

Abstract
Objective
There is a need to identify a cognitive composite that is sensitive to tracking preclinical AD decline to be used as a primary endpoint in treatment trials.
Method
We capitalized on longitudinal data, collected from 1995 to 2010, from cognitively unimpaired presenilin 1 (PSEN1) E280A mutation carriers from the world’s largest known early-onset autosomal dominant AD (ADAD) kindred to identify a composite cognitive test with the greatest statistical power to track preclinical AD decline and estimate the number of carriers age 30 and older needed to detect a treatment effect in the Alzheimer’s Prevention Initiative’s (API) preclinical AD treatment trial. The mean-to-standard-deviation ratios (MSDRs) of change over time were calculated in a search for the optimal combination of one to seven cognitive tests/sub-tests drawn from the neuropsychological test battery in cognitively unimpaired mutation carriers during a two and five year follow-up period, using data from non-carriers during the same time period to correct for aging and practice effects. Combinations that performed well were then evaluated for robustness across follow-up years, occurrence of selected items within top performing combinations and representation of relevant cognitive domains.
Results
This optimal test combination included CERAD Word List Recall, CERAD Boston Naming Test (high frequency items), MMSE Orientation to Time, CERAD Constructional Praxis and Ravens Progressive Matrices (Set A) with an MSDR of 1.62. This composite is more sensitive than using either the CERAD Word List Recall (MSDR=0.38) or the entire CERAD-Col battery (MSDR=0.76). A sample size of 75 cognitively normal PSEN1-E280A mutation carriers age 30 and older per treatment arm allows for a detectable treatment effect of 29% in a 60-month trial (80% power, p=0.05).
Conclusions
We have identified a composite cognitive test score representing multiple cognitive domains that has improved power compared to the most sensitive single test item to track preclinical AD decline in ADAD mutation carriers and evaluate preclinical AD treatments. This API composite cognitive test score will be used as the primary endpoint in the first API trial in cognitively unimpaired ADAD carriers within 15 years of their estimated age at clinical onset. We have independently confirmed our findings in a separate cohort of cognitively healthy older adults who progressed to the clinical stages of late-onset AD, described in a separate report, and continue to refine the composite in independent cohorts and compared with other analytical approaches.
Keywords: composite cognitive score, API, Alzheimer’s Prevention Initiative, E280A, PSEN1, presenilin1, sample size, preclinical, cognitively unimpaired, autosomal dominant, ADAD
INTRODUCTION
There is an urgent need to find effective preclinical Alzheimer’s disease (AD) treatments, which we have previously defined as “interventions that are started in the absence of mild cognitive impairment (MCI) or dementia and intended to postpone the onset, reduce the risk of, or completely prevent the clinical stages of AD”(1). Several such trials are recently launched or being planned including those with the strategy of testing therapies in people who are at the highest imminent risk of developing MCI or AD dementia due to factors such as age and genetic backgrounds or presence of biomarker evidence of AD (2–5). Detecting a treatment effect in a preclinical AD trial using clinical progression or cognitive outcome developed for studies in MCI or AD dementia as the primary endpoint may not be desirable due to a large sample size and lengthy follow-up required (6) or the psychometric properties of the tests themselves (7–9). Using multiple cognitive assessments that are sensitive to preclinical AD as potentially successful outcomes inflates Type-I error. Using an appropriate composite minimizes the number of outcomes employed and thus risk of Type-I error, it can be empirically derived and its sensitivity to detecting and tracking preclinical AD can be validated in independent datasets. As a result, it affords a measure of multiple cognitive domains that can serve as a primary endpoint in preclinical treatment trials (10).
Slight, but measureable cognitive decline have been reported during preclinical AD, retrospective and prospective studies of cognitively normal individuals who subsequently progressed to AD dementia have found episodic memory decline to be a defining feature of preclinical AD (11–15). Decline in other cognitive domains, such as executive (16), visual spatial (13), and global cognitive functioning (13;17) also occurs during the transition from normal aging to preclinical AD and into the clinical stages of AD. In cognitively unimpaired individuals with significant fibrillar amyloid burden, decline has been observed primarily in episodic memory, executive function and language (18–22). Studies of cognitively unimpaired autosomal dominant AD (ADAD) mutation carriers have reported subtle decline in memory, language, praxis, abstract reasoning and attention (23).
Recent research has focused on developing a measure of AD-related cognitive decline to track the progression of preclinical AD in order to evaluate investigational preclinical AD treatments with increased statistical power(24). A theoretically driven approach reasons that a composite (i.e., a test score derived from two or more different cognitive tests) should be constructed a priori from cognitive assessments known to decline relatively early in the disease progression. A related approach is to construct a composite score that summarizes the performance in a specific domain, such as memory (25) or executive functioning(26), believed to be preferentially affected by AD. An empirically driven approach employs computational modeling techniques to identify an endpoint or composite(24) based on its sensitivity to detect and track the outcome of interest, such as preclinical AD. Analyses methods that can be used for developing cognitive composites include, among others, latent variable analyses or partial least squares regression(24;27;28), Item response theory (IRT)(29) and principal components regression analysis(30), Rasch Measurement Theory or item-level analysis. Note that these approaches are not mutually exclusive (eg. theoretical knowledge of preclinical AD can be taken into account when empirically deriving a composite cognitive test score).
In the present study, we aimed to develop a composite cognitive test score that is highly sensitive to detecting and tracking preclinical cognitive decline, corresponding to an analysis of a change from baseline, rather than to optimize the discrimination between those who progress to clinical AD versus those who remain cognitively unimpaired. We examined longitudinal data from cognitively unimpaired presenilin 1 (PSEN1) E280A mutation carriers and non-carriers from the world’s largest known early-onset ADAD kindred to develop a composite cognitive test score most sensitive to detecting and tracking preclinical cognitive decline and calculate the number of cognitively unimpaired PSEN1 E280A mutation carriers within 15 years of their estimated age at clinical onset needed to evaluate an amyloid-modifying treatment in the Alzheimer’s Prevention Initiative (API)’s first preclinical AD trial(1;31–33). We hypothesize that the identified composite will have higher sensitivity and have greater statistical power to detect and track cognitive decline associated with preclinical AD compared to the most sensitive individual cognitive test/sub-test score or to the entire neuropsychological assessment battery given that the empirically driven approach allows for the addition of assessments that improve overall sensitivity despite perhaps being less sensitive individually to preclinical AD decline.
