

Abstract
Purpose
The growth and survival of neurofibromatosis type 2 (NF2)–deficient cells are enhanced by the activation of multiple signaling pathways including ErbBs/IGF-1R/Met, PI3K/Akt, and Ras/Raf/Mek/Erk1/2. The chaperone protein HSP90 is essential for the stabilization of these signaling molecules. The aim of the study was to characterize the effect of HSP90 inhibition in various NF2-deficient models.
Experimental Design
We tested efficacy of the small-molecule NXD30001, which has been shown to be a potent HSP90 inhibitor. The antiproliferative activity of NXD30001 was tested in NF2-deficient cell lines and in human primary schwannoma and meningioma cultures in vitro. The antitumor efficacy of HSP90 inhibition in vivo was verified in two allograft models and in one NF2 transgenic model. The underlying molecular alteration was further characterized by a global transcriptome approach.
Results
NXD30001 induced degradation of client proteins in and suppressed proliferation of NF2-deficient cells. Differential expression analysis identified subsets of genes implicated in cell proliferation, cell survival, vascularization, and Schwann cell differentiation whose expression was altered by NXD30001 treatment. The results showed that NXD30001 in NF2-deficient schwannoma suppressed multiple pathways necessary for tumorigenesis.
Conclusions
HSP90 inhibition showing significant antitumor activity against NF2-related tumor cells in vitro and in vivo represents a promising option for novel NF2 therapies.
Introduction
Vestibular schwannomas account for approximately 5% to 10% of all tumors inside the skull (1); about 1 out of every 100,000 individuals per year develops a vestibular schwannomas (2). Conventional treatment of this benign tumor includes surgical removal and radiotherapy but to date no validated chemotherapy is available due to poor response to tested interventions (3). Both sporadic and familial forms of vestibular schwannomas lack expression of a functional neurofibromatosis type 2 (NF2) protein, merlin/schwannomin (4, 5). Although sporadic vestibular schwannomas occur in later stages of life, the hallmark of familial NF2 is the development of early-onset, bilateral vestibular schwannomas, often associated with other cranial and spinal nerves schwannomas, meningiomas, and ependymomas that frequently requires repeated invasive surgeries. Hence, less invasive chemotherapy will be highly beneficial to the patients with NF2 for tumor control and prevention.
Merlin is a unique member of the ezrin–radixin–moesin gene family with tumor-suppressing activities (6). Its over-expression was shown to decrease cell growth concomitantly with cell-cycle arrest and apoptosis (7, 8), whereas its depletion by antisense oligonucleotides resulted in increased cell proliferation (9). Consistently, mice heterozygous for Nf2 inactivation (10), homozygous for tissue-specific Nf2 inactivation (11), and transgenic for expression of a human dominant-negative NF2 mutant (12) developed a range of tumors. Merlin is involved in cellular function by providing the link between the actin cytoskeleton and multiple membrane-associated proteins, which are essential for processing extracellular signals, cell adhesion, and cytoskeletal architecture (reviewed in refs. 13, 14). Various biologic pathways were suggested for merlin's involvement in cell proliferation control, including negatively regulation of Rac pathway necessary for Ras transformation (15), and contact inhibition of growth through interaction with CD44 (16). Recently, merlin's translocation into the nucleus was shown to suppress tumorigenesis by inhibiting the nuclear E3 ubiquitin ligase CRL4 (DCAF1) implicated in DNA replication and cell-cycle progression (17).
Multiple links between merlin and its interacting proteins suggested the activation of various signaling pathways in NF2-related tumors, which present a challenge for developing targeted therapeutics for NF2. HSP90 is a ubiquitous molecular chaperone that is responsible for maintaining a subset of proteins involved in cell proliferation and transformation (18). HSP90 inhibition induces proteasomal degradation of its client proteins, providing an attractive therapeutic strategy that can simultaneously suppress multiple signaling pathways. HSP90 is often found overexpressed in malignant tumors, and its elevated level was shown to correlate with poor survival among patients (19). A study showed that the HSP90 complexes in tumor cells possess greater affinity to an HSP90 inhibitor 17-AAG, thereby promising selectivity for targeting tumor cells over normal cells (20). There is single report suggesting the efficacy of HSP90 inhibition in NF2 (21). Many known client proteins of HSP90 were found to be coactivated in human NF2-related tumors, such as ERBBs, AKT, and MET (refs. 22, 23; and our unpublished observations). Studies also showed the role of platelet-derived growth factor receptor (PDGFR) and integrin–FAK pathways in the growth of schwannoma (24, 25). Accordingly, we hypothesize that targeting HSP90 will be efficacious for NF2 therapeutics.
In this study, we aimed to evaluate the efficacy of HSP90 inhibition in NF2-deficient cells in cell culture systems (hereafter referred to as “in vitro”) and in mouse allograft and in a genetically modified NF2 mouse model (“in vivo”). The novel small-molecule inhibitor series pochonin and pochoximes, developed on the radicicol scaffold, were shown to be highly potent and specific inhibitors of HSP90 with antiproliferative activity (26, 27). One of the lead compounds NXD30001 (pochoximeA) penetrated the blood-brain and blood-spinal cord barriers and accumulated in nervous tissues (28), a pre-requisite for treating tumors in the central/peripheral nervous system including some NF2-related tumors, such as ependymomas. To further characterize the underlying molecular mechanisms of action, we used the next-generation sequencing (NGS) approach for assessing the global abundance of transcripts. Treatment of NF2-deficient cells with NXD30001 in vitro resulted in the depletion of multiple signaling molecules implicated in NF2. Concomitantly, in vivo administration of NXD30001 reduced growth of NF2-deficient tumors, and further gene expression analysis identified multiple biologic pathways that may contribute the efficacy of HSP90 inhibition against NF2-related tumorigenesis.
Materials and Methods
Reagents
The structure of NXD30001 and its formulation have been described elsewhere (27). Details are given in Supplementary Materials and Methods.
Cell lines and primary cultures
Mouse Nf2−/− embryonic Schwann cells, ESF-FC1801, and mouse schwannoma cells expressing the human Sch-Δ(39–121) mutant, 08031-9, were derived in our laboratory. An immortalized human schwannoma cells HEI193 was a gift from Dr. Lim (House Research Institute, Los Angeles, CA). The human schwannoma and meningioma and the mouse Schwann cell (mSC) primary cultures were freshly prepared and used before exceeding 5 passages. Details are given in Supplementary Materials and Methods.
Cell viability and proliferation assay
MTS-based monolayer cell proliferation assay (Promega) and soft agar clonogenic assay were used to test cell viability and proliferation at 72 hours and at 3 weeks of drug administration in vitro, respectively. The drug concentration that inhibits cell growth by 50% (GI50) was determined using Prism 5.0 (GraphPad Software). Details are given in Supplementary Materials and Methods.
Allograft models and antitumor efficacy assay
All animal experiments were carried out in strict adherence to the NIH guidance in the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals. The ESC-FC1801 cells and the transplantable schwannoma homogenate, 08031-9, were grafted subcutaneously in nude and in FVB/N syngenic mice, respectively. Mice were subjected to intraperitoneal injection of NXD30001 at 100 mg/kg/d, 3 days a week for 4 weeks. Details are given in Supplementary Materials and Methods.
Pharmacokinetic analysis
The concentration of NXD30001 in the plasma and grafted tumors was measured following single dose and repeated dose administration. The assay was conducted at Cerep Inc.. Details are given in Supplementary Materials and Methods.
Morphometric analysis of Schwann cell tumorlets in NF2 transgenic mice
F1 mice of NF2 transgenic miceTgP0-Sch-Δ(39–121)-27, 5 weeks of age, were subjected to oral administration of 17-DMAG (Alvespimycin) at 10 mg/kg/d or 20 mg/kg/d, 3 days a week for 8 weeks. The development of Schwann cell tumorlets in the spinal nerve roots was histopathologically scored at the endpoint, and was compared between drug-treated and vehicle-treated mice. Details are given in Supplementary Materials and Methods.
Quantitative RT-PCR analysis
Total RNA was isolated, reverse transcribed, then subjected for TaqMan gene expression assay using the 7500 Real-Time PCR System (Applied Biosystems). The quantification was expressed relative to freshly dissected mouse sciatic nerve. Details are given in Supplementary Materials.
Western blotting
Cultured cells (5 × 105) and pulverized frozen tumor samples were lysed in radioimmunoprecipitation assay buffer and the total proteins were extracted, separated, and transferred using standard procedures. Antibodies used for Western blotting are given in Supplementary Materials and Methods.
Immunocytochemistry, immunohistochemistry, TUNEL, and BrdUrd staining
For the detection of S100β, cells were grown on cover glass, fixed, and stained using anti-S100β antibody and Alexa594-conjugated secondary antibody. The apoptotic cells were fixed and labeled using DeadEnd Fluorometric TUNEL System (Promega). The cells undergoing DNA replication were detected using bromodeoxyuridine (BrdUrd) Staining Kit (Invitrogen). For detection of vascularization, paraffin sections of grafted tumors were subjected for platelet/endothelial cell adhesion molecule (PECAM) immunohistochemistry followed by hematoxylin-eosin staining. All images were acquired using the AxioImager.M1 microscope (Carl Zeiss). Details are given in Supplementary Materials and Methods.
Cell-cycle analysis
The distribution of cells in different phases of cell cycle was measured by flow cytometry. The ESC-FC1801 cells (1 × 106/10-mm dish) were treated with NXD30001 for 0, 24, 48, and 72 hours, then fixed in 70% ethanol, and incubated in PBS-T containing 10 μg/mL propidium iodide and 100 μg/mL RNase for 1 hour. The cells were analyzed on the FACSAria II platform (BD Bioscience).
