

Abstract
Purpose
The major cause of morbidity in breast cancer is development of metastatic disease, for which few effective therapies exist. Since tumor cell dissemination is often an early event in breast cancer progression and can occur prior to diagnosis, new therapies need to focus on targeting established metastatic disease in secondary organs. We report an effective therapy based on targeting cell surface-localized glucose regulated protein 78 (GRP78). GRP78 is expressed normally in the endoplasmic reticulum, but many tumors and disseminated tumor cells are subjected to environmental stresses and exhibit elevated levels of GRP78, some of which is localized at the plasma membrane.
Experimental Design and Results
Here we show that matched primary tumors and metastases from patients who died from advanced breast cancer also express high levels of GRP78. We utilized a peptidomimetic targeting strategy that employs a known GRP78-binding peptide fused to a pro-apoptotic moiety (designated BMTP78) and show that it can selectively kill breast cancer cells that express surface-localized GRP78. Further, in preclinical metastasis models, we demonstrate that administration of BMTP78 can inhibit primary tumor growth as well as prolong overall survival by reducing the extent of outgrowth of established lung and bone micrometastases.
Conclusions
The data presented here provide strong evidence that it is possible to induce cell death in established micrometastases by peptide mediated targeting of cell surface localized GRP in advanced breast cancers. The significance to patients with advanced breast cancer of a therapy that can reduce established metastatic disease should not be underestimated.
Keywords: Breast cancer, metastasis, GRP78, targeted therapy, cell stress
INTRODUCTION
Breast cancer is a major cause of mortality in woman of all racial and ethnic backgrounds (1). Although advances in early detection, surgical techniques, chemotherapy and diagnostic imaging have improved overall survival, the prognosis for patients with metastatic breast cancer remains poor (2). There is now increasing evidence to support the observation that metastasis is an early event in breast cancer progression, with possibly up to 90% of patients already having disseminated tumor cells at diagnosis (3). Thus, therapies that aim to prevent cancer cell escape from the primary tumor may be less effective than those that focus on inhibition of the outgrowth of established micrometastases. Further, strategies that target the primary neoplasm often become ineffective in late stage disease as secondary tumors evolve drug resistance. The realization that circulating tumor cells (CTCs) in peripheral blood and disseminated tumor cells (DTCs) lodged in bone marrow are early predictors of clinical outcome in multiple cancers, now enables the stratification of patients who are candidates for therapeutic targeting of metastatic disease (4).
Tumor cell-targeted drug delivery can enhance the therapeutic value of chemotherapy by reducing systemic toxicity (5). One potential target for drug delivery is glucose regulated protein 78 (GRP78/BiP), which normally resides in the endoplasmic reticulum and acts as a molecular chaperone to assist in protein folding and assembly (6). GRP78 is instrumental to the initiation of the unfolded protein response (UPR), which is considered to be a pro-survival mechanism adopted by cells in response to various environmental stresses including acidosis, glucose deprivation and hypoxia (7). Due to the continual metabolic demand of growing tumors, often in a stressful environment with low blood supply, levels of GRP78 become elevated in many cancers (8–11), including both hormone receptor positive and negative breast cancer (12–14). Indeed, gene expression profiling indicates that high levels of GRP78 expression are associated with a shorter relapse-free survival in breast cancer patients (15). Further, in a cohort of 127 patients, two-thirds of the primary breast tumors were immunoreactive for GRP78, with high expression correlating negatively with response towards doxorubicin-based therapy (12).
GRP78 can localize to the plasma membrane in cancer cells and tumor-associated vasculature (16–19), offering an opportunity for disease-specific targeting. The mechanism by which it localizes to the plasma membrane remains unclear. Several suggestions with some supporting evidence have been proposed (20), but it is evident that stress conditions in the cell can increase GRP78 levels and result in re-localization of the protein to the plasma membrane (21). GRP78 binds to cell surface localized Cripto, leading to an inhibition of TGFβ and an enhancement of LIMK1 signaling that subsequently promotes tumor cell growth and motility (22).
Previously, we epitope-mapped, or “fingerprinted” the circulating pool of antibodies elicited against tumors in cancer patients, leading to the isolation of GRP78 as a target in prostate and breast cancer (17, 23). Multiple GRP78 binding peptides have been identified, with the WIFPWIQL (amino acid sequence) peptide displaying the highest efficacy (24). By coupling a pro-apoptotic moiety to this GRP78-targeting peptide, we created BMTP78 (bone metastasis targeting peptide-78) and demonstrated its ability to induce apoptosis following receptor-mediated internalization within cancer cells (17). Here we demonstrate using three different preclinical models that this targeting strategy can effectively reduce established metastatic disease as well as primary tumor burden, leading to prolonged survival in tumor-challenged mice. We also show the specificity of this therapy to tumor cells by comparing the response to the peptide of both normal and tumor cells when grown in oxygen concentrations similar to those found in tissues. Finally, we demonstrate that high levels of GRP78 are retained in metastatic tumors of our preclinical model and in secondary lesions derived from breast cancer patients.
MATERIALS AND METHODS
Cell culture
The neomycin resistant, mCherry fluorescent protein expressing mouse mammary tumor lines 67NR, 66cl4, EF43-fgf4 and 4T1.2 were cultured to 70% confluence in alpha minimal essential medium (αMEM) containing 5% fetal calf serum (FCS), 50U/ml penicillin G and 50μg/ml streptomycin sulphate (25). Normal NMuMG mammary epithelial cells were cultured to 70% confluence in Dulbecco’s MEM containing 10% FCS, 100IU/ml insulin, 50U/ml penicillin G and 50μg/ml streptomycin sulphate. Immortalized normal human breast epithelial MCF10A cells obtained from ATCC and used at early passage were cultured in MEM/F-12 containing 5% horse serum, insulin 100IU/ml, 20ng/ml epidermal growth factor (EGF) and 0.5μg/ml hydrocortisone. Human breast tumor lines MCF-7 and MDA-MB-231-luc (authenticated by CellBank Australia in 2011 by STR profiling) were cultured in αMEM containing 10% FCS, 1mM pyruvate and 100 IU/ml insulin. MDA-MB-468, SkBr3 and BT474 human breast carcinoma cells were cultured in RPMI 1640-HEPES containing 10% FCS, 50U/ml penicillin G and 50μg/ml streptomycin sulphate. Cells were incubated at 37°C with 5% CO2 and 95% air or with 5% CO2 and 10% O2 in N2.