METHODS
Participants
Descendants of patients with confirmed PSEN1 E280A mutations were enrolled into the E280A Antioquia cohort study between 1995 and 2010 conducted by the Neuroscience group at University of Antioquia and approved by the medical ethics board of the University of Antioquia, Colombia (34). Participants in the cohort study must be age 17 years or older; there are no exclusion criteria regarding medical and neuropsychological monitoring. The participants or their guardians provided their informed consent. Participants without signs of dementia are not provided their genetic status. The original dataset is available from Grupo de Neurociencias de Antioquia, Universidad de Antioquia, Medellín, Colombia.
For the present study, only data from cohort study participants who met the following criteria were used in the analyses: 1) age 30 or older at baseline (approximately 15 years prior to median age of clinical onset)(23), 2) were not diagnosed with MCI or dementia due to AD between the baseline and two- and five-year follow-up visits, 3) had a minimum of two or five years of longitudinal neuropsychological testing data, and 4) no report of retardation, cerebral paralysis, cerebral lesion, major psychiatric disease, serious systemic illness, uncontrolled seizures, or alcohol abuse, which would preclude participation in a typical clinical trial. The resulting dataset was comprised of 56 PSEN1 E280A carriers and 78 non-carriers for the 24-month analyses, 31 carriers and 57 non-carriers for the 60-month analyses (Figure 1).
Figure 1.

Participant selection
Cognitive and Clinical Evaluations
An initial interview and follow-up examination(s), including medical, psychological and neuropsychological assessments, were performed by neurologists or psychologists trained in neuropsychology who were masked to participants’ carrier status. The assessment protocol included the CERAD (Consortium to Establish a Registry for Alzheimer’s disease) neuropsychological battery as well as additional tests to further assess constructional abilities and abstraction which were translated to Spanish and adapted to the cultural and linguistic idiosyncrasies of the target population (referred to as the CERAD-Col)(23;34–36).
The CERAD-Col assessment battery and details of its administration have been previously described (23;34–36) and is shown in Table 1. Dementia functional scales were also administered.
Table 1.
Cognitive Assessments
| Cognitive Assessments | Cognitive Domain Tested |
|---|---|
| Visual A cancellation tests | attention |
| Trail making test-Part A | attention |
| Memory of three phrases tests | memory |
| Rey-Osterrieth Complex Figure-Recall | memory |
| CERAD Word List –Recall, - Recognition | memory |
| Recall of line drawing test | memory |
| Categorical verbal fluency | language ability |
| Boston naming test | language ability |
| Constructional praxis test | constructional abilities |
| Rey-Osterrieth Complex Figure-Copy | constructional abilities |
| Raven’s progressive matrices (Part A) | abstract reasoning |
| Wechsler intelligence scale-arithmetic (revised) | calculation abilities |
| Wisconsin card sorting test | executive function |
| Phonological verbal fluency-F test | executive function |
| Mini Mental State Examination | orientation, memory, attention and concentration, language ability |
Diagnostic classification followed a procedure previously described (23). Briefly, MCI criteria included 1) clinically significant cognitive decline as indicated by cognitive test scores of 2 standard deviations or more away from the mean normal value scores for non-carriers in at least one test on any cognitive domain, adjusted for age and education, 2) subjective memory impairment corroborated by an informant. Dementia criteria included 1) impaired instrumental activities of daily living, 2) impaired activities of daily living, and 3) meeting DSM-IV criteria for Dementia (American Psychiatric Association, 1994).
Data Analysis
We perform an search of every combination of one to seven cognitive assessments and calculate the corresponding annualized mean to standard deviation ratios (MSDR) of the standardized change over time for the cognitively unimpaired PSEN1 E280A mutation carriers age 30 and older during two (n=57) and five years (n=31) of follow-up. MSDR values were adjusted for practice effects using data from the kindred mutation non-carriers (two year follow-up n = 78; five year follow-up n=56) by calculating the mean change of the composite score in the non-carriers and subtracting this value from the composite score change calculated in the mutation carriers. The MSDR was chosen as a measurement of sensitivity to the longitudinal decline for a cognitive test combination, representing the coefficient of change (the mean of standardized change divided by the standard deviation of standardized change) and was calculated as:
Where xi is a change in standardized cognitive score i,
n is a number of cognitive scores in the composite
Xj is the change in composite score of subject j
σx is the standard deviation of the cognitive scores
The MSDR is quite similar to an effect size, as components of the MSDR are used to calculate it and, the larger the MSDR value, the greater the sensitivity to detecting and tracking cognitive decline over time. Prior to calculating the MSDRs, each cognitive assessment was standardized on a 0–1 scale, similar to a z-score. For assessments that did not have a predefined maximum score (such as Categorical Fluency), a value of 2 standard deviations above the mean was used as the maximum.
Results from these analyses were used as one way to assess the combinations and determine an optimal composite. Tests that were consistently represented in the combinations with the highest sensitivity and that also demonstrated consistency within separate years of the 2 and 5 year follow-up time period were identified as robust items for measuring change. The optimal combination was then evaluated for construct validity and was used to calculate the sample size required in a 60-month trial to detect a 25% treatment effect with 80% power and p=0.05, as well as the detectable treatment effect a 60-month trial with 75 PSEN1 E280A mutation carriers per treatment arm.
