

Abstract
Background
Deficient production of reactive oxygen species (ROS) by the phagocyte NADPH oxidase in Chronic Granulomatous Disease (CGD) results in susceptibility to certain pathogens secondary to impaired oxidative killing and mobilization of other phagocyte defenses. PPARγ agonists including pioglitazone (Pio), approved for Type 2 diabetes therapy, alter cellular metabolism and can heighten ROS production. It was hypothesized that Pio treatment of gp91phox−/− mice, a murine model of human CGD, would enhance phagocyte oxidant production and killing of S. aureus, a significant pathogen in this disorder.
Objectives
We sought to determine if Pio treatment of gp91phox−/− mice enhanced phagocyte oxidant production and host defense.
Methods
Wild type (WT) and gp91phox−/− mice were treated with the PPARγ agonist Pio, and phagocyte ROS and killing of S. aureus investigated.
Results
As demonstrated by three different ROS sensing probes, short-term treatment of gp91phox−/− mice with Pio enhanced stimulated ROS production in neutrophils and monocytes from blood and neutrophils and inflammatory macrophages recruited to tissues. Mitochondria were identified as the source of ROS (mtROS). Findings were replicated in human CGD monocytes following ex vivo Pio treatment. Importantly, while mtROS were deficient in gp91phox−/− phagocytes, their restoration with treatment significantly enabled killing of S. aureus both ex vivo and in vivo.
Conclusions
Together, the data support the hypothesis that signaling from the NADPH oxidase under normal circumstances governs phagocyte mtROS production, and that such signaling is lacking in the absence of a functioning phagocyte oxidase. PPARγ agonism appears to bypass the need for the NADPH oxidase for enhanced mtROS production and partially restores host defense in CGD.
Keywords: Chronic Granulomatous Disease, phagocytes, mitochondria, oxidants, thioglitazones
Introduction
Chronic Granulomatous Disease (CGD) is a rare disease attributed to mutations in the genes encoding components of the phagocyte NADPH oxidase. The inability of phagocytes1–3, to mount an oxidative burst predisposes to infections with certain bacterial and fungal pathogens, e.g. Staphylococcus aureus, and Aspergillus, most evident in the lung, skin, lymph nodes and liver4, and even low levels of residual ROS production by phagocytes are associated with improved survival5. In addition to direct oxidative killing of pathogens, phagocyte ROS orchestrate other antimicrobial defenses, e.g. activation of antimicrobial proteins within the phagolysosome6 and recruitment of antimicrobial autophagocytic machinery to phagolysosomes, These are deficient in CGD phagocytes7–9.
Pio, and other thioglitazones, are so-called nutrient restricting drugs approved for Type 2 diabetes. As PPARγ agonists, they slowly mimic the actions of insulin with increased peripheral glucose disposal. Additionally, their nutrient restriction signaling is associated with altered macrophage metabolism, e.g. activation of AMPK10, and various anti-inflammatory activities11, 12. Intriguingly, PPARγ agonism has also been associated with enhanced host defense against Candida albicans13–15, Staphylococcus aureus16 Klebsiella pneumoniae17, and Streptococcus pneumoniae18. Two mechanisms have been proposed: enhanced pathogen containment by phagocytosis and encapsulation and/or enhanced ROS production by phagocytes, though the source(s) was not identified. With regard to ROS production, PPARγ ligands/agonists are reported to decrease expression of NADPH oxidase subunits of 22 and 47 kDa in some cells19, 20, but have also been shown to increase mtROS output in others21,22. Recent data implicating nutrient restriction or “starvation signaling”, in the enhancement of mtROS production, and entrainment of mtROS for killing of intracellular and extracellular bacteria23, suggest a possible mechanism by which PPARγ agonists may contribute to host defense.
Given these observations, we asked whether Pio treatment of gp91phox−/− mice would enhance oxidant production by CGD phagocytes, and if so, bolster host defense. We found that Pio treatment induced mtROS production in stimulated neutrophils, monocytes and inflammatory macrophages of CGD as well as normal mice, and importantly, the restored phagocytes demonstrated significantly enhanced killing of S. aureus both in vitro and in vivo. Crosstalk between the NADPH oxidase and mtROS leading to optimal antimicrobial responses by normal phagocytes was also shown. Consequently, in the absence of a functional NADPH oxidase, this signaling is lost, but importantly, can be restored at the level of mtROS production by Pio treatment.
Methods
Animals
Male mice, C57BL/6 and gp91phox−/−, were purchased from JAX or bred in-house. They received care in accordance with the IACUC, and given Pio (10 mg/kg/d), Bisphenol A diglycidyl ether (BADGE) a PPARγ antagonist, or vehicle (carboxymethyl cellulose) via oral gavage for 5 days unless otherwise indicated. All agents were well tolerated. Mice were euthanized by CO2 inhalation.
Reagents
Pio, BADGE, MitoTEMPO, PMA, DPI, catalase and superoxide dismutase (SOD) were from Sigma (St. Louis, MO). Conjugated antibodies to CD115, F4/80, Ly6G and CD11b were from eBiosciences (San Diego, CA). Antibodies to Nox1, Nox4, Duox, gp91phox, p20phox, p22phox, p47phox, p67phox were from SCBT (Santa Cruz, CA). Zymosan, DHR, MitoTracker Green and MitoSOX Red were from Life Technologies (Grand Island, NY).
Isolation of blood and peritoneal leukocytes
Red blood cells were lysed (Pharmlyse (BD Biosciences)) whole blood from mice (terminal cardiac puncture), before staining with markers and analysis by flow cytometry (CyAn™ ADP analyzer, Becton Dickinson Biosciences). Peritoneal cells were harvested by lavage from mice injected i.p. with 1 mg/mL zymosan in PBS. Cells were washed, suspended in PBS (3% FBS), blocked with anti–mouse Fc (CD16/32; eBiosciences) for 30min and stained for 1h on ice with conjugated antibodies prior to flow cytometry.
ROS detection
DHR was performed using phagocytes (106), incubated for 15 min with 5 μM DHR (PBS, 0.05% gelatin, 0.09% glucose, 1mM EDTA), stimulated with PMA (200 ng/ml) for 15 min/37°C. Cells were washed and analyzed by flow cytometry.
Cytochrome c reduction measuring release of superoxide was performed as described24.
mtROS were detected with MitoSOX Red. Cells (106) were incubated (37°C) with 25 nM MitoTracker Green in the dark (DMEM, 10% FBS) followed by 4 μM MitoSox Red (15 min each), washed with PBS, and analyzed by flow cytometry or Zeiss LSM 700 confocal microscopy.
Bactericidal assays
In vitro assay
2 × 105 peritoneal phagocytes (10h post zymosan) were co-cultured with 2 × 106 CFU S. aureus [strain 502A (ATCC #27217) grown overnight in Lauryl Broth and washed twice with saline] in 100 μI RPMI (phenol red free, 1% mouse serum) at 37°C for 1h. Phagocytes were lysed (1 ml pH 11 water25), bacteria pelleted at 10,000 × g for 10 min (2X) and washed with 1 ml saline. Bacterial numbers were determined using a modified Alamar blue assay26. This assay was similarly adapted for killing of Burkholderia cepecia ATCC #15416.
In vivo assay
S. aureus peritonitis was induced as published by Pollock et al27 with minor modification. Briefly, mice were injected i.p. with 0.5 ml of 2×107 CFU/ml S. aureus (saline), peritonea lavaged at designated times, and cell counts and viable bacteria in lavage fluid and cells (following lysis as above) determined.
Human blood monocytes
Heparinized blood was obtained from X-linked CGD subjects and normal controls at the National Institutes of Health, Bethesda, MD with the approval of the Institutional Review Board. The blood was express shipped overnight to Denver, CO. PBMCs were isolated using percoll gradients28, plated in X-vivo10 to adhere monocytes for 2h, washed (5X) to remove non-adherent cells, and cultured at 37°C, 10% CO2 without and with Pio (10 μM). Cells were then stimulated with PMA and mtROS detected by flow cytometry as above.
