

Abstract
The replicative cycle of the Human Immunodeficiency Virus type-1 begins after fusion of the viral and target-cell membranes. The envelope glycoprotein gp41 transmembrane subunit contains conserved hydrophobic domains that engage and perturb the merging lipid bilayers. In this work, we have characterized the fusion-committed state generated in vesicles by CpreTM, a synthetic peptide derived from the sequence connecting the membrane-proximal external region (MPER) and the transmembrane domain (TMD) of gp41. Pre-loading cholesterol-rich vesicles with CpreTM rendered them competent for subsequent lipid-mixing with fluorescently-labeled target vesicles. Highlighting the physiological relevance of the lasting fusion-competent state, the broadly neutralizing antibody 4E10 bound to the CpreTM-primed vesicles and inhibited lipid-mixing. Heterotypic fusion assays disclosed dependence on the lipid composition of the vesicles that acted either as virus or cell membrane surrogates. Lipid-mixing exhibited above all a critical dependence on the cholesterol content in those experiments. We infer that the fusion-competent state described herein resembles bona-fide perturbations generated by the pre-hairpin MPER-TMD connection within the viral membrane.
Keywords: HIV gp41, HIV fusion, HIV MPER, 4E10 antibody, membrane fusion assay, Cholesterol
1-Introduction
Fusion of the surrounding envelope with the target cell membrane precedes the entry of the human immunodeficiency virus type-1 (HIV-1) into the host cell. The fusion reaction is mediated by the envelope glycoprotein (Env) transmembrane gp41 subunit (reviewed in [1–3]). Two distinct types of structural elements within the gp41 ectodomain are involved in the promotion of the process: the six-helix bundle (6-HB) or trimeric “hairpin”, a helical domain that closes by zippering, and two membrane-transferring hydrophobic regions, the N-terminal fusion peptide (FP) and the C-terminal membrane-proximal external region (MPER), the latter located preceding the transmembrane domain (TMD) (Fig. 1A).
Figure 1. Fusion-committed state generated in vesicles by CpreTM.

A) Designation of the HIV-1 gp41 MPER-TMD connecting sequence. The schematic diagram designates gp41 regions: FP, fusion peptide; HR1 and HR2, amino- and carboxy-terminal helical regions, respectively; MPER, membrane-proximal external region; TMD transmembrane domain. The CpreTM peptide sequence covers Env residues 671–693 (below). B) Left: kinetics of CpreTM incorporation into vesicles monitored by measuring energy transfer from tryptophans to membrane-residing d-DHPE. The peptide was added to a vesicle suspension (100 μM lipid) at the time indicated by the arrow (t = 50 s). The peptide-to-lipid ratio was 1:150 in this assay. Right: Lipid-mixing assay based on the use of fusion-committed vesicles. Unlabeled POPC:Chol (1:1) vesicles (90 μM lipid) were primed at t = 60 s with CpreTM under conditions allowing fast incorporation of the peptide (indicated by the 1st intensity spark). Further co-incubation with the fluorescently-labeled target vesicles devoid of peptide (10 μM lipid) resulted in the mixing of the constituent lipids of both types of vesicles, monitored as the dilution of the probes into the whole vesicle population (arrow at t = 180 s). The final CpreTM-to-lipid ratio was 1:300. Control blank vesicles without CpreTM underwent no lipid-mixing upon target vesicle addition (−CpreTM trace). The dotted trace follows lipid-mixing of CpreTM-primed vesicles that were treated with MAb4E10 (10 μg mL−1) prior to target vesicle addition (2nd intensity spark at t = 120 s). Lipid-mixing was strongly attenuated in this case.
The prevailing mechanistic model posits that upon fusion activation FP and MPER-TMD regions anchor the target cell and viral membranes, respectively. Subsequent formation of the 6-HB involves the packing of C-terminal (CHR) and N-terminal (NHR) helical regions in reverse direction [4, 5]. As a consequence, the anti-parallel helices place the FP and MPER-TMD region at the same end of the 6-HB. Thus, it is assumed that the conformational energy released upon 6-HB formation can be coupled to pull membranes together. Although not so detailed structural information exists, some of the “pre-hairpin” intermediate stages can be identified by functional experiments combining inhibitory peptides and lipids with temperature jumps [2]. These transient and reversible protein structures assist formation and stabilization of lipid intermediates evolving along the fusion pathway, i.e., apposed curved bilayers, hemifused bilayers and flickering fusion pores [2, 3]. In contrast, 6-HB completion would be coupled to the formation of an irreversible fusion pore, the proteolipid structure mediating the release of the luminal content of the virus into the cytoplasmic matrix [6].