Mutation analysis of NF2-related human schwannomas and meningiomas
NF2 mutations were screened on the genomic DNA isolated from patient-derived schwannoma and meningioma by direct sequencing of 17 exons and the adjacent splice sites (Supplementary Table S1). LOH was determined by SALSA Multiplex Ligation-dependent Probe Amplification (MLPA) kit for NF2 (#P044-B1, MRC-Holland). The automated sequencing and the MLPA analysis were conducted at Laragene Inc.
Transcriptome analysis by high-throughput mRNA sequencing
Total RNA from matched ESC-FC1801 cultured cells and allograft tumors (NXD30001 treated and untreated; n = 1) was used for global sequencing. The expression of 35,604 gene loci in the mouse genome was reported by Cufflinks in the unit of “fragments per kilobase of exon per million fragments mapped.”. The data have been submitted to Gene Expression Omnibus (GEO) with the accession number GSE40187. Differentially expressed genes were subjected to pathway analysis using MetaCore (Thomson Reuters). Details are given in Supplementary Materials and Methods.
Statistical analysis
Quantitative data are presented as mean ± SD when applicable. Statistical analysis was carried out using two-tailed unpaired t test on GraphPad Prism 5.0 Software. P < 0.05 was considered statistically significant. For the transcriptome analysis, genes were considered differentially expressed by the Cufflinks software when the false discovery rate (FDR) corrected P value after Benjamini–Hochberg correction for multiple testing was less than 0.05.
Results
Elevated HSP90 expression in NF2-related human schwannomas
We previously conducted an RNA expression study using microarrays comparing human schwannomas from patients with NF2 with human saphenous nerves from normal individuals (Manent, manuscript in preparation). We found that the expression of HSP90AA and HSP90AB (encoding HSP90) was elevated in schwannoma when compared with normal nerves (Fig. 1A): HSP90AA probes 1 and 2 were upregulated 2.7- and 2.1-folds, respectively. The HSP90AA probe 4 and the HSP90AB probe 4 were also increased in the schwannoma samples. These results indicated an increased requirement of HSP90 in NF2-related schwannomas.
Figure 1.
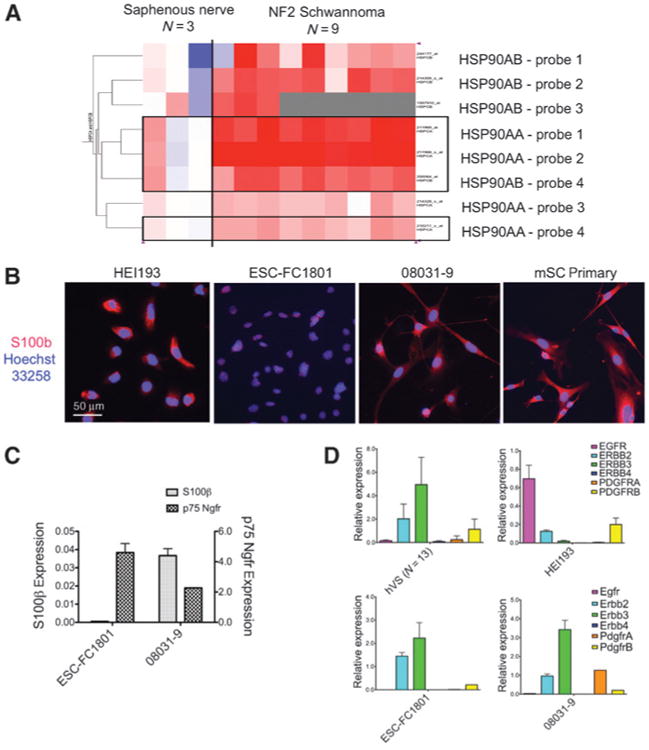
A, heatmap of HSP90 gene expression in the human schwannoma. Expression of HSP90AA and HSP90AB (each with 4 different probe sets) in NF2-related schwannoma (n = 9) were compared with that in human Schwann cells of saphenous nerves (n = 3). Of the 8 probe sets, 3 HSP90AA and 1 HSP90AB probes were statistically different from normal (shown in boxes; FDR = 0.005; 9,479 probe sets total). Red, upregulated; blue, downregulated. B, NF2-deficient cell lines HEI193, ESC-FC1801, and 08031-9, and the mSC primary culture, were shown with cytosolic Schwann cell marker S100β and nuclear marker Hoechst 33258. C, the differential expression of Schwann cell markers (S100β, p75 Ngfr) in ESC-FC1801 and 08031-9. The relative mRNA level was expressed against normal sciatic nerve. D, expression of receptor tyrosine kinases ErbBs and Pdgfrs in the hVS and in the NF2-deficient cell models. The relative mRNA level was expressed against normal nerve. Columns, mean; bars, SD.
Growth inhibition and client protein degradation by NXD30001 in NF2-deficient cells in vitro
Two NF2-deficient cell models were used in this study: mouse embryonic Schwann cells ESC-FC1801 lacking Nf2 exon 2, and mouse schwannoma cells 08031-9 expressing a human Sch-Δ(39–121) dominant-negative mutant of NF2. The expression of the Schwann cell marker S100β was positive in more than 99% of the 08031-9 cells, comparable with the mSC primary culture, but was found to be low in the ESC-FC1801 cells (Fig. 1B). Quantitative PCR (qPCR) confirmed the lower S100β expression in ESC-FC1801, whereas p75Ngfr was higher in ESC-FC1801 than that in 08031-9 (Fig. 1C). In human vestibular schwannoma (hVS), expression of ERBB2 and ERBB3 was upregulated compared with that in normal nerves, whereas EGF receptor (EGFR) was predominant in the immortalized schwannoma cells HEI193, which lacked ERBB3 (Fig. 1D, top). Consistent with their neuregulin dependency, ESC-FC1801 and 08031-9 expressed high levels of ErbB2 and ErbB3 but lacked Egfr (Fig. 1D, bottom), showing similar expression pattern to hVS.
HSP90 inhibition by NXD30001 effectively inhibited the growth of the NF2-deficient cells at low nanomolar concentration after 72 hours exposure in vitro (Fig. 2A). ESC-FC1801 was 10-fold more sensitive to the drug compared with the normal mSC, signifying the selectivity of the drug for the Schwann cells lacking NF2. NXD30001 at 100 nmol/L and 1,000 nmol/L induced nuclear DNA fragmentation, a hallmark of apoptosis, in a small percentage of the cells (Fig. 2B and Supplementary Fig. S1): Nf2−/− Schwann cells, ESC-FC1801, were more sensitive to apoptosis than NF2-deficient schwannoma cells, HEI193 and 08031-9, but less sensitive than normal mSC. No apoptotic cell was observed in the patient-derived schwannoma primary cultures (data not shown). The results indicated that NF2-deficient cells acquired resistance to apoptosis, and the primary effect of HSP90 inhibition in the schwannoma cells was cytostatic rather than cytotoxic.
Figure 2.
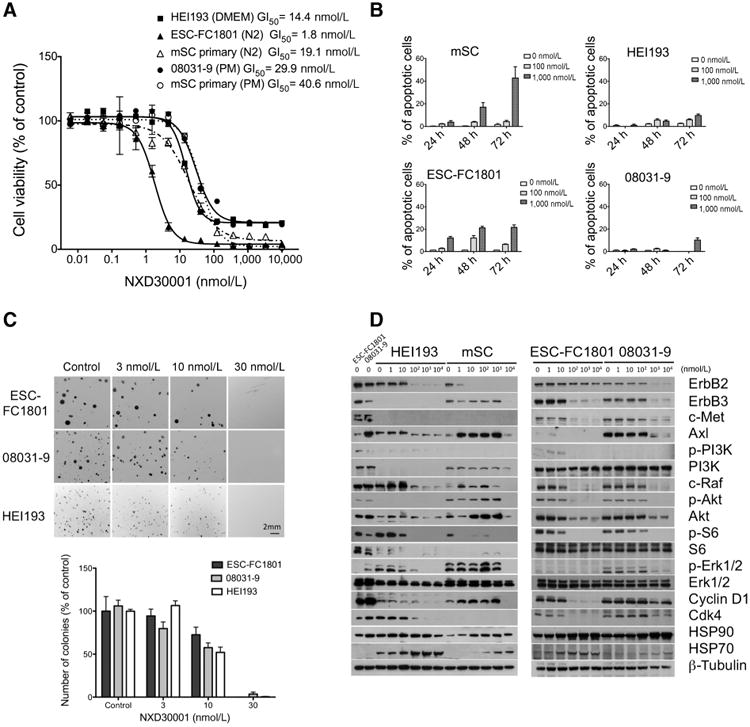
Antiproliferative effect of NXD30001 in NF2-deficient Schwann/schwannoma cell lines. A, dose response to NXD30001 was determined for NF2-deficient cells along with the mSC primary culture. Cells were treated with NXD30001 for 72 hours and the number of viable cells was quantified. B, NF2-deficient cells were treated with 100 nmol/L and 1,000 nmol/L NXD30001 in vitro and apoptotic cells were detected by fluorometric terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling (TUNEL) assay. Percentage of apoptotic cells was quantified from 500 to 1,000 cells. C, NF2-deficient cells were cultured as single cell suspension for 2 to 3 weeks in the soft agar media with or without NXD30001. Numbers of colonies (n = 3 per group) were quantified relative to the vehicle-treated control. D, NF2-deficient cells were treated with various concentrations of NXD30001 for 24 hours and the dose-dependent client protein degradation was displayed by Western blot analysis. Columns, mean; bars, SD.
Long-term effect of NXD30001 in the Nf2-deficient cells in vitro was evaluated by soft agar clonogenic assay. Because of their benign nature, primary cultures of mouse Schwann cells or human schwannoma cells do not form colonies in this assay, and were therefore excluded from the analysis. NXD30001 inhibited colony formation (both in number and in size) of ESC-FC1801, 08031-9, and HEI193 in a dose-dependent manner (Fig. 2C). In contrast to the 72-hour cell viability assay, the antiproliferative efficacy in the long-term culture was similar in the 3 cell lines tested, with GI50 = 11.8 nmol/L, 10.8 nmol/L, and 10.0 nmol/L, respectively. The result indicated that the pathway leading to growth inhibition was independent of the NF2 status because Nf2−/− ESC-FC1801, HEI193 with isoform 3, and 08031-9 carrying mutant NF2 responded similarly to the drug.