Statistics
Each experiment was performed 3–5 times unless otherwise indicated. Analysis and p value calculations were conducted using analysis of variance (JMP statistical program 4.0.1; SAS Institute). The Wilcoxon matched-pairs signed rank test was used for single and multiple comparisons, respectively. p ≤ 0.05 were considered significant. Data are reported as mean ± SEM.
Results
In vivo treatment with Pio enhances blood neutrophil and monocyte ROS production
WT and gp91phox−/− mice were treated with either Pio or vehicle by gavage for 5 days to test whether phagocyte oxidant production would be enhanced/restored. Previously, this treatment resulted in PPARγ activation in inflammatory macrophages and accelerated resolution of sterile peritonitis in CGD mice29. Following treatment, blood leukocytes were collected, incubated with DHR, stimulated with the PKC activator, PMA, and analyzed for conversion to rhodamine by flow cytometry. Using this classic test of stimulated ROS production, leukocytes from WT mice demonstrated enhanced DHR fluorescence as expected (Fig. 1A), and Pio treatment had minimal effect. Leukocytes from vehicle-treated gp91phox−/− mice showed no evidence of stimulated ROS production. In contrast, a subpopulation of leukocytes from Pio-treated gp91phox−/− mice showed significantly enhanced DHR fluorescence following stimulation, indicating some restoration of oxidant production in cells lacking a functional NADPH oxidase.
Figure 1. Pio pretreatment of mice enhances PMA-stimulated ROS production by gp91phox−/− neutrophils and monocytes.
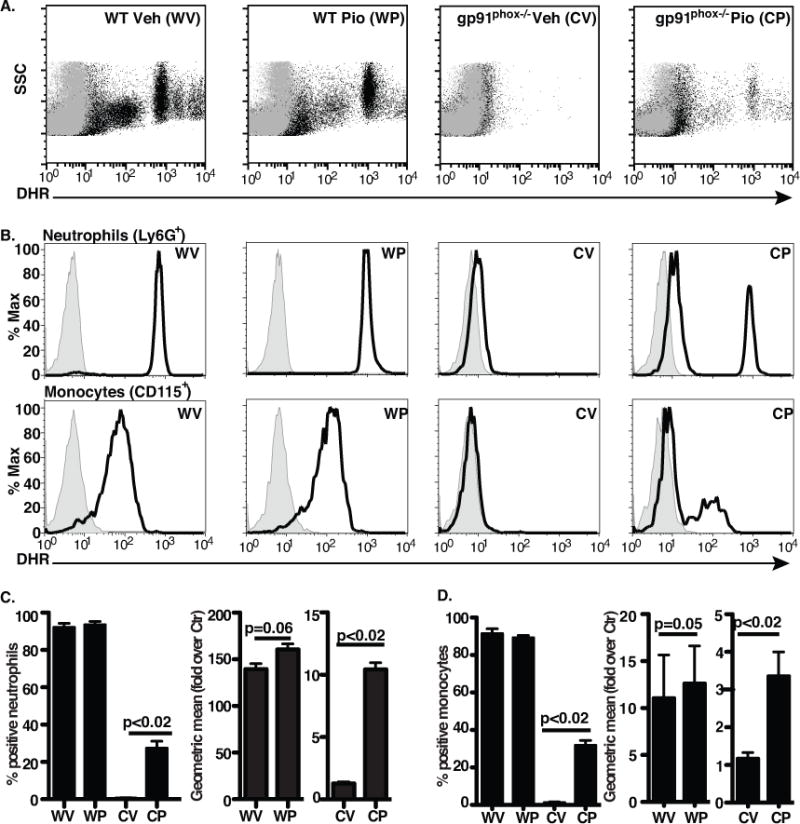
ROS measured by DHR in blood leukocytes from vehicle(V)- or Pio(P)-treated WT (WV, WP) and gp91phox−/− mice (CV, CP): A) Representative dot-plots, gray, no PMA; black, with PMA are shown. (B) Representative histograms for neutrophils and monocytes: gray, no PMA; black, with PMA. Aggregate data for neutrophils (C) and monocytes (D) are shown as percent of cells exhibiting enhanced fluorescence and change in geometric mean fluorescence with PMA. N = 8 mice/group.
To investigate cell types producing oxidants, blood leukocytes were stained with antibodies to surface markers prior to DHR loading and stimulation (Supp. Fig. 1, gating strategy). As expected, nearly all PMA-stimulated neutrophils (Ly6G+) and 90% of monocytes (CD115+) from WT mice, regardless of treatment, demonstrated stimulated ROS production, and these cells from vehicle-treated gp91phox−/− mice showed little enhancement (Fig. 1B–D). In contrast, 27% of neutrophils and 32% of monocytes from Pio-treated knockout mice demonstrated increased DHR fluorescence comparable to the respective populations of stimulated WT phagocytes. Lymphocytes of either genotype did not demonstrate increased DHR fluorescence following stimulation (data not shown).
To determine the optimal Pio dose and treatment duration for maximal ROS production, Pio doses ranging from 1–10 mg/kg/d were tested to simulate levels achieved during treatment of humans30,31. A dose-dependent decrease in the percentage of stimulated gp91phox−/− neutrophils and monocytes producing ROS was observed at 3 and 1 mg/kg/day Pio (Supp. Fig. 2). Mice were also treated with Pio (10 mg/kg/d) or vehicle for up to 14 days to ensure steady state levels before oxidant production was assessed. Maximal stimulated ROS production by gp91phox−/− leukocytes was evident by 5 days (Supp. Fig. 3). Thus, further experiments were conducted following 5 days of treatment at 10 mg/kg/d as in initial experiments.
Recruited phagocytes from Pio-treated gp91phox−/− mice show enhanced superoxide production
To further investigate oxidant production and potential sources of oxidants, greater numbers of phagocytes were needed than were available from blood, and so a sterile peritonitis model was used to recruit phagocytes. Recruited phagocytes from Pio or vehicle-treated gp91phox−/− and WT mice were quite comparable in numbers, types, and zymosan ingestion at 6–10h following zymosan injection (Supp. Fig. 4). Recruited neutrophils (Ly6G+) and inflammatory macrophages (F4/80lo+) derived from recruited monocytes18, were incubated with DHR, stimulated with PMA and analyzed by flow cytometry with results similar to those of the blood phagocytes from both genotypes and treatment groups (Supp. Fig. 4D).
The nature of ROS produced in recruited phagocytes was then investigated using cytochrome c reduction, a relatively specific assay for superoxide. Significant release of stimulated oxidants from vehicle- and Pio-treated WT phagocytes was demonstrated (Fig. 2). As in the DHR assay, partial restoration of stimulated ROS production from Pio-treated gp91phox−/− phagocytes was evident. Pretreatment with SOD, but not catalase, inhibited ROS production supporting superoxide production. The nonspecific flavochrome inhibitor, DPI32,33 ablated the production of superoxide by the NADPH oxidase of WT phagocytes, and interestingly, also inhibited oxidant production by Pio-restored gp91phox−/− leukocytes suggesting a flavochrome as the source.
Figure 2. Pio pretreatment enhances superoxide production by recruited phagocytes from gp91phox−/− mice.
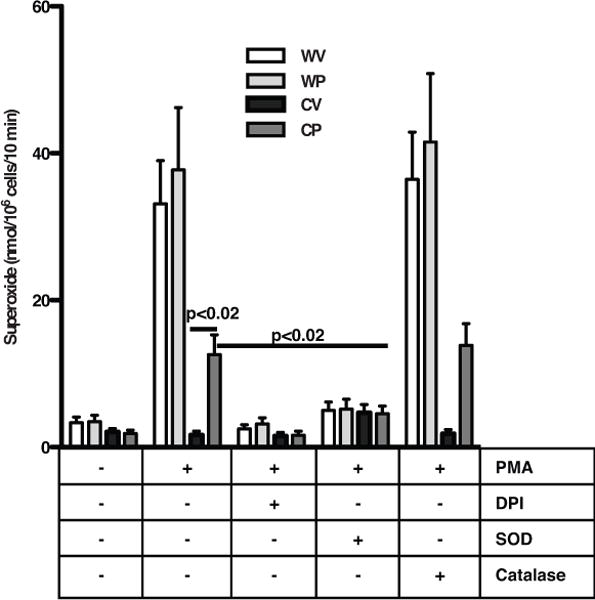
Ten hours after intraperitoneal zymosan injection, phagocytes were harvested from vehicle- or Pio-treated WT (WV, WP) or gp91phox−/− (CV, CP) mice. Phagocytes were stimulated with PMA in the presence and absence of inhibitors, and oxidants measured by the reduction of cytochrome c. N = 9 mice/group.