With regards to the entry process, it has been argued that HIV-1 productive infection predominantly occurs upon endocytosis of the viral particles and fusion with endosomes [7, 8]. According to this mechanism, fusion activation and initial exchange of lipids occur at the plasma membrane mediated by ‘fusogenic pre-bundles’, while eventual release of the capsid through the fusion pore depends on endosome-resident factors [8, 9]. Thus, initiation of fusion at the plasma membrane would not require the folding into the final 6-HB structure, implying that prior to their mechanical coupling, the bilayers must undergo focal perturbations to exchange their lipids [10].
Accumulating evidence indicates that insertion of MPER would be required to generate hydrophobic discontinuities within the rigid viral membrane [11–16]. MPER is highly enriched in conserved aromatic residues that promote partitioning from water into the membrane interface (reviewed in [17]). Mutational analyses provided early evidence supporting the involvement of this domain in HIV-1 fusion [11, 18], most likely by disrupting envelope lipid packing [19] and/or by establishing favorable interactions with the Chol-rich viral membrane through the “cholesterol recognition/interaction amino acid consensus” (CRAC) LWYIK sequence located at its C-terminus [20–22] (Fig. 1A). MPER function in viral entry is further supported by recent findings indicating that anti-MPER neutralizing antibodies, such as 4E10, have evolved mechanisms to recognize membrane-inserted epitopes and block their membrane activity [23–27].
To get further insight into the fusion-related properties of the MPER domain, we have characterized in recent work the interaction of the CpreTM peptide with Chol-containing membranes [16, 28, 29]. The CpreTM sequence includes N-terminal TMD residues that may contribute to MPER interactions with the viral membrane components, particularly with Chol [16, 30] (Fig. 1A). In addition, it was C-teminally truncated at a hydrophobicity minimum, which was shown later to encompass a putative metastable kink at the TMD [28, 31]. We have recently reported all-atom Molecular Dynamics Simulations (MDS) revealing the activity of CpreTM in Chol-rich membranes [32]. Those studies confirmed partitioning of the sequence into the membrane interface and establishment of phospholipid-aromatic interactions therein. These interactions resulted in head-group condensation and concomitant disorder of acyl chains, which in turn led to phospholipid protrusion and acyl-chain splay. Moreover, lipid vesicles treated with CpreTM acquired the capacity to fuse with a supplement of untreated vesicles.
Here we sought to characterize the fusion commitment state generated by CpreTM in Chol-rich membranes. The broadly neutralizing 4E10 antibody reverted the fusion-competent state by targeting CpreTM dimers. Moreover, in heterotypic vesicle fusion assays we found that transmission of the CpreTM-generated perturbation from the fusion-committed into the target membranes depended primarily on the Chol content. Notwithstanding the Chol effect, the presence sphyngomyelin (SM) significantly enhanced the susceptibility of target membranes to undergo fusion with vesicles primed by low CpreTM doses. Collectively, our findings support an adaptation of MPER-TMD connecting sequence for perturbing the highly rigid, Chol-rich HIV envelope during the first stages of the fusion process.
2-Materials and Methods
2.1 Materials
The peptide sequences derived from the gp41 MPER-TMD region, KKK-NWFDITNWLWYIKLFIMIVGGLV-KK (CpreTM), and KKK-NAADITNWLWYIKLFIMIVGGLV-KK (Cala), were produced by solid-phase synthesis using Fmoc chemistry as C-terminal carboxamides and purified by HPLC. To increase water-solubility both peptides incorporated 5 additional Lys residues (in italics) [33]. 1-palmitoyl-2-oleoyl-sn-glycero-3-phophocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), egg sphingomyelin (SM) and Cholesterol (Chol) were purchased from Avanti Polar Lipids (Birmingham, AL, USA). The N-(5-dimethylaminonaphtalene-1-sulfonyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (d-DHPE), N-(7-nitro-benz-2-oxa-1,3-diazol-4-yl)phosphatidylethanolamine (N-NBD-PE) and N-(lissamine Rhodamine B sulfonyl)phosphatidylethanolamine (N-Rh-PE) fluorescent probes were from Molecular Probes (Eugene, OR, USA). Rabbit anti-human IgG-HRP was obtained from Santa Cruz Biotechnology (Dallas, Texas, USA). Monoclonal 4E10 antibody (MAb4E10) was kindly donated by D. Katinger (Polynum Inc., Vienna, Austria).