Western blot analysis revealed higher baseline expression of HSP90 and its client proteins ErbB2, c-Met, Axl, c-Raf and Cdk4, in the mouse NF2-deficient cells than in the Nf2+/+ mSC (Fig. 2D). Upon HSP90 inhibition, HSP90 and HSP70 increased concomitantly, whereas client proteins showed dose-dependent degradation at 24 hours. Of the concentrations tested, the minimum dose sufficient to deplete client proteins was 100 nmol/L for ESC-FC1801, 1,000 nmol/L for 08031-9, and between 100 nmol/L and 1,000 nmol/L for HEI193, which corresponded with their GI50 in the cell viability assay. In mSC, on the other hand, some client proteins, such as Axl, c-Raf, and Akt, increased at lower doses, and the degradation only occurred at 10,000 nmol/L showing resistance to protein degradation by HSP90 inhibition in normal Schwann cells.
Given that drug efficacy can differ between different species, primary cultures of patient-derived schwannoma and meningioma were also tested in the cell viability assay. The GI50 for 10 human schwannoma and meningioma ranged from 20 nmol/L to 1 μmol/L (Table 1). The NF2 mutation analysis on the original tumor samples revealed no obvious correlation between drug sensitivity and the NF2 status. However, the mitotically active cells of the sporadic vestibular schwannoma-5, which carried biallelic nonsense/frameshift NF2 mutations, showed high sensitivity to the drug. Likewise, highly proliferating spinal schwannoma-1 and meningioma-2, which were derived from a single patient with NF2 with history of multiple lesions, responded significantly to the drug.
Table 1. Antiproliferative activity of NXD30001 in human schwannoma and meningioma primary cultures.
| Primary cells | GI50 (nmol/L) | Clinical status | Age | NF2 gene mutation | Predicted effect | LOH |
|---|---|---|---|---|---|---|
| HEI193 (immortalized) | 14.9 | NF2 | c.1575-1G>A | r.1575_1737del | Hemizygous loss of Ch 22 | |
| p.Val525_Leu595del | ||||||
| Vestibular schwannoma-1 | 113.4 | Sporadic | 60 | c.888delT | p.Ile296fs | N.A. |
| Vestibular schwannoma-5 | 50.0 | Sporadic | 48 | c.161_162delTG | p.Lys54fs | N.A. |
| c.784C>T | p.Arg262* | |||||
| Vestibular schwannoma-9 | 117.5 | Sporadic | 45 | c.592C>T | p.Arg198* | N.A. |
| Vestibular schwannoma-11 | 130.0 | NF2 | 23 | c.447+1G>T | p.Tyr150_Arg172del | Hemizygous loss of NF2 |
| Vestibular schwannoma-13 | 110.2 | Sporadic | 46 | c.115-5_115-51del47 | r.? | N.A. |
| Vestibular schwannoma-17 | 63.5 | Sporadic | 13 | c.478delC | p.Arg160fs | Hemizygous loss of Ch 22 |
| Spinal schwannoma-1 | 18.6 | NF2a | 22 | c.1445delCinsTGG | p.Pro482fs | Homologous duplication |
| Spinal meningioma-2 | 63.4 | NF2a | 22 | c.1445delCinsTGG | p.Pro482fs | Homologous duplication |
| Cranial meningioma-3 | 1050.0 | NF2 | 53 | NF2 mutation not identified | Normal | Hemizygous loss of Ch 22 |
| Cranial meningioma-4 | 734.0 | NF2 | 39 | c.1021C>T | p.Arg341* | Hemizygous loss of NF2 |
NOTE: Classification of sporadic or NF2-associated status was based on the diagnoses from referring physicians. Mutations are listed with the numbering of bases showing alterations relative to the cDNA sequence with the initiator ATG beginning at base = 1. GI50 is determined for 72 hours drug treatment.
Abbreviations: c., cDNA; r., mRNA; p., protein sequences; N.A., not analyzed.
The tumors were 2 spinal cord tumors from the same patient.
Antitumor efficacy of NXD30001 in NF2-deficient allograft models in vivo
NXD30001 was administered for 4 weeks in 2 subcutaneous allograft models; ESC-FC1801 cells inoculated in nude mice, and 08031-9 mouse schwannoma homogenate inoculated in syngenic FVB/N mice. In nude mice, NXD30001 exhibited severe toxicity when administered at 100 mg/kg/d, 3 days/week, which resulted in progressive weight loss (Fig. 3A, top left), compelling the change of schedule to 2 days/week after the first 2 weeks. By contrast, FVB/N mice tolerated the same regime without major weight loss (Fig. 3A, top right) or other clinical toxicities. In both models, the tumor growth was significantly inhibited in the drug-treated group (Fig. 3A, bottom, P < 0.001 and P < 0.0001, respectively).
Figure 3.
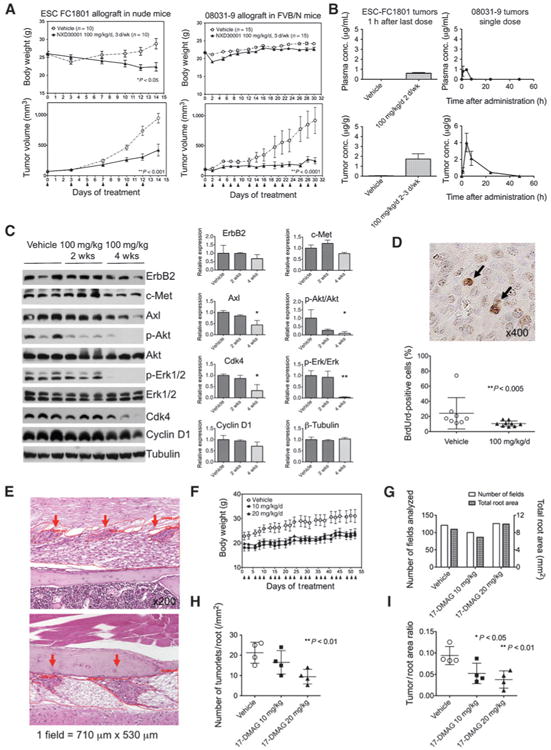
Antitumor efficacy and tolerability of NXD30001 in vivo NF2 models. A, tumor-bearing mice were injected with NXD30001 at the days indicated with arrows, and the body weight and the tumor volume were monitored. ESC-FC1801 allograft tumors in female nude mice (n = 10 per group) and 08031-9 allograft tumors in FVB/N syngenic mice (n = 15 per group) were treated with 100 mg/kg/d, 3 days/week. B, concentration of NXD30001 in the plasma (top) and tumor (bottom) was determined by liquid chromatography/tandem mass spectrometry. Left, measurement in the ESC-FC1801 tumors treated with 100 mg/kg/d, 2 to 3 days/week for 4 weeks and collected 1 hour after the last injection. Right, measurement in the 08031-9 tumors treated with a single dose of 100 mg/kg and collected at indicated time (n = 3 per group). C, pharmacodynamic analysis of ESC-FC1801 tumors treated with 100 mg/kg/d of NXD30001, 2 to 3 days/week for 2 and 4 weeks, showing downregulation of multiple HSP90 client proteins. *, P < 0.05; **, P < 0.01. D, the mitotic index of the grafted 08031-9 tumors was compared by the percentage of BrdUrd-positive cells (arrows) scored from 3 independent sections per tumor. E, morphometric analysis of Schwann cell tumorlets (red arrows) in NF2 transgenic mice. F, 5-week old transgenic mice were treated with 10 mg/kg/d or 20 mg/kg/d of 17-DMAG (n = 4 per group) at the days indicated with arrows for 8 weeks, and their body weight was monitored. G, number of microscopic fields and the total root area analyzed per group. Number of tumorlets per nerve root area (H), and the ratio of tumor area to root area (I), showing reduced development of tumorlets in 17–DMAG–treated mice. Points and columns, mean; bars, SD.
The concentration of NXD30001 in the ESC-FC1801 tumors after repeated administration reached above 1 μg/g, a dose equivalent to approximately 2 μmol/L in vitro (Fig. 3B, left). Furthermore, a single administration (100 mg/kg) to the 08031-9 tumor-bearing FVB/N mice showed selective drug accumulation in the tumors (Fig. 3B, right), resulting in the tumor/plasma ratio of area under curve [AUC(0-t)] as 8.9. HSP90 client and related proteins (Axl, p-Akt, p-Erk, and Cdk4) were reduced after 4-week drug administration in the ESC-FC1801 tumors (Fig. 3C). Consistently, cellular proliferation in the drug-treated tumors, evaluated by the BrdUrd incorporation, was suppressed in the drug-treated tumors (Fig. 3D).
Evaluation of HSP90 inhibition against orthotopic tumor development in NF2 transgenic mice
The NF2 transgenic mouse model expressing a human Sch-Δ(39–121) dominant-negative mutant protein was previously created and characterized (12). Hundred percent of the heterozygous transgenic mice in the (FVB/NCrlxC3H/HeNCrl)F1 background were found to develop multiple benign Schwann cell tumorlets in the spinal nerve roots by 3 months of age (Fig. 3E). To evaluate HSP90 inhibition as a preventive strategy in NF2, we carried out drug efficacy test against schwannoma development using a well-characterized HSP90 inhibitor 17-DMAG on our NF2 transgenic mice. No weight loss was observed during the 8-week treatment with 17-DMAG at 10 mg/kg/d and 20 mg/kg/d, 3 days/week (Fig. 3F). About a hundred microscopic fields per each group, covering equivalent nerve root areas, from the lumber and sacral spines were subjected to morphometric analysis (Fig. 3G). Both the number of tumorlets per root area and the ratio of tumorlet/root area were dose dependently reduced after HSP90 inhibition (Fig. 3H and I).