Flow cytometry and RT-PCR were used to investigate several flavochromes as potential sources of ROS (Supp. Fig. 5). These included the NADPH oxidases and associated components, the dual oxidases and other flavochromes. Absence of gp91phox protein was verified in the phagocytes of gp91phox−/− mice with and without Pio treatment. Pio did not decrease the expression of p22phox or p47phox as has been described in other systems19,20. Other NOXs and DUOXs were either undetectable or unchanged following Pio treatment. Pretreatment of the cells with inhibitors for iNOS, 12/15 lipoxygenase, cPLA2 and COX, used at standard doses, had no effect on stimulated oxidant production suggesting a source other than these for oxidant production (data not shown).
Pio treatment enhances mtROS production in activated phagocytes
PPARγ agonists have been shown to affect mitochondrial biogenesis and functions in various systems21,34. Mitochondria are rich sources of flavochrome enzymes and have several sites for superoxide production35–37. Using MitoTracker Green, a dye selectively taken up by mitochondria, mitochondrial content was investigated in blood leukocytes of Pio- – and vehicle-treated mice. Pio had little effect on mitochondrial content in blood phagocytes from either WT or gp91phox−/− mice (Supp. Fig. 6).
mtROS production was next assessed using the probe MitoSOX Red, a triphenylphosphonium (TPP+)-linked dihydroethidium compound that concentrates within mitochondria and fluoresces red when oxidized by ROS. Harvested peritoneal phagocytes were loaded with MitoTracker Green, then MitoSOX Red, and immediately analyzed by fluorescence microscopy without further stimulation. Recruited phagocytes harvested from vehicle- or Pio-treated WT mice showed heterogeneity in MitoSOX Red staining, with some cells having detectible colocalization of MitoSOX Red with MitoTracker Green and others not. This was irrespective of whether they contained detectible ingested zymosan particles (Fig. 3, top 2 rows, and insets). Notably, zymosan particles, free or within the phagolysosome, took up the stains and fluoresced brightly in the green channel and to a lesser degree in the red channel as previously reported38 (Supp. Fig. 7A, and Fig. 3). In contrast to WT phagocytes, recruited phagocytes of vehicle-treated gp91phox−/− mice showed almost no MitoSOX Red fluorescence (Fig. 3, third row). Pio treatment of gp91phox−/− mice resulted in clearly detectible MitoSOX Red staining of some leukocytes (Fig. 3, bottom row), again, irrespective of whether they contained obvious zymosan or not. Flow cytometric analysis of the inflammatory cells quantified and corroborated these findings (Fig. 4). First, heterogeneity in MitoSOX Red staining was evident for both recruited phagocyte populations, neutrophils and inflammatory macrophages, but was significantly higher for WT phagocytes than for gp91phox−/− phagocytes. Second, Pio, compared to vehicle treatment, enhanced MitoSOX Red fluorescence, and this was evident for both WT and gp91phox−/− neutrophils and inflammatory macrophages.
Figure 3. Pio pretreatment of gp91phox−/− mice enhances production of mtROS in recruited phagocytes.
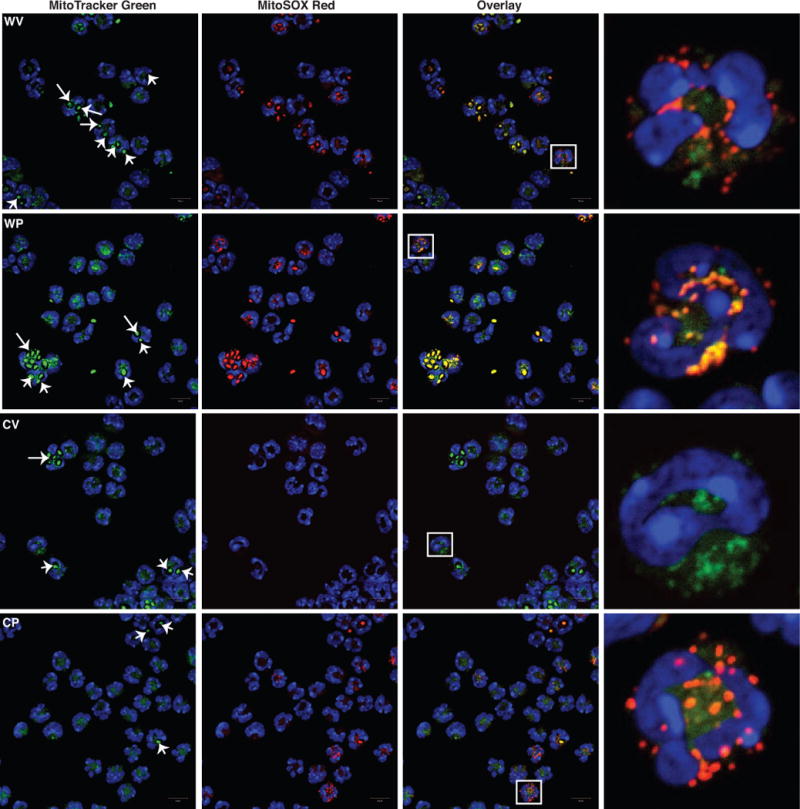
Recruited phagocytes as in Fig. 2 from vehicle- or Pio-treated WT (WV, WP) and gp91phox−/− (CV, CP) mice, were stained with MitoTracker Green, MitoSOX Red and DAPI, and analyzed by confocal microscopy (63X). Last panel: high-resolution images of a single cell (white box). Arrows denote ingested zymosan. Representative images are shown. N=3 mice/group.
Figure 4. Pio treatment enhances production of mtROS by neutrophils (A) and macrophages (B) harvested from inflamed peritonea of WT and gp91phox−/− mice.
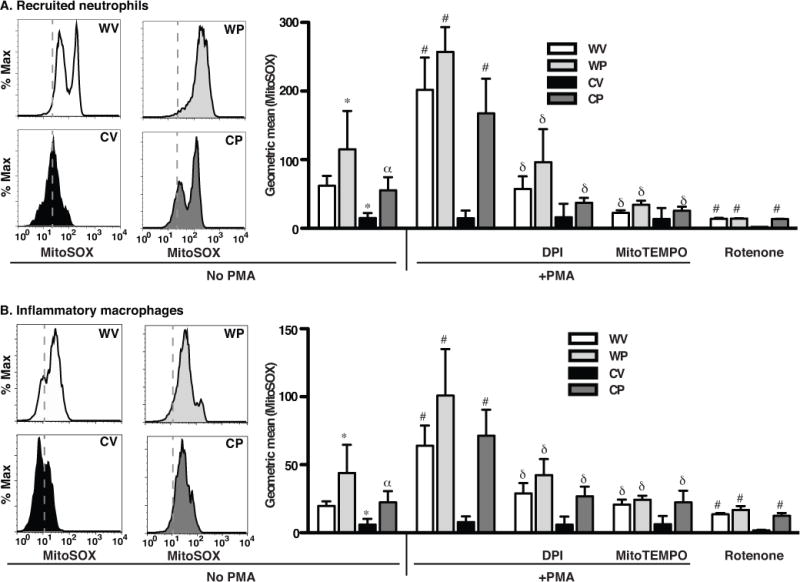
Recruited phagocytes from mice (WV, WP, CV, CP) as in Fig. 2 and 3 were treated with or without DPI or MitoTEMPO (15 min), stained with MitoSOX and then stimulated with or without PMA, and analyzed by flow cytometry. Representative histograms without PMA, left; line depicts relative fluorescence of CV neutrophils or macrophages. Aggregate data, right. N=8 mice/group. p ≤ 0.02 for comparisons with *WV without PMA, αCV without PMA, #respective genotype/treatment group without PMA, and δrespective genotype/treatment group with PMA alone.
