

Abstract

Seven crystal structures of alanyl aminopeptidase from Neisseria meningitides (the etiological agent of meningitis, NmAPN) complexed with organophosphorus compounds were resolved to determine the optimal inhibitor–enzyme interactions. The enantiomeric phosphonic acid analogs of Leu and hPhe, which correspond to the P1 amino acid residues of well-processed substrates, were used to assess the impact of the absolute configuration and the stereospecific hydrogen bond network formed between the aminophosphonate polar head and the active site residues on the binding affinity. For the hPhe analog, an imperfect stereochemical complementarity could be overcome by incorporating an appropriate P1 side chain. The constitution of P1′-extended structures was rationally designed and the lead, phosphinic dipeptide hPhePψ[CH2]Phe, was modified in a single position. Introducing a heteroatom/heteroatom-based fragment to either the P1 or P1′ residue required new synthetic pathways. The compounds in the refined structure were low nanomolar and subnanomolar inhibitors of N. meningitides, porcine and human APNs, and the reference leucine aminopeptidase (LAP). The unnatural phosphinic dipeptide analogs exhibited a high affinity for monozinc APNs associated with a reasonable selectivity versus dizinc LAP. Another set of crystal structures containing the NmAPN dipeptide ligand were used to verify and to confirm the predicted binding modes; furthermore, novel contacts, which were promising for inhibitor development, were identified, including a π–π stacking interaction between a pyridine ring and Tyr372.
Introduction
Phosphinic peptide analogues are potent, reversible, and competitive inhibitors of metalloproteases, the enzymes that catalyze the cleavage of an amide bond in the biologically active peptide through a metal-ion-assisted process.1−3 The tetrahedrally shaped phosphinate group mimics the structural and electronic features of the gem-diolate transition state intermediate from the hydrolysis process. After binding, the group is coordinated to the central metal ion, blocking its catalytic activity (Figure 1).
Figure 1.

Schematic representation showing the binding mode of phosphinic peptide inhibitors and zinc metalloproteases. The P1 and P1′ side chains specific to the S1 and S1′ binding pocket are colored in pink (left panel). Shown are the structure of hPhePψ[CH2]Phe 1, which is a recognized inhibitor of aminopeptidases and the lead compound for this study, and the structure of hPhePψ[CH2]Tyr 2, which is a unique P1′-modified compound with improved activity (right panel).25,26
The selectivity issues with the phosphinic peptide inhibitors are primarily attributed to the structural features of the Pn and Pn′ residues, which must access the corresponding (Sn and Sn′) enzyme binding pockets. These structural attributes can be optimized further for the inhibition of metalloendopeptidases, which cleave internal peptide bonds, by peptide elongating at the N- and C-termini. A fundamental P1–P1′ pseudodipeptide building block is typically incorporated into a sequence that corresponds to the privileged structures of well-studied substrates. Alternatively, the P2–Pn and P2′–Pn′ amino acid sequence is refined through combinatorial techniques.4,5 For exopeptidases, any modifications are limited to one side of the central ligand core. Therefore, the basic P1–P1′ scaffold must be more carefully optimized, which creates a challenging synthetic problem. Dedicated multistep preparations are required when residues designed to complement the most discriminating S1 and S1′ subsites are more complex than simple alkyl or aryl substituents and contain a functional group.6 Parallel approaches that provide access to diverse phosphinic dipeptides are valuable but still limited by certain starting materials and reaction types. This scenario is adequately illustrated by the successful parallel addition of C-, N-, and S-nucleophiles to P1′ dehydroalanine,7−9 the synthesis of P1′ isoxazole and isoxazoline derivatives through a 1,3-dipolar cycloaddition starting from the appropriate unsaturated systems,10,11 and the alkylation/arylation of an amino-modified P1′ side chain.12 Usually, a single heteroatom-containing structural modification requires a unique synthetic approach.
The function and substrate specificity of zinc-dependent aminopeptidases make them excellent model enzymes for validating the effectiveness of P1/P1′ modifications in phosphinic dipeptides. The most recognized representatives of these hydrolases are microsomal alanyl aminopeptidase (EC 3.4.11.2, APN/CD13) and cytosolic leucine aminopeptidase (EC 3.4.11.1, LAP), ubiquitous enzymes among kingdoms and species.13−18 They are the members of the metalloprotease superfamily (M) and are formally referred to as M1 and M17 aminopeptidases, respectively.13,14 Both peptidases preferentially release N-terminal hydrophobic amino acids from peptides or proteins; the substrate specificity of APN also includes basic residues. In mammals, this activity is involved in physiological metabolism of regulatory and bioactive peptides, antigen presentation, and angiogenesis control (APN).15−18 The medical potential of APN and LAP is connected with their functions in tumorigenesis and metastasis and with pathogenesis of hypertension (APN).16,19−22 M1 and M17 aminopeptidases found in human pathogens, e.g., Plasmodium falciparum, are responsible for digestive proteolysis and nutrition delivery. Since hemoglobin degradation in the host erythrocytes is directly responsible for the clinical symptoms of malaria, the proteases have been identified as molecular targets to treat this prevalent disease.23 Importantly, a comprehensive structure–activity relationship for the inhibition of APN and LAP from different sources with phosphorus-containing inhibitors is currently available.3,24 However, only one case illustrates the significance of a rational, heteroatom-involved modification in the phosphinic dipeptides structure. When a canonical inhibitor of aminopeptidases, specifically hPhePψ[CH2]Phe 1, was substituted with a p-hydroxyl group in the P1′ portion, generating hPhePψ[CH2]Tyr 2 (Figure 1), the affinity of the latter to APNs (of the porcine kidney and N. meningitidis) increased 6- to 8-fold compared to the lead.25,26 The preference for the modified side chain residue was explained using a mammalian homology model and involved formation of one specific hydrogen bond to the γ-carboxylate of Glu413.25
Consequently, we have planned to develop a series of single-point modified hPhePψ[CH2]Phe 1 derivatives, including both P1 and P1′ unnatural analogs. We have hypothesized that improving the activity and selectivity of the dipeptidic derivatives may be rationally controlled with single modifications of the side chain substituent structure. The huge advantage of using this approach is that neither an overall sequence expansion nor the production of large combinatorial libraries is required.
Here, we show studies on phosphinic compounds of the designed structures, which were synthesized using dedicated multistep approaches, and their structure–activity relationships against four aminopeptidases: N. meningitides APN, human (Homo sapiens) APN, porcine (Sous scrofa) APN, and porcine LAP. We have also investigated the binding mode of seven unique ligands to NmAPN by using the X-ray crystallography, which illustrated optimization of the inhibitor structure and gave insights into transition state analogue interactions with a monozinc aminopeptidase.