Weighting the optimal composite cognitive test score
After identifying the optimal composite cognitive test score, we examined whether the MSDR could be increased (and therefore, the sensitivity improved) by weighting the individual assessments included in the composite. An search of every potential weighting combination to optimize the sensitivity such that
where wij is the weight for test i and subject j and wij ≥ 0 and was conducted in PSEN1 E280A carriers during the two and five year follow-up period. Note that wij is the same for every subject (ie. though weights differ between the tests/sub-test, the weight for each test/sub-test is constant across all subjects). Data from the non-carriers were used to adjust for practice effects. The combination of weights that resulted in the largest adjusted MSDR was then used to calculate the sample size needed to detect a 25% treatment effect and estimate the treatment effect that could be detected in 75 PSEN1 E280A mutation carriers with 80% power and p = 0.05.
Evaluating the optimal composite cognitive test score
In order to confirm the sensitivity of the composite cognitive test score, the MSDR of the composite was compared to the MSDR of the CERAD Word List Recall, an episodic memory assessment, and the entire MSDR of the CERAD-Col battery. Additionally, to evaluate the stability of the composite cognitive test score, selected test items from the composite test score were replaced with a different test item from the same cognitive domain and the resulting MSDRs and required sample sizes were compared.
RESULTS
Participant Characteristics
At baseline, the PSEN1 E280A carriers and kindred non-carriers did not differ in terms of age or level of education. The carrier group included in the 2-year analysis had a higher ratio of males compared to the non-carrier group, but this difference was not present in the 5-year analysis (Table 2).
Table 2.
Baseline characteristics of participants included in the power analysis for 24-month and 60-month RCTa
|
|
||||||
|---|---|---|---|---|---|---|
| 2 years analysis | 5 years analysis | |||||
| Carrier (n=56) | Non-Carrier (n=78) | p-valueb | Carrier (n=31) | Non-Carrier (n=57) | p-valueb | |
| Age | 43.93±6.49 | 44.99±10.07 | 0.49 | 41.37±4.36 | 45.35±11.05 | 0.06 |
| Education | 7.84±4.57 | 7.29±3.97 | 0.92 | 6.74±4.93 | 6.86±4.96 | 0.92 |
| Sex (%M/F) | 39/61 | 22/78 | 0.03 | 35/65 | 37/63 | 0.89 |
| MMSE | 26.63±4.01 | 28.95±1.79 | 1.27e-5 | 26.97±2.36 | 28.53±2.10 | 2.01e-3 |
| Verbal Fluency (Animal) | 15.51±5.08 | 18.33±4.45 | 1.01e-3 | 17±4.77 | 18.82±3.91 | 0.06 |
| Boston Naming | 12.02±2.23 | 12.64±2.01 | 0.01 | 12.3±2.14 | 12.44±1.93 | 0.77 |
| CERAD Word List-Immediate | ||||||
| Correct | 13.49±5.90 | 17.78±4.83 | 1.18e-5 | 15.43±4.88 | 17.24±4.81 | 0.11 |
| Intrusions | 3.55±4.54 | 1.45±2.09 | 5.06e-4 | 2.87±2.75 | 1.56±3.27 | 0.07 |
| CERAD Word List-Recall | ||||||
| Correct | 3.75±2.89 | 6.49±2.22 | 1.08e-8 | 4.23±2.74 | 6.47±2.07 | 5.73e-5 |
| Intrusions | 1.34±1.62 | 0.6±1.44 | 7.20e-3 | 1.03±1.50 | 0.56±1.40 | 0.15 |
| CERAD Recognition of words | ||||||
| Correct “yes” | 8.6±1.70 | 9.55±1.03 | 1.19e-4 | 9.13±1.11 | 9.35±1.44 | 0.49 |
| Correct “no” | 8.81±1.82 | 9.9±0.41 | 1.23e-6 | 9.3±1.18 | 9.73±1.10 | 0.01 |
| Constructional Praxis | 9.26±1.57 | 9.51±1.6 | 0.38 | 9.7±1.21 | 9.6±1.62 | 0.77 |
| Trail Making Test-A | ||||||
| Errors | 1.11±3.63 | 0.28±1.39 | 0.08 | 0.37±1.55 | 0.13±0.49 | 0.31 |
| Time | 115.43±80 | 81.39±61.55 | 0.01 | 93.07±50.94 | 86.96±50.95 | 0.62 |
| Recall of drawings | 7.2±7.01 | 14.06±7.15 | 3.08e-7 | 8.18±7.76 | 13.63±7.51 | 2.16e-3 |
| Raven’s Progressive Matrices | 7.92±2.28 | 8.41±2.08 | 0.21 | 8.03±2.06 | 8.36±2.23 | 0.51 |
MMSE, Mini-Mental State examination
All data are mean±SD unless otherwise stated.
Baseline group differences were compared using two-tailed T-Test, and Chi Square test.
Individual Cognitive Assessment Properties
The most sensitive individual neuropsychological tests for differentiating PSEN1 E280A carriers from non-carriers at baseline included the CERAD Word List Recall, CERAD Word List Recognition, Recall of Drawings, MMSE Total, and MMSE Orientation to Time (p<=0.05) (Table 2). The individual neuropsychological tests most sensitive to longitudinal decline during the five-year follow-up period (unadjusted for practice effect) include Memory of Three Phrases, Wechsler-Arithmetic, Recall of Drawings, Rey-Osterrieth Complex Figure-Copy, Constructional Praxis (cube), Raven Progressive Matrices (Set A), CERAD Word List Recognition-Total Correct, Constructional Praxis, MMSE Orientation to Time (Table 3). After adjusting for practice effects using data from the non-carriers, the individual neuropsychological tests most sensitive to longitudinal decline during the five-year follow-up were nearly identical to those from the unadjusted analysis (Table 3).
Table 3.