Peptides
The peptide WIFPWIQL-GG-D(KLAKLAK)2 (designated BMTP78) and the control peptide WIFPWIQL-GG-D(KLAKLAK), which contains an ineffective apoptotic moiety fused to the GRP78-homing domain, were synthesized by GL Biochemical Ltd (Shanghai, China) or by PolyPeptide (CA, USA). All peptides were prepared to 95% purity and the quality was assessed by the manufacturers using HPLC.
In vitro peptide-mediated cell killing assays
Cells (10,000) were seeded into wells of a 24 well plate and allowed to establish overnight prior to treatment with either a control peptide or BMTP78 at the specified concentration for 20 hours. In some experiments, the peptide incubation followed a pre-treatment for one hour with 1μg/ml goat anti-GRP78 polyclonal antibody (#sc-1050, Santa Cruz Biotechnology) or with an isomatched control IgG antibody. To determine the effects of lowered oxygen concentration on surface GRP78 levels and on the efficacy of BMTP78 treatment, cells were grown for several passages in 1%, 5% or 10% O2 prior to analysis. Following treatment, WST-1 reagent (Roche) was added to the cultures and cell viability was measured using a spectrophotometric assay, according to manufacturer’s instructions. Viability was scored in triplicate for all time points, doses and treatments.
Flow cytometry
Cells were incubated with primary anti-GRP78 antibody (#sc-1050 Santa Cruz) diluted to 1:200 for one hour followed by FITC-conjugated donkey anti-goat IgG (#5-095-147 Jackson Laboratories) diluted to 1:200 for 30 min. Between incubations, cells were washed twice with blocking solution (2% FCS in PBS). Cells were analyzed on a Becton Dickinson FACS DiVa flow cytometer using FCS3 software. For analysis of tumor cells isolated from tissues, we used 4T1.2-mCherry cells. Excised organs and tumors were disaggregated in 10 ml DMEM containing 1mg/ml collagenase A (Roche) for 30min at 37°C, followed by filtration through a 40μm nylon gauze (BD Falcon, MA, USA). Erythrocytes were removed by a brief incubation in red blood cell lysis buffer (1M NH4Cl, 100mM KHCO3, 0.5M EDTA) and the remaining cells were suspended in blocking solution containing a viability dye. The proportion of mCherry-positive tumor cells that expressed GRP78 was assessed as described above. In the case of non-tumor burdened tissues, cell surface-localized GRP78 was assessed on all viable dissociated cells, as assessed by propidium iodide staining.
BMTP78 therapy in tumor bearing mice
Female Balb/c and SCID mice (6–8 weeks) were obtained from the Walter and Eliza Hall Institute (Melbourne, Australia). All animal work was performed following approval from the animal ethics committee of the Peter MacCallum Cancer Centre. 67NR, 66cl4 and 4T1.2 tumor cells (1×105) were implanted into the fourth mammary gland of Balb/c mice. MDA-MB-231-luc (2×106) and EF43-fgf4-mCherry cells (2×105) were injected intravenously (lateral tail vein) into SCID and Balb/c mice respectively. Mice were treated at weekly intervals by intraperitoneal injection with 15mg/kg BMTP78 or control peptides or saline vehicle following confirmation of established growth of the lesions. Primary tumor growth was measured with electronic calipers. Metastatic burden of 4T1.2 tumors in lung and spine was analyzed by multiplexed, TaqMan-based quantitative genomic PCR (qPCR) of the neomycin resistance gene present in the tumor cells, as described previously (25). Similarly, metastatic burden of EF43-fgf4 cells was measured by qPCR of the mCherry reporter gene present in these cells. Data are expressed as the relative metastatic burden, which is calculated based on the difference in the number of PCR cycles necessary to amplify the neomycin gene to a threshold value compared to a ubiquitous gene (vimentin) that is present in all cells. Lung burden in mice bearing MDA-MB-231-luc tumors was scored by bioluminescence. For Kaplan-Meier analysis of disease-free survival, the 4T1.2 mammary tumors were excised surgically ten days after implantation in the mammary gland. Intraperitoneal administration of BMTP78 peptide or control peptide (15mg/kg) or saline commenced 24 hours following primary tumor excision and was repeated at weekly intervals. Mice were monitored daily and culled at the first sign of stress due to metastatic disease, which was confirmed at autopsy.
Immunohistochemistry
Levels of GRP78 were measured on formalin-fixed, paraffin-embedded (FFPE) sections on five tissue arrays comprising matched human primary breast cancers and metastases (26). GRP78 was also measured in FFPE tissues obtained from tumor-bearing Balb/c mice. Sections were treated with 1μg/ml primary anti-GRP78 antibody (#sc-1050 Santa Cruz Biotechnology) or with isotype control antibody overnight at 4°C. Following several washes and incubation in anti-goat HRP-conjugated secondary antibody (#sc-2020 Santa Cruz Biotechnology), GRP78 was detected with diaminobenzidine chromogenic substrate (DAKO).
BMTP78 toxicity and stability
BMTP78 is remarkably stable, necessitating treatment only once per week to achieve efficacy. Indeed, we have determined by MALDI-TOF that these peptides are stable for up to 7 days at 37°C and can be detected in the circulation of mice for at least 4 hours following intravenous injection. Preliminary studies using Cynomolgus monkeys indicate that BMTP78 has an estimated half-life of 90 minutes at a dose of 6mg/kg (unpublished data). BMTP78 was administered to mice at 15mg/kg as based on a dose finding study in rodents (Study number 02-07-01741-1 at the Michael E. Keeling Center for Comparative Medicine and Research, UTMDACC). At this dose, a mild, but reversible nephrosis was observed. Structurally similar compounds containing the D(KLAKLAK)2 motif have previously been administered to mice (17, 27) and primates (28) with similar effects in the kidneys.
Statistical Analyses
Empirical data were compiled and analyzed using Prism 5 software. Results were considered significantly different if p<0.05, according to the specific statistical analysis used, as described in each figure legend.