Results and Discussion
P1 Amino Acid Analogs
The phosphonic analogues of amino acids are among the simplest transition state inhibitors of aminopeptidases. The acidic group holds the shape and charge necessary to mimic the high-energy gem-diolate structure, while the constituents of the P1 side chain typically correspond to the preferentially bound N-terminal residues of the peptidic substrates. Accordingly, the substrate specificity of these enzymes, which is expressed using the lowest values of the Michaelis constant, can be translated into the structure of phosphonic ligands after designing an appropriate P1 fragment.27 For alanyl aminopeptidases, the hydrophobic and basic substituents of Leu, Phe, Arg, and Lys are clearly favorable.13 Nevertheless, it has been found that fluorogenic substrates comprising extended substituents of noncoded amino acids are bound tighter than the derivatives of naturally occurring counerparts.27,28 The α-aminoalkylphosphonic acids based on these bulkier residues exhibit inhibition constants within the low micromolar or submicromolar range, making them an attractive scaffold for further elongation at the P1′ position.3,24,26,29,30
To analyze the binding of the P1 phosphorus ligands to the APNs, we selected two prototypical aminophosphonic acids: analogues of Leu (3) and hPhe (4) that exhibited promising kinetic parameters (Table 1). These parameters were correlated with the stereochemistry of the ligand and the formation of a specific hydrogen bond network between the polar head of the compounds and the NmAPN active site, as revealed by the crystal structures. The activity of LeuP toward meningococcal and human APNs depended strongly on the absolute configuration of the α-carbon atom. The natural l analog (R-3) was a low micromolar inhibitor (Ki = 1.92 μM for NmAPN and Ki = 2.62 μM for HsAPN), approximately 2 orders of magnitude more potent than the d enantiomer (S-3). An even more pronounced difference was found previously for mammalian leucine aminopeptidase.31,32 Unpredictably, the absolute configuration of the ligand was not critical for SsAPN. However, the binding to that enzyme was clearly weaker (Ki = 53 μM)32 possibly because of a shortage of stereospecific contacts. The phosphonic acid analog of hPhe (R-4) is slightly more active toward NmAPN than LeuP, and its potency does not depend on the stereochemical arrangement. Ki is 0.97 μM for the l enantiomer, and the same value, within the experimental error, is displayed for the racemic mixture 4. This observation is valuable because the optically pure forms of hPheP are not readily available. The hPheP enantiomers cannot be obtained/separated through typical 1-phenylethylamine-mediated approaches: convenient stereoselective syntheses33 or recrystallization of the diastereomeric salt of their phosphinic precursors (Cbz-5).34 The only reported enantio-enriching method for R-4 and S-4 involves a careful stereoselective separation of Cbz-5 using chiral chromatography,35 followed by N-deprotection and P-oxidation.34 Apparently, the flexible 2-phenylethyl side chain of hPhe is particularly well accommodated inside the S1 cavity, making the absolute configuration less critical. A similar situation is revealed for three peptidases tested in this work (NmAPN, SsAPN, and SsLAP): the activity of the enantiomeric and racemic hPhe is practically equal, remaining at a good level (Ki < 5 μM). For the human APN, the inhibition reaches a nanomolar level and stereodiscrimination of hPheP becomes slightly more pronounced (Ki = 230 nM for the enantiomeric inhibitor and Ki = 790 nM for the racemic one). Interestingly, H-phosphinic acid (5) reproduces the potency of its oxidized counterpart, which contains a three-oxygen-atom phosphorus group. This observation reveals that the presence of one oxygen atom in the inhibitors was not critical for binding; therefore, this position could be used to elongate the P1′ fragment further.
Table 1. Inhibitory Activity of Enantiomeric and Racemic Phosphorus-Containing Analogs of Amino Acids: Phosphonic Acid Analog of Leucine (3), Phosphonic (4) and H-Phosphinic (5) Acid Analogs of Homophenylalanine toward N. meningitides APN, Human (H. sapiens) APN, and Porcine (S. scrofa) APN and Porcine LAP.

The binding mode of 1-aminoalkylphosphonic acids LeuP (3) and hPheP (4) to the active site of NmAPN was revealed through crystallographic studies and molecular modeling. The crystals containing the recombinant selenomethionine-labeled protein soaked with an inhibitor do not significantly change the overall arrangement of the binding site architecture compared to the native protein36 and the corresponding Escherichia coli enzyme.37−41 The inhibitors dock to the S1 cleft, mimicking the P1 fragment of a substrate in its transition state (Figure 2). The two negatively charged oxygen atoms of the phosphonate moiety bidentately coordinate with the central zinc cation. The O1–Zn and O2–Zn distances are almost equal for both enantiomers of leucine analog 3, specifically 2.10 and 2.04 Å for the R enantiomer and 2.10 and 2.06 Å for the S enantiomer (Figure 2A and Figure 2B, respectively). Each of these oxygen atoms forms an additional hydrogen bond: one with Tyr377 (2.42 and 2.48 Å for R-3 and S-3, respectively) and the other with Glu294 (2.97 and 2.80 Å for R-3 and S-3, respectively). The third oxygen atom is exposed to the solvent. The amino moiety forms three hydrogen bonds with the carboxylates of Glu117, Glu260, and Glu316. The N–H···O distances for Glu117 and Glu260 are similar for both enantiomers (2.73 and 2.89 Å for R-3 and 2.73 and 2.86 Å for S-3). The corresponding distance to Glu316 differs significantly between the enantiomers. For natural analog R-3, the length is 2.92 Å, while for S-3, the heteroatoms are 3.15 Å apart. This structural disparity may explain the weaker interaction between S-3 and the enzyme and its inhibitory constant, which is 2 orders of magnitude higher. The inclusion of the hydrophobic isobutyl fragment in the cavity is facilitated by the Met256 residue, which adopts a conformation suitable for sticking to the inhibitor portion through lipophilic interactions.
Figure 2.
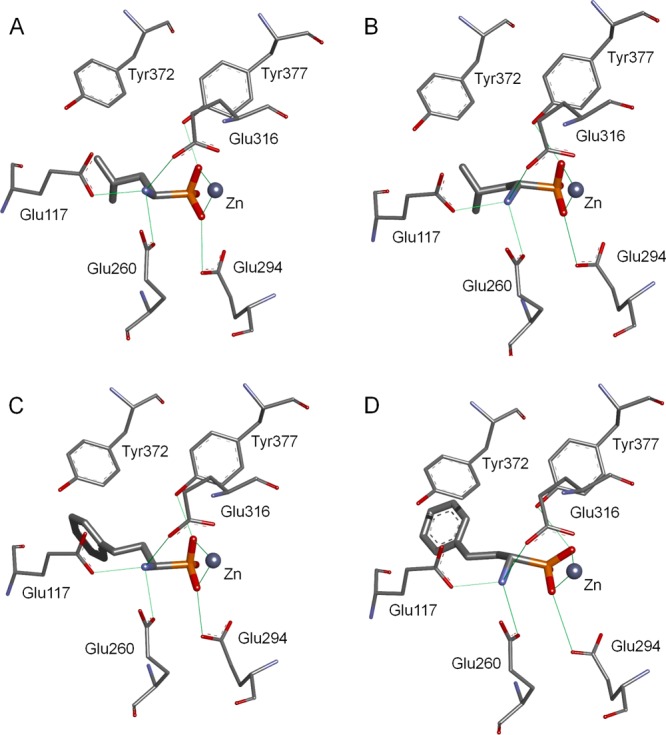
Crystal structures of the complexes containing enantiomeric phosphonic acid analogs of leucine and homophenylalanine with NmAPN: (R)-LeuP-NmAPN (A), (S)-LeuP-NmAPN (B), and (R)-hPheP-NmAPN (C), and a modeled complex of (S)-hPheP-NmAPN (D). The hydrogen bonds and ligand–metal interactions are marked as green lines.