mtROS staining was also assessed following ex vivo PMA stimulation (Fig. 4 and Supp. Fig. 7B). MitoSOX Red fluorescence was enhanced following stimulation of WT leukocytes regardless of treatment group, and for leukocytes of Pio-treated gp91phox−/− mice. No enhancement was noted following PMA stimulation of leukocytes from vehicle-treated gp91phox−/− mice (Fig. 4, and Supp. Fig. 7B). The enhanced MitoSOX Red fluorescence was ablated when leukocytes were pretreated with DPI for 15 min prior to PMA-stimulation. Pretreatment with MitoTEMPO, a mitochondrial antioxidant, also inhibited PMA-stimulated enhancement, and in fact, suppressed MitoSOX Red fluorescence in recruited cells at baseline (Fig. 4 and Supp. Fig. 7C). Finally, rotenone, a mitochondrial complex 1 inhibitor, also suppressed MitoSOX Red staining in recruited neutrophils and inflammatory macrophages, regardless of genotype or treatment group, indicating complex 1 as a key source of mtROS production (Fig. 4).
Because zymosan itself fluoresced with the mitochondrial stains (Supp. Fig. 7A), these findings were confirmed in a zymosan-free system using blood neutrophils and monocytes. Baseline fluorescence of blood leukocytes was generally lower than that of recruited leukocytes (Fig. 5 compared with Fig. 4). Following PMA stimulation, marked enhancement of mtROS production was noted for both WT leukocytes and Pio-restored gp91phox−/− leukocytes compared to leukocytes from vehicle-treated gp91phox−/− mice. Monocytes stained less brightly with MitoSOX Red than neutrophils. Notably, minimal heterogeneity was seen for the phagocyte populations under these conditions: essentially all WT and the Pio-restored gp91phox−/− phagocytes responded to PMA with mtROS production. As with the recruited peritoneal leukocytes, stimulated mtROS were largely ablated by pretreating the blood cells with either DPI or MitoTEMPO. Taken together, these data demonstrate that Pio treatment appears to largely restore stimulated mtROS production of gp91phox−/− leukocytes, both neutrophils and monocytes/inflammatory macrophages, whether circulating in the blood or recruited to inflamed tissues.
Figure 5. Pio treatment of gp91phox−/− mice enhances production of mtROS by stimulated blood neutrophils (A) and monocytes (B).
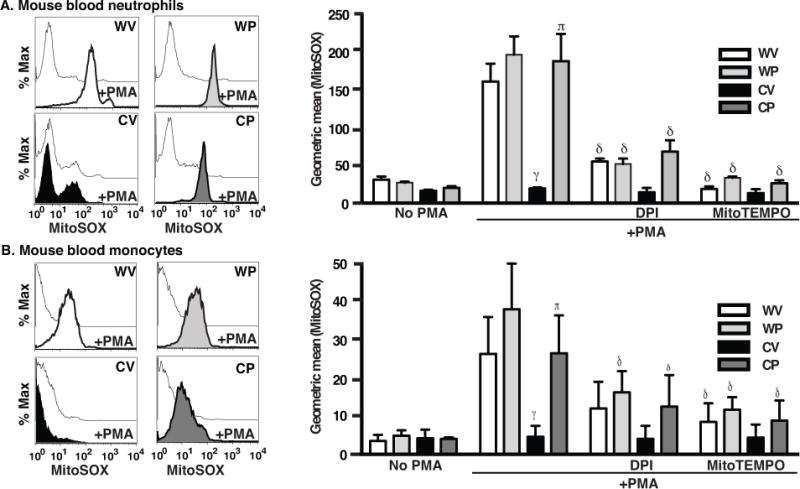
Blood leukocytes from WT (WV, WP) and gp91phox−/− (CV, CP) mice as in Fig. 1 were treated with and without DPI or MitoTEMPO for 15 min, stained with MitoSOX Red, stimulated or not with PMA and analyzed by flow cytometry. Representative histograms, left, and aggregate data, right. N=8 mice/group. p ≤ 0.02 for comparisons with γWV with PMA, πCV with PMA and δrespective genotype/treatment group with PMA alone.
Monocytes from humans with X-linked CGD and normal controls were similarly tested for stimulated mtROS production with and without ex vivo treatment with Pio. Untreated normal human monocytes, freshly isolated or after several days of culture, showed marked enhancement of mtROS following PMA stimulation (Fig. 6 and Supp. Fig. 8). X-linked CGD monocytes, however, did not show enhanced mtROS production in response to PMA (Fig. 6), replicating the findings for murine monocytes. After establishing optimal culture conditions for ex vivo Pio treatment (10 μM for 2 days (Supp. Fig. 8)), human CGD monocytes were then cultured under these conditions and showed marked enhancement of mtROS production following PMA stimulation, with restoration to levels comparable to those of stimulated normal monocytes (Fig. 6). As with murine cells, provision of mitoTEMPO prior to PMA stimulation largely ablated the detection of mtROS. Of note, in the absence of phagocyte stimulation, mtROS production was only minimally enhanced by ex vivo culture with Pio in either normal or X-CGD monocytes (Fig. 6), similar to findings following in vivo treatment in the murine model.
Figure 6. Ex vivo treatment of human CGD monocytes with Pio enhances stimulated mtROS production.
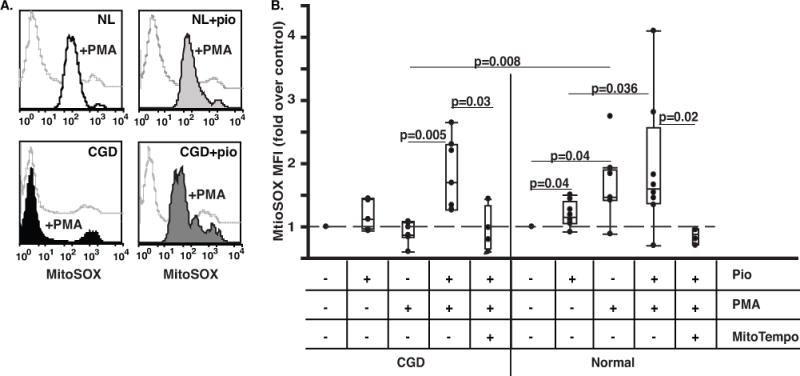
Human monocytes from X-linked CGD and normal subjects were isolated from heparinized blood, plated, and treated with 10 μM Pio for 2 days. The cells were then stained with MitoSOX Red and stimulated with PMA in the presence or absence of MitoTEMPO and analyzed by flow cytometry; Representative histograms (A) and aggregate data normalized to each individual untreated control (B) are shown. N=9 and 7 for normal subjects (NL) and CGD patients, respectively.
Pio enhancement of mtROS production is PPARγ dependent
Pio has been shown to have PPARγ dependent and independent effects on mitochondria21,22,34. PPARγ expression is ordinarily low in monocytes and neutrophils39, but is upregulated in monocytes upon recruitment into inflammatory tissues18,29 or following thioglitazone treatment40. As shown in Supp. Fig. 9A and in our earlier observations29, PPARγ mRNA expression was lower in recruited gp91phox−/− phagocytes compared to WT, and expression increased in both genotypes following Pio treatment, confirming that Pio activates PPARγ in these cells29. Dependence on PPARγ for Pio-enhanced mtROS production was tested using transgenic mice with myeloid specific ablation of PPARγ (LysMCre × PPARγfl/fl). As shown in Supp. Fig. 9B, neutrophils and inflammatory macrophages from LysMCre × PPARγfl/fl mice, unlike WT, did not show enhanced MitoSOX Red fluorescence following Pio treatment supporting PPARγ dependence. WT and gp91phox−/− mice were also treated with the PPARγ antagonist, BADGE, concurrently with Pio or vehicle. BADGE dose-dependently inhibited the enhancement in mtROS production following Pio treatment in both genotypes (Supp. Fig. 9C). Finally, rapid (within minutes), PPARγ-independent mtROS production has been reported for Pio employed ex vivo41. Therefore, peritoneal phagocytes of untreated WT and gp91phox−/− mice were incubated for 1h ex vivo with Pio (10 μM) and assessed for mtROS. As with human monocytes (Supp. Fig 8), no enhancement in mtROS production was detected following this brief exposure to Pio (data not shown). Taken together, these data support the hypothesis that enhancement of mtROS production by Pio treatment is PPARγ dependent.
