

Abstract
Spore photoproduct lyase (SP lyase), a member of the radical S-adenosylmethionine superfamily of enzymes, catalyzes the repair of 5-thyminyl-5,6-dihydrothymine [spore photoproduct (SP)], a type of UV-induced DNA damage unique to bacterial spores. The anaerobic purification and characterization of Clostridium acetobutylicum SP lyase heterologously expressed in Escherichia coli, and its catalytic activity in repairing stereochemically defined synthetic dinucleotide SPs was investigated. The purified enzyme contains between 2.3 and 3.1 iron atoms per protein. Electron paramagnetic resonance (EPR) spectroscopy reveals an isotropic signal centered at g = 1.99, characteristic of a [3Fe–4S]+ cluster accounting for 3–4% of the iron in the sample. Upon reduction, a nearly axial signal (g = 2.03, 1.93 and 1.92) characteristic of a [4Fe–4S]+cluster is observed that accounts for 34–45% of total iron. Addition of S-adenosyl-methionine to the reduced enzyme produces a rhombic signal (g = 2.02, 1.93, 1.82) unique to the S-adenosyl-L-methionine complex while decreasing the overall EPR intensity. This reduced enzyme is shown to rapidly and completely repair the 5R diastereomer of a synthetic dinucleotide SP with a specific activity of 7.1 ± 0.6 nmol min−1 mg−1, whereas no repair was observed for the 5S diastereomer.
Keywords: Spore photoproduct lyase, Spore photoproduct, S-Adenosylmethionine, Radical S-adenosyl-L-methionine, Dinucleotides
Introduction
Bacterial spores exhibit significant resistance to a variety of environmental stresses, including UV radiation. This remarkable resistance to UV radiation is due to both the intriguing photochemistry of spore DNA as well as the efficient repair of DNA damage during spore germination. The primary UV photoproduct in spore DNA is unique; rather than the cyclobutane pyrimidine dimers and pyrimidine (6–4) photoproducts formed in vegetative cells, spores accumulate 5-thyminyl-5,6-dihydrothymine [spore photoproduct (SP), Fig. 1] upon UV irradiation [1–3]. During germination, SP is repaired by the enzyme spore photoproduct lyase (SP lyase) in a reaction involving radical-mediated direct reversal of damage without base excision [4, 5].
Fig. 1.

The formation of spore photoproduct (SP) and the repair reaction catalyzed by SP lyase
SP lyase is a member of the radical S-adenosyl-L-methionine (AdoMet) superfamily of enzymes and contains the characteristic three-cysteine motif (CX3CX2C) responsible for coordinating the essential [4Fe–4S] cluster [6–8]. The fourth ligand to the [4Fe–4S] cluster in members of this superfamily is AdoMet, which coordinates the unique iron of the cluster through the methionyl amino and carboxylate groups [9–19]. In the absence of AdoMet the fourth ligand to the [4Fe–4S] cluster is currently unknown. Common mechanistic features of these enzymes include the one-electron reductive cleavage of AdoMet by a reduced [4Fe–4S]+ cluster to generate methionine and a 5′-deoxyadenosyl radical intermediate, the latter of which abstracts a hydrogen atom from the substrate. We have previously presented evidence indicating that in SP lyase, the highly reactive adenosyl radical intermediate initiates catalysis by abstracting a hydrogen atom from C-6 of SP [20, 21]. The resulting substrate radical is then suggested to undergo radical-mediated β-scission to produce two thymines, reforming AdoMet in the process [20–22].
When the SP lesion is generated in bacterial spores, a chiral center is formed at C-5a, resulting in two possible stereoisomers, either (5R)-SP or (5S)-SP (Fig. 2). Until recently, little was known regarding the stereospecificity of SP formation in vivo, or the stereospecificity of repair by SP lyase. Previous work has resulted in a variety of SP substrates being made both by chemical synthesis [23–28] and by UV irradiation [21, 28–30]. Whereas the UV irradiation method of preparing substrates results in the formation of a variety of photoproducts and thus is not ideal for detailed mechanistic studies, che0mical synthesis can provide pure SP substrates but is difficult, time-consuming, and expensive. Further complicating both types of SP formation are the low yields for chemical characterization of the substrates.
Fig. 2.
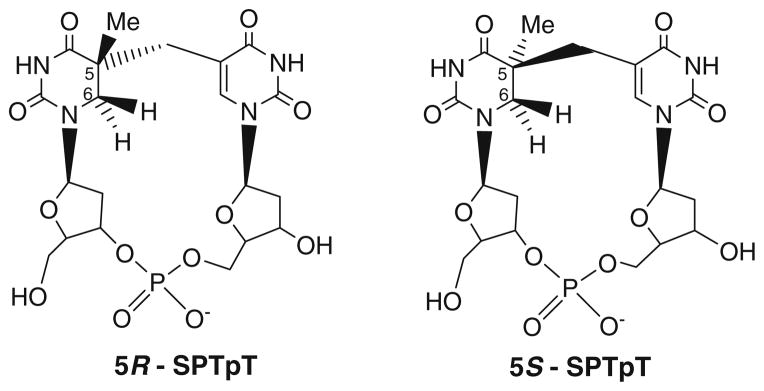
Structures of (5R)-SPTpT and (5S)-SPTpT synthetic substrates
The most recent progress toward defining SP lyase substrates has resulted in the conclusion that (5R)-SP is formed in vivo and that this stereoisomer is specifically repaired by SP lyase [27, 31]. These results were surprising given earlier work reporting that only the (5S)-SP stereoisomer was a substrate for SP lyase [26, 30]. These contradicting findings were obtained for the open dinucleoside forms of SP, which could take on structures quite distinct from that for SP in DNA. We were therefore interested in characterizing the repair of both stereoisomers of a more biologically relevant substrate. We report here the anaerobic purification and characterization of SP lyase from Clostridium acetobutylicum. We also present the first results of enzymatic repair assays of the pure synthetic 5R stereoisomer of the SPTpT substrate ((5R)-SPTpT) and 5S stereoisomer of the SPTpT substrate ((5S)-SPTpT) dinucleotides by the C. acetobutylicum SP lyase. Together, the results reported herein support our conclusion that only (5R)-SP is repaired by SP lyase. Further, the results show that the more physiologically relevant dinucleotide SPTpT is a significantly better substrate for SP lyase than the open dinucleoside SP.
Materials and methods
Biochemical methods
Materials
C. acetobutylicum genomic DNA was obtained from the ATCC (ATCC824D), Escherichia coli Tuner(DE3)pLysS and the pET-14b expression vector were obtained from Novagen, and the primers were obtained from Integrated DNA Technologies. All enzymes were obtained from Promega or Invitrogen. Gel extraction and plasmid DNA purification kits were obtained from QIAGEN. All chemicals were of the highest purity commercially available, unless indicated otherwise.