The most sensitive individual item MSDRs during 5-year analysis
| Cognitive Assessment | Unadjusted MSDR | Cognitive Assessment | Adjusted MSDR |
|---|---|---|---|
| Memory of Three | |||
| Phrases | 0.99 | Wechsler-Arithmetic Memory of Three | 1.09 |
| Wechsler-Arithmetic | 0.96 | Phrases | 0.98 |
| Recall of Drawings | 0.85 | Recall of Drawings | 0.94 |
| Rey-Osterrieth Complex Figure-Copy | 0.85 | Rey-Osterrieth Complex Figure-Copy | 0.92 |
| Constructional Praxis (cube) | 0.84 | Raven Progressive Matrices | 0.85 |
| Raven Progressive Matrices | 0.81 | MMSE Orientation to Time | 0.77 |
| CERAD Word List Recognition-Total Correct | 0.73 | Constructional Praxis (cube) | 0.72 |
| Constructional Praxis | 0.73 | Constructional Praxis | 0.68 |
| MMSE Orientation to Time | 0.73 | Trail Making Test A-Time | 0.67 |
Empirically Deriving the Alzheimer’s Prevention Initiative (API) Composite Cognitive Test Score
The combination most sensitive to detecting preclinical cognitive decline related to ADAD, adjusting for aging and practice effects, that has construct validity and also include tests/sub-tests that are robust across follow-up time periods consisted of: MMSE Orientation to Time, CERAD Boston Naming Test (high frequency items), CERAD Word List Recall, Constructional Praxis and Ravens Progressive Matrices (Set A). Based on the five-year follow-up data, the total 60-month MSDR of the composite cognitive test score is 1.62. In comparison, the most sensitive individual cognitive assessment is the Memory of Three Phrases, with a total 60-month MSDR of 0.99, making the composite cognitive test score considerably more sensitive to tracking preclinical cognitive decline in ADAD mutation carriers. Based on the two-year longitudinal data, a shorter study with the same composite cognitive test score would result in total MSDR of 1.06. This is important to consider as the MSDR is a coefficient of change (the mean change divided by the standard deviation of change), in which a larger value indicates the sensitivity of the measure, thereby impacting the required sample size and detectable treatment effect(24).
Based on the MSDR of the API Composite Cognitive Test Score, 97 PSEN1 E280A mutation carriers who complete the trial per group age 30 and older are needed to detect a 25% treatment effect in a 60-month RCT (Table 4). In contrast, if using the CERAD Word List Recall, 1223 mutation carriers per group age 30 and older are needed to detect a 25% treatment effect in a 60-month RCT, while using the entire CERAD-Col battery would require 355 carriers per group (Figure 2).
Table 4.
Estimated sample size (completers) required to detect 25% treatment effect with 80% power and alpha=0.05
| Composite measure* | Month | Total n | Adjusted MSDR | Estimated sample size (completers)/group |
|---|---|---|---|---|
| Unweighted | 60 | 56 | 1.62 | 97 |
| Unweighted | 24 | 95 | 1.06 | 225 |
| Weighted | 60 | 56 | 1.93 | 69 |
| Weighted | 24 | 95 | 1.19 | 179 |
Composite measure includes: CERAD Word List Recall, CERAD Boston Naming Test-High, MMSE Orientation to Time, Constructional Praxis, and Raven’s Progressive Matrices
CERAD = Consortium to establish a Registry for Alzheimer’s Disease
MMSE = Minimental State Exam
Figure 2.
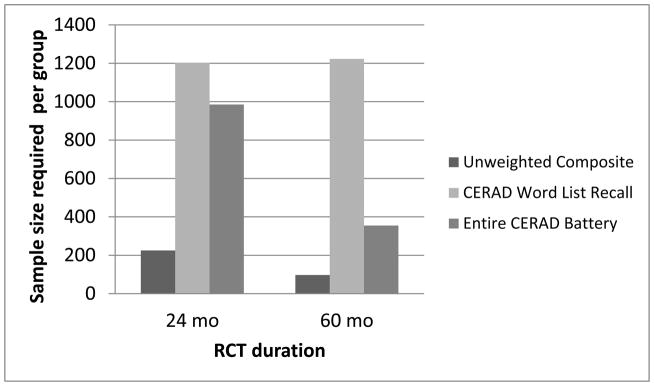
PSEN1 E280A mutation carriers per group over the age of 30 required to detect a 25% treatment effect in a 24/60-month RCT using unweighted composite, CERAD Word List Recall or Entire CERAD battery endpoints with 80% statistical power and 0.05 type-I error.
Using the API Composite Cognitive Test Score, we estimate that a trial of 75 mutation carriers who complete the trial per treatment arm age 30 and older would permit us to detect a 29% treatment effect in a 60-month trial (Table 5). In comparison, using the CERAD World List Recall would permit us to detect a 103% treatment effect, while the CERAD-Col would permit us to detect a 55% treatment effect.
Table 5.
Estimated minimal detectable treatment effect required using with 75 completers per group, 80% power and alpha=0.05
| Composite measure* | Month | Total n | Adjusted MSDR | Detectable treatment effect |
|---|---|---|---|---|
| Unweighted | 60 | 56 | 1.62 | 29 |
| Unweighted | 24 | 95 | 1.06 | 44 |
| Weighted | 60 | 56 | 1.93 | 24 |
| Weighted | 24 | 95 | 1.19 | 39 |
Composite measure includes: CERAD Word List Recall, CERAD Boston Naming Test-High, MMSE Orientation to Time, Constructional Praxis, and Raven’s Progressive Matrices
CERAD = Consortium to establish a Registry for Alzheimer’s Disease
MMSE = Minimental State Exam
Results from the sensitivity analyses suggest that substituting either the CERAD Boston Naming Test (total score) or Categorical Fluency Test (animals) for the CERAD Boston Naming Test (high frequency items) requires a larger sample size to detect a 25% treatment effect in a 24-month trial. Substituting the CERAD Boston Naming Test(total score) for the high frequency items would require 398 participants per group, while replacing the CERAD Boston Naming Test (high frequency items) with Category Fluency Test (animals), requires 404 participants per group. Similarly, but to a lesser degree, in a 60-month trial, substituting the CERAD Boston Naming Test(total score) for the high frequency items would require a larger sample size of 98 participants per group, while replacing the CERAD Boston Naming Test (high frequency items) with Category Fluency Test (animals), requires 102 participants per group.