The strength of binding for inhibitor R-3 is 1 order of magnitude higher for LAP than for NmAPNs. Indeed, the mode of interaction with (R)-LeuP, which was revealed using the crystal structure of the monozinc NmAPN complex, is quite different from that observed for dinuclear aminopeptidases.42−44 When the bovine lens LAP is cocrystallized with R-3, the phosphonic acid is a tridentate ligand for zinc ions.42 One zinc ion is bound by two phosphonic oxygen atoms, while the other is bound by one of the oxygens (the metal-bridging O atom) and an amino group. Therefore, one metal site binds the N-terminal NH2 group on the inhibitors and substrates. In APN, this role is entirely fulfilled by the glutamate-rich region (Glu117, Glu260, and Glu316) of the S1 pocket. When the amount of metal–inhibitor contact between aminophosphonic acids and a two-metal system (LAP) is increased relative to a one-metal site (APN), the development of selective APN inhibitors versus LAP seems difficult. This challenge can be addressed by optimizing the structure of the side chain to explore the specificity of the binding pocket.
Although phosphonic acid analogs of homophenylalanine have arisen as important inhibitors of aminopeptidases,3,24,26,29,30 the structural and stereochemical context and the nature of favorable binding to these proteins have not been disclosed. The crystal structure of the R-4-NmAPN complex (Figure 2C) and the molecular model of S-4-NmAPN (Figure 2D) imply that the binding mode between the aminophosphonate head of the enantiomeric phosphonic acid analogs of homophenylalanine and the protein is identical to that with LeuP. However, the aromatic portion binds to the S1 cavity slightly differently, depending on the configuration of the α carbon atom. The crystal structure of the R-4 complex reveals the free arrangement of 2-phenylethyl without any of the characteristic hydrophobic contacts between the aromatic ring and the surrounding residues (Figure 2C). In contrast, the docking model of the S enantiomer implies that a defined pointing direction, specifically toward the position needed for π–π stacking with the aromatic ring of Tyr372 (Figure 2D), is necessary. This favorable interaction may decrease the difference between the free energies of binding that arise from the different hydrogen bond networks formed by the enantiomers. The sum of these contradictory effects makes the measured inhibition constants equal.
Phosphinic Dipeptides Design
The novel inhibitors were rationally designed based on the structures of NmAPN, porcine kidney APN, and human APN complexed with a canonical phosphinate ligand that showed a high affinity for M1 and M17 aminopeptidases.25,26,45,46 Diastereomerically pure hPhePψ[CH2]Phe (1) of an ll (R,S) stereochemistry,47 which corresponds to the natural amino acid configuration, was cocrystallized with the bacterial protein. An analogous complex containing the porcine and human orthologs were modeled based on the crystal structures of bestatin-bound SsAPN48 and amastatin-bound HsAPN.49 The overall mode of the inhibitor binding was similar between the complexes (Figure 3 and Supporting Information Figure S1A and Figure S1B, respectively) and comparable to that of the previously determined complex structure of tripeptide lll (R,S,S)-AlaPψ[CH2]PhePhe and E. coli APN.40 This binding mode involved the bidentate coordination of negatively charged phosphinate oxygens to the zinc metal ion, P–O···H bonds to the hydroxylate of tyrosine (Tyr477 for HsAPN and Tyr472 for SsAPN) and the carboxylate of glutamic acid (Glu389 for HsAPN and Glu350 for SsAPN), and a hydrogen network formed between the amino group and the neighboring carboxylates/carboxyamide of the glutamic acids/glutamine (Glu355, Glu411, and Gln213 for HsAPN and Glu350, Glu406, and Gln208 for SsAPN). These interactions corresponded to those defined for phosphonic acids and NmAPN (Figure 2). Additionally, the C-terminal carboxylate of the inhibitor fell within the hydrogen bond distance to the N–H of Gly257 in the structures of NmAPN (Gly352 and Arg381 for HsAPN, and Ala348 for SsAPN). These fundamental contacts between the inhibitor backbone and the enzyme residues should have been reproduced in the modified target structures, while the fit of the hydrophobic side chains within the binding pockets would require the appropriate adjustments.
Figure 3.

Crystal structure of the complex containing R,S-1 with NmAPN showing the potential interactions between a modified or substituted P1 and P1′ phenyl rings and the active site residues (those involved are highlighted in bold). Intermolecular hydrogen bonds and ligand–metal interactions are marked in green. The potential contact sites are marked as blue-gray, red-dark gray, or gray spheres, indicating hydrogen bond donors, acceptors, or lipophilic fragments, respectively.
An analysis of the potential interactions, which was performed using the Pharmacophore module of the Discovery Studio software package (Accelrys, Inc., San Diego, CA, USA), revealed several options for modifying the core structure of compound 1, particularly the phenyl fragments located in both the S1 and S1′ cavities of the meningococcal and mammalian (human and porcine) APNs (Figure 3 and Supporting Information Figure S1A and Figure S1B, respectively). The N-terminal phenyl ring of (R,S)-hPhePψ[CH2]Phe is located close to several hydrogen bond-accepting carbonyl groups (Gln115, Glu117, Gln818, and Ala258 for NmAPN, Gln211, Asn350, and Ala351 for HsAPN, and Leu185, Gln208, and Ala346 for SsAPN). Most of the proposed N–H structural fragments (to be introduced), which complement the carbonyls, either overlap with the phenyl ring (in particular for NmAPN) or hold a position in proximity to the ring. Therefore, a pyridyl ring seemed to be a consensus structure comprising the favorable changes of the phenyl on the lead. In addition, other hydrogen bond donors (Asn255, Met256, and Asn369 for NmAPN, Ser895 for HsAPN, and Gln206, Ser209, and Ser464 for SsAPN) are positioned near the distal portion of the aromatic fragment. The OH and CH2OH groups used to replace the phenyl meta or para hydrogen atom should have potential contacts with these groups. When modifying the C-terminal phenyl ring, the proximity of carboxylates (Glu378 and Asp323 for NmAPN, Glu418 for HsAPN, and Glu413 and Asp434 for SsAPN) seemed the most beneficial. A positively charged methylamino substituent introduced in position 3 or 4 of the P1′ aromatic ring of an inhibitor should form a salt bridge with one of these acidic groups. Finally, both the S1 and S1′ cavities are spacious, indicating that a phenyl substituent extension may increase the surface area available for lipophilic interactions.