Expression and purification of SP lyase
The splB gene was cloned from C. acetobutylicum using ATCC824D genomic DNA as a template. Amplification of the splB gene was accomplished with the synthetic oligonucleotide primers 5′-GAGCGCGCGCCATATGGAAA ATATGTTTAGAAGAGTTATATTTG-3′ (containing an NdeI site) and 5′-GCGCGCGCGGATCCTTAAATTATA TACTTAATTGTTGCCTTG-3′ (containing a BamHI site) using standard PCR techniques. The NdeI/BamHI-digested PCR product was cloned into the same sites in pET14b, in-frame with an N-terminal hexahistidine tag. The resulting construct (pET14b/C.a.spl) was then transformed into NovaBlue E. coli for isolation and purification of the plasmid DNA. pET14b/C.a.spl was then transformed into Tuner(DE3)pLysS E. coli for protein overexpression. The fidelity of the PCR product was verified by sequencing.
A single colony of the resulting overexpression strain was used to inoculate 50 mL of Luria–Bertani medium containing 50 μg mL−1 ampicillin. This culture was grown to saturation at 310 K and used to inoculate a 10-L flask of a defined 3-(N-morpholino)propanesulfonic acid medium containing 50 μg mL−1 ampicillin [32]. The 10-L culture was grown at 310 K in a New Brunswick Scientific fermentor (250 rpm, 5 psi O2). When the culture reached an optical density at 600 nm of 0.6, isopropyl β-D-thiogalactopyranoside (IPTG) was added to a final concentration of 0.5 mM, and the medium was supplemented with 750 mg of Fe(NH4)2(SO4)2. The culture was grown for an additional 2 h, and then was cooled to 298 K and placed under nitrogen. The culture was further cooled to 277 K, supplemented with an additional 750 mg of Fe(NH4)2(SO4)2, and left under nitrogen for 12 h. The cells were then harvested by centrifugation and stored at 193 K until used for purification.
SP lyase was purified under anaerobic conditions as previously described [21] with the following changes: pelleted cells (12–16 g) were resuspended in lysis buffer containing 20 mM sodium phosphate, pH 7.5, 350 mM NaCl, 5% glycerol, 10 mM imidazole, 1% Triton X-100, 10 mM MgCl2, 1 mM phenylmethylsulfonyl fluoride, 0.5 mg lysozyme per gram of cells, less than 1 mg of DNase I and RNase A. SP lyase was purified using a HisTrap HP 5-mL column (GE Healthcare) that had previously been equilibrated with buffer A (20 mM sodium phosphate, 350 mM NaCl, 5% glycerol, 10 mM imidazole, pH 7.5). The column was washed with 35 mL of buffer A, followed by a step gradient of 5% buffer B (20 mM sodium phosphate, 350 mM NaCl, 5% glycerol, 500 mM imidazole, pH 7.5) for 25 mL, and then a linear gradient from 5% buffer B to 50% buffer B for 45 mL. The collected fractions were analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and those determined to be 95% or more pure were pooled. The protein was then dialyzed against 20 mM sodium phosphate, 350 mM NaCl, 5% glycerol, pH 7.5 and concentrated using an Amicon concentrator fitted with a YM-10 membrane. The concentrated protein was placed in O-ring-sealed tubes, flash-frozen, and stored at 193 K.
Protein and iron assays
Protein concentrations were determined by the Bradford method [33] using a kit sold by Bio-Rad and bovine serum albumin as a standard. Iron assays were performed using the method of Beinert [34].
Assay of SP lyase activity
All solutions were prepared anaerobically prior to use. Assays with and without SP lyase (50 μM) were set up in parallel in an MBRAUN glove box maintained at less than 1 ppm O2. Reaction mixtures (565-μL total volume) contained 50 μM SP lyase, 1 mM AdoMet, 500 μM SPTpT substrate, 5 mM dithiothreitol (DTT), and 1 mM dithionite all in 17 mM sodium phosphate, 100 mM NaCl, 6 mM KCl, pH 7.5. All reaction mixtures were incubated at 303 K with 80-μL aliquots taken every 10 min for 60 min. The reaction was stopped by flash-freezing in liquid nitrogen and protein was removed by boiling all samples for 1 min followed by centrifugation at top speed. The supernatant of each sample was then diluted 1:3 in H2O and analyzed for thymidylyl-(3′–5′)-thymidine (TpT) and SPTpT content by high-performance liquid chromatography (HPLC). All assays were run at least in duplicate, most often triplicate. Error bars represent the standard deviation of the product concentration.
HPLC analysis
Assays for repair of the SP dinucleotides (SPTpT) to a thymine dinucleotide monophosphate (TpT) were analyzed by HPLC (monitored at 260 nm) and were further confirmed by co-injection of authentic TpT and by mass spectrometry (MS). Samples (20 μL of a 1:3 dilution of each assay) were run over a C18 Waters Spherisorb 5 μm ODS2 4.6 mm × 150 mm analytical column at 1 mL min−1 with a linear gradient of acetonitrile in 2 mM triethylammonium acetate (0–10% from 0 to 20 min, then 10–20% from 20 to 30 min). Integration of the TpT and SPTpT peaks allowed for quantification of repair as compared with control samples.
Mass spectrometry
TpT formation in repair assays was confirmed by subjecting the HPLC fractions corresponding to the isolated TpT peak to electrospray ionization (ESI) MS using a Quattro II mass spectrometer.
Spectroscopic measurements
Low-temperature X-band continuous-wave electron paramagnetic resonance (EPR) spectra were recorded using a Varian E-109 spectrometer modified with a National Instruments computer interface (for data collection and field control) and equipped with an Air Products and Chemicals (Allentown, PA, USA) LTD-3-110 Heli-Tran liquid helium transfer refrigerator. The EPR parameters were as follows: sample temperature 12 K; microwave frequency 9.24 GHz; microwave power 2 mW; time constant 0.50. Each spectrum shown is the average of two scans. Samples were prepared in an MBRAUN glove box maintained at less than 1 ppm O2 with protein concentrations between 560 and 640 μM. Reduced samples were prepared by adding 5 mM DTT, 50 mM tris(hydroxymethyl)aminomethane, 100 μM 5-deazariboflavin and illuminating the sample with a 300-W halogen lamp for 1 h in an EPR tube placed in an ice–water bath. Spin concentration was determined by double integration of the EPR signals arising from samples and from a Cu(II)-EDTA standard of known concentration under identical conditions. UV–vis spectra were recorded at room temperature with a Cary 6000i UV–vis-near IR (Varian) spectrophotometer. Samples were prepared inside an anaerobic MBRAUN chamber and transferred to anaerobic Spectrosil quartz 1.4-mL cuvettes (Starna Cells, Atascadero, CA, USA).