Results from the weighting analyses indicated that applying a higher weighting to the CERAD Boston Naming Test-High improved the MSDR. Although the weightings had similar patterns at two and five years, they were not identical. Compared to the original, non-weighted composite cognitive test score, the weighted composite cognitive test score requires 69 PSEN1 E280A mutation carriers per group age 30 and older to detect a 25% treatment effect in 60-month trial. Using the weighted composite in a trial of 75 mutation carriers per group age 30 and older would permit us to detect a 24% treatment effect in a 60-month trial (Table 5).
DISCUSSION
We empirically identified an API composite cognitive test score sensitive to preclinical cognitive decline in ADAD mutation carriers within 15 years of their estimated age at clinical onset. We propose that this composite is well-suited for preclinical ADAD trials to evaluate treatment effects with smaller sample sizes and improved statistical power compared to the most sensitive individual cognitive assessment or larger test batteries, and in a manner that is reasonably likely to predict a treatment’s clinical benefit. The API composite cognitive test score and the analytic approach used in its development appears to fit into the Food and Drug Administration’s framework in the recent draft guidance regarding a cognitive assessment being a primary efficacy measure in preclinical AD trials (37). Moreover, the optimal combination of assessments empirically identified in preclinical ADAD mutation carriers is quite similar to the composite cognitive test score identified in older adults who progressed to clinical stages of LOAD(38).
The empirically identified composite cognitive test score consisted of 5 test items targeting several different cognitive domains. The composite has greater statistical power to detect a treatment effect compared to that of a single test item (CERAD Word List Recall), supporting the notion that combining test items can result in better captured variance for tracking preclinical cognitive decline. The optimal composite cognitive test score is more sensitive than that of the entire neuropsychological test battery. This is consistent with the hypothesis that including test items that capture overlapping variation or that are not sensitive to preclinical AD into the test score can lower the overall sensitivity of the score to track preclinical cognitive decline. The other reason the entire battery may be less sensitive than a subset is that the entire test battery may include assessments that are psychometrically noisy as well as tests that have excellent psychometric properties, thus increasing the overall variability.
Our optimal composite cognitive test scores incorporates cognitive assessments from several difference domains complementing those of recent studies, which suggest that preclinical AD cognitive decline presents in multiple domains, (13;39), in addition to decline in episodic memory (though it remains a defining trait of preclinical AD)(11–15;23), along with other studies focusing on cognitive domain specific composite scores based on data from the Alzheimer’s Disease Neuroimaging Initiative(25;26). These research results suggest that composite endpoints may offer greater power and sensitivity to detect cognitive changes.
Confirming the findings in this study, we obtained a very similar composite cognitive test score from an independent analysis performed in older adults who later progressed to clinical stages of LOAD. Both optimal cognitive composite test scores consisted of assessments from the same domains/assessments with the exception of the present study included a test of Constructional Praxis whereas the other included visual spatial ability test -Symbol Digits Modalities, despite substantial differences in the cohorts’ neuropsychological test batteries (38). In addition, the results from the API efforts complement a recent study, that suggested that multiple cognitive domains decline in preclinical AD, including verbal and working memory, visuospatial, and global functioning(13).The significant overlap between the two optimal composite test scores suggests the similar patterns of cognitive decline between LOAD and ADAD, despite evidently different ages of onset, and possible different time courses and underlying etiologies and biological processes Likewise, researchers preparing the Alzheimer’s Disease Cooperative Study (ADCS) “A4” trial in cognitively healthy older adults with amyloid burden pathology have implemented a similar approach using other datasets and found comparable results to those reported in this study(4).
Despite the similarity of the composite cognitive test scores, the MSDR of the API composite cognitive test score empirically derived using the PSEN1 E280A cohort data is considerably higher than that from older adults who progress to the clinical stages of LOAD described in a separate report(38). This is consistent with the fact that PSEN1 E280A mutation carriers are certain to develop symptomatic dementia in a predictable timeline and clinical course. Additionally, since the PSEN1 E280A carriers are relatively young, the cognitive decline observed is likely only due to the predisposition to AD, that is, preclinical AD decline, as opposed to a confounding aging effect that is observed in older adults who progress to clinical stages of LOAD.
Although individual neuropsychological tests have varying levels of sensitivity to detecting and tracking preclinical AD decline (measured by their MSDRs), this analytic approach allowed us to empirically characterize the composite cognitive test score resulting in the high overall sensitivity to track preclinical decline by simultaneously determining a combination of individual tests that complement each other to capture as much variability as possible. As a result, more sensitive cognitive tests/sub-tests may not be included in the composite endpoint, since these items may correlate with another assessment that captures the same information and has a higher MSDR. The tests/sub-tests that are included and have smaller MSDR may measure variability not captured by other assessments in the combination. This is different from the approach in which each sensitive test is determined individually at a time and then simply combined to form a composite. The result of the latter approach may be a composite with a lower sensitivity due to overlap in elements of variability captured by the tests, making them redundant to each other and in turn weaken overall sensitivity of the composite. Similarly, the latter approach may result in the loss of opportunity to identify tests that may be less sensitive on their own, but add to the composite by capturing additional aspect of variability not captured by other assessments. Another possible reason our proposed analytic approach resulted in increased sensitivity is that it helps reduce the impact of error due to other idiosyncratic single test items or sub-domains in the composite. Moreover, weighting the individual tests in the composite allowed the tests that capture additional variability or are more sensitive to preclinical ADAD decline to have a greater effect on the composite test score, resulting in even higher statistical power to detect preclinical cognitive decline.
In this study, we aimed to characterize the aspects of the disease that decline consistently across individuals in order to assess effectiveness of a treatment in slowing decline in a preclinical AD trial, rather than discrimination between those who subsequently progressed and those that did not, or the neuropathological underpinnings of AD that result in a change in cognitive functioning. This approach also allows for the incorporation of data from various points along the preclinical AD stages, just as in a preclinical trial, some participants may progress to cognitive impairment within months while others remain cognitively healthy for many years. In addition, we chose to adjust for practice effects (40) to better capture the cognitive decline specific to AD. The non-carrier group showed an increase on the API composite cognitive score, while the carrier group showed a reduction. This suggests that, unlike the non-carrier group, the carrier group was not able to benefit as much from prior exposure to the tests. Although it is important to account for differences in study participants’ baseline cognitive function, the present study did not adjust for such differences, given that they can be accounted for when analyzing the trial data to determine whether a treatment is effective at slowing cognitive decline.