Synthesis
The α,α′-phosphinic pseudodipeptides were obtained through alternative synthetic approaches that involved formation of two C–P bonds starting from hypophosphorus acid (H3PO2).6 To prepare the P1-diversified set of phosphinic pseudodipeptides (6–12), the C → N strategy seemed more convenient (Scheme 1). This route usually involves a three-component amidoalkylation reaction between a carbamate, an aldehyde and an H-phosphinic acid,50 such as 26 (which is obtained through a phospha-Michael addition of α-benzylacrylate to H3PO2, as described elsewhere51). In our case, the synthetic challenge involved preparing aldehydes 13–19, which are key substrates for the condensation. To achieve this goal, two types of ester reductions were used: a Fukuyama reduction with a thioester using a silyl hydride in the presence of a palladium catalyst52 and a DIBAL-H reduction of a carboxylic ester.53 The utility of both versions can be exemplified by obtaining 3-pyridyl (18) and 4-pirydyl-3-propanal (19) from commercially available starting materials (Scheme 1, insert). Therefore, acid 20 was converted to the corresponding thioester (21) through an EDC-mediated esterification with thioethanol. The reduction using triethylsilane and Pd/C proceeded smoothly, providing aldehyde 18. The preparation of another isomer began by elongating 4-pyridinecarboxaldehyde 22 in a Knoevenagel condensation/decarboxylation. Acrylic acid 23 was esterified to 24 quantitatively in refluxing methanol. Afterward, the double bond was reduced using a standard hydrogenation over Pd/C to give 25. The reduction of aldehyde 19 proceeded smoothly when using DIBAL-H. The product was not stable; therefore, it was purified and used immediately in the amidoalkylation reaction. Detailed procedures are included in the Supporting Information.
Scheme 1. Synthesis of the P1-Modified Phosphinic Pseudodipeptides with the 3-Pyridyl and 4-Pirydyl-3-propanal Substrates.

Reagents and conditions: (a) EDC·HCl, EtSH, CH2Cl2, 2 h; (b) Et3Si, 10% Pd/C, CH2Cl2, 1 h; (c) malonic acid, pyridine, piperidine, 100 °C, 30 min; (d) MeOH, conc H2SO4, reflux, 3 h; (e) H2, 10% Pd/C, MeOH, 24 h; (f) DIBAL-H, −78 °C, Et2O, 1 h; (g) AcOH/AcCl 5/1, 48 h; (h) 6 M HCl, reflux, 12 h; (i) Dowex AG 50W-X4 (H+).
The aldehydes were used in the subsequent amidoalkylation reaction with benzyl carbamate and H-phosphinic acid 26 in acetic acid/acetyl chloride for 48 h, generating the desired phosphinic dipeptides 27–33. Free phosphinic compounds 6–12 were isolated in their pure forms after hydrolysis and reversed phase silica gel column chromatography.
The N → C approach is a typical P1′ divergent route (Scheme 2) involving a phospha-Michael addition of a protected H-phosphinic analog of an amino acid (which is easily obtained from a Kabachnik–Fields reaction with H3PO2) to the appropriate acrylates (α-substituted α,β-unsaturated esters). Accordingly, to synthesize compounds 34−36, we utilized the P–H analog of homophenylalanine 37 as the phosphorus-containing component. Obtaining the acrylates from commercially available materials was a separate and demanding challenge, as illustrated by the multistep preparation of α-(aminomethylbenzyl)acrylate precursors of the P1′ fragments (39 and 40, Scheme 2, insert). Starting from bromomethylbenzonitriles 41 and 42, the corresponding amino alcohols 45 and 46 were obtained after substituting the bromides using aqueous BaCO3, followed by Pd-mediated catalytic hydrogenation of intermediate nitriles 43 and 44. A significant improvement in the reaction yield was evident compared to the previously described LiAlH4 reduction.12 The bromide was reintroduced by heating the amino alcohols with 46% aqueous HBr. The amino group was Boc-protected without separating 47 and 48. Bromides 49 and 50 were used for the C-alkylation of diethyl malonate in the presence of NaH, which proceeded in very good yield. Finally, monoacids 53 and 54, which were obtained from substituted malonates 51 and 52 after monosaponification, were transformed to the desired acrylates (39 and 40) through a consecutive decarboxylation/Mannich reaction using diethylamine and formaldehyde at room temperature.54
Scheme 2. Synthesis of P1′-Modified Phosphinic Pseudodipeptides with the Detailed Preparation of α-(Aminomethylbenzyl)acrylate Substrates.

Reagents and conditions: (a) BaCO3, H2O, 4 h, reflux; (b) H2, 10% Pd/C, MeOH, 2 days; (c) 46% HBr/H2O, 3.5 h, reflux; (d) (Boc)2O, NaHCO3, H2O, dioxane, 18 h; (e) NaH, CH2(CO2Et)2, DMF, 20 min, then 49 or 50, 60 °C, 19 h; (f) KOH, EtOH, 18 h, then HCl 0.5 N; (g) Et2NH, HCHO, CH2Cl2, 18 h, (h) BSA, CH2Cl2, 48 h; (i) 6 M HCl, reflux, 18 h; (j) Dowex AG 50W-X4 (H+).
Acrylates 38–40 were subsequently added to 37, which was activated to more nucleophilic tervalent ester with N,O-bistrimethylsilylacetamide (BSA) as the silylating agent. The BSA-mediated phospha-Michael reaction proceeded under mild conditions and was fully compatible with the relatively sensitive Boc group, generating N-protected phosphinic dipeptide analogs 55–57 in good yield after purification. Global deprotection was achieved easily by heating the phosphinates with 6 M HCl, followed by ion exchange chromatography purification to obtain compounds 34–36. For further details, see the Supporting Information.
Structure–Activity Study
The newly synthesized phosphinic dipeptide analogs were tested for inhibition against the four aminopeptidases (one bacterial and three mammalian) and compared to the reference compounds: hPhePψ[CH2]Phe (1) and hPhePψ[CH2]Tyr (2). Pseudodipeptides 6–12 contain a modification of the P1 portion, and 34–36 are P1′-modified. Finally, two refined structures (58 and 59) (synthesized according to the general procedure depicted in Scheme 1 and described in detail in the Supporting Information) combine both privileged optimization variants. For the best P1 and P1′ single-point options, the biological activity is supplemented with the crystal structures, which show the binding mode of theses ligands to the NmAPN active site.