Synthetic methods
All reactions were carried out in oven- or flame-dried glassware under a nitrogen atmosphere. Solvents were distilled prior to use. Dichloromethane and pyridine were distilled from calcium hydride. Tetrahydrofuran was distilled over sodium/benzophenone prior to use. Purification of reaction products was carried out by flash chromatography using silica gel (230–400 mesh). All reagents were commercially available and used without further purification. All reactions were monitored by thin-layer chromatography using silica gel 60, F-254. 1H-NMR and 13C-NMR spectra were recorded with Bruker DPX-300, Bruker DRX-500, or Bruker DRX-600 instruments. NMR spectra were recorded on solutions in deuterated chloroform (CDCl3), with residual chloroform (δ 7.27 ppm for 1H-NMR and δ 77.0 ppm for 13C-NMR) or deuterated dimethyl sulfoxide (dimethyl-d6 sulfoxide), with residual dimethyl sulfoxide (δ 2.50 ppm for 1H-NMR and δ 35.0 ppm for 13C-NMR) used as the standard, and were reported in parts per million. The abbreviations for signal coupling are as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. The multiplicities of the 13C-NMR signals were determined by heteronuclear multiple quantum correlation and distortionless enhancement by polarization transfer techniques. Mass spectra (high-resolution fast atom bombardment or ESI) were recorded with a quadrupole time of flight (TOF) and a 70-VSE spectrometer at the Mass Spectrometry Laboratory, Noyes Laboratory, University of Illinois Urbana-Champaign.
Synthesis and stereochemical assignment of (5R)-SPTpT and (5S)-SPTpT
The synthesis of 5R and 5S SPs with the phosphate backbone (SPTpT) was achieved from their corresponding pure 5R and 5S dinucleoside substrates [27] using published procedures [23] after separation of the diastereomers following the first coupling step of the synthesis as described in [27]. The structures of synthesized (5R)-SPTpT and (5S)-SPTpT were fully characterized using NMR techniques. The stereochemistry of the R and S diastereomers was assigned on the basis of nuclear Overhauser effect spectroscopy and rotating-frame nuclear Overhauser effect spectroscopy cross-peaks of precursors as well as the pure phosphotriester [27]. The structures of the fully deprotected (5R)-SPTpT and (5S)-SPTpT were confirmed by 1H-NMR, 31P-NMR, 13C-NMR, and MS.
(5R)-SPTpT
(5R)-SPTpT was synthesized as a white solid; Rf. 0.1; 1H-NMR (CD3OD): δ 1.24 (s, 3H, Me), 2.1 (dd, 1H, CH2, 2′), 2.2–2.9 (m, 1H, CH, 2′), 2.3–2.38 (m, 1H, CH, 2′), 2.4–2.49 (m, 1H, CH, 2′), 2.60 (d, J = 14 Hz, 1H, CH2, bridge), 2.8 (d, J = 14 Hz, 1H, CH2, bridge), 3.3 (d, J = 14 Hz, 1H, CH2, base), 3.4 (d, J = 13.5 Hz, 1H, CH2, base), 3.62–3.67 (m, 2H, CH2, 5′), 3.7 (dd, J = 3.5, 10 Hz, 2H, CH2, 5′), 3.9 (bt, 1H, CH, 4′), 4.0 (bs, 1H, 4′), 4.5–4.53 (m, 1H, 3′), 4.6–4.67 (m, 1H, 3′), 6.1–6.17 (m, 2H, 1′), 7.8 (s, 1H, base); 13C-NMR (CD3OD): δ 24.2 (CH3, C5), 36.5 (CH2 bridge), 37.2 (CH2), 39.9 (CH2), 41.9, 47.3 (CH2-base), 61.5 (CH2), 65.2 (CH2), 71.2 (C 3′), 73.2 (C 3′), 83.8 (C 4′), 84.3 (C, 1′), 85.2 (C 1′), 86.5 (C 4′), 112.0 (Cquat), 140.1 (CH), 152.5 (Cquat), 155.5 (Cquat), 165.4 (Cquat), 176.9 (Cquat); 31P-NMR (CD3OD): 0.064; high-resolution MS: m/z: 547.1420 (calcd. for C20H28N4O12P; 547.1441) (M + H); MS: (TOF, MS, ESI) m/z 547, 449, 362, 314.
(5S)-SPTpT
(5S)-SPTpT was synthesized as a white solid; Rf. 0.1; 1H-NMR (D2O): δ 1.02 (s, 3H, Me), 1.93–1.95 (m, 1H, CH2, 2′), 2.0–2.08 (m, 1H, CH, 2′), 2.12–2.22 (m, 2H, CH, 2′), 2.5 (d, J = 15 Hz, 1H, CH2, bridge), 2.6 (d, J = 15 Hz, 1H, CH2, bridge), 3.2 (d, J = 13.5 Hz, 1H, CH2, base), 3.3 (d, J = 13.5 Hz, 1H, CH2, base), 3.83–4.0 (m, 6H, CH2, 5′ and 3′), 4.2 (bt, 1H, 3′), 4.4 (bt, 1H, 3′), 6.1 (t, 1H, 1′), 6.2 (t, 1H, 1′), 7.6 (s, 1H, base); 31P-NMR (D2O): 0.91; high-resolution MS: m/z: 547.1438 (calcd. for C20H28N4O12P; 547.1441) (M + H); MS: (TOF, MS, ESI) m/z 547, 569.
Results
Overexpression and purification of SP lyase
The C. acetobutylicum splB gene was cloned into pET-14b containing an N-terminal hexahistidine tag. The resulting construct was used to transform E. coli Tuner(DE3)pLysS for overproduction of histidine-tagged SP lyase. Under this IPTG-inducible system, C. acetobutylicum SP lyase over-expression can be easily observed (Fig. 3, lane 3); the overproduced protein migrates at around 41 kDa on SDS-PAGE, consistent with the amino acid sequence of SP lyase. The crude extract (Fig. 3, lane 4) was loaded onto a Ni HisTrap™ HP affinity column and eluted with an imidazole gradient. SP lyase was eluted between 150 and 250 mM imidazole as a dark-brown band, consistent with the presence of an iron–sulfur cluster in the protein. Pure fractions (Fig. 3, lane 5) were identified by SDS-PAGE, pooled, dialyzed to remove the imidazole, and then concentrated. About 80–100 mg of pure SP lyase could be obtained from 10 L of culture, which is 3–4 times the amount we previously obtained for the growth and purification of His6-SP lyase from Bacillus subtilis [21].
Fig. 3.