RESULTS
GRP78 is expressed in primary human breast cancers and matched metastases
Using tissue microarrays, we assessed the levels of GRP78 by immunohistochemistry in matched primary and secondary tumors obtained posthumously from breast cancer patients (26). We detected strong immunostaining of GRP78 in both primary tumors and metastases in a variety of organs in all patients (Figure 1), indicating that GRP78-targeted therapeutics could be valuable for treating breast cancer and associated metastases. It is important to note that given the abundant cytoplasmic levels of GRP78, it is not possible to distinguish cell surface localized GRP78 by immunohistochemistry. However, the point is made that tumors from patients who died from metastatic disease express abundant GRP78, some of which is likely to be surface localized.
Figure 1. GRP78 staining of primary breast carcinomas and their matched metastases.
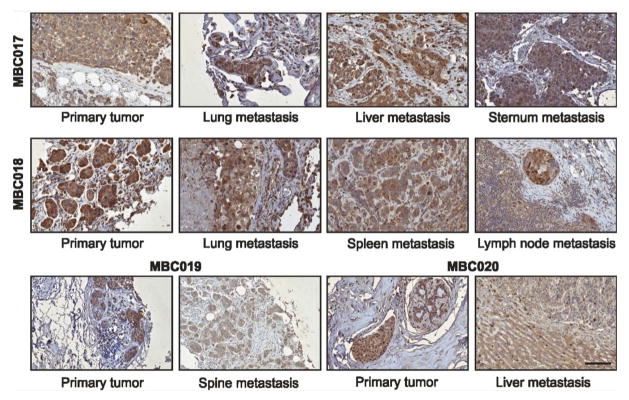
Primary tumors and matched metastases immunostained for GRP78 are shown for four of the five patients, MBC017, MBC018, MBC019 and MBC20. Scale bar is 100μm and applies to all images.
GRP78 is expressed in metastatic mouse mammary tumors
The 4T1 mammary tumor model is a syngeneic model consisting of cell lines with varying metastatic capacities, ranging from the non- or weakly metastatic lines, 67NR and 66cl4, to the highly metastatic 4T1.2 that spreads to several sites including bone after primary tumor growth in the mammary gland (25, 29, 30). To determine the utility of this model for GRP78-targeted therapy, we assessed GRP78 levels in primary tumors and matched distant metastases by immunohistochemistry. Minimal GRP78 protein was observed in epithelial cancer cells of 67NR and 66cl4 primary tumors (Figure 2A). In contrast, high levels of GRP78 were detected in the aggressive 4T1.2 primary tumors, which persisted in matched, spontaneous metastases within the lung, heart, kidney and bone (Figure 2A).
Figure 2. GRP78 expression is elevated in highly metastatic tumors and is available for therapeutic targeting.

A) Immunolocalization of GRP78 in a panel of syngeneic mouse mammary tumors and in spontaneous kidney, femur, heart and lung metastases from 4T1.2 tumors. Elevated GRP78 levels are observed in cancer cells relative to host stroma. m, metastatic cancer cells, k, kidney epithelium, g, glomeruli, b, bone, c, cardiac muscle, e, lung epithelium. Scale bars represent 50μm except on lung metastasis image (25μm). B) FACS analysis of cell surface GRP78 in cells isolated from normal mouse organs and from 4T1.2 primary and secondary lesions (n=3 mice). C) Viability of metastatic 4T1.2 cells after exposure to BMTP78. The control peptide is WIFPWIQL-GG-D(KLAKLAK). D) The ability of BMTP78 to kill 4T1.2 cells following a 1h pretreatment with 1μg/ml of anti-GRP78 antibody or with the isotype matched IgG control. All results are expressed relative to the zero BMTP78 value, set at 100%. Mean values are shown, error bars represent SEM. *P<0.05, **P<0.01, two-tailed t-test.
Localization of GRP78 to the surface of tumor cells is a prerequisite for the success of GRP78-targeted therapy in metastatic breast cancer. mCherry fluorescent tumor cells were isolated from several 4T1.2 primary tumors and matched metastases, and the level of surface localized GRP78 was assessed by flow cytometry. The number of cells positive for cell surface GRP78 was significantly higher in the primary tumor cells than in cells isolated from normal non-cancerous organs (Figure 2B & Supplementary Figure 1). The number of GRP78 positive tumor cells further increased in matched metastases in the spine, lung, heart and axillary lymph nodes (ALN) (Figure 2B), indicating that GRP78 is indeed a rational therapeutic target in both primary and secondary breast tumors.
Peptide-mediated cell killing of human and mouse mammary cell lines in vitro
WIFPWIQL was identified previously as a GRP78-binding peptide (17) (24). BMTP78, a GRP78-targeted therapeutic, was generated by coupling WIFPWIQL to the pro-apoptotic peptide D(KLAKLAK)2 (31) that kills cells by disrupting mitochondrial membrane permeability following internalization. We first tested the efficacy of BMTP78 on mouse mammary tumor cells in vitro. BMTP78 caused a dose-dependent loss of viability of 4T1.2 cells, while control peptides lacking either a functional D(KLAKLAK)2 motif (WIFPWIQL-GG-D(KLAKLAK) or the GRP78-targeting peptide alone, had no effect (Figure 2C and data not shown). Pretreatment of the cells with an anti-GRP78 antibody, which in itself had no effect on viability (Supplementary Figure 2), prevented BMTP78-mediated cytotoxicity (Figure 2D). Thus, the WIFPWIQL peptide selectively targets the tumor cell surface-localized GRP78, enabling internalization of BMTP78 and cell death.
The response of five human breast tumor lines and one non-tumorigenic line (MCF10A) to BMTP78 was also evaluated. As we reported previously (17), all lines were sensitive to 20μM BMTP78 regardless of tumorigenic status, and all were protected by pretreatment with the anti-GRP78 antibody (Supplementary Figure 3). We hypothesized that the lack of differential response between transformed and non-transformed cells was due to the oxidative stress of tissue culture in atmospheric levels of oxygen (20%). To explore this, we cultured normal and transformed human breast and mouse mammary epithelial cells in 1%, 5%, 10% or 20% O2 for at least 24h. Physiological O2 levels in tissues range from 2% in the brain to 3–6% for most tissues apart from lung, which is around 13%. Growth in 5% or 10% O2 significantly reduced the number of surface GRP78-positive cells in two non-transformed lines, MCF10A and NMuMG (Figure 3A&D and Supplementary Figure 4) and significantly decreased their sensitivity to BMTP78 (Figure 3B&E). In contrast, the number of surface GRP78 positive cells (Figure 3A&D and Supplementary Figure 4) and the response to BMTP78 (Figure 3C&F) of the two tumorigenic lines, MDA-MB-231 and 4T1.2, was not altered by the oxygen concentration in which they were grown. Thus, under more physiological oxygen concentrations, normal cells present minimal GRP78 on the cell surface and remain unresponsive to BMTP78, whereas tumor cells are in a constant state of stress, regardless of their environment (32). As reported by others (33, 34), growth under hypoxic conditions (1% oxygen) induced a stress response and an increase in surface GRP78 in the two non-transformed lines but had no effect on the two tumor lines (Supplementary Figure 4).