All of the studied compounds exhibited strong, frequently slow-binding (for NmAPN and HsAPN exclusively), competitive inhibition of the aminopeptidases (Ki values given in Table 2; for further details see Supporting Information). Nearly all of the Ki constants were in a nanomolar range. Tested against human APN, several phosphinic pseudodipeptides (10–12, 35, 36, 58, and 59) showed an exceptional inhibition level (0.2–2.0 nM), falling into a picomolar range in two particular cases. The kinetic studies and the crystal structures of selected ligand–NmAPN complexes show that all of the phosphinates interact with the enzymes in a manner analogous to the lead compound. Consequently, the backbone of the novel pseudopeptide reproduces the contacts shown in the skeleton of hPhePψ[CH2]Phe 1 (Figure 3 and Figure S1, and Figure 4A): bidentate Zn complexation with the phosphinate group and a set of hydrogen bonds between the ligand functional groups and the enzyme. Thus, the observed difference in the inhibitory activity should likely result from the mode and free energy of binding of the modified P1 and P1′ side chain residues.
Table 2. Inhibitory Activity of the Newly Designed and Obtained Phosphinic Dipeptide Analogs toward N. meningitides APN and Mammalian Aminopeptidases: Human APN, Porcine APN, and Porcine LAP, Compared to Lead Compounds hPhePψ[CH2]Phe (1) and hPhePψ[CH2]Tyr (2)a.


The most significant inhibition is highlighted in bold. Measurements were made after 30–60 min of incubation and calculated using Morrison’s equation for tight binding inhibitors (for details see the Supporting Information).
Increasing the size of the P1 and P1′ hydrophobic fragments of the inhibitors by substituting a 3,5-dimethylphenyl group was not beneficial. The affinity of compounds 6 and 34 is particularly diminished for LAP. For APNs, the inhibition constant values do not differ much from the data obtained for nonsubstituted phosphinate 1. Therefore, more spatial and less sterically restrictive S1 and S1′ cavities are present in the latter enzymes compared to LAP. The strongly electron-withdrawing p-nitro substitution at P1 exerts an ambiguous influence on the affinity of the ligand 7. No change in potency was observed with NmAPN and HsAPN. Pig APN accommodates well enlarged hydrophobic fragments and altered aromatic systems. This behavior manifests as a 4-fold improvement in the Ki value to 69 nM for 7 (compared to 276 nM measured for 1). However, the opposite effect was measured for LAP (3-fold drop).
Substituting the P1 phenyl ring with meta and para hydroxyl groups (compounds 8 and 9) decreased the inhibitory activity compared to 1. Therefore, electron-donating functionalities are not well tolerated; the expected hydrogen bonds were not formed with Asn255, Met256, and Asn369 of NmAPN (or corresponding residues of the human and porcine orthologs). However, when the same functional group (OH) is not conjugated with the aromatic system (instead, it is separated with a methylene linker (CH2OH)), a highly active inhibitor (10) of porcine enzymes was obtained. The Ki improved 7-fold compared to 1 for SsAPN and 3-fold for LAP. Therefore, compound 10 is the most active organophosphorus inhibitor of leucine aminopeptidase reported so far.
In accordance with the molecular models, replacing the phenyl ring with a pyridyl moiety (compounds 11 and 12) appeared the most advantageous when optimizing the P1 position. The affinity of these phosphinates toward all four aminopeptidases is enhanced, particularly toward porcine APN (Ki = 19 nM for 3-pyridyl and Ki = 23 nM for 4-pirydyl). The reason for this effectiveness was unexpected. As revealed by the crystal structure of NmAPN-11 (Figure 4B), the pyridyl group does not form any of the predicted hydrogen bonds with the residues lining the proximal portion of the S1 pocket (Gln115, Glu117, Gln818, Ala258); instead, it is favorably stacked against the phenol group of Tyr372. The π–π stacking, which is mediated by the reversed polarization of the interacting rings, produced high inhibition levels, although the optimal binding of the α-amino group was visibly distorted. The distance covered by one of the hydrogen bonds formed by NH3+ is extended to 3.2 Å. A similar effect of compensation of the amino group distortion by favorable π–π stacking provided by the P1 aromatic ring was suggested in the model of the (S)-hPheP-NmAPN complex (Figure 3D). Therefore, Tyr372 emerged as a primary target for optimizing the inhibitor structure.
Figure 4.

Crystal structures of NmAPN complexes with phosphinic dipeptides. Hydrogen bonds and ligand–metal interactions are marked in green. All of the interactions with the pseudopeptidic backbone are marked for (R,S)-hPhePψ[CH2]Phe (A). For P1 and P1′-modified compounds only the zinc coordination and specific contacts are shown: NmAPN-11 B (B), NmAPN-35 (C), and NmAPN-36 (D).
Impressive data were obtained when aminomethylene was substituted at the P1′ position (compounds 35 and 36). Para derivative 36 exhibited an inhibition constant below 10 nM for all APN orthologs, specifically 9.0 nM for NmAPN, 0.69 nM for the human enzyme, and 4.0 nM for the porcine one. Meta-substituted compound 35 was only slightly less active (10–20 nM for NmAPN and SsAPN). To illustrate the structural context, complexes involving NmAPN and both of the active inhibitors were resolved (Figure 4C and Figure 4D). The predicted significance of the salt bridge between the added amino group of the inhibitor and the carboxylate of Asp323 was confirmed for compound 36. The distance between the heteroatoms is 2.65 Å, revealing a tight hydrogen bonding interaction (Figure 4D). For phosphinate 35, the P1′ aminomethylbenzyl residue is partially disordered. Two equally populated conformations are visible (Figure 4C). The predicted conformation shows that the amino group was directed toward Asp323, while the symmetrically rotated molecule displays the same group exposed to the solvent. Both 35 and 36 are reasonably selective versus LAP by at least a factor of 15, which is difficult to achieve for organophosphorus inhibitors. LAP is a prototypical two-zinc aminopeptidase, and the interactions of the aminophosphonate fragment with both metals are typically much stronger for these types of molecules (N-C-PO2-; see discussion of (R)-LeuP binding).
Finally, two combinations (compounds 58 and 59) containing privileged substituents at P1 (pyridylethyl) and P1′ (aminomethylbenzyl) were verified and high anti-aminopeptidase activity was revealed. In particular, the top potency was achieved with the human APN (Ki = 0.21 nM for 58 and Ki = 1.1 nM for 59) and the porcine kidney APN (Ki = 1.5 nM for 58 and Ki = 3.9 nM for 59). To our knowledge compound 58 is the most active inhibitor of HsAPN described to date. For SsAPN, additional extended phosphinic analogs, which were at least tripeptides, reportedly exhibited similar levels of inhibition.55,56 Doubled modifications were less favorable for NmAPN than for SsAPN, possibly indicating that the adverse constraints were caused by two strong interactions.