Sodium dodecyl sulfate polyacrylamide gel electrophoresis analysis of Clostridium acetobutylicum SP lyase expression and purification. SP lyase was expressed in Escherichia coli Tuner(DE3)-pLysS/pET14b as described in the text. Samples were run on a 12% tris(hydroxymethyl)aminomethane–HCl gel. Lane 1 molecular mass standards, lane 2 preinduced culture, lane 3 postinduced culture, lane 4 clarified crude lysate, lane 5 purified SP lyase
Spectroscopic characterization of C. acetobutylicum SP lyase
The UV–vis spectrum of the purified enzyme (Fig. 4, solid line) is characteristic of the presence of an iron–sulfur cluster. The spectrum exhibits a broad shoulder with a maximum at 413 nm, similar to what has previously been observed for B. subtilis SP lyase [21], as well as for anaerobically purified pyruvate formatelyase activating enzyme and lipoyl synthase [35, 36]. Anaerobically purified SP lyase contains iron [between 2.3 and 3.1 (±0.2) depending on the preparation] and is stable at concentrations greater than 1 mM in the absence of AdoMet. This is in contrast to B. subtilis SP lyase, which was highly susceptible to precipitation at concentrations greater than 250 μM [21]. SP lyase can be photoreduced under anaerobic conditions in the presence of DTT, tris(hydroxymethyl)aminomethane, and 5-deazariboflavin, and reduction is accompanied by a decrease in the visible absorption to produce a spectrum more characteristic of a [4Fe–4S]+ cluster (Fig. 4, dashed line).
Fig. 4.

UV–vis absorption spectra of SP lyase. Spectra of as-isolated enzyme (solid line) and enzyme reduced with 5-deazariboflavin (dashed line) are presented. For both spectra, the protein was 150 μM in 20 mM sodium phosphate, 350 mM NaCl, 5 mM dithiothreitol (DTT), 5% glycerol, pH 7.5. The reduced protein also contained 100 μM 5-deazariboflavin. The spectra were recorded in a 1 cm path length cuvette under anaerobic conditions at room temperature
The as-isolated SP lyase EPR spectrum exhibits an isotropic signal that is centered at a g value of 1.99 (Fig. 5, spectrum A). This signal is similar to what has previously been observed for B. subtilis SP lyase and is consistent with the assignment of a [3Fe–4S]+ cluster being present in the as-isolated form of the enzyme. Spin quantification of this signal, however, shows that it accounts for only about 3–4% of the total iron, a very small amount compared with the 25–35% [3Fe–4S]+ we observed for B. subtilis SP lyase [21]. Upon photoreduction of SP lyase, significant changes in the EPR spectral properties are observed (Fig. 5, spectrum B). The reduced enzyme displays a nearly axial signal with g values of 2.03, 1.93, and 1.92, similar to what has previously been reported for B. subtilis SP lyase reduced with sodium dithionite [21, 30] and for reconstituted C. acetobutylicum SP lyase reduced with sodium dithionite [29], although the signal shown here is significantly stronger than those reported previously. This signal in Fig. 5, spectrum B is consistent with the presence of a [4Fe–4S]+ cluster in the reduced form of the enzyme which accounts for between 32 and 45% of the total iron, indicating incomplete reduction. When AdoMet is added to the reduced enzyme (Fig. 5, spectrum C), a similar signal is observed but with much lower intensity (between 5 and 13% of total iron as [4Fe–4S]+) and slightly altered g values (2.03, 1.92, and 1.82). Both the reduced signal intensity and the altered g values are presumably indicative of the interaction of AdoMet with the active-site [4Fe–4S]+ cluster, as has been observed for other radical AdoMet enzymes.
Fig. 5.

X-band EPR spectra of SP lyase. Spectra of the as-isolated enzyme (A), enzyme photoreduced with 5-deazariboflavin (B), and photoreduced enzyme to which S-adenosyl-L-methionine (AdoMet) has been added (C) are presented. The protein was in 20 mM sodium phosphate, 350 mM NaCl, 5% glycerol, pH 7.5. The as-isolated protein was 640 μM; the reduced sample was 600 μM and was prepared with the addition of 5 mM DTT and 100 μM 5-deazariboflavin. The reduced plus AdoMet sample was 560 μM and was prepared by adding AdoMet to 3 mM final concentration after reduction with 5-deazariboflavin. Conditions of measurement: temperature 12 K; microwave power 2 mW; microwave frequency 9.24 GHz; modulation amplitude 10.00; receiver gain 1.6 × 103. The average of two scans is presented. SPL SP lyase
To further probe the nature of the EPR spectral changes observed in the presence of AdoMet, we investigated the effects on the EPR spectrum of the [4Fe–4S]+ state of SP lyase in the presence of AdoMet cleavage products and an AdoMet analog (Fig. 6). The enzyme was subjected to deazariboflavin-mediated photoreduction and was then mixed with AdoMet, L-methionine, 5′-deoxyadenosine (dAdo), or S-adenosyl-L-homocysteine (SAH) and the 12 K EPR spectrum was recorded. To accurately compare the spectra, the samples were prepared from the same stock of SP lyase. The reduced enzyme and reduced enzyme in the presence of methionine display similar features with a nearly axial signal with g values of 2.03 and 1.92 (Fig. 6, spectra A, C). In the presence of dAdo and SAH, the enzyme displays an axial signal with sharpening of the spectral lines, but maintains g values of 2.03 and 1.92 (Fig. 6, spectra D, E). In the presence of AdoMet (Fig. 6, spectrum B and inset), the spectrum changes once again to a rhombic signal with g values of 2.03, 1.92, and 1.82. It is fascinating that the rhombic signal observed for the reduced enzyme in the presence of AdoMet is not observed with any of these similar molecules, and suggests that this spectral change is specific to the AdoMet–cluster interaction.
Fig. 6.

X-band EPR spectral evidence for a novel interaction of SP lyase with AdoMet. Spectra of SP lyase acquired at 12 K normalized to 570 μM are presented: enzyme photoreduced with 5-deazariboflavin (A); photoreduced enzyme plus 1.5 mM AdoMet (B); photoreduced enzyme plus 1.5 mM methionine (C); photoreduced enzyme plus 1.5 mM 5′-deoxyadenosine (dAdo) (D); photoreduced enzyme plus 1.5 mM S-adenosyl-L-homocysteine (SAH) (E). Inset: Magnification of spectrum B. The protein was in 20 mM sodium phosphate, 350 mM NaCl, 5% glycerol, pH 7.5. Conditions of measurement: temperature 12 K; microwave power 2 mW; microwave frequency 9.24 GHz; modulation amplitude 10.00; receiver gain 1.6 × 103. The average of two scans is presented. SPL SP lyase
Characterization of synthetic dinucleotide SP
The structures of the fully deprotected 5R and 5S dinucleotide SPs were confirmed by 1H-NMR (Fig. 7a), 13C-NMR, 31P-NMR (Fig. 7b), and MS. In addition, the structures of these two closely related products were based on our recent report where we used 2D rotating-frame nuclear Overhauser effect spectroscopy and nuclear Overhauser effect spectroscopy of open and closed forms, as well as of the 5R and 5S dinucleosides, to confirm the stereochemistry [27, 37].
Fig. 7.