There are some limitations to the present study. For instance, development of the optimal composite cognitive test score was constrained by the starting neuropsychological test battery used in the Antioquia Cohort study and the composite development sample size available. That said, we achieved remarkably similar results with independent efforts to empirically deriving a composite cognitive test score based on data from individuals who progress to the clinical stages of LOAD(38) despite differences in the cohorts’ starting neuropsychological test battery. Likewise, scientists preparing for the Alzheimer’s Disease Cooperative Study (ADCS) “A4” trial in cognitively healthy individuals with significant fibrillar amyloid burden have undertaken a similar effort using other datasets and have produced results comparable to those reported here(4). The generalizability and sensitivity of the composite cognitive test score to other ADAD mutations remains unknown, given the limited preclinical longitudinal data available. That said, recent evidence has suggested that there is no significant difference in cognitive measures when comparing PSEN1 mutation carriers to PSEN2 and APP mutation carriers (41). Additional efforts are underway to confirm the generalizability and power of the API composite cognitive test score in other populations followed to clinical progression (which may include different assessment batteries), and to estimate the statistical power in different preclinical AD participant groups (e.g., APOE ε4 homozygotes or heterozygotes at different ages, older adults with our without biomarker evidence of AD). The results from these analyses, along with sample size estimates, will be reported separately.
In summary, we examined longitudinal data from cognitively unimpaired PSEN1 E280A mutation carriers within 15 years of their estimated mean age of dementia onset and conducted an search of every combination of one to seven cognitive assessments to identify the optimal combination that is sensitive to tracking preclinical AD decline over a two and five year time period, while controlling for practice effects using data from kindred mutation non-carriers. The empirically identified API composite cognitive test score is being used as the primary endpoint in the first API trial in cognitively unimpaired ADAD carriers within 15 years of their estimated age at clinical onset. This composite endpoint requires fewer participants to detect a treatment effect compared to using the most sensitive individual cognitive test or the entire CERAD-Col neuropsychological test battery. A similar composite cognitive test score was independently derived in cognitively unimpaired older adults who subsequently progressed to clinical stages of LOAD(38). As a result of these efforts, other preclinical trial investigators are extending the API composite cognitive test score development strategy for use in their planned trials and studies.
CLINICAL POINTS.
We have identified a composite cognitive test score representing multiple cognitive domains that has improved power to track preclinical AD decline in ADAD mutation carriers and evaluate preclinical AD treatments.
This API composite cognitive test score will be used as the primary endpoint in the first API trial in cognitively unimpaired ADAD carriers within 15 years of their estimated age at clinical onset.
We have independently confirmed our findings in a separate cohort of cognitively healthy older adults who progressed to the clinical stages of late-onset AD, and continue to refine the composite.
Acknowledgments
We thank Madelyn Gutierrez, BA, Eliana Henao, MS and Adriana Ruiz, MS (Grupo de Neurociencias de Antioquia, Universidad de Antioquia, Medellín, Colombia) for their assistance and support in coordination and data collection.
We also thank Laura Jakimovich, RN, MS, Carolyn Langlois, MA, Auttawut Roontiva, MS, (Banner Alzheimer’s Institute, Phoenix AZ and Arizona Alzheimer’s Consortium, Phoenix AZ) for their support and assistance in coordination, and data analysis.
This work was supported by: the National Institute on Aging (R01AG031581 and P30AG19610 to EMR), National Institutes of Health (RF1 AG041705), the state of Arizona, Arizona Alzheimer’s Consortium (to NA) and contributions from the Banner Alzheimer’s Foundation, the Nomis Foundation (salary support for NA, JBSL, SBH, KC, ASF, MF, MW, PNT and EMR), and Colciencias (1115-408-20543 and 1115-408-20512) (for data collection, data maintenance and salary support for CA, ANB, LM, CM, VT, SM and FL).
SBH is a consultant for Banner Alzheimer’s Institute. MF and MW are stock shareholders of F Hoffmann-La Roche, Ltd. PNT receives a) consulting fees from Abbott Laboratories, AbbVie, AC Immune, Boehringer-Ingelheim, California Pacific Medical Center, Chase Pharmaceuticals, CME Inc., Medavante, Otsuka, Sanofi-Aventis; b) consulting fees and research support from Avanir, Bristol Meyers Squibb, Cognoptix, Janssen, Merck and Company, F Hoffmann-La Roche, Ltd; c) research support from AstraZeneca, Baxter Healthcare Corp., Functional Neuromodulation (f(nm)), GE, Genentech, Pfizer, Targacept; d) other research support from NIA, Arizona Department of Health Services, owns stock options in Adamas and is listed as a contributor to a patent owned by the University of Rochester, “Biomarkers of Alzheimer’s Disease”.
Footnotes
Portions of this study were presented at the 2011 Alzheimer’s Association International Conference, Paris, France and the 2011 Clinical Trials on Alzheimer’s Disease (CTAD), San Diego, CA.
They have no conflict of interest to report.