Figure 3. Atmospheric oxygen concentrations increase surface localized GRP78 in non-tumorigenic cells.

The effect of oxygen on cell surface expression of GRP78 in human (A) or mouse (D) normal breast epithelial or tumorigenic cells was determined by flow cytometry. Human cells (B, C) or mouse cells (E, F) grown in either 10% or 20% O2 were pre-incubated with 1μg/ml of anti-GRP78 antibody or the isotype matched IgG control for 1h before challenge with 20μM BMTP78 or the control peptide (WIFPWIQL-GG-D(KLAKLAK). Results are expressed as percent viable cells compared to the control peptide. Mean values are depicted; error bars, SEM. *P<0.05, **P<0.01. # P<0.01 for cells in 10% O2 compared to 20% O2, two-tailed t-test.
BMTP78 therapy suppresses primary tumor growth and metastasis leading to prolonged disease-free survival
To assess the anti-tumor activity of BMTP78 in vivo, mice bearing 67NR, 66cl4 or 4T1.2 mammary tumors were treated once weekly with either 15mg/kg BMTP78 or control peptide. The administration of four doses of these peptides did not cause any lasting weight loss or general health issues in the tumor-bearing mice (Supplementary Figure 5). BMTP78 did not alter the growth rate of low GRP78 expressing 67NR or 66cl4 tumors (Figure 4A&B) but delayed 4T1.2 primary tumor growth (data not shown), resulting in significantly smaller tumors at endpoint (p<0.05) (Figure 4C). Spontaneous metastatic tumor burden in lung and spine was also decreased significantly (p<0.05) (Figures 4D&E). However, since primary tumor growth was altered, a specific effect on metastasis could not be delineated from these data.
Figure 4. GRP78-mediated suppression of tumor growth and metastasis following BMTP78 treatment.
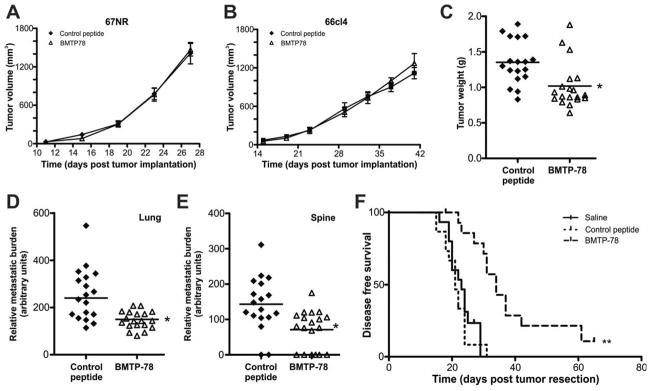
Mice bearing 67NR (n=4) (A), 66cl4 (n=4) (B) or 4T1.2 (n=18) (C) mammary tumors were treated once weekly with 15mg/Kg BMTP78 or control peptide, commencing the day after tumor cell inoculation. Primary tumor growth was measured using electronic calipers. 4T1.2 primary tumors were weighed upon excision at the end of the experiment (C). Metastatic burden from 4T1.2 tumors in lung (D) and spine (E) was measured by qPCR. (F) Metastasis-free survival of mice treated with BMTP78, the control peptide or saline (n=15/group) administered once weekly, commencing the day after surgical resection of the primary 4T1.2 tumor on day ten. Mice were monitored daily and culled at the first signs of distress or ill-health due to manifestation of visible lung metastasis (confirmed at autopsy). For panels A, B, each point represents the mean+/−SEM. For panels C, D and E, each point represents the primary tumor weight (C) or metastatic burden (D, E) of an individual mouse, with the bar indicating the mean for the group. *p<0.05, two-tailed t-test. For panel F, **p<0.01, Log-rank test.
To address this, we tested the ability of BMTP78 to prolong disease-free survival in mice bearing established micrometastatic disease. 4T1.2 mammary tumors were excised surgically from all mice at ten days post-implantation prior to randomization into three groups (n=15) to receive weekly doses of BMTP78, control peptide or saline. Metastatic cells are present in the lungs at this time (33). BMTP78 treatment significantly extended disease-free survival of the mice (median survival 34 days), compared to either the control peptide (median survival 21 days) or the saline-treated controls (median survival 23 days) (Figure 4F) (p<0.01). While there was a significant delay in the onset of disease in the BMTP78 treated group, there were no visible differences at autopsy in the distribution of metastases between the three groups.
To extend these findings, we tested BMTP78 therapy in two experimental lung metastasis models using human MDA-MB-231 cells or mouse EF43-fgf4 mammary tumor cells, both of which are sensitive to BMTP78 (Figure 3, and (17)). Mice were administered two doses of BMTP78 at weekly intervals after allowing tumor cells to commence growth in the lungs. In both models, a significant reduction in the expansion of experimental lung metastases was demonstrated (Figure 5). Taken together, these data demonstrate that BMTP78 can reduce the growth of aggressive breast tumors and promote survival through the targeted killing of metastatic cells.
Figure 5. BMTP78 reduces growth of established lung metastases.