NMR spectroscopic characterization of the purified synthetic dinucleotide SPs. a 1H-NMR spectra of (5S)-SPTpT in D2O (top) and (5R)-SPTpT in CD3OD (bottom). b 31P-NMR spectra of (5R)-SPTpT (top) and (5S)-SPTpT (bottom) in CD3OD/D2O
Enzymatic repair of the dinucleotide SPs
We have previously demonstrated that SP lyase repairs the (5R)-SP synthetic dinucleoside but not the (5S)-SP synthetic dinucleoside; turnover of (5R)-SP was, however, quite slow (0.4 nmol min−1 mg−1) and did not proceed to completion [27]. Using the techniques described herein, we have now synthesized both the (5R)-SP and the (5S)-SP dinucleotide monophosphate (SPTpT) and for the first time have performed repair assays on both diastereomers. These synthetic dinucleotide substrates contain a phosphodiester bond linking the sugar groups of the thymidine dimer, providing a more biologically relevant substrate for the enzyme to act upon (Fig. 2). Activity assays were carried out anaerobically and analysis by HPLC allowed for the quantification of TpT product formation over time to obtain a rate of repair.
These assays clearly demonstrate the complete repair of (5R)-SPTpT to form the TpT product with ten turnovers in about 40 min (Fig. 8a). The complete repair of the (5R)-SPTpT substrate to form the TpT product is indicated by a significant decrease in the (5R)-SPTpT peak (eluted at 8.5 min) and a significant increase in the TpT peak (eluted at 16.2 min). Formation of TpT was confirmed by co-injection with authentic TpT as well as by MS analysis [m/z = 547 (M + H); 569 (M + Na)] of the HPLC fractions corresponding to the TpT peak (Fig. 9b). There was no detectable (5R)-SPTpT substrate in the reaction at 40 min or later time points. That this conversion of (5R)-SPTpT to TpT was dependent on SP lyase and AdoMet was demonstrated by control reactions lacking either enzyme or cofactor; in neither case was any formation of TpT detected (data not shown).
Fig. 8.

High performance liquid chromatography (HPLC) chromatograms demonstrating the time-dependent formation of thymidylyl-(3′–5′)-thymidine (TpT) due to repair of (5R)-SPTpT (a) but not (5S)-SPTpT (b), upon incubation of 500 μM SP with SP lyase (50 μM), AdoMet (1 mM), DTT (5 mM) and dithionite (1 mM) in 17 mM sodium phosphate, 100 mM NaCl, 6 mM KCl, pH 7.5 at 303 K. SPTpT is eluted at 8.5 min (5R) or 8.1 min (5S) and TpT is eluted at 16.2 min under these conditions. HPLC chromatograms of the negative control (lacking substrate) (c) demonstrating the time-dependent cleavage of AdoMet by SP lyase (50 μM) incubated with AdoMet (1 mM), DTT (5 mM) and dithionite (1 mM) in 17 mM sodium phosphate, 100 mM NaCl, 6 mM KCl, pH 7.5 at 303 K. Integration of the TpT, SPTpT, AdoMet, and dAdo peaks allowed for the quantification of each isomer of SP and AdoMet cleavage (d). The repair of (5R)-SPTpT (solid squares) demonstrates the repair over the course of 40 min, whereas assays of (5S)-SPTpT (solid triangles) resulted in no detectable formation of TpT. Formation of dAdo is shown for (5R)-SPTpT (open squares) as well as for (5S)-SPTpT (open triangles)
Fig. 9.

Extended electrospray ionization mass spectrometry of (5R)-SPTpT a before repair by SP lyase and b after repair by SP lyase as isolated from the assay mixture (TpT) (M + H = 457; M + Na = 569; M + K = 585)
In contrast to these results with (5R)-SPTpT, in assays with (5S)-SPTpT no significant decrease in peak intensity for the substrate (eluted at 8.1 min) or increase in peak intensity for the product TpT was observed (Fig. 8b). Co-injection of the (5S)-SPTpT reaction mix with authentic TpT results in the formation of a new peak not previously observed in the reaction mix. These results demonstrate that (5R)-SPTpT is a substrate for SP lyase, whereas (5S)-SPTpT is not. For assays with either diastereomer of the synthetic dinucleotide SP, dAdo formation is observed (Fig. 8a, b); such dAdo production is also observed under these assay conditions in the absence of the synthetic substrates, indicating an uncoupling between substrate turnover and the reductive cleavage of AdoMet (Fig. 8c). Such uncoupling is common among radical AdoMet enzymes in the presence of nonphysiological reducing agents [38]. Similar quantities of dAdo are observed for the experiments shown in Fig. 8b [with the nonsubstrate (5S)-SPTpT] and in Fig. 8c (with no substrate), indicating that the nonsubstrate synthetic dinucleotide has no effect on the rate of uncoupled AdoMet cleavage. The dAdo production in the presence of the synthetic substrate (5R)-SPTpT, however, is greater than in these other two cases (Figs. 8a, d). A comparison between the rates of turnover of the synthetic dinucleotides and the rates of reductive cleavage of AdoMet in each assay is provided in Fig. 8d. Whereas the AdoMet cleavage in the assays of (5S)-SPTpT is clearly uncoupled, as no substrate turnover is observed, the observation of only a small excess of dAdo produced over repaired (5R)-SPTpT could lead one to believe that Ado-Met is acting as a substrate [accounting for one dAdo per (5R)-SPTpT repaired], with the remaining dAdo produced as a result of uncoupling. We have, however, demonstrated previously with “native” substrate that SP lyase uses AdoMet catalytically and not as a substrate [21]. We therefore conclude that the approximately 1.3:1 ratio of dAdo to TpT in the assays of (5R)-SPTpT is simply a result of enhanced uncoupled AdoMet cleavage in the presence of a “poor” substrate for SP lyase.
Under the given assay conditions, SP lyase was found to have a specific activity for the repair of (5R)-SPTpT of 7.1 ± 0.6 nmol min−1 mg−1 (or 0.29 ± 0.02 mol mol−1 min−1), comparable to the 0.24 mol mol−1 min−1 previously observed for repair of an SPTpT substrate produced by irradiation [30]. Furthermore, the kcat for the repair of the dinucleotide (5R)-SPTpT substrate by SP lyase was 0.30 ± 0.01 min−1, significantly higher than the 0.021 ± 0.004 min−1 determined for the repair of the (5R)-SP dinucleoside. Together, these results illustrate that whereas the stereochemical requirement for the R configuration is maintained as the substrate gains both complexity and physiological relevance in going from dinucleoside to dinucleotide, significantly higher rates of catalysis are achieved by the simple introduction of a phosphodiester bridge.
Discussion
We have provided here the first detailed investigation of the repair activity of SP lyase toward stereochemically defined dinucleotide SPs. We expressed C. acetobutylicum SP lyase in E. coli and following anaerobic purification, an active enzyme with an iron–sulfur cluster was obtained. This enzyme has proved to be much more stable and soluble than the enzyme from B. subtilis [21]. Furthermore, the C. acetobutylicum SP lyase yields significantly more protein, about 80–100 mg from a 10-L culture, as compared with about 25 mg per 10 L for the B. subtilis SP lyase.