Global transcriptional alterations induced by NXD30001 in the NF2-deficient cells
NGS was used to characterize the transcriptional consequences of HSP90 inhibition in the NF2-deficient cells. The expression profiles of cultured ESC-FC1801 cells (control vs. treated with 1 μmol/L NXD30001 for 24 hours) were further compared with those of ESC-FC1801 tumors (control vs. treated with 100 mg/kg/d, 3 days/week for 2 weeks) to provide the added context and contribution of the microenvironment for growing tumors (Supplementary Fig. S2A). About 27 million paired-end reads of RNA sequences per sample (100 bp in size, Illumina HiSeq 2000) from the in vitro experimental groups, and about 10 million single-end reads (76 bp in size, Illumina GAIIx) from in vivo experimental groups, were obtained from TopHat. The absolute values of gene expression for 35,604 gene loci were determined by Cufflinks and Cufflinks' differential expression analysis (Cuffdiff; Supplementary Fig. S2B). The differential expression in fold change was highly reproducible by qPCR analysis conducted on selected genes (Supplementary Table S2).
Alterations of HSP90 and its interacting proteins (www.picard.ch/downloads/HSP90interactors) upon HSP90 inhibition involved both up and downregulation (Supplementary Fig. S2C; individual data in the GEO database). In vitro, significant increase of Hsp90aa1, Hsp90ab1, Hspa8 (HSC70), Hspa5 (GRP-78), and Hsf1 showed the well-known response to HSP90 inhibition and heat shock. Subsets of upregulated transcripts also indicated various cellular responses, including induction of apoptosis (Dedd2, Dapk3), autophagy (Map1lc3a, Map1lc3b, Gabarapl2), oxidative stress response (Pink1), inflammatory response (F2r), and antiapoptotic response (Sgk1). On the other hand, survivin (Birk5), an apoptosis inhibitor and essential regulator of mitosis, was among the most significantly downregulated transcripts whose expression was reduced to less than 5% of control. Similarly, transcripts of centrioles-associated tubulins (Tubd1 and Tube1), majority of mitotic kinases (Cdk1, Cdk6, Cdk11, Aurkb, Plk1, Plk4), DNA polymerases (Pola1, Pola2, Polh), DNA helicase (Blm), DNA damage checkpoint and repair proteins (Chek1&2, Rad50, Mre11a), cell-cycle proteins (Ccnb1&2), and some histone proteins were concomitantly silenced after the HSP90 inhibition in vitro. In contrast, transcriptional alteration in vivo included different gene sets: most of the chaperone proteins, mitotic components, and DNA modulators remained unchanged. Instead, modification was observed in many cytoskeletal (actin, tubulin, myosin) and histone components. However, the increase of apoptosis-related genes (Dedd, Ask1) was in accordance with the apoptotic response in vitro. Transcriptional changes in the HSP90 clients implicated in NF2 included upregulation of Pdgfrb, Erbb3, and Lats2, downregulation in Egfr, Vegfr2, Esr1, and Akt3. The results showed that the overall consequences are complex and involve multiple responses, such as possible feedback required for the growth and survival of ESC-FC1801 tumor graft.
Pathway analysis of HSP90 inhibition in NF2-deficient cells
Next, we aimed to systematically identify enriched pathways and regulatory networks that were affected by NXD30001. Cuffdiff revealed changes in approximately 3% of the total genes; 1,383 and 965 genes in vitro and in vivo, respectively, with 128 genes in common (Supplementary Fig. S2B). Remarkably, there was almost no overlap in the affected biologic processes between in vitro and in vivo conditions (Table 2).
Table 2. Transcriptional alteration caused by HSP90 inhibition in NF2-deficient Schwann cells ESC-FC1801.
| GO ID | Description | P | Genes (fold change up) | Genes (fold change down) |
|---|---|---|---|---|
| In vitro | ||||
| GO:0007049/GO:0022402 | Cell cycle/Cell cycle process | 1.60E-46 | Arhgap8, Camk2a, Cdc2l1, Cgref1, Chfr, Dusp1, Gadd45a, Mtus1, Pmp22, Ppp1cc, Rgs2, Sept4, Tex15, Usp16 | 2610039C10Rik, 4632434l11Rik, AC102494.1, Anapc1, Anapc7, Anln, Appl2, Aspm, Aurka, Aurkb, B230120H23Rik, Birc5, Brca1, Brca2, Bub1, Bub1b, C79407, Ccdc99, Ccna2, Ccnb1, Ccnb2, Ccnd1, Ccnf, Cdc123, Cdc14a, Cdc23, Cdc25c, Cdc45l, Cdc6, Cdc7, Cdca2, Cdca3, Cdca5, Cdca8, Cdkn3, Cenpe, Cenpf, Cenph, Cep110, Cep55, Chaf1a, Chaf1b, Chek2, Cit, Ckap2, Ckap5, Cks1b, Clspn, Cntrob, Ddx11, Dlgap5, Dscc1, E2f8, Ercc6l, Esco2, Espl1, Exo1, F630043A04Rik, Fam83d, Fancd2, Fanci, Gas2l3, Gsg2, Haus1, Haus5, Hells, Hjurp, Incenp, Kif11, Kif18a, Kif20b, Kntc1, Lig1, Lin9, Mcm8, Mdc1, Mki67, Mlh1, Mre11a, Myh10, Nasp, Ncapd2, Ncapd3, Ncapg2, Ndc80, Nde1, Nek1, Nek2, Nsl1, Nuf2, Nup43, Nusap1, Oip5, Plk1, Pms2, Pola1, Prc1, Prdm9, Psrc1, Pttg1, Racgap1, Rad50, Rad51, Rad51c, Rbbp8, Rbl1, Sass6, Sept10, Sept3, Sept6, Sept9, Sgol1, Sgol2, Ska1, Smc2, Spag5, Spc25, Steap3, Stmn1, Suv39h1, Suv39h2, Tacc3, Tial1, Timeless, Tpx2, Top2a, Trip13, Ube2c, Uhrf1, Wee1, Zfp318, Zwilch |
| GO:0051301 | Cell division | |||
| GO:0007059 | Chromosome segregation | |||
| GO:0048285 | Organelle fission | 3.77E-29 | Chfr, Fis1, Usp16 | 2610039C10Rik, AC102494.1, Anapc1, Anapc7, Anln, Aspm, Aurkb, Birc5, Bub1, Bub1b, C79407, Ccdc99, Ccna2, Ccnb1, Ccnb2, Ccnf, Cdc23, Cdc25c, Cdc6, Cdca2, Cdca3, Cdca5, Cdca8, Cenpe, Cenpf, Cenph, Cep55, Cit, Ckap5, Ercc6l, F630043A04Rik, Fam83d, Haus1, Haus5, Hells, Incenp, Kif11, Kif18a, Kif20b, Kntc1, Ncapd2, Mlh1, Myh10, Myo5a, Ncapd3, Ncapg2, Ndc80, Nde1, Nek1, Nek2, Nsl1, Nuf2, Nup43, Nusap1, Oip5, Plk1, Plxna2, Pttg1, Sema6a, Sgol1, Ska1, Smc2, Spag5, Spc25, Tacc3, Timeless, Ube2c, Wee1, Zwilch |
| GO:0051640 | Organelle localization | |||
| GO:0051303 | Establishment of chromosome localization | |||
| GO:0006259 | DNA metabolic process | 3.51E-19 | Atrip, Gadd45a, Giyd2, Sgk1 | 0610007P08Rik, Atad5, Atr, Bard1, Blm, Bnip3, Brca1, Brca2, Brip1, Ccdc111, Cdc45l, Cdc6, Chaf1a, Chaf1b, Chek2, Clspn, Dclre1b, Dclre1c, Dna2, Dscc1, Dtl, Eme1, Endog, Ercc2, Esco2, Exo1, Fam175a, Fancb, Fancd2, Fancg, Fanci, Gen1, Gins2, Hells, Hmgb2, Kif22, Lig1, Lin9, Mcm10, Mcm8, Mdc1, Mlh1, Mpg, Mre11a, Nasp, Neil3, Nek1, Orc3l, Parp2, Pms2, Pola1, Pola2, Pold3, Pole, Pole2, Polg, Poli, Polm, Polq, Pot1a, Prim1, Prim2, Pttg1, Rad18, Rad50, Rad51, Rad51ap1, Rad51c, Recql4, Rfc3, Rfc4, Rtel1, Timeless, Top2a, Trip13, Ttn, Uhrf1, Xrcc2, Xrcc5 |
| GO:0006974 | Response to DNA damage stimulus | |||
| GO:0006260 | DNA replication | |||
| GO:0007017 | Microtubule-based process | 1.47E-08 | Gadd45a, Klc1 | Birc5, Brca1, Ccdc99, Cenpe, Ckap5, Cntrob, Espl1, Haus1, Haus5, Kif11, Kif14, Kif15, Kif16b, Kif18a, Kif18b, Kif20a, Kif20b, Kif22, Kif23, Kif2c, Kif4, Kif9, Mlh1, Nav1, Ndc80, Nde1, Nuf2, Nusap1, Psrc1, Spc25, Stmn1, Tacc3, Tpx2, Tubd1, Tube1 |