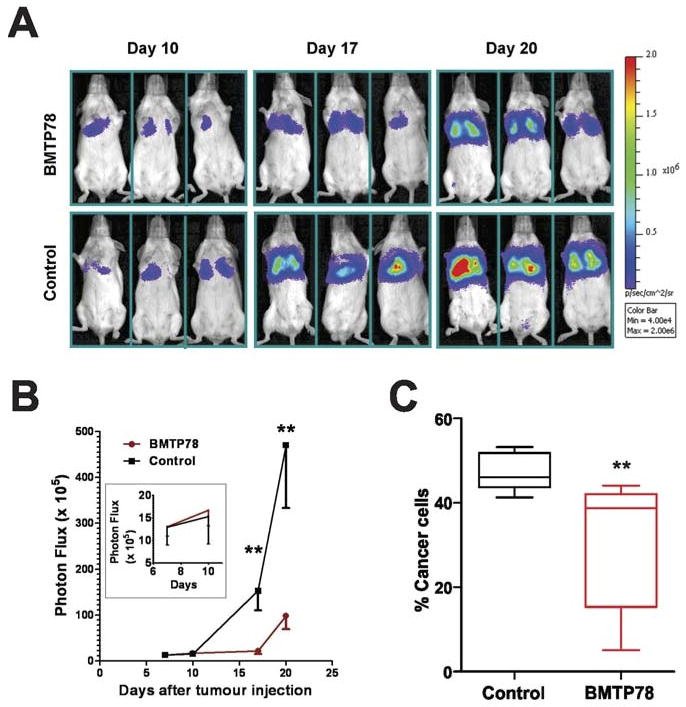
(A) Mice bearing established MDA-MB-231-luc lung metastases (n=7/group) were treated with BMTP78 on days 10 and 17 after tumor cell inoculation and monitored by in vivo bioluminescence imaging. (B) Assessment of lung tumor burden by bioluminescence. The inset shows bioluminescence prior to the first treatment on day 10, demonstrating that the tumors were growing in the lung prior to therapy. (C) Assessment of EF43-fgf4-mCherry lung tumor burden (n=5/group) on day 16 following treatment on days 3 and 10. The number of tumor cells in the lung was determined by flow cytometry comparing the proportion of viable mCherry-positive cells relative to viable mCherry-negative cells. **P<0.05 analysed by two-way Anova.
DISCUSSION
Successful therapy for established metastatic disease in breast cancer remains a major challenge in the clinic. Five-year survival drops from 90% for early stage, non-metastatic breast cancer to 20% once tumor progression has occurred (2). In one study, GRP78 was found to be positive in 76% of primary breast cancer cases, but did not correlate with tumor size, grade, lymph node metastases, vascular invasion, histological type or Nottingham Prognostic Index (13). However, expression of GRP78 has value in the prediction of tumor response to doxorubicin and to taxanes (12, 35). While high levels of GRP78 predict a poor response to doxorubicin, expression of GRP78 predicts a better response to doxorubicin followed by treatment with paclitaxel or docetaxel (12, 35). The mechanism behind these differential responses is not well understood.
In recent years, GRP78 has emerged as a prime target for anti-cancer treatment because of its elevated expression in tumor cells as a result of the constant stress to which they are subjected (6, 11, 12, 14, 36–38). Mice transgenic for MMTV-PyMT on a heterozygous GRP78 background have provided strong evidence that a partial reduction of GRP78 is sufficient to retard tumor proliferation, survival and angiogenesis (38). A genetic reduction in global GRP78 levels can delay early primary tumor growth and experimental metastasis to lung, with a selective reduction of GRP78 only in tumor vasculature being sufficient to suppress lung metastasis (19).
There are several methods by which GRP78 can be targeted. Some studies have used therapies that block the activity of GRP78 (36, 37, 39–41) or utilize AB5 subtilase cytotoxin that specifically cleaves GRP78 (42). Antibodies against surface GRP78 can also induce pro-apoptotic signaling (43, 44), although antibodies selectively targeting the amino terminal of surface GRP78 have been shown to enhance proliferation (10). The other approach is to utilize the selective surface expression of GRP78 in tumor cells for targeted delivery of a therapeutic directly to the tumor, thereby minimizing systemic toxicity (5). A 13-mer peptide that targets GRP78 on melanoma cells induced apoptosis when coupled to either taxol (45) or D(KLAKLAK)2 (18). The GRP78 ligand we describe here has been coupled to N-(2-hydroxypropyl)methacrylamide copolymers containing aminohexylgeldanamycin, resulting in apoptosis in prostate cancer cells in vitro (46). Another peptide that targets GRP78 on irradiated tumors, coupled to paclitaxel-encapsulating nanoparticles, extended tumor doubling time in mice bearing MDA-MB-231 or GL261 tumors (47).
However, none of these reports has addressed the role of GRP78 in metastatic disease, despite the fact that metastasis remains the most prominent cause of death in patients with most types of solid tumors. In this study, we tested a peptide therapy comprising a GRP78 binding peptide linked to the pro-apoptotic KLAKLAK peptide. This peptide, BMTP78, was shown previously to bind cell surface GRP78, become internalized and induce apoptosis (17). Here we tested the efficacy of this therapy against established metastatic disease. Using a highly aggressive mammary tumor model, we have demonstrated that BMTP78 is active not only against primary mammary tumors but also against established metastatic disease (Figure 4). BMTP78 therapy was well tolerated by the mice, despite a temporary reduction in body weight, thereby indicating that normal tissues are not internalizing significant levels of this toxic peptide. Additional experiments using two different breast tumor lines, the human MDA-MB-231 and the murine EF43-fgf4 in experimental lung metastasis assays, confirmed that BMTP78 therapy is able to suppress the growth of established micrometastases (Figure 5). While the use of fluorescence imaging has great value in tracking metastases in vivo (48), caution should be exercised with immune competent mice, where the fluorescent protein can sometimes be immunogenic. However, we have found that mCherry fluorescence has no impact on metastasis in Balb/c mice and was therefore used in the EF43-fgf4 experiment (Figure 5C).
The association of surface GRP78 expression with stress was evident in all our cell lines grown in 20% oxygen. It should be noted that not all tumour lines are reported to express surface GRP78, including some prostate cancer lines and, in contrast to our data, MDA-MB-231 cells (10). Regardless of their tumorigenic status, cells in normoxic conditions expressed GRP78 on the plasma membrane and were sensitive to BMTP78. However, in oxygen levels closer to those experienced by cells in tissues, only the tumorigenic cells remained in a stressed state with surface-localized GRP78. Thus, BMTP78 can be efficacious against tumor cells in vivo without significant normal tissue toxicity. Despite the detection of GRP78 on only 5–20% of tumor cells isolated from mice (Figure 2C), BMTP78 was able to effectively suppress metastatic outgrowth. It is possible that cells with low surface GRP78 are still killed effectively by BMTP78 and/or that surface GRP78 is internalized rapidly, such that the acute administration of anti-GRP78 antibody as shown in Figure 2B does not detect all positive cells.