Phagocytes of Pio -treated gp91phox−/− mice show enhanced killing of S. aureus
S. aureus is a significant pathogen in CGD and efficient killing of this bacteria by phagocytes is dependent on ROS production27,42. The ability of Pio treatment to enhance phagocyte killing of S. aureus was next investigated in vitro. Leukocytes were harvested from inflamed peritonea of vehicle-and Pio-treated WT and gp91phox−/− mice 10h following zymosan injection, co-incubated ex vivo with viable S. aureus for 1h and the percentage of bacteria killed assessed. As shown, S. aureus were readily killed by WT leukocytes, but not by leukocytes from vehicle-treated gp91phox−/− mice (Fig. 7). Pio treatment of the gp91phox−/− mice enhanced leukocyte killing to approximately 36% seen for WT. In all cases, pretreatment of leukocytes with SOD nearly abolished killing or even slightly enhanced survival of the bacteria. Finally, pretreatment of leukocytes obtained from Pio-treated gp91phox−/− mice with MitoTEMPO ablated most of their ability to kill S. aureus. Interestingly, MitoTEMPO pretreatment of WT leukocytes reduced S. aureus killing by almost half, suggesting that mitochondria play a significant role in the provision of oxidants for bacterial killing in normal phagocytes. Killing assays were also performed using B. cepacia, another pathogen relevant to CGD. As with S. aureus, leukocytes from vehicle-treated gp91phox−/− mice were unable to kill this pathogen while killing by leukocytes of Pio-treated gp91phox−/− mice was restored to approximately 47% of WT killing (Supp. Fig. 10). Again, killing by all phagocytes was ablated in the presence of SOD and MitoTEMPO.
Figure 7. Recruited phagocytes from Pio-treated gp91phox−/− mice show enhanced killing of S. aureus ex vivo.
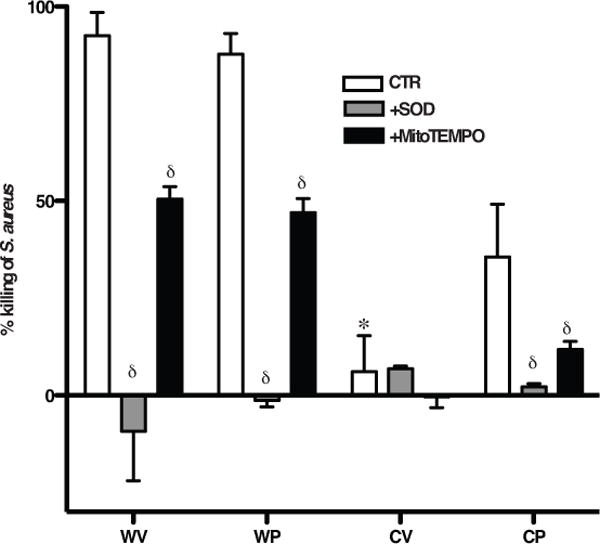
Recruited, peritoneal phagocytes from WV, WP, CV, CP mice as in Fig. 2 were treated with or without SOD or MitoTEMPO for 30 min, co-incubated with S. aureus (1×106 CFU) at 37°C for 1h. The percent of bacteria killed was determined. p≤0.02 *compared to phagocytes from WV and δcompared to phagocytes from respective treatment group in the absence of inhibitors. N=6 mice/group.
Finally, the ability of Pio treatment to enhance host defense against S. aureus in vivo was tested utilizing a well-described, non-lethal model of peritonitis27. WT and gp91phox−/− mice treated with either Pio or vehicle were injected i.p. with viable S. aureus, and peritonea lavaged at 24h or 48h for CFU enumeration. Similar numbers of inflammatory cells were recruited to the peritoneum for both genotypes and treatment groups at both time points (Supp. Fig. 11). WT mice readily cleared bacteria over 48h regardless of treatment. In contrast, vehicle treated gp91phox−/− mice showed clearance that was significantly delayed as has been described previously (Fig. 8). Importantly, Pio treatment of the gp91phox−/− mice resulted in an enhanced clearance of S. aureus from peritonea at 24h, demonstrating that Pio treatment restored early host defenses.
Figure 8. Pio treatment of gp91phox−/− mice enhances clearance of S. aureus in vivo.
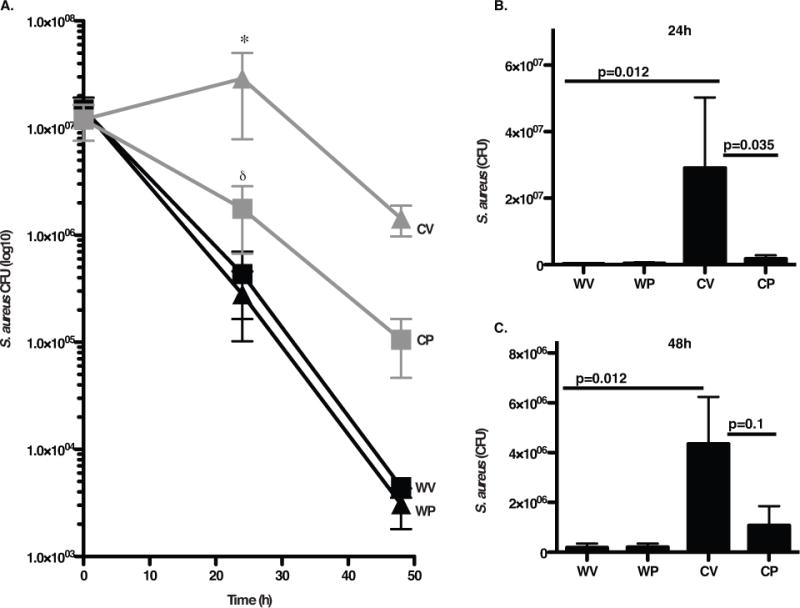
Vehicle or Pio-treated WT (WV, WP) and gp91phox−/− (CV, CP) mice were injected intraperitoneally with S. aureus, and peritonea lavaged at 24h and 48h. A). Bacterial numbers in lavage were determined. p ≤ 0.02 for comparisons with *WV and δCV. B–C). CFU data are depicted on a linear scale for clarity. N=16 and 11 mice/group at 24h and 48h, respectively.
Discussion
We have demonstrated that Pio treatment of gp91phox−/− mice enhances stimulated oxidant production from mitochondria of activated neutrophils, monocytes and inflammatory macrophages. Ex vivo treatment of human CGD monocytes with Pio similarly enhanced mtROS in response to stimulation (Fig. 6). Even phagocytes of Pio treated WT mice showed some enhancement in mtROS production (Fig. 5). Mitochondrial oxidant production in each case was shown using MitoSOX Red, a DHE probe that preferentially localizes to mitochondria and reacts with various ROS43. ROS production was also demonstrated using two other indicators, DHR and cytochrome c, each differing in detection of ROS. DHR, an intracellular probe that is not directed specifically to mitochondria, is capable of reacting with a number of oxidizing species and can be autoamplifying43 likely explaining the heterogeneity in cellular fluorescence with this reagent. Cytochrome c reduction with SOD inhibition is most specific for detecting superoxide, but requires extracellular ROS production following a robust stimulus such as PMA for detection24. A key finding of this investigation was that Pio treatment enabled gp91phox−/− phagocytes to kill S. aureus and B. cepacia, significant pathogens in CGD, in a mtROS dependent manner (Fig. 7–8, Supp Fig. 10). Clinical trials in human CGD will be required to determine whether Pio has efficacy as an adjunctive treatment to traditional antimicrobials for the enhancement of host defense.