2.2 Lipid vesicle preparation
Large unilamellar vesicles (LUV) were prepared according to the extrusion method in 5 mM Hepes, 100 mM NaCl (pH 7.4) using membranes with a nominal pore-size of 0.1 μm. Distributions of vesicle sizes were determined by quasielastic light scattering using a Malvern Zeta-Sizer Nano ZS instrument (Malvern Instruments, Malvern, UK). The mean diameter of POPC:Chol 1:1 (mol:mol) vesicles was 118 nm. After extrusion, phospholipid and Chol concentration of liposome suspensions were respectively determined by phosphate analysis and the cholesterol oxidase/peroxidase method (BioSystems, Barcelona, Spain). In the case of POPC:Chol 1:1 (mol:mol) vesicles, Chol mol % was found to be 48.6±2.6 (mean±S.D., n=12).
2.3 Membrane binding assays
Corrected Trp spectra were recorded using a FluoroMax-3 (Jobin Ybon, Horiba) with excitation set at 280 nm and 2-nm slits. Degree of peptide association with the vesicles was estimated from the shifts in the maximum emission wavelength and the fractional changes in emitted fluorescence. Kinetics of partitioning was measured by energy transfer from the Trp peptide to the surface d-DHPE fluorescent probe as in [15]. In brief, 6 mol % of the d-DHPE probe was included in the target vesicle composition and its fluorescence was measured at an emission wavelength of 510 nm, while the excitation wavelength was that of the Trp residue (280 nm).
Vesicle flotation in sucrose gradients was performed following the method described by Yethon et al. [34]. 100 μl of a sample containing N-Rh-PE-labeled liposomes (1.5 mM lipid concentration) was adjusted to a sucrose concentration of 1.4 M in a final volume of 300 μl, and subsequently overlaid with 400 and 300 μl-layers of 0.8 and 0.5 M sucrose, respectively. The gradient was centrifuged at 436,000 x g for 3 h in a TLA 120.2 rotor (Beckman Coulter, Brea CA, USA). After centrifugation, four 250 μl-fractions were collected. Material adhered to the tubes was collected into a 5th fraction by washing with 250 μl of hot (100o C) 1% (w/v) SDS. The presence of CpreTM and/or Mab4E10 in the different fractions was revealed by Western Blot analysis after SDS-PAGE separation.
2.4 Lipid-mixing with fusion-committed vesicles
Membrane lipid mixing was monitored using the resonance energy transfer (RET) assay, described by Struck et al. [35]. The assay is based on the dilution of N-NBD-PE and N-Rh-PE. Dilution due to membrane mixing results in an increased N-NBD-PE fluorescence. Vesicles containing 0.6 mol% of each probe (target vesicles) were added at 1:10 ratio to unlabeled vesicles (routinely CpreTM-primed vesicles). The final lipid concentration in the mixture was 100 μM. The ensuing increase in NBD emission upon mixing of target-labeled and primed-unlabeled lipid bilayers was monitored at 530 nm with the excitation wavelength set at 465 nm. A cutoff filter at 515 nm was used between the sample and the emission monochromator to avoid scattering interferences. The fluorescence scale was calibrated such that the zero level corresponded to the initial residual fluorescence of the labeled vesicles and the 100 % value to complete mixing of all the lipids in the system (i.e., the fluorescence intensity of vesicles containing 0.06 mol% of each probe).
3-Results
3.1 Fusion-competent state generated in vesicles by CpreTM
Our recent work supports the involvement of the MPER-TMD region spanning Env residues 671–693 (Fig 1A) in the focal disruption of the Chol-enriched viral membrane [16, 32]. Particularly, MDS revealed that CpreTM, a peptide based on the MPER-TMD linker, inserted subtly into the membrane-interface and induced therein phospolipid and acyl-chain extraction, a lasting perturbation that could prime Chol-rich membranes for fusion [32]. The goal of the present study is to get further insights into the fusion-committed state generated by CpreTM, and on the lipid requirements that govern its propagation into target membranes.
Results shown in Fig. 1B evidence lipid exchange between CpreTM-primed vesicles and fluorescently-labeled target vesicles devoid of peptide. The kinetics of energy transfer from Trp residues to the dansyl moieties at the membrane interface demonstrated that CpreTM partitioned from water into membranes in less than 10 seconds (left panel). The right panel discloses the time course of the assay using the fusion-committed vesicles. At time 60 sec (indicated by the fluorescence intensity spark labeled as “+CpreTM”), peptide (0.33 μM) was added to the unlabeled-vesicle solution (90 μM lipid), and, after 120 sec (indicated by the arrow), the mixture was supplemented with N-NBD-PE/N-Rh-PE-labeled vesicles (10 μM lipid). The resulting increase in NBD fluorescence over time was consistent with the dilution of the probes upon mixing of lipids of target and primed vesicles (+CpreTM trace). Such increase was strongly attenuated upon addition of the MAb4E10 antibody prior to the target vesicles (+4E10, dotted trace), and not observed at all upon incubation of vesicles following the same protocol but in the absence of peptide (−CpreTM trace). Together, these results support that perturbations specifically induced by CpreTM on one vesicle population could be transmitted into target vesicles devoid of peptide, a phenomenon that resulted in the mixing of their constituent lipids.