Conclusions
The phosphinic moiety is commonly believed to imitate the geometry, electron distribution, and metal complexation ability of the gem-diolate transition state formed during peptide bond hydrolysis. Accordingly, phosphinic-based pseudopeptide analogues are effective inhibitors of zinc-containing biomedically significant proteases, including representatives of the M1 alanyl and M17 leucine families of aminopeptidases. In this work, we showed that a significant improvement in the activity and selectivity of the dipeptidic phosphinate inhibitors could be achieved after optimizing the P1 and P1′ substituent rationally; however, this process required the development of synthetically challenging, multistep procedures. The derivatives of the refined structure were low nanomolar inhibitors of NmAPN, human APN, and aminopeptidases APN and LAP from porcine kidney, which are the most potent compounds of this type reported to date. Merging the favorable P1 and P1′ modifications was particularly effective for the mammalian alanyl aminopeptidase, yielding compound 58 with Ki = 0.2 nM (human) and Ki = 1.5 nM (porcine). The functions of this enzyme linked to angiogenesis, tumorigenesis, and invasion19−22 make this class of compounds potential anticancer drugs that may exhibit a reasonable selectivity toward molecular targets with related substrate specificity (e.g., LAP, as proven here). The structural aspects of the binding of the ligand to NmAPN provided unique insight into metalloaminopeptidase inhibition using transition state analogues.
Acknowledgments
The work was supported by a grant from the Polish National Science Centre (Grant 2013/09/B/ST5/00090). The Discovery Studio package was used under a Polish country-wide license. The use of software resources (Discovery Studio program package) of the Wrocław Centre for Networking and Supercomputing is also kindly acknowledged. The Structural Biology Center beamlines at APS are supported by U.S. Department of Energy Office of Biological and Environmental Research program under Contract DE-AC02-06CH11357. The structural studies were performed at the Midwest Center for Structural Genomics supported by the National Institutes of Health Grant GM094585.
Glossary
Abbreviations Used
- AaP
phosphonic analog of an amino acid
- AaPH
H-phosphinic analog of an amino acid
- Aa1Pψ[CH2]Aa2
a phosphinic dipeptide analogue
- DIBAL-H
diisobutylaluminum hydride
- Boc
tert-butyloxycarbonyl
- BSA
N,O-bis(trimethylsilyl)acetamide, Cbz: benzyloxycarbonyl, EDC, 1-ethyl-3-(3′-dimethylaminopropyl)carbodiimide
- hPhe
homophenylalanine
Supporting Information Available
Full details regarding the design, preparation, purification, and characterization (experimental procedures and NMR, MS, and HPLC data) of the compounds (purity of the final compounds 6–12, 34–36, 58, and 59 assessed at >95% by analytical reverse-phase HPLC using gradient elution), as well as the enzyme preparation, the kinetic data with the methodology used to calculate the inhibition constants, and the mechanism of inhibition, crystallographic data collection, and structural determination; molecular formula strings in csv format. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
PDB codes for Neisseria meningitides alanyl aminopeptidase complexed with organophosphorus inhibitors are as follows: 4PU2 (compound R-3), 4PVB (S-3), 4PW4 (R-4), 4QME (R,S-1), 4QIR (11), 4QUO (35), and 4QHP (36).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Yiotakis A.; Georgiadis D.; Matziari M.; Makaritis A.; Dive V. Phosphinic Peptides: Synthetic Approaches and Biochemical Evaluation as Zn-Metalloprotease Inhibitors. Curr. Org. Chem. 2004, 8, 1135–1158. [Google Scholar]
- Collinsova M.; Jiracek J. Phosphinic Acid Compounds in Biochemistry, Biology and Medicine. Curr. Med. Chem. 2000, 7, 629–647. [DOI] [PubMed] [Google Scholar]
- Mucha A.; Kafarski P.; Berlicki L. Remarkable Potential of the α-Aminophosphonate/Phosphinate Structural Motif in Medicinal Chemistry. J. Med. Chem. 2011, 54, 5955–5980. [DOI] [PubMed] [Google Scholar]
- Jiracek J.; Yiotakis A.; Vincent B.; Lecoq A.; Nicolaou A.; Checler F.; Dive V. Development of Highly Potent and Selective Phosphinic Peptide Inhibitors of Zinc Endopeptidase 24-15 Using Combinatorial Chemistry. J. Biol. Chem. 1995, 270, 21701–21706. [DOI] [PubMed] [Google Scholar]
- Jiracek J.; Yiotakis A.; Vincent B.; Checler F.; Dive V. Development of the First Potent and Selective Inhibitor of the Zinc Endopeptidase Neurolysin Using a Systematic Approach Based on Combinatorial Chemistry of Phosphinic Peptides. J. Biol. Chem. 1996, 271, 19606–19611. [DOI] [PubMed] [Google Scholar]
- Mucha A. Synthesis and Modifications of Phosphinic Dipeptide Analogues. Molecules 2012, 17, 13530–13568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matziari M.; Georgiadis D.; Dive V.; Yiotakis A. Convenient Synthesis and Diversification of Dehydroalaninyl Phosphinic Peptide Analogues. Org. Lett. 2001, 3, 659–662. [DOI] [PubMed] [Google Scholar]
- Matziari M.; Beau F.; Cuniasse P.; Dive V.; Yiotakis A. Evaluation of P1′-Diversified Phosphinic Peptides Leads to the Development of Highly Selective Inhibitors of MMP-11. J. Med. Chem. 2004, 47, 325–336. [DOI] [PubMed] [Google Scholar]
- Matziari M.; Nasopoulou M.; Yiotakis A. Active Methylene Phosphinic Peptides: A New Diversification Approach. Org. Lett. 2006, 8, 2317–2319. [DOI] [PubMed] [Google Scholar]
- Makaritis A.; Georgiadis D.; Dive V.; Yiotakis A. Diastereoselective Solution and Multipin-Based Combinatorial Array Synthesis of a Novel Class of Potent Phosphinic Metalloprotease Inhibitors. Chem.—Eur. J. 2003, 9, 2079–2094. [DOI] [PubMed] [Google Scholar]
- Jullien N.; Makaritis A.; Georgiadis D.; Beau F.; Yiotakis A.; Dive V. Phosphinic Tripeptides as Dual Angiotensin-Converting Enzyme C-Domain and Endothelin-Converting Enzyme-1 Inhibitors. J. Med. Chem. 2010, 53, 208–220. [DOI] [PubMed] [Google Scholar]