Reference List
- 1.Reiman EM, Langbaum JBS, Tariot PN. Alzheimer’s Prevention Initiative: a proposal to evaluate presymptomatic treatments as quickly as possible. Biomarkers in Medicine. 2010 Feb 1;4(1):3–14. doi: 10.2217/bmm.09.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reiman EM, Langbaum JB, Fleisher AS, Caselli RJ, Chen K, Ayutyanont N, et al. Alzheimer’s Prevention Initiative: a plan to accelerate the evaluation of presymptomatic treatments. J Alzheimers Dis. 2011 Jan 1;26( Suppl 3):321–9. doi: 10.3233/JAD-2011-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mills SM, Mallmann J, Santacruz AM, Fuqua A, Carril M, Aisen PS, et al. Preclinical trials in autosomal dominant AD: implementation of the DIAN-TU trial. Rev Neurol (Paris) 2013 Oct;169(10):737–43. doi: 10.1016/j.neurol.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sperling R, Donohue M, Aisen P. The A4 Trial: Anti-amyloid treatment of asymptomatic Alzheimer’s disease. Alzheimers Dement. 2012 Jul;8(4 supplement):p425–p426. [Google Scholar]
- 5.Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM, Crenshaw DG, Sundseth SS, et al. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenomics J. 2010 Oct;10(5):375–84. doi: 10.1038/tpj.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dekosky ST. Maintaining adherence and retention in dementia prevention trials. Neurology. 2006 Nov 14;67(9 Suppl 3):S14–S16. doi: 10.1212/wnl.67.9_suppl_3.s14. [DOI] [PubMed] [Google Scholar]
- 7.ADAPT Research Group. Cognitive Function Over Time in the Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT): Results of a Randomized, Controlled Trial of Naproxen and Celecoxib. Arch Neurol. 2013;65(7):896–905. doi: 10.1001/archneur.2008.65.7.nct70006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Silverberg NB, Ryan LM, Carrillo MC, Sperling R, Petersen RC, Posner HB, et al. Assessment of cognition in early dementia. Alzheimers Dement. 2011 May 1;7(3):e60–e76. doi: 10.1016/j.jalz.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cano SJ, Posner HB, Moline ML, Hurt SW, Swartz J, Hsu T, et al. The ADAS-cog in Alzheimer’s disease clinical trials: psychometric evaluation of the sum and its parts. J Neurol Neurosurg Psychiatry. 2010 Dec;81(12):1363–8. doi: 10.1136/jnnp.2009.204008. [DOI] [PubMed] [Google Scholar]
- 10.Aisen PS, Andrieu S, Sampaio C, Carrillo M, Khachaturian ZS, Dubois B, et al. Report of the task force on designing clinical trials in early (predementia) AD. Neurology. 2011 Jan 18;76(3):280–6. doi: 10.1212/WNL.0b013e318207b1b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elias MF, Beiser A, Wolf PA, Au R, White RF, D’Agostino RB. The preclinical phase of alzheimer disease: A 22-year prospective study of the Framingham Cohort. Arch Neurol. 2000 Jun;57(6):808–13. doi: 10.1001/archneur.57.6.808. [DOI] [PubMed] [Google Scholar]
- 12.Saxton J, Lopez OL, Ratcliff G, Dulberg C, Fried LP, Carlson MC, et al. Preclinical Alzheimer disease: neuropsychological test performance 1.5 to 8 years prior to onset. Neurology. 2004 Dec 28;63(12):2341–7. doi: 10.1212/01.wnl.0000147470.58328.50. [DOI] [PubMed] [Google Scholar]
- 13.Johnson DK, Storandt M, Morris JC, Galvin JE. Longitudinal study of the transition from healthy aging to Alzheimer disease. Arch Neurol. 2009 Oct;66(10):1254–9. doi: 10.1001/archneurol.2009.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilson RS, Leurgans SE, Boyle PA, Bennett DA. Cognitive decline in prodromal Alzheimer disease and mild cognitive impairment. Arch Neurol. 2011 Mar;68(3):351–6. doi: 10.1001/archneurol.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amieva H, Le GM, Millet X, Orgogozo JM, Peres K, Barberger-Gateau P, et al. Prodromal Alzheimer’s disease: successive emergence of the clinical symptoms. Ann Neurol. 2008 Nov;64(5):492–8. doi: 10.1002/ana.21509. [DOI] [PubMed] [Google Scholar]
- 16.Grober E, Hall CB, Lipton RB, Zonderman AB, Resnick SM, Kawas C. Memory impairment, executive dysfunction, and intellectual decline in preclinical Alzheimer’s disease. J Int Neuropsychol Soc. 2008 Mar;14(2):266–78. doi: 10.1017/S1355617708080302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Small BJ, Backman L. Longitudinal trajectories of cognitive change in preclinical Alzheimer’s disease: a growth mixture modeling analysis. Cortex. 2007 Oct;43(7):826–34. doi: 10.1016/s0010-9452(08)70682-8. [DOI] [PubMed] [Google Scholar]
- 18.Stonnington CM, Chen K, Lee W, Locke DE, Dueck AC, Liu X, et al. Fibrillar amyloid correlates of preclinical cognitive decline. Alzheimers Dement. 2013 Apr 11; doi: 10.1016/j.jalz.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pike KE, Ellis KA, Villemagne VL, Good N, Chetelat G, Ames D, et al. Cognition and beta-amyloid in preclinical Alzheimer’s disease: data from the AIBL study. Neuropsychologia. 2011 Jul;49(9):2384–90. doi: 10.1016/j.neuropsychologia.2011.04.012. [DOI] [PubMed] [Google Scholar]
- 20.Ellis KA, Lim YY, Harrington K, Ames D, Bush AI, Darby D, et al. Decline in cognitive function over 18 months in healthy older adults with high amyloid-beta. J Alzheimers Dis. 2013 Jan 1;34(4):861–71. doi: 10.3233/JAD-122170. [DOI] [PubMed] [Google Scholar]
- 21.Lim YY, Maruff P, Pietrzak RH, Ames D, Ellis KA, Harrington K, et al. Effect of amyloid on memory and non-memory decline from preclinical to clinical Alzheimer’s disease. Brain. 2013 Oct 30; doi: 10.1093/brain/awt286. [DOI] [PubMed] [Google Scholar]