The clone of C. acetobutylicum SP lyase used for these studies included an N-terminal six-histidine tag to aid in purification on a nickel affinity column. Our strictly anaerobic isolation of SP lyase yielded a brown protein containing between 2.3 and 3.1 iron atoms per protein monomer, depending on the preparation. The observation that SP lyase is being isolated with less than four iron atoms per monomer indicates the enzyme does not contain a fully loaded [4Fe–4S] cluster, an observation not uncommon among radical AdoMet enzymes. This purified protein has UV–vis spectroscopic features characteristic of an iron–sulfur cluster (Fig. 4), although they are not indicative of which type of cluster the enzyme contains. EPR spectroscopy reveals only a minor amount (about 3–4% of total iron) being present as a [3Fe–4S]+ cluster (Fig. 5, spectrum A), which suggests the isolated enzyme contains a mixture of EPR-silent clusters (most likely [2Fe–2S]2+ and [4Fe–4S]2+) in addition to the minor [3Fe–4S]+ component giving rise to the characteristic EPR signal. EPR spectroscopy of the reduced enzyme shows that a significant proportion of the iron is present in EPR-active [4Fe–4S]+ clusters (Fig. 5, spectrum B), with the remainder being EPR-silent and presumably in a [4Fe–4S]2+ state from observations of other radical AdoMet enzymes. Reductive cluster conversions (from [3Fe–4S]+ and [2Fe–2S]+ states to [4Fe–4S]2+/+) have previously been reported for several enzymes in the radical AdoMet superfamily, including pyruvate formatelyase activating enzyme [32, 39–41], biotin synthase [42–44], lysine 2,3-aminomutase [45, 46], lipoyl synthase [47, 48] and anaerobic ribonucleotide reductase activating enzyme [49], although the physiological relevance of these cluster conversions remains unknown.
Addition of AdoMet to the reduced enzyme produces a decreased signal intensity in the EPR spectrum, similar to previous observations with B. subtilis SP lyase [21]. However, we also observed a change in the line shape to a rhombic signal not previously observed for SP lyase in the presence of AdoMet (Fig. 5, spectrum C). This novel signal appears to be unique to the interaction of AdoMet with the reduced enzyme, as the addition of AdoMet cleavage products or AdoMet analogs to the reduced SP lyase did not produce the rhombic signal observed when AdoMet is present (Fig. 6). The decrease in [4Fe–4S]+ signal intensity upon addition of AdoMet, which is also not observed upon addition of AdoMet analogs or cleavage products, has previously been suggested to be due to the reductive cleavage of AdoMet (which would be accompanied by cluster oxidation to the EPR-silent [4Fe–4S]2+ state) [50]; we did not, however, observe significant AdoMet cleavage in the absence of substrate and dithionite while being incubated in an ice bath (conditions in which the EPR sample is prepared; data not shown), suggesting that the cluster is not being oxidized. Although a small amount (30 μM) of dAdo is observed to form, this amount does not fully account for the change in signal intensity upon the addition of AdoMet. Another possibility is that the addition of AdoMet results in conversion of the [4Fe–4S]+ cluster from S = 1/2 to a higher spin state which we are not observing under our current EPR conditions. The reason behind this dramatic reduction in signal intensity is currently under active investigation.
We have recently demonstrated the repair of the synthetic (5R)-SP, but not the (5S)-SP, dinucleoside (lacking a phosphodiester bridge) by SP lyase [27]. This rudimentary substrate provided the first definitive evidence for the stereochemical requirements for SP repair by SP lyase, although repair was quite slow (0.4 nmol min−1 mg−1) and did not proceed to completion. In the present work, we characterized for the first time the ability of SP lyase to repair the pure stereochemically defined dinucleotide substrates. The (5R)-SPTpT and (5S)-SPTpT synthetic dinucleotides, which include a phosphodiester bridge linking the sugars, provide more complete and biologically relevant SPs with which to further investigate the stereochemical requirements for repair by SP lyase. Our results clearly demonstrate that only (5R)-SPTpT acts as a substrate for SP lyase, and that repair is relatively rapid, with a specific activity 18-fold greater (7.1 nmol min−1 mg−1) than that observed when the 5R dinucleoside SP was used as a substrate. The turnover rate is lower, however, than that observed with “native” substrate (0.33 μmol -min−1 mg−1), generated by irradiating DNA with UV light under appropriate conditions, indicating that the incorporation of SPTpT into a DNA strand is necessary for optimal substrate efficiency. We observe multiple turnovers of our (5R)-SPTpT substrate, with 100% repair (ten turnovers) of (5R)-SPTpT over the course of 40 min, consistent with more efficient repair, and with less enzyme inactivation occurring during turnover, relative to assays with the dinucleoside substrate.
Our results provide the most convincing evidence to date for the stereospecificity of the repair reaction catalyzed by SP lyase, and support our previous conclusions based on assays with the minimal dinucleoside substrate. Our results, however, contradict earlier reports that (5S)-SP but not (5R)-SP is a substrate for SP lyase [26, 28]. Although it is not possible for us to determine precisely what led to a different conclusion in this previously published work, the turnover of (5R)-SPTpT reported here is both rapid and complete, allowing for complete characterization of turnover and providing definitive evidence for our conclusion that SP lyase is stereospecific for (5R)-SP (Fig. 8). Our conclusion that the 5R isomer of SP is the only one repaired by SP lyase is further supported by a recent report that UV irradiation of TpT produced only (5R)-SPTpT [31].
A comparison of specific activities for SP lyase from different sources with different synthetic and naturally occurring substrates is provided in Table 1. Assays in which the synthetic SP dinucleoside is used as substrate show the lowest activity, consistent with this being a minimal substrate lacking structural features required for efficient turnover. The addition of the phosphodiester bridge to make the SPTpT dinucleotide substrate, whether generated synthetically (this work) or by UV irradiation of TpT [29, 30], considerably enhances the observed activity for SP lyase, increasing kcat 14-fold. Substrates in which the SP lesion is generated by UV irradiation of a single-stranded oligo [28] or double-stranded DNA [21] give rise to the highest specific activities measured for SP lyase. Comparison of these rates indicates that both the phosphodiester bridge between the sugars of the SP lesion and the nucleotides adjacent to the SP lesion, and possibly the overall structure and conformation of the DNA in which the SP lesion resides, contribute to rates of catalysis. Further insight into these effects will require investigation of binding thermodynamics with different substrates, as well as full kinetic analysis of each substrate, both of which are currently under way.
Table 1.