| GO:0000910 | Cytokinesis | 2.42E-06 | – | Anln, Aurkb, Birc5, Brca2, Cit, Cntrob, Incenp, Myh10, Nusap1 Prc1, Racgap1 |
| GO:0048168 | Regulation of neuronal synaptic plasticity | 1.48E-05 | Camk2a, Hras1, Snca | Bhlhe40, Dlg4, Egr2, Grik2, Nf1, Rims1, Syngr1 |
| GO:0006796 | Phosphate metabolic process | 5.74E-05 | Bmp2k, Camk2a, Cdc2l1, Dclk3, Dusp1, Dusp14, Epha4, F2r, Gadd45g, Gnptab, Lpar3, Lyn, Map3k14, Mast2, Nrbp2, Pink1, Ppm1l, Ppp1cc, Prkci, Prkcq, Prkd1, Ptpn14, Ptprm, Sgk1, Snca, Snrk, Styk1, Tek, Tgfa, Tgfb2 | AC102494.1, Acvrl1, Adrbk2, Atr, Aurka, Aurkb, B230120H23Rik, BC030499, Bub1, Bub1b, Ccnb1, Ccnd1, Cdc14a, Cdc25c, Cdc7, Cdkn3, Chek2, Cit, Dusp6, Dusp7, Fgf2, Fgfr1, Gsg2, Gyk, Ick, Map2k3, Mapk9, Mastl, Melk, Mertk, Mtm1, Nek1, Nek2, Pask, Pbk, Pdik1l, Pdk1, Pdp1, Plk1, Plk4, Prkd3, Ptpn6, Rps6ka1, Rps6ka6, Tnk2, Ttn, Ulk4, Vrk1, Vrk3, Wee1, Wnk3, Wnk4, Wnt5a |
| GO:0006323 | DNA packaging | 9.23E-05 | Aifm2 | Asf1b, Cdca5, Cenpa, H2afz, Hells, Hist1h1b, Hist1h2bl, Hist1h4c, Hist1h4d, Hjurp, Ncapd2, Ncapd3, Ncapg2, Nusap1, Smarca2, Smc2, Suv39h1, Top2a |
|
| ||||
| In vivo | ||||
| GO:0007155 | Cell adhesion | 6.90E-11 | Adam23, Arvcf, Cadm4, Cdh13, Cdh19, Cdh26, Chl1, Col16a1, Col20a1, Col27a1, Col5a3, Ddr1, Dlg1, Fat4, Gpnmb, Hes1, Hspg2, Itga6, Itga7, Itgav, Itgb8, Lama2, Lamb1-1, Mcam, Megf10, Msln, Nid1, Npnt, Nrcam, Pcdh10, Pcdh20, Ptprf, Sorbs1, Sympk, Thbs4, Tnc, Ttyh1, Wnt7b | Acan, Adam12, Amigo2, App, Cd34, Cd36, Cd44, Cd93, Cdh3, Cdh5, Col12a1, Dchs1, Fbln5, Fn1, Lgals1, Lgals3bp, Pecam1, Ror2, Spp1, Tek, Wisp1 |
| GO:0022610 | Biologic adhesion | |||
| GO:0043062 | Extracellular structure organization | 2.68E-07 | Adamts14, Cacna1a, Chrna1, Col5a3, Hspg2, Lrp4, Nid1, Npnt, Tnc, Vwa1, Wnt7b | Acan, Apbb2, App, Atp7a, Ccdc80, Col3a1, Fbln5, Lgals3, Mmp9, Nrg1, Serpinh1, Tfip11 |
| GO:0030198 | Extracellular matrix organization | |||
| GO:0042552 | Myelination | 1.87E-05 | Erbb3, Gal3st1, Gjc3, Lgi4, Mbp, Nab2, Plp1, Pmp22, Pou3f1, Scd2, Sox10 | Nrg1 |
| GO:0007422 | Peripheral nervous system dev. | |||
| GO:0030030 | Cell projection organization | 3.28E-05 | Cacna1a, Ccdc88a, Chl1, Dclk1, Dnaic2, Fgd3, Fgd4, Gas7, Itga6, Kif3a, Lamb1-1, Mtap2, Nefl, Ngfr, Nrcam, Numb, Prkg1 Ptprz1, Rpl24, Rtn4rl2, Sema4f, Sema6c, Ttyh1, Tubb4 | Apbb2, App, Atp7a, Clu, Lpar1, Slc11a2, Vav3 |
| GO:0030031 | Cell projection assembly | |||
| GO:0042325 | Phosphorylation | 5.68E-04 | Ccdc88a, Cenpe, Dlg1, Fgd4, Gnai2, Lats2, Madd, Mdfic, Pdgfrb, Sh3bp5 | App, Atp7a, Cd74, Dusp9, Egfr, Fabp4, Ghr, Hbb-b1, Hbb-b2, Lpar1, Map2k6, Map3k5, Nrg1, Prkce, Rgma, Spry1, Vav3 |
| GO:0051338 | Transferase activity | |||
| GO:0046578 | Regulation of Ras protein signal transduction | 1.91E-03 | Arap3, Arhgef10l, Fgd3, Fgd4, Kalrn, Plekhg2, Tbc1d24 | Evi5, Farp1, Git2, Iqsec2, Kras, Lpar1, Nrg1, Rabgap1, Spry1, Tbc1d1, Vav3 |
| GO:0044271 | Cellular nitrogen compound biosynthetic process | 2.21 E-03 | Adc, Adcy1, Atp6v0b, Hdc, Mipol1, Nmnat2, Ppox, Prodh, Qtrtd1, Rp2h | Adcy7, Asns, Atp1b1, Atp2a1, Atp5o, Atp7a, Bcat1, Ldhd, Nos3, Paics, Prps1l1, Psph, Qprt, Rora, Rrm2b, Slc11a2 |
| GO:0006029 | Proteoglycan metabolic process | 2.43E-03 | AC127554.1, Sgsh | Acan, Bgn, Csgalnact2, Glce, Spock3 |
| GO:0021675 | Nerve development | 3.01 E-03 | Erbb3, Hes1, Ngfr, Rpl24 | Hoxb2, Hoxb3 |
| GO:0021545 | Cranial nerve development | |||
| GO:0001501 | Skeletal system development | 3.92E-03 | Dlg1, Gja5, Gpnmb, Hspg2, Nab2, Pdgfrb, Smad3, Sox6 | Acan, Acd, Atp7a, Col1a1, Fmn1, Hoxb2, Hoxb3, Lgals3, Mmp13, Mmp14, Mmp9, Mn1, Osr2, Ror2, Spp1 |
| GO:0009628 | Response to abiotic stimulus | 4.12E-03 | Bcl2l1, Brcc3, Cryab, Hspb7, Ngfr, Otop1 Ric8, Scara5, Sox2, Strbp, Tsc22d3 | App, Col1a1, Dclre1c, Hmgn1, Kras, Mmp13, Mmp14, Obfc2a, Stx1a, Usp1 |
| GO:0001568 | Blood vessel development | 8.31 E-03 | Apoe, Ccbe1, Gja5, Hdac7, Itga7, Itgav, Plcd3, Prl2c2, Tnfsf12 | AC158680.2, Anpep, Atp7a, Cdh5, Col1a1, Col3a1, Mmp14, Nos3, Sema3c, Sox18, Zfpm2 |
| GO:0008088 | Axon cargo transport | 9.86E-03 | Nefm, Klc1, Klc2 | App |
In vitro, there were significant changes exclusively in a gene set associated with cell-cycle and DNA damage (Table 2, top). NXD30001 increased the expression of genes known to negatively regulate cell proliferation (Cgref1, Chfr, Dusp1), whereas it depleted the expression of genes essential for cell-cycle progression, DNA replication, and DNA repair. These include phase-specific cyclins (Ccna2, Ccnb1&2, Ccnd1), cell division cycle proteins (Cdc14a, Cdc25c), initiator of DNA replication (Prim1&2), DNA polymerases (Pola1&2, Polh), DNA methyltransferase (Dnmt1), mitotic kinases (Aurka,b, Bub1, Bub1b), and anaphase-promoting complex (APC; Anapc1,7). The major DNA repair checkpoint proteins ataxia telangiectasia mutated (Atm) and ATM and RAD3 related (Atr), as well as DNA repair proteins (Brca1&2) were also significantly downregulated. These changes potentially affect all phases of the cell cycle, thereby inhibiting cell proliferation.
In vivo, on the other hand, there were changes in genes implicated in diverse biologic pathways (Table 2, bottom): cell adhesion promoting genes Adam11&23, Cadm4, Mcam, Chl1, Cdh13, Dlg1, integrins, laminins, and collagens, some of which were shown to be absent in certain cancers, were found to be upregulated. Conversely, merlin-interacting hyaluronate (HA) receptor CD44 whose expression is higher in Schwannoma (29) was downregulated. Many genes involved in blood vessel development were also deregulated; the reduced expression of vascular endothelial-specific cadherin (Cdh5) and endothelial marker Pecam1, together with complete lack of hemoglobin expression, indicated the overall suppression of vascularization. Remarkably, genes associated with myelination and peripheral nervous system development, such as Ngfr, Erbb3, Egr2, Sox10, Pmp22, and Plp1, were concomitantly upregulated in the NXD30001-treated tumor.
Experimental validation of gene expression profiles
To validate the transcriptome analysis, we examined the selected biologic processes altered by NXD30001. We identified severe downregulation of genes essential for DNA synthesis, cell-cycle progression, and cell division in vitro. Consistently, cell-cycle analysis revealed depletion of cells in S phase and accumulation in G2–M phase after NXD30001 treatment (Fig. 4A). The increasing population of propidium iodide-unstained cells over time corresponded with the induction of apoptosis. Immunocytochemical staining confirmed that BrdUrd incorporation was significantly suppressed after NXD30001 treatment (Fig. 4B). The results concluded that NXD30001 induced cell-cycle arrest at multiple phases followed by apoptosis in ESC-FC1801 cells in vitro.
Figure 4.