In the 4T1.2 metastasis model, spontaneous dissemination occurred in all mice by 10 days after tumor cell inoculation, as evidenced by their rapid death in the control groups (Figure 4F). However, BMTP78 therapy provided a significant extension in the metastasis-free period following primary tumor resection. Consistent with this finding, disseminated tumor cells (DTC) isolated from breast cancer patients have increased levels of stress proteins related to the unfolded protein response (UPR), presumably due to the microenvironmental stresses to which they are subjected in foreign tissues (49).
It has been reported that a third of breast cancer patients, following primary tumor resection and without clinical evidence of disease, who were examined between 7 and 22 years after diagnosis, had detectable levels of circulating tumor cells (CTC) that remained at a steady state level of 1–2 cells per 12 ml of blood, despite their existence for less than 3 hours in circulation (50). CTC and DTC are often resistant to conventional cancer therapies and can persist for many years after the initial therapy, and high levels are associated with reduced disease-free survival (51). The implication from these studies is that the DTC are dividing and replenishing the CTC, even though the DTC do not reach a clinically detectable size for many years.
Rapid advances in the ability to detect CTC and DTC, and the realization that the number of these cells can be a predictor of recurrence, are improving our ability to predict patients at risk of developing secondary disease. Patients with detectable CTC or DTC could be candidates for a therapy designed to kill CTC/DTC and micrometastases by virtue of their elevated surface expression of GRP78 (49). Thus, BMTP78 could be a potent therapy for patients with elevated levels of CTC, who are known to be at increased risk of developing distant metastases. As it is likely that BMTP78 would be provided as an adjuvant therapy, future preclinical studies should investigate the optimal regimens for combination with currently used chemotherapies, including doxorubicin and taxanes, given the differential importance of GRP78 in the response to these therapies (12, 35).
Upon screening of tissue microarrays, each containing the primary breast tumor and multiple metastases from an individual patient, we found diffuse staining for GRP78 in all samples, an expected result since all these patients died from metastatic breast cancer. The maintenance of GRP78 protein in metastatic nodules validates the concept of targeting this protein in patients with advanced disease and offers the possibility of killing established micrometastases, as opposed to other therapies that are aimed at blocking the initial stages of metastasis. Since the onset of metastasis has probably occurred before diagnosis, therapies that target established metastatic disease are of greater clinical significance.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
STATEMENT OF TRANSLATIONAL RELEVANCE.
The major cause of death in patients with solid tumors is metastasis to distant sites, resulting in organ failure. Since metastasis has often commenced prior to diagnosis, the focus of therapy should be on eliminating metastatic disease. In this study, we demonstrate a method to directly target metastatic tumour cells with toxic agents by virtue of their selective cell surface expression of GRP78. Unlike normal cells, tumor cells are under constant stress, leading to up-regulation of GRP78 both in the endoplasmic reticulum and on the cell surface. In this study, we demonstrate that a toxin coupled to a GRP78 binding peptide can cause a significant delay in the growth of established micrometastatic disease in three models of breast cancer metastasis. Further, we show that primary tumors and matched metastases from patients who died from breast cancer express high levels of GRP78.
Acknowledgments
GRANT SUPPORT
This project was supported by grants from the Association for International Cancer Research (AICR) (RLA, RP), the National Health and Medical Research Council (NHMRC) program (RGR), The Department of Defense Congressionally Directed Medical Research Programs: Breast Cancer Research Program Impact Award (RP) and the Marcus Foundation (WA, RP). Fellowship support from the National Breast Cancer Foundation of Australia (RLA), from the NHMRC (RGR), The Susan G. Komen for the Cure (BLE) and The University of Melbourne Postgraduate Scholarships (YRM and YC) are gratefully acknowledged.
We thank Connie Sun, Lea Bitner and Christina Restall for technical support.
Footnotes
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
The University of Texas M.D. Anderson Cancer Center and two of its researchers (WA and RP) have equity positions in and are paid consultants for Alvos Therapeutics and Ablaris Therapeutics, which are subjected to certain restrictions under university policy; the university manages and monitors the terms of these arrangements in accordance with its conflict-of-interest policy.
References
- 1.Montazeri A. Health-related quality of life in breast cancer patients: a bibliographic review of the literature from 1974 to 2007. J Exp Clin Cancer Res. 2008;27:32. doi: 10.1186/1756-9966-27-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones SE. Metastatic breast cancer: the treatment challenge. Clin Breast Cancer. 2008;8:224–33. doi: 10.3816/CBC.2008.n.025. [DOI] [PubMed] [Google Scholar]
- 3.Klein CA. Gene expression sigantures, cancer cell evolution and metastatic progression. Cell Cycle. 2004;3:29–31. [PubMed] [Google Scholar]
- 4.Giuliano M, Giordano A, Jackson S, Hess KR, De Giorgi U, Mego M, et al. Circulating tumor cells as prognostic and predictive markers in metastatic breast cancer patients receiving first-line systemic treatment. Breast Cancer Res. 2011;13:R67. doi: 10.1186/bcr2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sato M, Yao VJ, Arap W, Pasqualini R. GRP78 signaling hub a receptor for targeted tumor therapy. Adv Genet. 2010;69:97–114. doi: 10.1016/S0065-2660(10)69006-2. [DOI] [PubMed] [Google Scholar]