Catalytic activities of spore photoproduct (SP) lyase with a series of synthetic and natural substrates
| Substrate | Specific activity (mol mol−1 min−1)a,b | kcat (min−1) | Temperature (K) | Source of SP lyase | Reference |
|---|---|---|---|---|---|
| Dinucleoside | 0.02c | 0.021e | 303 | Clostridium acetobutylicum | [27] |
| 0.007c | NR | NR | Bacillus subtilis | [26] | |
| Dinucleotide | 0.29c | 0.30 | 303 | C. acetobutylicum | This study |
| 0.24d | NR | 293 or 310 | B. subtilis | [30] | |
| 0.2d | NR | NR | C. acetobutylicum | [29] | |
| Single-stranded 6-mer oligo | 100d | NR | NR | Geobacillus stearothermophilus | [28] |
| DNA | 14d | NR | 310 | B. subtilis | [21] |
NR not reported
Activities for SP lyase observed in our laboratory have been converted to the same units for previously published results to allow for direct comparisons
The activity for G. stearothermophilus SP lyase was converted to the same units to allow direct comparison using the literature value of 40.5 kDa as the molecular mass
Stereochemically defined synthetic substrates
Substrates produced by UV irradiation
kcat calculated and reported in this study
In conclusion, we reported here the anaerobic purification and characterization of SP lyase from C. acetobutylicum, showing that it contains a reducible iron–sulfur cluster that reacts in a very specific manner with AdoMet. The spectroscopic signatures of C. acetobutylicum SP lyase are consistent with its membership in the radical AdoMet superfamily, and are similar to observations for SP lyase from other sources. However, we observed a novel change in the EPR signal with the addition of AdoMet to the reduced enzyme not previously observed for SP lyase from other sources. We also synthesized the pure stereochemically defined dinucleotide lesions (5R)-SPTpT and (5S)-SPTpT, and demonstrated that the 5R isomer is efficiently repaired to TpT, whereas no repair of the 5S isomer was detected. The rapid and complete repair of the more physiologically relevant (5R)-SPTpT substrate, relative to the earlier report of the 5R dinucleoside substrate, provides a convincing demonstration of not only the stereospecificity of SP lyase, but also the importance of the phosphodiester bridge of the lesion for efficient enzymatic repair.
Acknowledgments
We would like to thank David Schwab and Eric Shepard for assistance in running EPR samples, and David Singel for use of his EPR instrument. This research was supported by the National Institutes of Health (GM67804 to J.B.B.). The Astrobiology Biogeocatalysis Research Center is supported by the NASA Astrobiology Institute (NAI05-19). The authors acknowledge support for the Mass Spectrometry Laboratory at the University of Illinois at Urbana-Champaign. The 70-VSE mass spectrometer was purchased in part with a grant from the Division of Research Resources, National Institutes of Health (RR 04648). The Ultima quadrupole TOF mass spectrometer was purchased in part with a grant from the National Science Foundation, Division of Biological Infrastructure (DBI-0100085).
Abbreviations
- SP lyase
Spore photoproduct lyase
- SP
Spore photoproduct
- AdoMet
S-Adenosyl-L-methionine
- dAdo
5′-Deoxyadenosine
- SAH
S-Adenosyl-L-homocysteine
- TpT
Thymidylyl-(3′–5′)-thymidine
- (5R)-SPTpT
5R stereoisomer of the SPTpT substrate
- (5S)-SPTpT
5S stereoisomer of the SPTpT substrate
- EPR
Electron paramagnetic resonance
References
- 1.Donnellan JE, Jr, Setlow RB. Science. 1965;149:308–310. doi: 10.1126/science.149.3681.308. [DOI] [PubMed] [Google Scholar]
- 2.Varghese AJ. Biochem Biophys Res Commun. 1970;38:484–490. doi: 10.1016/0006-291x(70)90739-4. [DOI] [PubMed] [Google Scholar]
- 3.Nicholson WL, Setlow B, Setlow P. Proc Natl Acad Sci USA. 1991;88:8288–8292. doi: 10.1073/pnas.88.19.8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Munakata N, Rupert CS. J Bacteriol. 1972;111:192–198. doi: 10.1128/jb.111.1.192-198.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Munakata N, Rupert CS. Mol Gen Genet. 1974;130:239–250. doi: 10.1007/BF00268802. [DOI] [PubMed] [Google Scholar]
- 6.Fajardo-Cavazos P, Salazar C, Nicholson WL. J Bacteriol. 1993;175:1735–1744. doi: 10.1128/jb.175.6.1735-1744.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nicholson WL, Chooback L, Fajardo-Cavazos P. Mol Gen Genet. 1997;255:587–594. doi: 10.1007/s004380050532. [DOI] [PubMed] [Google Scholar]
- 8.Sofia HJ, Chen G, Hetzler BG, Reyes-Spindola JF, Miller NE. Nucleic Acids Res. 2001;29:1097–1106. doi: 10.1093/nar/29.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walsby C, Ortillo D, Yang J, Nnyepi M, Broderick WE, Hoffman BM, Broderick JB. Inorg Chem. 2005;44:727–741. doi: 10.1021/ic0484811. [DOI] [PubMed] [Google Scholar]
- 10.Walsby CJ, Hong W, Broderick WE, Cheek J, Ortillo D, Broderick JB, Hoffman BM. J Am Chem Soc. 2002;124:3143–3151. doi: 10.1021/ja012034s. [DOI] [PubMed] [Google Scholar]
- 11.Walsby CJ, Ortillo D, Broderick WE, Broderick JB, Hoffman BM. J Am Chem Soc. 2002;124:11270–11271. doi: 10.1021/ja027078v. [DOI] [PubMed] [Google Scholar]
- 12.Layer G, Moser J, Heinz DW, Jahn D, Schubert W-D. EMBO J. 2003;22:6214–6224. doi: 10.1093/emboj/cdg598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen D, Walsby C, Hoffman BM, Frey PA. J Am Chem Soc. 2003;125:11788–11789. doi: 10.1021/ja036120z. [DOI] [PubMed] [Google Scholar]