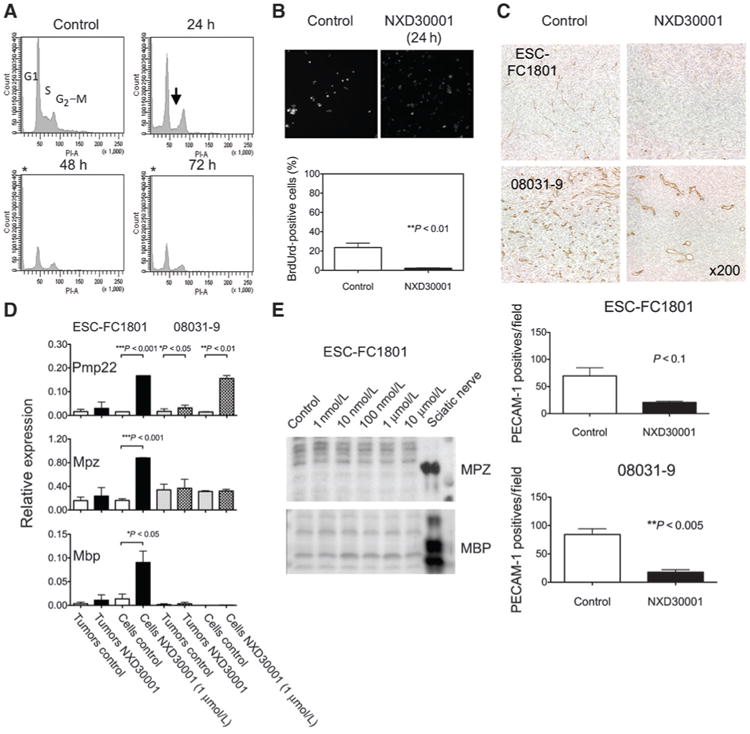
Experimental validation of transcriptome analysis. A, effects of NXD30001 on cell cycle in ESC-FC1801 cells treated with dimethyl sulfoxide or 1 μmol/L NXD30001. DNA content was measured by flow cytometry. An arrow indicates depletion of cells in S phase, and unstained cells (asterisk) correspond with apoptotic cells. B, DNA synthesis in ESC-FC1801 cells was shown by immunocytochemistry using anti-BrdUrd antibody. C, vascular regression caused by NXD30001, 100 mg/kg/d, 3 times/week for 4 weeks, in ESC-FC1801 and 08031-9 tumors. Immunohistochemical images and the mean number of PECAM-positive blood vessels in the representative sections of the tumors sampled on day 14 (ESC-FC1801) and day 28 (08031-9) were shown. D, upregulation of myelin-specific markers by NXD30001 in NF2-deficient cells. qPCR was conducted on the total RNA isolated from the vehicle or drug-treated tumors (n = 3–8) and cultured cells (n = 3) of FC-1801 and 08031-9. The relative values were expressed against normal sciatic nerve. E, Western blot analysis of myelin-specific proteins in the ESF-FC1801 cells treated with NXD30001. Columns, mean; bars, SD.
In our in vivo model, genes involved in angiogenesis were found to be deregulated in the drug-treated tumor. Accordingly, we conducted immunohistochemistry of endothelial marker PECAM-1, which revealed that the ESC-FC1801 tumors contained less blood vessels than the 08031-9 tumors (Fig. 4C). Upon HSP90 inhibition, the number of blood vessels significantly decreased in the drug-treated tumors.
Many genes that encode major components of myelin sheath, Pmp22, Mbp, Mpz, and Prx, were concomitantly upregulated in the drug-treated tumor, indicating that HSP90 inhibition in the NF2-deficient cells induced redifferentiation toward Schwann cell lineage. To test this hypothesis, we examined the expression of Pmp22, Mpz, and Mbp mRNA without or with NXD30001 in the ESC-FC1801 and 08031-9 cells (1 μmol/L for 24 hours), and their grafted tumors (100 mg/kg/d, 2 to 3 days/week for 4 weeks). Both tumors and cells showed upregulation of the myelin-specific genes, particularly Pmp22 (Fig. 4D). The effect was broader in the ESC-FC1801 cells, as no significant induction of Mpz was detected in 08031-9 cells. Remarkably, the proteins of these genes were not detectable in the drug-treated cells despite the induction of their mRNA in vitro (Fig. 4E).
Discussion
In this study, we validated the efficacy of a novel HSP90 inhibitor NXD30001 against NF2-related tumors in vitro and in vivo. Exposure to NXD30001 resulted in degradation of major client proteins activated in NF2 concomitantly with growth suppression in the NF2-deficient cells. Degradation occurred at lower doses in all NF2-deficient cells tested than in the normal Schwann cells in vitro. On the other hand, cells derived from schwannoma (08031-9, HEI-193, and human tumor primary cultures) manifested partial resistance to apoptosis. Hence, HSP90 inhibition in NF2-deficient cells involves both cytostatic and cytotoxic events, which together determined the overall antiproliferative efficacy. NXD30001 accumulated selectively in the tumors well above the GI50 in vitro, hence, we conclude that the observed antitumor activity in vivo is primarily attributable to the HSP90 inhibition.
The NF2-deficient cells used in this study displayed differential baseline gene expression, reflecting their difference of origin and adaptation to the in vitro culture. For example, absence of S100β and presence of Ngfrp75 in the ESC-FC1801 cells indicated their immature nature as compared with that of 08031-9 (30). Remarkably, the predominant expression of Erbb2 and Erbb3 in ESC-FC1801 resembled that of human schwannomas, which may have contributed to its selective sensitivity to the drug, as exemplified by the study on melanoma cells where response to HSP90 inhibition partly correlated with the expression of ERBB2 (31). Nonetheless, the long-term exposure to the NXD30001 in the clonogenic assay and in the allograft models showed comparable antiproliferative effect in all cell lines tested, implying that HSP90 inhibition by repeated dose may be cumulative, and the effect is independent of the NF2 status.
Our allograft models of 2 different NF2-deficient cells, ESC-FC1801 and 08031-9, and the NF2 transgenic mouse model, TgP0-Sch-Δ(39–121)-27, provide valuable tools essential for preclinical study for NF2. In view of immune system as body's major defense mechanism against cancer development, the use of immunocompetent FVB/N mice is beneficial for the evaluation of antitumor activity of a drug. Furthermore, the preclinical use of a genetically modified mouse model of NF2 schwannomagenesis allows us to address the drug delivery to the peripheral nerves for the efficacy studies. Because of their extra-axial nature, schwannomas are not protected by the blood–brain barrier (32). By using different HSP90 inhibitors in multiple NF2 models in vivo, we ascertained the efficacy of HSP90 inhibition in the NF2 tumorigenesis.
The drug sensitivity to NXD30001 in the patient-derived schwannoma and meningioma primary cells in vitro was variable. This implied that the clinical response of human tumors to HSP90 inhibition may also differ significantly between patients. We identified a few schwannomas/meningioma cells with significant response to the HSP90 inhibition, which carry truncating mutations and/or are of spinal origin from patients with severe clinical phenotypes (i.e., younger age of onset, existence of multiple lesions, and larger tumor size). These primary cells sustained advanced proliferation over time despite that most human schwannoma/meningioma cells undergo progressive senescence in a short period in vitro. Nonsense or frameshift NF2 mutations have been associated to more severe disease phenotypes in NF2 (33). Also, spinal tumors are associated with severe NF2 phenotypes (34). Further analysis with larger number of cases will be necessary to evaluate whether the patients with NF2 malignant phenotypes have therapeutic advantage in HSP90 inhibition.
The molecular consequences caused by HSP90 inhibition in NF2-related tumors are of great interest, also in the context of understanding NF2 tumorigenesis and therapeutic targets. Our transcriptome analysis recapitulated many biologic responses reported for HSP90 inhibition, including cell-cycle arrest, induction of apoptosis, modification of cell adhesion and cytoskeletons, inhibition of angiogenesis, and induction of cell differentiation. Furthermore, our data showed striking differences in gene expression between the short-term HSP90 inhibition in cell culture environment and the long-term administration in the microenvironment of solid tumor. Although many of the cell-cycle–associated genes and DNA damage response genes are the known HSP90 clients, the primary effect of NXD30001 in vitro was exclusively enriched in suppression of cell-cycle transit, DNA replication and repair, spindle assembly, and chromosome separation. HSP90 protein was shown to be concentrated on the centrosomes whose inhibition caused abnormal centrosome segregation and maturation, aberrant cell division spindles, and impaired chromosome segregation (35). Recently, Lyman and colleagues systematically showed 3 types of perturbation in the cell cycle, at M-phase, G2-phase, and G1-phase, by different HSP90 inhibitors in various cancer cells, signifying the multiple involvement of HSP90 in cell proliferation (36). NXD30001 also downregulated the expression of antiapoptotic survivin (37) whose expression is positively regulated by ErbB2 signaling (38). Our experimental validation showing cell-cycle arrest and apoptosis was in good agreement with these results, which further indicated the potential of HSP90 inhibition for sensitizing NF2-deficient cells to radiotherapy through suppression of DNA damage repair mechanisms and cell survival.
The effect of NXD30001 in vivo involved complex biologic changes including cell adhesion, extra-ellular matrix remodeling, vascularization, and differentiation, where much of merlin's function is also implicated. Although NXD30001 did not necessarily reverse the individual gene expression deregulated in human schwannoma (39, 40), the transcriptional alteration induced by NXD30001 and by the restoration of wild-type merlin in the Nf2−/− ESC-FC1801 cells overlapped strikingly (17), including downregulation of survivin, Aurkb, Brca2, Ccna2, Ccnd1, and Dedd2. Moreover, a number of molecular markers for tumorigenesis were altered toward expression profile preferable for therapeutic outcome. These results confirmed the beneficial effect of targeting HSP90 in NF2.