- 22.Sperling RA, Johnson KA, Doraiswamy PM, Reiman EM, Fleisher AS, Sabbagh MN, et al. Amyloid deposition detected with florbetapir F 18 ((18)F-AV-45) is related to lower episodic memory performance in clinically normal older individuals. Neurobiol Aging. 2013 Mar;34(3):822–31. doi: 10.1016/j.neurobiolaging.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Acosta-Baena N, Sepulveda-Falla D, Lopera-Gomez CM, Jaramillo-Elorza MC, Moreno S, Aguirre-Acevedo DC, et al. Pre-dementia clinical stages in presenilin 1 E280A familial early-onset Alzheimer’s disease: a retrospective cohort study. Lancet Neurol. 2011 Mar;10(3):213–20. doi: 10.1016/S1474-4422(10)70323-9. [DOI] [PubMed] [Google Scholar]
- 24.Hendrix SB. Measuring clinical progression in MCI and pre-MCI populations: enrichment and optimizing clinical outcomes over time. Alzheimers Res Ther. 2012 Jul 13;4(4):24. doi: 10.1186/alzrt127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crane PK, Carle A, Gibbons LE, Insel P, Mackin RS, Gross A, et al. Development and assessment of a composite score for memory in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) Brain Imaging Behav. 2012 Dec;6(4):502–16. doi: 10.1007/s11682-012-9186-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gibbons LE, Carle AC, Mackin RS, Harvey D, Mukherjee S, Insel P, et al. A composite score for executive functioning, validated in Alzheimer’s Disease Neuroimaging Initiative (ADNI) participants with baseline mild cognitive impairment. Brain Imaging Behav. 2012 Dec;6(4):517–27. doi: 10.1007/s11682-012-9176-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dowling NM, Hermann B, La RA, Sager MA. Latent structure and factorial invariance of a neuropsychological test battery for the study of preclinical Alzheimer’s disease. Neuropsychology. 2010 Nov;24(6):742–56. doi: 10.1037/a0020176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayden KM, Kuchibhatla M, Romero HR, Plassman BL, Burke JR, Browndyke JN, et al. Pre-clinical Cognitive Phenotypes for Alzheimer Disease: A Latent Profile Approach. Am J Geriatr Psychiatry. 2013 Sep 27; doi: 10.1016/j.jagp.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Balsis S, Unger AA, Benge JF, Geraci L, Doody RS. Gaining precision on the Alzheimer’s Disease Assessment Scale-cognitive: a comparison of item response theory-based scores and total scores. Alzheimers Dement. 2012 Jul;8(4):288–94. doi: 10.1016/j.jalz.2011.05.2409. [DOI] [PubMed] [Google Scholar]
- 30.Xiong C, van Belle G, Chen K, Tian L, Luo J, Gao F, et al. Combining Multiple Markers to Improve the Longitudinal Rate of Progression: Application to Clinical Trials on the Early Stage of Alzheimer’s Disease. Statistics in Biopharmaceutical Research. 2013 Feb 1;5(1):54–66. doi: 10.1080/19466315.2012.756662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reiman EM, Langbaum JBS. Brain imaging in the evaluation of putative Alzheimer’s disease slowing, risk-reducing and prevention therapies. In: Jagust WJ, D’Esposito M, editors. Imaging the Aging Brain. New York: Oxford University Press; 2009. pp. 319–50. [Google Scholar]
- 32.Reiman EM, Langbaum JBS, Fleisher AS, Caselli RJ, Chen K, Ayutyanont N, et al. Alzheimer’s Prevention Initiative: a plan to accelerate the evaluation of presymptomatic treatments. In: Ashford JW, editor. Handbook of Imaging the Alzheimer Brain. Amsterdam: IOS Press; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ayutyanont N, Lopera F, Hendrix S, Langbaum J, Madrigal La, Moreno S, et al. Composite cognitive endpoints with improved power to detect presymptomatic Alzheimer’s disease treatment effects: Findings in the Colombian kindred with the E280A Presenilin 1 mutation and the Alzheimer’s Prevention Initiative. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2011 Jul 1;7(4):S608. [Google Scholar]
- 34.Lopera F, Ardilla A, Martinez A, Madrigal L, Arango-Viana JC, Lemere CA, et al. Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. JAMA. 1997 Mar 12;277(10):793–9. [PubMed] [Google Scholar]
- 35.Arango Lasprilla JC, Iglesias J, Lopera F. Neuropsychological study of familial Alzheimer’s disease caused by mutation E280A in the presenilin 1 gene. Am J Alzheimers Dis Other Demen. 2003 May;18(3):137–46. doi: 10.1177/153331750301800306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosselli MC, Ardila AC, Moreno SC, Standish VC, Arango-Lasprilla JC, Tirado VM, et al. Cognitive decline in patients with familial Alzheimer’s disease associated with E280a presenilin-1 mutation: a longitudinal study. J Clin Exp Neuropsychol. 2000 Aug;22(4):483–95. doi: 10.1076/1380-3395(200008)22:4;1-0;FT483. [DOI] [PubMed] [Google Scholar]
- 37.Food and Drug Administration. Draft Guidance. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER); 2013. Guidance for industry Alzheimer’s disease: Developing drugs for the treatment of early stage disease. [Google Scholar]
- 38.Langbaum JB, Hendrix SB, Ayutyanont N, Chen K, Fleisher AS, Shah RC, Barnes LL, Bennett DA, Tariot PN, Reiman EM. An empirically derived composite cognitive endpoint with improved power to track and evaluate treatments for preclinical Alzheimer’s disease. Alzheimer’s and Dementia. doi: 10.1016/j.jalz.2014.02.002. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Riley KP, Jicha GA, Davis D, Abner EL, Cooper GE, Stiles N, et al. Prediction of preclinical Alzheimer’s disease: longitudinal rates of change in cognition. J Alzheimers Dis. 2011;25(4):707–17. doi: 10.3233/JAD-2011-102133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilson RS, Li Y, Bienias JL, Bennett DA. Cognitive decline in old age: separating retest effects from the effects of growing older. Psychol Aging. 2006 Dec;21(4):774–89. doi: 10.1037/0882-7974.21.4.774. [DOI] [PubMed] [Google Scholar]
- 41.Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012 Aug 30;367(9):795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]