- 14.Berkovitch F, Nicolet Y, Wan JT, Jarrett JT, Drennan CL. Science. 2004;303:76–79. doi: 10.1126/science.1088493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hänzelmann P, Schindelin H. Proc Natl Acad Sci USA. 2004;101:12870–12875. doi: 10.1073/pnas.0404624101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hänzelmann P, Schindelin H. Proc Natl Acad Sci USA. 2006;103:6829–6834. doi: 10.1073/pnas.0510711103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lepore BW, Ruzicka FJ, Frey PA, Ringe D. Proc Natl Acad Sci USA. 2005;102:13819–13824. doi: 10.1073/pnas.0505726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nicolet Y, Rubach JK, Posewitz MC, Amara P, Mathevon C, Atta M, Fontecave M, Fontecilla-Camps JC. J Biol Chem. 2008;283:18861–18872. doi: 10.1074/jbc.M801161200. [DOI] [PubMed] [Google Scholar]
- 19.Vey JL, Yang J, Li M, Broderick WE, Broderick JB, Drennan CL. Proc Natl Acad Sci USA. 2008;105:16137–16141. doi: 10.1073/pnas.0806640105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheek J, Broderick JB. J Am Chem Soc. 2002;124:2860–2861. doi: 10.1021/ja017784g. [DOI] [PubMed] [Google Scholar]
- 21.Buis JM, Cheek J, Kalliri E, Broderick JB. J Biol Chem. 2006;281:25994–26003. doi: 10.1074/jbc.M603931200. [DOI] [PubMed] [Google Scholar]
- 22.Mehl RA, Begley TP. Org Lett. 1999;1:1065–1066. doi: 10.1021/ol9908676. [DOI] [PubMed] [Google Scholar]
- 23.Kim SJ, Lester C, Begley TP. J Org Chem. 1995;60:6256–6257. [Google Scholar]
- 24.Friedel MG, Pieck JC, Klages J, Dauth C, Kessler H, Carell T. Chem Eur J. 2006;12:6081–6094. doi: 10.1002/chem.200600169. [DOI] [PubMed] [Google Scholar]
- 25.Bürckstümmer E, Carell T. Chem Commun. 2008:4037–4038. doi: 10.1039/b810008j. [DOI] [PubMed] [Google Scholar]
- 26.Friedel MG, Berteau O, Pieck JC, Atta M, Ollagnier-de-Choudens S, Fontecave M, Carell T. Chem Commun. 2006:445–447. doi: 10.1039/b514103f. [DOI] [PubMed] [Google Scholar]
- 27.Chandra T, Silver SC, Zilinskas E, Shepard EM, Broderick WE, Broderick JB. J Am Chem Soc. 2009;131:2420–2421. doi: 10.1021/ja807375c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pieck JC, Hennecke U, Pierik AJ, Friedel MG, Carell T. J Biol Chem. 2006;281:36317–36326. doi: 10.1074/jbc.M607053200. [DOI] [PubMed] [Google Scholar]
- 29.Chandor A, Douki T, Gasparutto D, Gambarelli S, Sanakis Y, Nicolet Y, Ollagnier-de-Choudens S, Atta M, Fontecave M. C R Chim. 2007;10:756–765. [Google Scholar]
- 30.Chandor A, Berteau O, Douki T, Gasparutto D, Sanakis Y, Ol-lagnier-de-Choudens S, Atta M, Fontecave M. J Biol Chem. 2006;281:26922–26931. doi: 10.1074/jbc.M602297200. [DOI] [PubMed] [Google Scholar]
- 31.Mantel C, Chandor A, Gasparutto D, Douki T, Atta M, Fontecave M, Bayle P-A, Mouesca J-M, Bardet M. J Am Chem Soc. 2008;130:16978–16984. doi: 10.1021/ja805032r. [DOI] [PubMed] [Google Scholar]
- 32.Krebs C, Henshaw TF, Cheek J, Huynh B-H, Broderick JB. J Am Chem Soc. 2000;122:12497–12506. [Google Scholar]
- 33.Bradford M. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 34.Beinert H. Methods Enzymol. 1978;54:435–445. doi: 10.1016/s0076-6879(78)54027-5. [DOI] [PubMed] [Google Scholar]
- 35.Miller JR, Busby RW, Jordan SW, Cheek J, Henshaw TF, Ashley GW, Broderick JB, Cronan JE, Jr, Marletta MA. Biochemistry. 2000;39:15166–15178. doi: 10.1021/bi002060n. [DOI] [PubMed] [Google Scholar]
- 36.Broderick J, Henshaw T, Cheek J, Wojtuszewski K, Smith S, Trojan M, McGhan R, Kopf A, Kibbey M, Broderick W. Biochem Biophys Res Commun. 2000;269:451–456. doi: 10.1006/bbrc.2000.2313. [DOI] [PubMed] [Google Scholar]
- 37.Chandra T, Broderick WE, Broderick JB. Nucleosides Nucleotides Nucleic Acids. 2009;28:1016–1029. doi: 10.1080/15257770903362172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duschene KS, Veneziano SE, Silver SC, Broderick JB. Curr Opin Chem Biol. 2009;13:74–83. doi: 10.1016/j.cbpa.2009.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Broderick JB, Duderstadt RE, Fernandez DC, Wojtuszewski K, Henshaw TF, Johnson MK. J Am Chem Soc. 1997;119:7396–7397. [Google Scholar]
- 40.Krebs C, Broderick WE, Henshaw TF, Broderick JB, Huynh BH. J Am Chem Soc. 2002;124:912–913. doi: 10.1021/ja017562i. [DOI] [PubMed] [Google Scholar]
- 41.Yang J, Naik SG, Ortillo DO, Garcia-Serres R, Li M, Broderick WE, Huynh BH, Broderick JB. Biochemistry. 2009;48:9234–9241. doi: 10.1021/bi9010286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ugulava NB, Gibney BR, Jarrett JT. Biochemistry. 2000;39:5206–5214. doi: 10.1021/bi9926227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duin EC, Lafferty ME, Crouse BR, Allen RM, Sanyal I, Flint DH, Johnson MK. Biochemistry. 1997;36:11811–11820. doi: 10.1021/bi9706430. [DOI] [PubMed] [Google Scholar]
- 44.Tse Sum Bui B, Florentin D, Marquet A, Benda R, Trautwein AX. FEBS Lett. 1999;459:411–414. doi: 10.1016/s0014-5793(99)01300-9. [DOI] [PubMed] [Google Scholar]
- 45.Lieder KW, Booker S, Ruzicka FJ, Beinert H, Reed GH, Frey PA. Biochemistry. 1998;37:2578–2585. doi: 10.1021/bi972417w. [DOI] [PubMed] [Google Scholar]
- 46.Petrovich R, Ruzicka F, Reed G, Frey P. Biochemistry. 1992;31:10774–10781. doi: 10.1021/bi00159a019. [DOI] [PubMed] [Google Scholar]
- 47.Busby RW, Schelvis JPM, Yu DS, Babcock GT, Marletta MA. J Am Chem Soc. 1999;121:4706–4707. [Google Scholar]
- 48.Ollagnier-de Choudens S, Fontecave M. FEBS Lett. 1999;453:25–28. doi: 10.1016/s0014-5793(99)00694-8. [DOI] [PubMed] [Google Scholar]
- 49.Ollagnier S, Meier C, Mulliez E, Gaillard J, Schuenemann V, Trautwein A, Mattioli T, Lutz M, Fontecave M. J Am Chem Soc. 1999;121:6344–6350. [Google Scholar]
- 50.Rebeil R, Nicholson WL. Proc Natl Acad Sci USA. 2001;98:9038–9043. doi: 10.1073/pnas.161278998. [DOI] [PMC free article] [PubMed] [Google Scholar]