There is no evidence that merlin directly regulates the expression of HSP90. However, a recent study reported the coimmunoprecipitation of merlin with HSP70, which forms a multichaperone complex with HSP90 (21), implying that merlin may be regulated by or involved in the function of this complex. Identification of potential specificity of HSP90 inhibition in the NF2-deficient tumors will be the next step of the study. CD44 is one of the few transcripts that responded to HSP90 inhibition with significant decrease both in vitro and in vivo. Merlin functions in contact inhibition of growth by inhibiting CD44 binding of its ligand HA (16, 41). On the other hand, CD44–HA interaction was shown to be essential for the activation of ErbB2 in cancerous cells, inhibition of which caused disassembly of a constitutive, lipidraft-associated, signaling complex, containing phosphorylated-ErbB2, CD44, ezrin, phosphoinositide 3-kinase, Hsp90, and cdc37 (42). Hence, it is conceivable that merlin restoration and Hsp90 inhibition share a common molecular effect to suppress ErbB2 signaling complex in Nf2−/− cells through disruption of CD44–HA interaction and complex assembly.
The present study also showed that the HSP90 inhibition in NF2-deficient cells simultaneously increased the transcription of myelin sheath-specific genes as well as the core transcription factors for Schwann cell differentiation, which are downregulated in the human vestibular schwannomas (43). Recently, the mTOR inhibitor, rapamycin, was shown to improve myelination of Schwann cells of neuropathic mice by increasing the expression of myelin-specific genes (44). Loss of NF2 constitutively activates mTOR complex 1 (45), whereas geldanamycin suppresses the mTOR-raptor signaling pathway (46), hence, the upregulation of myelin-specific genes by NXD30001 may be accomplished through inhibition of the mTOR signaling pathway. Property of HSP90 inhibitors to induce differentiation has been reported in different tumor cells: ansamycin induced functional differentiation in ERBB2-positive breast cancer cells (47). Likewise, radicicol increased expression of erythroid differentiation marker in myelogenous leukemia cells (48). Our transcriptome data may facilitate the identification of common mechanisms shared in the HSP90 inhibition and differentiation of tumor cells of different origins.
Taken together, HSP90 inhibition showed significant antitumor efficacy in NF2-deficient cells in vitro and in vivo. Our findings provide sufficient preclinical evidence for the benefit of the HSP90 inhibition against NF2-related tumors in human patients.
Supplementary Material
Supplementary Figure 1: Induction of Apoptosis by NXD30001 in NF2-deficient cells
Supplementary Figure 2: Global transcriptome profiling of NXD30001 efficacy
Supplemental Table 1. PCR primers for NF2 mutation screening
Supplemental Table 2. qPCR validation of transcriptome
Translational Relevance.
For sporadic neurofibromatosis type 2 (NF2)–deficient vestibular schwannoma, surgical removal is both an effective and sufficient intervention in most cases. However, patients with NF2 have high risks of developing cranial, spinal, and peripheral nerve schwannoma and meningioma at a younger age, the majority of whom present with hearing loss and deafness. Moreover, a significant proportion of meningiomas is inoperable. Therefore, development of effective chemotherapy would be most beneficial for overall NF2 tumor prevention. HSP90 inhibitors have acquired significant importance in the field of cancer chemotherapy in recent years. In particular, the response of ERBB2 (HER2)-positive breast cancer has offered promise for the use of HSP90 inhibitors as a novel therapeutic alternative, a strategy that is currently in clinical trials. Given their partial dependence on ErbB signaling, NF2-related tumors are readily amenable with this strategy.
Acknowledgments
The authors thank Dr. Mark Schwartz for providing surgical human NF2 tumor samples, Drs. Rick Friedman and Jeffrey Ohmen for coordination in transcriptome analysis, and Ms. Benedicte Chareyre and Ms. Tyan Ly for technical support.
Grant Support: This work was supported by a Drug Discovery Initiative award, Children's Tumor Foundation (to M. Giovannini), and by the House Research Institute.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest: R. Chen is employed (other than primary affiliation; e.g., consulting) as vice president, preclinical research by NexGenix Pharmaceuticals and has ownership interest (including patents) in NexGenix Pharmaceuticals. No potential conflicts of interest were disclosed by the other authors.
Authors' Contributions: Conception and design: M. Giovannini, K. Tanaka, R. Chen, Z.M. Jaffer, A.E. Rubenstein
Development of methodology: K. Tanaka, F. Chareyre, J. Manent, M. Niwa-Kawakita, C.H. White, J. Vitte, Z.M. Jaffer, S.F. Nelson, A.E. Rubenstein
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): K. Tanaka, F. Chareyre, J. Manent, M. Niwa-Kawakita, A.E. Rubenstein
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): K. Tanaka, A. Eskin, F. Chareyre, W.J. Jessen, M. Niwa-Kawakita, R. Chen, C.H. White, S.F. Nelson, A.E. Rubenstein
Writing, review, and/or revision of the manuscript: M. Giovannini, K. Tanaka, A. Eskin, F. Chareyre, J. Manent, R. Chen, Z.M. Jaffer, S.F. Nelson, A.E. Rubenstein
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): K. Tanaka, F. Chareyre, C.H. White
Study supervision: M. Giovannini
References
- 1.Lanser MJ, Sussman SA, Frazer K. Epidemiology, pathogenesis, and genetics of acoustic tumors. Otolaryngol Clin North Am. 1992;25:499–520. [PubMed] [Google Scholar]
- 2.Evans DG, Moran A, King A, Saeed S, Gurusinghe N, Ramsden R. Incidence of vestibular schwannoma and neurofibromatosis 2 in the North West of England over a 10-year period: higher incidence than previously thought. Otol Neurotol. 2005;26:93–7. doi: 10.1097/00129492-200501000-00016. [DOI] [PubMed] [Google Scholar]
- 3.Evans DG, Kalamarides M, Hunter-Schaedle K, Blakeley J, Allen J, Babovic-Vuskanovic D, et al. Consensus recommendations to accelerate clinical trials for neurofibromatosis type 2. Clin Cancer Res. 2009;15:5032–9. doi: 10.1158/1078-0432.CCR-08-3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trofatter JA, MacCollin MM, Rutter JL, Murrell JR, Duyao MP, Parry DM, et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell. 1993;72:791–800. doi: 10.1016/0092-8674(93)90406-g. [DOI] [PubMed] [Google Scholar]
- 5.Rouleau GA, Merel P, Lutchman M, Sanson M, Zucman J, Marineau C, et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature. 1993;363:515–21. doi: 10.1038/363515a0. [DOI] [PubMed] [Google Scholar]
- 6.Bretscher A, Edwards K, Fehon RG. ERM proteins and merlin: integrators at the cell cortex. Nat Rev. 2002;3:586–99. doi: 10.1038/nrm882. [DOI] [PubMed] [Google Scholar]
- 7.Lutchman M, Rouleau GA. The neurofibromatosis type 2 gene product, schwannomin, suppresses growth of NIH 3T3 cells. Cancer Res. 1995;55:2270–4. [PubMed] [Google Scholar]
- 8.Schulze KM, Hanemann CO, Muller HW, Hanenberg H. Transduction of wild-type merlin into human schwannoma cells decreases schwannoma cell growth and induces apoptosis. Hum Mol Genet. 2002;11:69–76. doi: 10.1093/hmg/11.1.69. [DOI] [PubMed] [Google Scholar]
- 9.Huynh DP, Pulst SM. Neurofibromatosis 2 antisense oligodeoxynucleotides induce reversible inhibition of schwannomin synthesis and cell adhesion in STS26T and T98G cells. Oncogene. 1996;13:73–84. [PubMed] [Google Scholar]
- 10.McClatchey AI, Saotome I, Mercer K, Crowley D, Gusella JF, Bronson RT, et al. Mice heterozygous for a mutation at the Nf2 tumor suppressor locus develop a range of highly metastatic tumors. Genes Dev. 1998;12:1121–33. doi: 10.1101/gad.12.8.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giovannini M, Robanus-Maandag E, van der Valk M, Niwa-Kawakita M, Abramowski V, Goutebroze L, et al. Conditional biallelic Nf2 mutation in the mouse promotes manifestations of human neurofibromatosis type 2. Genes Dev. 2000;14:1617–30. [PMC free article] [PubMed] [Google Scholar]
- 12.Giovannini M, Robanus-Maandag E, Niwa-Kawakita M, van der Valk M, Woodruff JM, Goutebroze L, et al. Schwann cell hyperplasia and tumors in transgenic mice expressing a naturally occurring mutant NF2 protein. Genes Dev. 1999;13:978–86. doi: 10.1101/gad.13.8.978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scoles DR. The merlin interacting proteins reveal multiple targets for NF2 therapy. Biochim Biophys Acta. 2008;1785:32–54. doi: 10.1016/j.bbcan.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 14.Stamenkovic I, Yu Q. Merlin, a “magic” linker between extracellular cues and intracellular signaling pathways that regulate cell motility, proliferation, and survival. Curr Protein Pept Sci. 2010;11:471–84. doi: 10.2174/138920310791824011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shaw RJ, Paez JG, Curto M, Yaktine A, Pruitt WM, Saotome I, et al. The Nf2 tumor suppressor, merlin, functions in Rac-dependent signaling. Dev Cell. 2001;1:63–72. doi: 10.1016/s1534-5807(01)00009-0. [DOI] [PubMed] [Google Scholar]