Activated neutrophils and monocytes are highly glycolytic cells with limited mitochondrial respiration44,45, nonetheless, electrons are still shuttled to maintain mitochondrial membrane potential and integrity46. Somewhat surprising was our evidence that enhanced mtROS production in stimulated phagocytes appeared to depend on the NADPH oxidase, and was lacking in gp91phox−/− phagocytes. Findings from other studies support these observations. First, reduced oxygen consumption by mitochondria was demonstrated in human CGD alveolar macrophages during phagocytosis of S. aureus1. Secondly, crosstalk between the NADPH oxidase for production of mtROS has been demonstrated experimentally: knockdown of p47phox in stimulated glioma cells ablated mtROS production47, and activation of the NADPH oxidase resulted in mitochondrial membrane depolarization, respiratory dysfunction and mtROS production48. From our investigation it appears that PPARγ is a signaling intermediary between the NADPH oxidase and mitochondria during phagocyte activation (Supp. Fig. 12). Oxidized lipids are ligands of this nuclear receptor49,50, and, as such, PPARγ activation can be a downstream target of ROS. In turn, its activation appears to enhance mtROS production21,41 suggesting a feedforward signaling loop between cellular ROS and PPARγ. Pio activation of PPARγ, therefore, appears to bypass the requirement for NADPH oxidase-derived ROS in gp91phox−/− phagocytes and results in mtROS production and enhanced bactericidal activity.
Mobilization of mitochondria to phagolysosomes has been suggested by studies showing localization of mitochondrial enzymes in isolated maturing phagosomes of neutrophils51. Similarly, West et al. have recently shown that stimulation of macrophages with TLR ligands on beads enhanced mtROS production and delivery of mitochondria to phagolysosomes, and that inhibition of these events reduced killing of intracellular bacteria in vitro52. Our data demonstrating zymosan-containing phagolysosomes surrounded by mitochondria (Fig. 4) also suggest direct delivery of mtROS. How mitochondria are routed to phagolysosomes is not entirely clear but likely involves assembly of autophagocytic machinery. mtROS, as well as thioglitazones, drive “starvation-induced” autophagy whereby intracellular organelles are delivered to autophagolysosomes (e.g. damaged mitochondria in so-called mitophagy) for degradation and to provide energy to the cell53,54. This autophagocytic process has been demonstrated to require ROS, is inhibitable with DPI, and is deficient in CGD neutrophils7,55,56. Precise dissection of molecular events leading to Pio restoration of phagocyte mtROS production and enhanced killing of bacteria will require cellular models that allow genetic manipulation and are the focus of future investigation.
Deficient activity of the NADPH oxidase is also associated with exaggerated sterile inflammation, and autoimmune consequences frequent in CGD (e.g. colitis, granuloma, poor wound healing) and systemic lupus erythematosus, as well as altered tissue and cellular functioning57–59. For instance, in the absence of a functional NADPH oxidase, macrophages are poorly programmed for inflammation resolving functions including efferocytosis, the clearance of activated and dying neutrophils, and production of anti-inflammatory mediators29,60–62. Oxidant-dependent PPARγ activation is critical in programming recruited monocytes and inflammatory macrophages derived from them18, and its activation is delayed in CGD macrophages with exaggerated inflammation as a consequence29. Oxidants and mitochondrial disruption are also critical to drive neutrophil apoptosis44, and for the development of appropriate “eat me” signals on activated and dying neutrophils63. Lacking these, CGD neutrophils accumulate in great numbers, ultimately die, deteriorate, enhance inflammation, and may promote autoimmunity. These observations, together with several in vivo models support the hypothesis that oxidants likely play increasingly recognized and important anti-inflammatory signaling roles64–66.
Thioglitazones are potent PPARγ agonists currently approved for use in Type 2 diabetes and result in production of IL-10, suppression of systemic inflammatory cytokine production (IL-6 and TNFα), and improved insulin resistance11,67. While cardiac toxicity in Type 2 diabetics raised safety concerns for rosiglitazone, Pio appears safer in this regard68. Short-term or intermittent Pio therapy is less likely to be associated with potential side effects69. Thioglitazones are currently in clinical trials for treatment of metabolic syndrome, obesity, end-stage renal disease, autoimmunity, rheumatoid arthritis, cystic fibrosis, and severe asthma (http://www.clinicaltrials.gov). Recently, we showed that treatment of gp91phox−/− mice with Pio reversed exaggerated sterile inflammation by enhancing clearance of neutrophils and suppression of cytokine production29. Whether PPARγ-mediated enhanced mtROS production was responsible for the restored macrophage resolving activities, and/or enhanced development of clearance signals on CGD neutrophils are open questions currently under investigation. These earlier observations regarding anti-inflammatory effects, along with the demonstration, here, that Pio treatment restores mtROS production by murine CGD phagocytes and enhances host defense against a relevant pathogen, make a compelling case for the investigation of this and possibly other on-the-shelf “starvation signaling” agents10 in patients with CGD.
Supplementary Material
Key Messages.
The NADPH oxidase governs mitochondrial oxidant production in activated phagocytes, and such crosstalk is absent in CGD.
Pioglitazone, a “nutrient restriction” therapeutic, restores mitochondrial ROS production and enhances host defense in murine CGD.
Acknowledgments
The authors would like to thank Mary C. Dinauer M.D., Ph.D., Brian Day, Ph.D. Robert Keith, M.D. and Heather Brechbhul, Ph.D. for many helpful discussions, and Ms. Brenda Sebern for assistance in preparing the manuscript.
Funding sources:
This investigation was entirely funded by the NIH grants AI058228 and HL34303 and the Chronic Granulomatous Disorder Society (UK).
Abbreviations
- CGD
chronic granulomatous disease
- Pio
pioglitazone
- PPAR
peroxisome proliferator activated receptor
- NADPH
nicotinamide adenine dinucleotide
- phox
phagocyte oxidase
- ROS
reactive oxygen species
- mt
mitochondria
- DHR
dihydrorhodamine
- PMA
phorbol 12-myristate 13-acetate
- DPI
diphenylene iodonium
- DAPI
4′-6-diamidino-2-phenylindole, dihydrochloride
- CFU
colony forming unit
- i.p.
intraperitoneal
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors have no conflict of interest to declare.
