paalberends ER, Ferrara C, Scellini B, Piroddi N, Montag J, Tesi C, Stienen GJ, Michels M, Ho CY, Kraft T, et al. Faster cross-bridge detachment and increased tension cost in human hypertrophic cardiomyopathy with the R403Q MYH7 mutation. The Journal of physiology. 2014a;592:3257–3272. doi: 10.1113/jphysiol.2014.274571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witjas-Paalberends ER, Guclu A, Germans T, Knaapen P, Harms HJ, Vermeer AM, Christiaans I, Wilde AA, Dos Remedios C, Lammertsma AA, et al. Gene-specific increase in the energetic cost of contraction in hypertrophic cardiomyopathy caused by thick filament mutations. Cardiovascular research. 2014b;103:248–257. doi: 10.1093/cvr/cvu127. [DOI] [PubMed] [Google Scholar]
- Xie F, Ye L, Chang JC, Beyer AI, Wang J, Muench MO, Kan YW. Seamless gene correction of beta-thalassemia mutations in patient-specific iPSCs using CRISPR/Cas9 and piggyBac. Genome Res. 2014;24:1526–1533. doi: 10.1101/gr.173427.114. [DOI] [PMC free article] [PubMed] [Google Scholar]