- 6.Li J, Lee AS. Stress induction of GRP78/BiP and its role in cancer. Curr Mol Med. 2006;6:45–54. doi: 10.2174/156652406775574523. [DOI] [PubMed] [Google Scholar]
- 7.Pfaffenbach KT, Lee AS. The critical role of GRP78 in physiologic and pathologic stress. Curr Opin Cell Biol. 2011;23:150–6. doi: 10.1016/j.ceb.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daneshmand S, Quek ML, Lin E, Lee C, Cote RJ, Hawes D, et al. Glucose-regulated protein GRP78 is up-regulated in prostate cancer and correlates with recurrence and survival. Hum Pathol. 2007;38:1547–52. doi: 10.1016/j.humpath.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 9.Zhuang L, Scolyer RA, Lee CS, McCarthy SW, Cooper WA, Zhang XD, et al. Expression of glucose-regulated stress protein GRP78 is related to progression of melanoma. Histopathology. 2009;54:462–70. doi: 10.1111/j.1365-2559.2009.03242.x. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez-Gronow M, Cuchacovich M, Llanos C, Urzua C, Gawdi G, Pizzo SV. Prostate cancer cell proliferation in vitro is modulated by antibodies against glucose-regulated protein 78 isolated from patient serum. Cancer Res. 2006;66:11424–31. doi: 10.1158/0008-5472.CAN-06-1721. [DOI] [PubMed] [Google Scholar]
- 11.Zhang J, Jiang Y, Jia Z, Li Q, Gong W, Wang L, et al. Association of elevated GRP78 expression with increased lymph node metastasis and poor prognosis in patients with gastric cancer. Clin Exp Metastasis. 2006;23:401–10. doi: 10.1007/s10585-006-9051-9. [DOI] [PubMed] [Google Scholar]
- 12.Lee E, Nichols P, Spicer D, Groshen S, Yu MC, Lee AS. GRP78 as a novel predictor of responsiveness to chemotherapy in breast cancer. Cancer Res. 2006;66:7849–53. doi: 10.1158/0008-5472.CAN-06-1660. [DOI] [PubMed] [Google Scholar]
- 13.Scriven P, Coulson S, Haines R, Balasubramanian S, Cross S, Wyld L. Activation and clinical significance of the unfolded protein response in breast cancer. Br J Cancer. 2009;101:1692–8. doi: 10.1038/sj.bjc.6605365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fernandez PM, Tabbara SO, Jacobs LK, Manning FC, Tsangaris TN, Schwartz AM, et al. Overexpression of the glucose-regulated stress gene GRP78 in malignant but not benign human breast lesions. Breast Cancer Res Treat. 2000;59:15–26. doi: 10.1023/a:1006332011207. [DOI] [PubMed] [Google Scholar]
- 15.Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, et al. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010;123:725–31. doi: 10.1007/s10549-009-0674-9. [DOI] [PubMed] [Google Scholar]
- 16.Shin BK, Wang H, Yim AM, Le Naour F, Brichory F, Jang JH, et al. Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J Biol Chem. 2003;278:7607–16. doi: 10.1074/jbc.M210455200. [DOI] [PubMed] [Google Scholar]
- 17.Arap MA, Lahdenranta J, Mintz PJ, Hajitou A, Sarkis AS, Arap W, et al. Cell surface expression of the stress response chaperone GRP78 enables tumor targeting by circulating ligands. Cancer Cell. 2004;6:275–84. doi: 10.1016/j.ccr.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y, Steiniger SC, Kim Y, Kaufmann GF, Felding-Habermann B, Janda KD. Mechanistic studies of a peptidic GRP78 ligand for cancer cell-specific drug delivery. Mol Pharm. 2007;4:435–47. doi: 10.1021/mp060122j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dong D, Stapleton C, Luo B, Xiong S, Ye W, Zhang Y, et al. A critical role for GRP78/BiP in the tumor microenvironment for neovascularization during tumor growth and metastasis. Cancer Res. 2011;71:2848–57. doi: 10.1158/0008-5472.CAN-10-3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: atypical GRP78 in cell viability, signalling and therapeutic targeting. Biochem J. 2011;434:181–8. doi: 10.1042/BJ20101569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Liu R, Ni M, Gill P, Lee AS. Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP. J Biol Chem. 2010;285:15065–75. doi: 10.1074/jbc.M109.087445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shani G, Fischer WH, Justice NJ, Kelber JA, Vale W, Gray PC. GRP78 and Cripto form a complex at the cell surface and collaborate to inhibit transforming growth factor beta signaling and enhance cell growth. Mol Cell Biol. 2008;28:666–77. doi: 10.1128/MCB.01716-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mintz PJ, Kim J, Do KA, Wang X, Zinner RG, Cristofanilli M, et al. Fingerprinting the circulating repertoire of antibodies from cancer patients. Nat Biotechnol. 2003;21:57–63. doi: 10.1038/nbt774. [DOI] [PubMed] [Google Scholar]
- 24.Blond-Elguindi S, Cwirla SE, Dower WJ, Lipshutz RJ, Sprang SR, Sambrook JF, et al. Affinity panning of a library of peptides displayed on bacteriophages reveals the binding specificity of BiP. Cell. 1993;75:717–28. doi: 10.1016/0092-8674(93)90492-9. [DOI] [PubMed] [Google Scholar]
- 25.Eckhardt BL, Parker BS, van Laar RK, Restall CM, Natoli AL, Tavaria MD, et al. Genomic analysis of a spontaneous model of breast cancer metastasis to bone reveals a role for the extracellular matrix. Mol Cancer Res. 2005;3:1–13. [PubMed] [Google Scholar]
- 26.Cimino A, Halushka M, Illei P, Wu X, Sukumar S, Argani P. Epithelial cell adhesion molecule (EpCAM) is overexpressed in breast cancer metastases. Breast Cancer Res Treat. 2010;123:701–8. doi: 10.1007/s10549-009-0671-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kolonin MG, Saha PK, Chan L, Pasqualini R, Arap W. Reversal of obesity by targeted ablation of adipose tissue. Nat Med. 2004;10:625–32. doi: 10.1038/nm1048. [DOI] [PubMed] [Google Scholar]