References
- 1.Hoidal JR, Fox RB, Repine JE. Defective oxidative metabolic responses in vitro of alveolar macrophages in chronic granulomatous disease. Am Rev Respir Dis. 1979;120:613–8. doi: 10.1164/arrd.1979.120.3.613. [DOI] [PubMed] [Google Scholar]
- 2.Johnston RB, Jr, Keele BB, Jr, Misra HP, Lehmeyer JE, Webb LS, Baehner RL, et al. The role of superoxide anion generation in phagocytic bactericidal activity. Studies with normal and chronic granulomatous disease leukocytes. J Clin Invest. 1975;55:1357–72. doi: 10.1172/JCI108055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine (Baltimore) 2000;79:170–200. doi: 10.1097/00005792-200005000-00004. [DOI] [PubMed] [Google Scholar]
- 4.Holland SM. Chronic granulomatous disease. Clin Rev Allergy Immunol. 2010;38:3–10. doi: 10.1007/s12016-009-8136-z. [DOI] [PubMed] [Google Scholar]
- 5.Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363:2600–10. doi: 10.1056/NEJMoa1007097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, et al. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. 2002;416:291–7. doi: 10.1038/416291a. [DOI] [PubMed] [Google Scholar]
- 7.Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MA, et al. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci U S A. 2009;106:6226–31. doi: 10.1073/pnas.0811045106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kyrmizi I, Gresnigt MS, Akoumianaki T, Samonis G, Sidiropoulos P, Boumpas D, et al. Corticosteroids block autophagy protein recruitment in Aspergillus fumigatus phagosomes via targeting dectin-1/Syk kinase signaling. J Immunol. 2013;191:1287–99. doi: 10.4049/jimmunol.1300132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011;21:290–304. doi: 10.1038/cr.2010.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Neill LA, Hardie DG. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature. 2013;493:346–55. doi: 10.1038/nature11862. [DOI] [PubMed] [Google Scholar]
- 11.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447:1116–20. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–46. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 13.Coste A, Dubourdeau M, Linas MD, Cassaing S, Lepert JC, Balard P, et al. PPARgamma promotes mannose receptor gene expression in murine macrophages and contributes to the induction of this receptor by IL-13. Immunity. 2003;19:329–39. doi: 10.1016/s1074-7613(03)00229-2. [DOI] [PubMed] [Google Scholar]
- 14.Coste A, Lagane C, Filipe C, Authier H, Gales A, Bernad J, et al. IL-13 attenuates gastrointestinal candidiasis in normal and immunodeficient RAG-2(−/−) mice via peroxisome proliferator-activated receptor-gamma activation. J Immunol. 2008;180:4939–47. doi: 10.4049/jimmunol.180.7.4939. [DOI] [PubMed] [Google Scholar]
- 15.Lefevre L, Gales A, Olagnier D, Bernad J, Perez L, Burcelin R, et al. PPARgamma ligands switched high fat diet-induced macrophage M2b polarization toward M2a thereby improving intestinal Candida elimination. PLoS One. 2010;5:e12828. doi: 10.1371/journal.pone.0012828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kielian T, Syed MM, Liu S, Phulwani NK, Phillips N, Wagoner G, et al. The synthetic peroxisome proliferator-activated receptor-gamma agonist ciglitazone attenuates neuroinflammation and accelerates encapsulation in bacterial brain abscesses. J Immunol. 2008;180:5004–16. doi: 10.4049/jimmunol.180.7.5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aronoff DM, Serezani CH, Carstens JK, Marshall T, Gangireddy SR, Peters-Golden M, et al. Stimulatory effects of peroxisome proliferator-activated receptor-gamma on Fcgamma receptor-mediated phagocytosis by alveolar macrophages. PPAR Res. 2007;2007:52546. doi: 10.1155/2007/52546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gautier EL, Chow A, Spanbroek R, Marcelin G, Greter M, Jakubzick C, et al. Systemic analysis of PPARgamma in mouse macrophage populations reveals marked diversity in expression with critical roles in resolution of inflammation and airway immunity. J Immunol. 2012;189:2614–24. doi: 10.4049/jimmunol.1200495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Inoue I, Goto S, Matsunaga T, Nakajima T, Awata T, Hokari S, et al. The ligands/activators for peroxisome proliferator-activated receptor alpha (PPARalpha) and PPARgamma increase Cu2+,Zn2+-superoxide dismutase and decrease p22phox message expressions in primary endothelial cells. Metabolism. 2001;50:3–11. doi: 10.1053/meta.2001.19415. [DOI] [PubMed] [Google Scholar]
- 20.Wang SP, Wu JW, Bourdages H, Lefebvre JF, Casavant S, Leavitt BR, et al. The Catalytic Function of Hormone-Sensitive Lipase is Essential for Fertility in Male Mice. Endocrinology. 2014:en20141031. doi: 10.1210/en.2014-1031. [DOI] [PubMed] [Google Scholar]
- 21.Feinstein DL, Spagnolo A, Akar C, Weinberg G, Murphy P, Gavrilyuk V, et al. Receptor-independent actions of PPAR thiazolidinedione agonists: is mitochondrial function the key? Biochem Pharmacol. 2005;70:177–88. doi: 10.1016/j.bcp.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 22.Omar HA, Salama SA, Arafa el SA, Weng JR. Antitumor effects of energy restriction-mimetic agents: thiazolidinediones. Biol Chem. 2013;394:865–70. doi: 10.1515/hsz-2013-0139. [DOI] [PubMed] [Google Scholar]
- 23.Martinet W, Schrijvers DM, Timmermans JP, Herman AG, De Meyer GR. Phagocytosis of bacteria is enhanced in macrophages undergoing nutrient deprivation. FEBS J. 2009;276:2227–40. doi: 10.1111/j.1742-4658.2009.06951.x. [DOI] [PubMed] [Google Scholar]
- 24.Guthrie LA, McPhail LC, Henson PM, Johnston RB., Jr Priming of neutrophils for enhanced release of oxygen metabolites by bacterial lipopolysaccharide evidence for increased activity of the superoxide-producing enzyme. J Exp Med. 1984;160:1656–71. doi: 10.1084/jem.160.6.1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Decleva E, Menegazzi R, Busetto S, Patriarca P, Dri P. Common methodology is inadequate for studies on the microbicidal activity of neutrophils. J Leukoc Biol. 2006;79:87–94. doi: 10.1189/jlb.0605338. [DOI] [PubMed] [Google Scholar]
- 26.Shiloh MU, Ruan J, Nathan C. Evaluation of bacterial survival and phagocyte function with a fluorescence-based microplate assay. Infect Immun. 1997;65:3193–8. doi: 10.1128/iai.65.8.3193-3198.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, et al. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet. 1995;9:202–9. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- 28.Haslett C, Guthrie LA, Kopaniak MM, Johnston RB, Jr, Henson PM. Modulation of multiple neutrophil functions by preparative methods and trace amounts of bacterial lipopolysaccharide. Am J Pathol. 1985;119:101–10. [PMC free article] [PubMed] [Google Scholar]
- 29.Fernandez-Boyanapalli R, Frasch SC, Riches DW, Vandivier RW, Henson PM, Bratton DL. PPARgamma activation normalizes resolution of acute sterile inflammation in murine chronic granulomatous disease. Blood. 2010;116:4512–22. doi: 10.1182/blood-2010-02-272005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shiomi T, Tsutsui H, Hayashidani S, Suematsu N, Ikeuchi M, Wen J, et al. Pioglitazone, a peroxisome proliferator-activated receptor-gamma agonist, attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation. 2002;106:3126–32. doi: 10.1161/01.cir.0000039346.31538.2c. [DOI] [PubMed] [Google Scholar]
- 31.Zhao W, Thacker SG, Hodgin JB, Zhang H, Wang JH, Park JL, et al. The peroxisome proliferator-activated receptor gamma agonist pioglitazone improves cardiometabolic risk and renal inflammation in murine lupus. J Immunol. 2009;183:2729–40. doi: 10.4049/jimmunol.0804341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Trush MA. Diphenyleneiodonium, an NAD(P)H oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochem Biophys Res Commun. 1998;253:295–9. doi: 10.1006/bbrc.1998.9729. [DOI] [PubMed] [Google Scholar]
- 33.Miramar MD, Costantini P, Ravagnan L, Saraiva LM, Haouzi D, Brothers G, et al. NADH oxidase activity of mitochondrial apoptosis-inducing factor. J Biol Chem. 2001;276:16391–8. doi: 10.1074/jbc.M010498200. [DOI] [PubMed] [Google Scholar]