- 16.Morrison H, Sherman LS, Legg J, Banine F, Isacke C, Haipek CA, et al. The NF2 tumor suppressor gene product, merlin, mediates contact inhibition of growth through interactions with CD44. Genes Dev. 2001;15:968–80. doi: 10.1101/gad.189601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li W, You L, Cooper J, Schiavon G, Pepe-Caprio A, Zhou L, et al. Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4(DCAF1) in the nucleus. Cell. 2010;140:477–90. doi: 10.1016/j.cell.2010.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–72. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 19.Pick E, Kluger Y, Giltnane JM, Moeder C, Camp RL, Rimm DL, et al. High HSP90 expression is associated with decreased survival in breast cancer. Cancer Res. 2007;67:2932–7. doi: 10.1158/0008-5472.CAN-06-4511. [DOI] [PubMed] [Google Scholar]
- 20.Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–10. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- 21.Angelo LS, Wu JY, Meng F, Sun M, Kopetz S, McCutcheon IE, et al. Combining curcumin (diferuloylmethane) and heat shock protein inhibition for neurofibromatosis 2 treatment: analysis of response and resistance pathways. Mol Cancer Ther. 2011;10:2094–103. doi: 10.1158/1535-7163.MCT-11-0243. [DOI] [PubMed] [Google Scholar]
- 22.Stonecypher MS, Chaudhury AR, Byer SJ, Carroll SL. Neuregulin growth factors and their ErbB receptors form a potential signaling network for schwannoma tumorigenesis. J Neuropathol Exp Neurol. 2006;65:162–75. doi: 10.1097/01.jnen.0000199575.93794.2f. [DOI] [PubMed] [Google Scholar]
- 23.Jacob A, Lee TX, Neff BA, Miller S, Welling B, Chang LS. Phosphatidylinositol 3-kinase/AKT pathway activation in human vestibular schwannoma. Otol Neurotol. 2008;29:58–68. doi: 10.1097/mao.0b013e31816021f7. [DOI] [PubMed] [Google Scholar]
- 24.Mukherjee J, Kamnasaran D, Balasubramaniam A, Radovanovic I, Zadeh G, Kiehl TR, et al. Human schwannomas express activated platelet-derived growth factor receptors and c-kit and are growth inhibited by Gleevec (Imatinib Mesylate) Cancer Res. 2009;69:5099–107. doi: 10.1158/0008-5472.CAN-08-4475. [DOI] [PubMed] [Google Scholar]
- 25.Ammoun S, Flaiz C, Ristic N, Schuldt J, Hanemann CO. Dissecting and targeting the growth factor-dependent and growth factor-independent extracellular signal-regulated kinase pathway in human schwannoma. Cancer Res. 2008;68:5236–45. doi: 10.1158/0008-5472.CAN-07-5849. [DOI] [PubMed] [Google Scholar]
- 26.Barluenga S, Wang C, Fontaine JG, Aouadi K, Beebe K, Tsutsumi S, et al. Divergent synthesis of a pochonin library targeting HSP90 and in vivo efficacy of an identified inhibitor. Angew Chem Int Ed Engl. 2008;47:4432–5. doi: 10.1002/anie.200800233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang C, Barluenga S, Koripelly GK, Fontaine JG, Chen R, Yu JC, et al. Synthesis of pochoxime prodrugs as potent HSP90 inhibitors. Bioorg Med Chem Lett. 2009;19:3836–40. doi: 10.1016/j.bmcl.2009.04.030. [DOI] [PubMed] [Google Scholar]
- 28.Zhu H, Woolfenden S, Bronson RT, Jaffer ZM, Barluenga S, Winssinger N, et al. The novel Hsp90 inhibitor NXD30001 induces tumor regression in a genetically engineered mouse model of glioblastoma multiforme. Mol Cancer Ther. 2010;9:2618–26. doi: 10.1158/1535-7163.MCT-10-0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sherman L, Jacoby LB, Lampe J, Pelton P, Aguzzi A, Herrlich P, et al. CD44 expression is aberrant in benign Schwann cell tumors possessing mutations in the neurofibromatosis type 2, but not type 1, gene. Cancer Res. 1997;57:4889–97. [PubMed] [Google Scholar]
- 30.Jessen KR, Brennan A, Morgan L, Mirsky R, Kent A, Hashimoto Y, et al. The Schwann cell precursor and its fate: a study of cell death and differentiation during gliogenesis in rat embryonic nerves. Neuron. 1994;12:509–27. doi: 10.1016/0896-6273(94)90209-7. [DOI] [PubMed] [Google Scholar]
- 31.Smith V, Sausville EA, Camalier RF, Fiebig HH, Burger AM. Comparison of 17-dimethylaminoethylamino-17-demethoxy-geldanamycin (17DMAG) and 17-allylamino-17-demethoxygeldanamycin (17AAG) in vitro: effects on Hsp90 and client proteins in melanoma models. Cancer Chemother Pharmacol. 2005;56:126–37. doi: 10.1007/s00280-004-0947-2. [DOI] [PubMed] [Google Scholar]
- 32.Wong HK, Shimizu A, Kirkpatrick ND, Garkavtsev I, Chan AW, di Tomaso E, et al. Merlin/NF2 regulates angiogenesis in schwannomas through a Rac1/semaphorin 3F-dependent mechanism. Neoplasia. 2012;14:84–94. doi: 10.1593/neo.111600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baser ME, Kuramoto L, Joe H, Friedman JM, Wallace AJ, Gillespie JE, et al. Genotype-phenotype correlations for nervous system tumors in neurofibromatosis 2: a population-based study. Am J Hum Genet. 2004;75:231–9. doi: 10.1086/422700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patronas NJ, Courcoutsakis N, Bromley CM, Katzman GL, MacCollin M, Parry DM. Intramedullary and spinal canal tumors in patients with neurofibromatosis 2: MR imaging findings and correlation with genotype. Radiology. 2001;218:434–42. doi: 10.1148/radiology.218.2.r01fe40434. [DOI] [PubMed] [Google Scholar]
- 35.Lange BM, Bachi A, Wilm M, Gonzalez C. Hsp90 is a core centrosomal component and is required at different stages of the centrosome cycle in Drosophila and vertebrates. EMBO J. 2000;19:1252–62. doi: 10.1093/emboj/19.6.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lyman SK, Crawley SC, Gong R, Adamkewicz JI, McGrath G, Chew JY, et al. High-content, high-throughput analysis of cell cycle perturbations induced by the HSP90 inhibitor XL888. PLoS ONE. 2011;6:e17692. doi: 10.1371/journal.pone.0017692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fortugno P, Beltrami E, Plescia J, Fontana J, Pradhan D, Marchisio PC, et al. Regulation of survivin function by Hsp90. Proc Natl Acad Sci U S A. 2003;100:13791–6. doi: 10.1073/pnas.2434345100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siddiqa A, Long LM, Li L, Marciniak RA, Kazhdan I. Expression of HER-2 in MCF-7 breast cancer cells modulates anti-apoptotic proteins Survivin and Bcl-2 via the extracellular signal-related kinase (ERK) and phosphoinositide-3 kinase (PI3K) signalling pathways. BMC Cancer. 2008;8:129. doi: 10.1186/1471-2407-8-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aarhus M, Bruland O, Saetran HA, Mork SJ, Lund-Johansen M, Knappskog PM. Global gene expression profiling and tissue microarray reveal novel candidate genes and down-regulation of the tumor suppressor gene CAV1 in sporadic vestibular schwannomas. Neurosurgery. 2010;67:998–1019. doi: 10.1227/NEU.0b013e3181ec7b71. [DOI] [PubMed] [Google Scholar]
- 40.Caye-Thomasen P, Borup R, Stangerup SE, Thomsen J, Nielsen FC. Deregulated genes in sporadic vestibular schwannomas. Otol Neurotol. 2010;31:256–66. doi: 10.1097/MAO.0b013e3181be6478. [DOI] [PubMed] [Google Scholar]
- 41.Bai Y, Liu YJ, Wang H, Xu Y, Stamenkovic I, Yu Q. Inhibition of the hyaluronan-CD44 interaction by merlin contributes to the tumor-suppressor activity of merlin. Oncogene. 2007;26:836–50. doi: 10.1038/sj.onc.1209849. [DOI] [PubMed] [Google Scholar]
- 42.Ghatak S, Misra S, Toole BP. Hyaluronan constitutively regulates ErbB2 phosphorylation and signaling complex formation in carcinoma cells. J Biol Chem. 2005;280:8875–83. doi: 10.1074/jbc.M410882200. [DOI] [PubMed] [Google Scholar]
- 43.Hung G, Colton J, Fisher L, Oppenheimer M, Faudoa R, Slattery W, et al. Immunohistochemistry study of human vestibular nerve schwannoma differentiation. Glia. 2002;38:363–70. doi: 10.1002/glia.10077. [DOI] [PubMed] [Google Scholar]
- 44.Rangaraju S, Verrier JD, Madorsky I, Nicks J, Dunn WA, Jr, Notterpek L. Rapamycin activates autophagy and improves myelination in explant cultures from neuropathic mice. J Neurosci. 2010;30:11388–97. doi: 10.1523/JNEUROSCI.1356-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lopez-Lago MA, Okada T, Murillo MM, Socci N, Giancotti FG. Loss of the tumor suppressor gene NF2, encoding merlin, constitutively activates integrin-dependent mTORC1 signaling. Mol Cell Biol. 2009;29:4235–49. doi: 10.1128/MCB.01578-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohji G, Hidayat S, Nakashima A, Tokunaga C, Oshiro N, Yoshino K, et al. Suppression of the mTOR-raptor signaling pathway by the inhibitor of heat shock protein 90 geldanamycin. J Biochem. 2006;139:129–35. doi: 10.1093/jb/mvj008. [DOI] [PubMed] [Google Scholar]
- 47.Münster PN, Srethapakdi M, Moasser MM, Rosen N. Inhibition of heat shock protein 90 function by ansamycins causes the morphological and functional differentiation of breast cancer cells. Cancer Res. 2001;61:2945–52. [PubMed] [Google Scholar]
- 48.Shiotsu Y, Neckers LM, Wortman I, An WG, Schulte TW, Soga S, et al. Novel oxime derivatives of radicicol induce erythroid differentiation associated with preferential G(1) phase accumulation against chronic myelogenous leukemia cells through destabilization of Bcr-Abl with Hsp90 complex. Blood. 2000;96:2284–91. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Induction of Apoptosis by NXD30001 in NF2-deficient cells
Supplementary Figure 2: Global transcriptome profiling of NXD30001 efficacy
Supplemental Table 1. PCR primers for NF2 mutation screening
Supplemental Table 2. qPCR validation of transcriptome