- Vassiliou S.; Xeilari M.; Yiotakis A.; Grembecka J.; Pawelczak M.; Kafarski P.; Mucha A. A Synthetic Method for Diversification of the P1′ Substituent in Phosphinic Dipeptides as a Tool for Exploration of the Specificity of the S1′ Binding Pockets of Leucine Aminopeptidases. Bioorg. Med. Chem. 2007, 15, 3187–3200. [DOI] [PubMed] [Google Scholar]
- a Turner A. J.Aminopeptidase N. In Handbook of Proteolytic Enzymes, 3rd ed.; Rawlings N. D., Salvesen G., Eds.; Academic Press: Amsterdam, 2013; pp 397–403. [Google Scholar]; b Turner A. J.PfA-M1 Aminopeptidase (Plasmodium falciparum). In Handbook of Proteolytic Enzymes, 3rd ed.; Rawlings N. D., Salvesen G., Eds.; Academic Press: Amsterdam, 2013; pp 445–448. [Google Scholar]; c Bhosale M.Alanyl Aminopeptidase (Bacterial-Type). In Handbook of Proteolytic Enzymes, 3rd ed.; Rawlings N. D., Salvesen G., Eds.; Academic Press: Amsterdam, 2013; pp 456–462. [Google Scholar]
- a Strater N.; Lipscomb W. N. Leucyl Aminopeptidase (Animal). In Handbook of Proteolytic Enzymes, 3rd ed.; Rawlings N. D., Salvesen G., Eds.; Academic Press: Amsterdam, 2013; pp 1465–1470. [Google Scholar]; b Walling L. L.Leucyl Aminopeptidase (Plant). In Handbook of Proteolytic Enzymes, 3rd ed.; Rawlings N. D., Salvesen G., Eds.; Academic Press: Amsterdam, 2013; pp 1471–1476. [Google Scholar]; c Nsangu D. M. M.; Mathew R. T.; Thivierge K.; Gardiner D. L.; Dalton J. P.. Leucyl Aminopeptidase of Plasmodium falciparum. In Handbook of Proteolytic Enzymes, 3rd ed.; Rawlings N. D., Salvesen G., Eds.; Academic Press: Amsterdam, 2013; pp 1481–1484. [Google Scholar]
- Matsui M.; Fowler J. H.; Walling L. L. Leucine Aminopeptidases: Diversity in Structure and Function. Biol. Chem. 2006, 387, 1535–1544. [DOI] [PubMed] [Google Scholar]
- Grembecka J.; Kafarski P. Leucine Aminopeptidase as a Target for Inhibitor Design. Mini-Rev. Med. Chem. 2001, 1, 133–144. [DOI] [PubMed] [Google Scholar]
- Luan Y.; Xu W. The Structure and Main Functions of Aminopeptidase N. Curr. Med. Chem. 2007, 14, 639–647. [DOI] [PubMed] [Google Scholar]
- Mina-Osorio P. The Moonlighting Enzyme CD13: Old and New Functions To Target. Trends Mol. Med. 2008, 14, 361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauvois B.; Dauzonne D. Aminopeptidase-N/CD13 (EC 3.4.11.2) Inhibitors: Chemistry, Biological Evaluations, and Therapeutic Prospects. Med. Res. Rev. 2006, 26, 88–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Xu W.; Aminopeptidase N. (APN/CD13) as a Target for Anti-Cancer Agent Design. Curr. Med. Chem. 2008, 15, 2850–2865. [DOI] [PubMed] [Google Scholar]
- Wickström M.; Larsson R.; Nygren P.; Gullbo J. Aminopeptidase N (CD13) as a Target for Cancer Chemotherapy. Cancer Sci. 2011, 102, 501–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitzerd S. M.; Verbrugge S. E.; Ossenkoppele G.; Jansen G.; Peters G. J. Positioning of Aminopeptidase Inhibitors in Next Generation Cancer Therapy. Amino Acids 2014, 46, 793–808. [DOI] [PubMed] [Google Scholar]
- Skinner-Adams T. S.; Stack C. M.; Trenholme K. R.; Brown C. L.; Grembecka J.; Lowther J.; Mucha A.; Drag M.; Kafarski P.; McGowan S.; Whisstock J. C.; Gardiner D. L.; Dalton J. P. Plasmodium falciparum Neutral Aminopeptidases: New Targets for Anti-Malarials. Trends Biochem. Sci. 2010, 35, 53–61. [DOI] [PubMed] [Google Scholar]
- Mucha A.; Drag M.; Dalton J. P.; Kafarski P. Metallo-aminopeptidase Inhibitors. Biochimie 2010, 92, 1509–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grembecka J.; Mucha A.; Cierpicki T.; Kafarski P. The Most Potent Organophosphorus Inhibitors of Leucine Aminopeptidase. Structure-Based Design, Chemistry, and Activity. J. Med. Chem. 2003, 46, 2641–2655. [DOI] [PubMed] [Google Scholar]
- Weglarz-Tomczak E.; Poreba M.; Byzia A.; Berlicki L.; Nocek B.; Mulligan R.; Joachimiak A.; Drag M.; Mucha A. An Integrated Approach to the Ligand Binding Specificity of Neisseria Meningitidis M1 Alanine Aminopeptidase by Fluorogenic Substrate Profiling, Inhibitory Studies and Molecular Modeling. Biochimie 2013, 95, 419–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drag M.; Bogyo M.; Ellman J. A.; Salvesen G. S. Aminopeptidase Fingerprints. An Integrated Approach for Identification of Good Substrates and Optimal Inhibitors. J. Biol. Chem. 2010, 285, 3310–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poreba M.; McGowan S.; Skinner-Adams T. S.; Trenholme K. R.; Gardiner D. L.; Whisstock J. C.; To J.; Salvesen G. S.; Dalton J. P.; Drag M. Fingerprinting the Substrate Specificity of M1 and M17 Aminopeptidases of Human Malaria, Plasmodium falciparum. PLoS One 2012, 7, e31938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drag M.; Grembecka J.; Pawelczak M.; Kafarski P. α-Aminoalkylphosphonates as a Tool in Experimental Optimisation of P1 Side Chain Shape of Potential Inhibitors in S1 Pocket of Leucine- and Neutral Aminopeptidases. Eur. J. Med. Chem. 2005, 40, 764–771. [DOI] [PubMed] [Google Scholar]
- Kannan Sivaraman K.; Paiardini A.; Sienczyk M.; Ruggeri C.; Oellig C. A.; Dalton J. P.; Scammells P. J.; Drag M.; McGowan S. Synthesis and Structure–Activity Relationships of Phosphonic Arginine Mimetics as Inhibitors of the M1 and M17 Aminopeptidases from Plasmodium falciparum. J. Med. Chem. 2013, 56, 5213–5217. [DOI] [PubMed] [Google Scholar]
- Giannousis P. P.; Bartlett P. A. Phosphorus Amino Acid Analogues as Inhibitors of Leucine Aminopeptidase. J. Med. Chem. 1987, 30, 1603–1609. [DOI] [PubMed] [Google Scholar]
- Lejczak B.; Kafarski P.; Zygmunt J. Inhibition of Aminopeptidases by Aminophosphonates. Biochemistry 1989, 28, 3549–3555. [DOI] [PubMed] [Google Scholar]
- Hamilton R.; Walker B.; Walker B. J. A Highly Convenient Route to Optically Pure α-Aminophosphonic Acids. Tetrahedron Lett. 1995, 36, 4451–4454. [Google Scholar]
- Baylis E. K.; Campbell C. D.; Dingwall J. G. 1-Aminoalkylphosphonous Acids. Part 1. Isosteres of the Protein Amino Acids. J. Chem. Soc., Perkin Trans. 1 1984, 2845–2853. [Google Scholar]