- 28.Barnhart KF, Christianson DR, Hanley PW, Driessen WH, Bernacky BJ, Baze WB, et al. A peptidomimetic targeting white fat causes weight loss and improved insulin resistance in obese monkeys. Sci Transl Med. 2011;3:108ra12. doi: 10.1126/scitranslmed.3002621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992;52:1399–405. [PubMed] [Google Scholar]
- 30.Lelekakis M, Moseley JM, Martin TJ, Hards D, Williams E, Ho P, et al. A novel orthotopic model of breast cancer metastasis to bone. Clinical and Experimental Metastasis. 1999;17:163–70. doi: 10.1023/a:1006689719505. [DOI] [PubMed] [Google Scholar]
- 31.Javadpour MM, Juban MM, Lo WC, Bishop SM, Alberty JB, Cowell SM, et al. De novo antimicrobial peptides with low mammalian cell toxicity. J Med Chem. 1996;39:3107–13. doi: 10.1021/jm9509410. [DOI] [PubMed] [Google Scholar]
- 32.Balin AK, Pratt L, Allen RG. Effects of ambient oxygen concentration on the growth and antioxidant defenses of of human cell cultures established from fetal and postnatal skin. Free Radic Biol Med. 2002;32:257–67. doi: 10.1016/s0891-5849(01)00807-3. [DOI] [PubMed] [Google Scholar]
- 33.Goldenberg-Cohen N, Raiter A, Gaydar V, Dratviman-Storobinsky O, Goldstein T, Weizman A, et al. Peptide-binding GRP78 protects neurons from hypoxia-induced apoptosis. Apoptosis. 2012;17:278–88. doi: 10.1007/s10495-011-0678-x. [DOI] [PubMed] [Google Scholar]
- 34.Hardy B, Raiter A. Peptide-binding heat shock protein GRP78 protects cardiomyocytes from hypoxia-induced apoptosis. J Mol Med (Berl) 2010;88:1157–67. doi: 10.1007/s00109-010-0657-7. [DOI] [PubMed] [Google Scholar]
- 35.Lee E, Nichols P, Groshen S, Spicer D, Lee AS. GRP78 as potential predictor for breast cancer response to adjuvant taxane therapy. Int J Cancer. 2011;128:726–31. doi: 10.1002/ijc.25370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martin S, Hill DS, Paton JC, Paton AW, Birch-Machin MA, Lovat PE, et al. Targeting GRP78 to enhance melanoma cell death. Pigment Cell Melanoma Res. 2010;23:675–82. doi: 10.1111/j.1755-148X.2010.00731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li M, Wang J, Jing J, Hua H, Luo T, Xu L, et al. Synergistic promotion of breast cancer cells death by targeting molecular chaperone GRP78 and heat shock protein 70. J Cell Mol Med. 2009;13:4540–50. doi: 10.1111/j.1582-4934.2008.00575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dong D, Ni M, Li J, Xiong S, Ye W, Virrey JJ, et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008;68:498–505. doi: 10.1158/0008-5472.CAN-07-2950. [DOI] [PubMed] [Google Scholar]
- 39.Kim JY, Hwang JH, Cha MR, Yoon MY, Son ES, Tomida A, et al. Arctigenin blocks the unfolded protein response and shows therapeutic antitumor activity. J Cell Physiol. 2010;224:33–40. doi: 10.1002/jcp.22085. [DOI] [PubMed] [Google Scholar]
- 40.Backer JM, Krivoshein AV, Hamby CV, Pizzonia J, Gilbert KS, Ray YS, et al. Chaperone-targeting cytotoxin and endoplasmic reticulum stress-inducing drug synergize to kill cancer cells. Neoplasia. 2009;11:1165–73. doi: 10.1593/neo.09878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J, Yin Y, Hua H, Li M, Luo T, Xu L, et al. Blockade of GRP78 sensitizes breast cancer cells to microtubules-interfering agents that induce the unfolded protein response. J Cell Mol Med. 2009;13:3888–97. doi: 10.1111/j.1582-4934.2009.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paton AW, Beddoe T, Thorpe CM, Whisstock JC, Wilce MC, Rossjohn J, et al. AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature. 2006;443:548–52. doi: 10.1038/nature05124. [DOI] [PubMed] [Google Scholar]
- 43.Rauschert N, Brandlein S, Holzinger E, Hensel F, Muller-Hermelink HK, Vollmers HP. A new tumor-specific variant of GRP78 as target for antibody-based therapy. Lab Invest. 2008;88:375–86. doi: 10.1038/labinvest.2008.2. [DOI] [PubMed] [Google Scholar]
- 44.de Ridder GG, Ray R, Pizzo SV. A murine monoclonal antibody directed against the carboxyl-terminal domain of GRP78 suppresses melanoma growth in mice. Melanoma Res. 2012;22:225–35. doi: 10.1097/CMR.0b013e32835312fd. [DOI] [PubMed] [Google Scholar]
- 45.Kim Y, Lillo AM, Steiniger SC, Liu Y, Ballatore C, Anichini A, et al. Targeting heat shock proteins on cancer cells: selection, characterization, and cell-penetrating properties of a peptidic GRP78 ligand. Biochemistry. 2006;45:9434–44. doi: 10.1021/bi060264j. [DOI] [PubMed] [Google Scholar]
- 46.Larson N, Ray A, Malugin A, Pike DB, Ghandehari H. HPMA copolymer-aminohexylgeldanamycin conjugates targeting cell surface expressed GRP78 in prostate cancer. Pharm Res. 2010;27:2683–93. doi: 10.1007/s11095-010-0267-7. [DOI] [PubMed] [Google Scholar]
- 47.Passarella RJ, Spratt DE, van der Ende AE, Phillips JG, Wu H, Sathiyakumar V, et al. Targeted nanoparticles that deliver a sustained, specific release of Paclitaxel to irradiated tumors. Cancer Res. 2010;70:4550–9. doi: 10.1158/0008-5472.CAN-10-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoffman RM. The multiple uses of fluorescent proteins to visualize cancer in vivo. Nat Rev Cancer. 2005;5:796–806. doi: 10.1038/nrc1717. [DOI] [PubMed] [Google Scholar]
- 49.Bartkowiak K, Effenberger KE, Harder S, Andreas A, Buck F, Peter-Katalinic J, et al. Discovery of a novel unfolded protein response phenotype of cancer stem/progenitor cells from the bone marrow of breast cancer patients. J Proteome Res. 2010;9:3158–68. doi: 10.1021/pr100039d. [DOI] [PubMed] [Google Scholar]
- 50.Meng S, Tripathy D, Frenkel EP, Shete S, Naftalis EZ, Huth JF, et al. Circulating tumor cells in patients with breast cancer dormancy. Clin Cancer Res. 2004;10:8152–62. doi: 10.1158/1078-0432.CCR-04-1110. [DOI] [PubMed] [Google Scholar]
- 51.Muller V, Stahmann N, Riethdorf S, Rau T, Zabel T, Goetz A, et al. Circulating tumor cells in breast cancer: correlation to bone marrow micrometastases, heterogeneous response to systemic therapy and low proliferative activity. Clin Cancer Res. 2005;11:3678–85. doi: 10.1158/1078-0432.CCR-04-2469. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.