- 34.Garcia-Ruiz I, Solis-Munoz P, Fernandez-Moreira D, Munoz-Yague T, Solis-Herruzo JA. Pioglitazone leads to an inactivation and disassembly of complex I of the mitochondrial respiratory chain. BMC Biol. 2013;11:88. doi: 10.1186/1741-7007-11-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brand MD. The sites and topology of mitochondrial superoxide production. Exp Gerontol. 2010;45:466–72. doi: 10.1016/j.exger.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2010;35:505–13. doi: 10.1016/j.tibs.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim KY, Ahn JH, Cheon HG. Apoptotic action of peroxisome proliferator-activated receptor-gamma activation in human non small-cell lung cancer is mediated via proline oxidase-induced reactive oxygen species formation. Mol Pharmacol. 2007;72:674–85. doi: 10.1124/mol.107.035584. [DOI] [PubMed] [Google Scholar]
- 38.Bassoe CF, Li N, Ragheb K, Lawler G, Sturgis J, Robinson JP. Investigations of phagosomes, mitochondria, and acidic granules in human neutrophils using fluorescent probes. Cytometry B Clin Cytom. 2003;51:21–9. doi: 10.1002/cyto.b.10003. [DOI] [PubMed] [Google Scholar]
- 39.Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, et al. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood. 2010;116:857. doi: 10.1182/blood-2009-07-235028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bouhlel MA, Derudas B, Rigamonti E, Dievart R, Brozek J, Haulon S, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–43. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 41.Perez-Ortiz JM, Tranque P, Burgos M, Vaquero CF, Llopis J. Glitazones induce astroglioma cell death by releasing reactive oxygen species from mitochondria: modulation of cytotoxicity by nitric oxide. Mol Pharmacol. 2007;72:407–17. doi: 10.1124/mol.106.032458. [DOI] [PubMed] [Google Scholar]
- 42.Hampton MB, Vanags DM, Porn-Ares I, Orrenius S. Involvement of extracellular calcium in phosphatidylserine exposure during apoptosis. FEBS Lett. 1996;399:277–82. doi: 10.1016/s0014-5793(96)01341-5. [DOI] [PubMed] [Google Scholar]
- 43.Kalyanaraman B. Oxidative chemistry of fluorescent dyes: implications in the detection of reactive oxygen and nitrogen species. Biochem Soc Trans. 2011;39:1221–5. doi: 10.1042/BST0391221. [DOI] [PubMed] [Google Scholar]
- 44.Maianski NA, Geissler J, Srinivasula SM, Alnemri ES, Roos D, Kuijpers TW. Functional characterization of mitochondria in neutrophils: a role restricted to apoptosis. Cell Death Differ. 2004;11:143–53. doi: 10.1038/sj.cdd.4401320. [DOI] [PubMed] [Google Scholar]
- 45.Oren R, Farnham AE, Saito K, Milofsky E, Karnovsky ML. Metabolic patterns in three types of phagocytizing cells. J Cell Biol. 1963;17:487–501. doi: 10.1083/jcb.17.3.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Raam BJ, Sluiter W, de Wit E, Roos D, Verhoeven AJ, Kuijpers TW. Mitochondrial membrane potential in human neutrophils is maintained by complex III activity in the absence of supercomplex organisation. PLoS One. 2008;3:e2013. doi: 10.1371/journal.pone.0002013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ahmed KA, Sawa T, Ihara H, Kasamatsu S, Yoshitake J, Rahaman MM, et al. Regulation by mitochondrial superoxide and NADPH oxidase of cellular formation of nitrated cyclic GMP: potential implications for ROS signalling. Biochem J. 2012;441:719–30. doi: 10.1042/BJ20111130. [DOI] [PubMed] [Google Scholar]
- 48.Daiber A. Redox signaling (cross-talk) from and to mitochondria involves mitochondrial pores and reactive oxygen species. Biochim Biophys Acta. 2010;1797:897–906. doi: 10.1016/j.bbabio.2010.01.032. [DOI] [PubMed] [Google Scholar]
- 49.Straus DS, Glass CK. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol. 2007;28:551–8. doi: 10.1016/j.it.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 50.Zhang R, Brennan ML, Shen Z, MacPherson JC, Schmitt D, Molenda CE, et al. Myeloperoxidase functions as a major enzymatic catalyst for initiation of lipid peroxidation at sites of inflammation. J Biol Chem. 2002;277:46116–22. doi: 10.1074/jbc.M209124200. [DOI] [PubMed] [Google Scholar]
- 51.Burlak C, Whitney AR, Mead DJ, Hackstadt T, Deleo FR. Maturation of human neutrophil phagosomes includes incorporation of molecular chaperones and endoplasmic reticulum quality control machinery. Mol Cell Proteomics. 2006;5:620–34. doi: 10.1074/mcp.M500336-MCP200. [DOI] [PubMed] [Google Scholar]
- 52.West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472:476–80. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species. J Cell Sci. 2007;120:4155–66. doi: 10.1242/jcs.011163. [DOI] [PubMed] [Google Scholar]
- 54.Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, et al. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature. 2013;501:512–6. doi: 10.1038/nature12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang J, Brumell JH. NADPH oxidases contribute to autophagy regulation. Autophagy. 2009;5:887–9. doi: 10.4161/auto.9125. [DOI] [PubMed] [Google Scholar]
- 56.Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–7. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 57.Campbell AM, Kashgarian M, Shlomchik MJ. NADPH oxidase inhibits the pathogenesis of systemic lupus erythematosus. Sci Transl Med. 2012;4:157ra41. doi: 10.1126/scitranslmed.3004801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jacob CO, Eisenstein M, Dinauer MC, Ming W, Liu Q, John S, et al. Lupus-associated causal mutation in neutrophil cytosolic factor 2 (NCF2) brings unique insights to the structure and function of NADPH oxidase. Proc Natl Acad Sci U S A. 2012;109:E59–67. doi: 10.1073/pnas.1113251108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Violi F, Sanguigni V, Carnevale R, Plebani A, Rossi P, Finocchi A, et al. Hereditary deficiency of gp91(phox) is associated with enhanced arterial dilatation: results of a multicenter study. Circulation. 2009;120:1616–22. doi: 10.1161/CIRCULATIONAHA.109.877191. [DOI] [PubMed] [Google Scholar]
- 60.Brown JR, Goldblatt D, Buddle J, Morton L, Thrasher AJ. Diminished production of anti-inflammatory mediators during neutrophil apoptosis and macrophage phagocytosis in chronic granulomatous disease (CGD) J Leukoc Biol. 2003;73:591–9. doi: 10.1189/jlb.1202599. [DOI] [PubMed] [Google Scholar]
- 61.Frasch SC, Fernandez-Boyanapalli RF, Berry KA, Murphy RC, Leslie CC, Nick JA, et al. Neutrophils regulate tissue neutrophilia in inflammation via the oxidant-modified lipid lysophosphatidylserine. Journal of Biological Chemistry. 2013;288:4583–93. doi: 10.1074/jbc.M112.438507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sanmun D, Witasp E, Jitkaew S, Tyurina YY, Kagan VE, Ahlin A, et al. Involvement of a functional NADPH oxidase in neutrophils and macrophages during programmed cell clearance: implications for chronic granulomatous disease. Am J Physiol Cell Physiol. 2009;297:C621–31. doi: 10.1152/ajpcell.00651.2008. [DOI] [PubMed] [Google Scholar]
- 63.Frasch SC, Berry KZ, Fernandez-Boyanapalli R, Jin HS, Leslie C, Henson PM, et al. NADPH oxidase-dependent generation of lysophosphatidylserine enhances clearance of activated and dying neutrophils via G2A. J Biol Chem. 2008;283:33736–49. doi: 10.1074/jbc.M807047200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pizzolla A, Gelderman KA, Hultqvist M, Vestberg M, Gustafsson K, Mattsson R, et al. CD68-expressing cells can prime T cells and initiate autoimmune arthritis in the absence of reactive oxygen species. Eur J Immunol. 2011;41:403–12. doi: 10.1002/eji.201040598. [DOI] [PubMed] [Google Scholar]
- 65.Lopes F, Coelho FM, Costa VV, Vieira EL, Sousa LP, Silva TA, et al. Resolution of neutrophilic inflammation by H2O2 in antigen-induced arthritis. Arthritis and rheumatism. 2011;63:2651–60. doi: 10.1002/art.30448. [DOI] [PubMed] [Google Scholar]
- 66.Voltan S, Martines D, Elli M, Brun P, Longo S, Porzionato A, et al. Lactobacillus crispatus M247-derived H2O2 acts as a signal transducing molecule activating peroxisome proliferator activated receptor-gamma in the intestinal mucosa. Gastroenterology. 2008;135:1216–27. doi: 10.1053/j.gastro.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 67.Consoli A, Devangelio E. Thiazolidinediones and inflammation. Lupus. 2005;14:794–7. doi: 10.1191/0961203305lu2223oa. [DOI] [PubMed] [Google Scholar]
- 68.Shah P, Mudaliar S. Pioglitazone: side effect and safety profile. Expert Opin Drug Saf. 2010;9:347–54. doi: 10.1517/14740331003623218. [DOI] [PubMed] [Google Scholar]
- 69.Wei L, MacDonald TM, Mackenzie IS. Pioglitazone and bladder cancer: a propensity score matched cohort study. British Journal of Clinical Pharmacology. 2013;75:254–9. doi: 10.1111/j.1365-2125.2012.04325.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.