- Lämmerhofer M.; Hebenstreit D.; Gavioli E.; Lindner W.; Mucha A.; Kafarski P.; Wieczorek P. High-Performance Liquid Chromatographic Enantiomer Separation and Determination of Absolute Configurations of Phosphinic acid Analogues of Dipeptides and Their α-Aminophosphinic Acid Precursors. Tetrahedron: Asymmetry 2003, 14, 2557–2565. [Google Scholar]
- Nocek B.; Mulligan R.; Bargassa M.; Collart F.; Joachimiak A. Crystal Structure of Aminopeptidase N from Human Pathogen Neisseria meningitides. Proteins 2008, 70, 273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addlagatta A.; Gay L.; Matthews B. W. Structure of Aminopeptidase N from Escherichia coli Suggests a Compartmentalized, Gated Active Site. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 13339–13344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K.; Nakajima Y.; Onohara Y.; Takeo M.; Nakashima K.; Matsubara F.; Ito T.; Yoshimoto T. Crystal Structure of Aminopeptidase N (Proteobacteria Alanyl Aminopeptidase) from Escherichia coli and Conformational Change of Methionine 260 Involved in Substrate Recognition. J. Biol. Chem. 2006, 281, 33664–33676. [DOI] [PubMed] [Google Scholar]
- Addlagatta A.; Gay L.; Matthews B. W. Structural Basis for the Unusual Specificity of Escherichia coli Aminopeptidase N. Biochemistry 2008, 47, 5303–5311. [DOI] [PubMed] [Google Scholar]
- Fournié-Zaluski M. C.; Poras H.; Roques B. P.; Nakajima Y.; Ito K.; Yoshimoto T. Structure of Aminopeptidase N from Escherichia coli Complexed with the Transition-State Analogue Aminophosphinic Inhibitor PL250. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2009, 65, 814–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumpena R.; Kishor C.; Ganji R. J.; Addlagatta A. Discovery of α,β- and α,γ-Diamino Acid Scaffolds for the Inhibition of M1 Family Aminopeptidases. Chem. Med. Chem. 2011, 6, 1971–1976. [DOI] [PubMed] [Google Scholar]
- Sträter N.; Lipscomb W. N. Transition State Analogue l-Leucinephosphonic Acid Bound to Bovine Lens Leucine Aminopeptidase: X-ray Structure at 1.65 Å Resolution in a New Crystal Form. Biochemistry 1995, 34, 9200–9210. [DOI] [PubMed] [Google Scholar]
- Stamper C.; Bennett B.; Edwards T.; Holz R. C.; Ringe D.; Petsko G. Inhibition of the Aminopeptidase from Aeromonas proteolytica by l-Leucinephosphonic Acid. Spectroscopic and Crystallographic Characterization of the Transition State of Peptide Hydrolysis. Biochemistry 2001, 40, 7035–7046. [DOI] [PubMed] [Google Scholar]
- Ataie N. J.; Hoang Q. Q.; Zahniser M. P.; Tu Y.; Milne A.; Petsko G. A.; Ringe D. Zinc Coordination Geometry and Ligand Binding Affinity: The Structural and Kinetic Analysis of the Second-Shell Serine 228 Residue and the Methionine 180 Residue of the Aminopeptidase from Vibrio proteolyticus. Biochemistry 2008, 47, 7673–7683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner-Adams T. S.; Lowther J.; Teuscher F.; Stack C. M.; Grembecka J.; Mucha A.; Kafarski P.; Trenholme K. R.; Dalton J. P.; Gardiner D. L. Identification of Phosphinate Dipeptide Analog Inhibitors Directed against the Plasmodium falciparum M17 Leucine Aminopeptidase as Lead Antimalarial Compounds. J. Med. Chem. 2007, 50, 6024–6031. [DOI] [PubMed] [Google Scholar]
- McGowan S.; Porter C. J.; Lowther J.; Stack C. M.; Golding S. J.; Skinner-Adams T. S.; Trenholme K. R.; Teuscher F.; Donnelly S. M.; Grembecka J.; Mucha A.; Kafarski P.; Degori R.; Buckle A. M.; Gardiner D. L.; Whisstock J. C.; Dalton J. P. Structural Basis for the Inhibition of the Essential Plasmodium falciparum M1 Neutral Aminopeptidase. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 2537–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucha A.; Lämmerhofer M.; Lindner W.; Pawelczak M.; Kafarski P. Individual Stereoisomers of Phosphinic Dipeptide Inhibitor of Leucine Aminopeptidase. Bioorg. Med. Chem. Lett. 2008, 18, 1550–1554. [DOI] [PubMed] [Google Scholar]
- Chen L.; Lin Y. L.; Peng G.; Li F. Structural Basis for Multifunctional Roles of Mammalian Aminopeptidase N. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 17966–17971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong A. H. M.; Zhou D.; Rini J. M. The X-ray Crystal Structure of Human Aminopeptidase N Reveals a Novel Dimer and the Basis for Peptide Processing. J. Biol. Chem. 2012, 287, 36804–36813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dmitriev E. M.; Rossinets A. E.; Ragulin V. V. Amidoalkylation of Hydrophosphoryl Compounds. Russ. J. Gen. Chem. 2011, 81, 1092–1104. [Google Scholar]
- Matziari M.; Yiotakis A. Shortcut to Fmoc-Protected Phosphinic Pseudodipeptidic Blocks. Org. Lett. 2005, 7, 4049–4052. [DOI] [PubMed] [Google Scholar]
- Fukuyama T.; Cheng Lin S.; Li L. Facile Reduction of Ethyl Thiol Esters to Aldehydes: Application to a Total Synthesis of (+)-Neothramycin A Methyl Ether. J. Am. Chem. Soc. 1990, 112, 7050–7051. [Google Scholar]
- Zakharkin L. I.; Khorlina I. M. Reduction of Esters of Carboxylic Acids into Aldehydes with Diisobutylaluminium Hydride. Tetrahedron Lett. 1962, 3, 619–620. [Google Scholar]
- Polla M. O.; Tottie L.; Nordén C.; Linschoten M.; Müsil D.; Trumpp-Kallmeyer S.; Aukrust I. R.; Ringom R.; Holm K. H.; Neset S. M.; Sandberg M.; Thurmond J.; Yu P.; Hategan G.; Anderson H. Design and Synthesis of Potent, Orally Active, Inhibitors of Carboxypeptidase U (TAFIa). Bioorg. Med. Chem. 2004, 12, 1151–1175. [DOI] [PubMed] [Google Scholar]
- Chen H.; Roques B. P.; Fournié-Zaluski M.-C. Design of the First Highly Potent and Selective Aminopeptidase N (EC 3.4.11.2) Inhibitor. Bioorg. Med. Chem. Lett. 1999, 9, 1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Noble F.; Mothé A.; Meudal H.; Coric P.; Danascimento S.; Roques B. P.; George P.; Fournié-Zaluski M.-C. Phosphinic Derivatives as New Dual Enkephalin-Degrading Enzyme Inhibitors: Synthesis, Biological Properties, and Antinociceptive Activities. J. Med. Chem. 2000, 43, 1398–1408. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.