However, the fusion phenomenon described in the previous figure could be elicited by a fraction of unbound peptide, and/or evolve as a consequence of peptide-induced vesicle solubilization. Thus, experiments were conducted to ensure that CpreTM bound quantitatively and irreversibly under the conditions selected to prime membranes (Fig. 2), and that its activity did not cause vesicle disintegration following a detergent-like mechanism (Fig. 3). Changes in CpreTM Trp emission intensity indicated that partitioning into POPC:Chol (1:1) vesicles reached saturation with ca. 50 μM lipid (Fig. 2A). Therefore, peptide binding was considered quantitative at the lipid concentration used to generate fusion-committed vesicles (90 μM lipid). The shift of the maximum emission wavelength was subtle at those concentrations (ca. 5 nm), consistent with a peptide interaction restricted to the most polar, peripheral region of the bilayer. Even though, the peptide interaction with vesicles was tight as put forward by flotation experiments, which disclosed the added peptide co-floating with vesicles upon physical separation of bound and unbound species by ultracentrifugation (Fig. 2B-top). In these experiments the peptide incubated in the absence of vesicles was recovered from the pellets, mostly in the SDS fraction (Fig. 2B-bottom). Thus, the fact that upon incubation with vesicles unbound peptide was not detected in solution, supports the irreversible CpreTM insertion into lipid bilayers. Finally, results displayed in Fig. 3 confirmed that perturbations induced by CpreTM did not cause membrane micellization, but rather a discrete increase of the vesicle mean diameter from ca. 120 to ca. 550 nm.
Figure 2. CpreTM peptide binding to vesicles.

A) Partitioning of CpreTM peptide into POPC:Chol (1:1) vesicles. Left: intrinsic fluorescence emission spectra (excitation wavelength: 280 nm) of the peptide incubated with increasing concentrations of vesicular lipid. Peptide concentration was 0.5 μM. Lipid concentrations were (from bottom to top): 0, 5, 10, 20, 40, 60 and 100 μM. Right: Changes in maximum emission wavelength (top) and fractional intensity (bottom) upon incubation with vesicles. B) Top: vesicle flotation experiments in sucrose gradients. CpreTM (15 μM) was added to POPC:Chol (1:1) vesicles at a peptide-to-lipid ratio of 1:100. The presence of the peptide in the different fractions was probed with MAb4E10 in Western Blot. Virtually all input peptide co-floated with liposomes indicating quantitative partitioning into membranes. Rhodamine emission (below) denoted the presence of vesicles in the fractions. Bottom: the peptide incubation was made in the absence of vesicles. In this case CpreTM was mainly recovered from the SDS fraction after centrifugation.
Figure 3. Size distribution of the vesicles determined by light scattering.
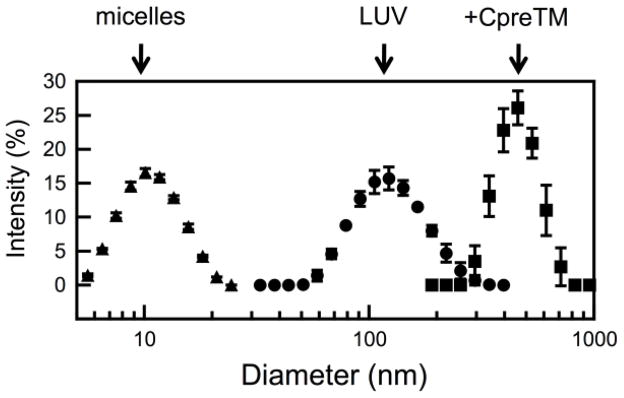
POPC:Chol (1:1) vesicles were measured before (LUV, circles) and after 15 min incubation with CpreTM (peptide-to-lipid ratio of 1:100) (+CpreTM, squares). Vesicles treated with Triton X-100 (10 % v/v) are shown as a reference for the size of micelles (micelles, triangles).
3.2 Association of MAb4E10 to fusion-committed vesicles
Results shown in the previous figures suggest that CpreTM was capable of inducing a lasting fusion commitment state in POPC:Chol (1:1) vesicles, which otherwise maintained their integrity. Data displayed in Figs. 4 and 5 further demonstrate that the HIV neutralizing MAb4E10 could associate to these vesicles and reverse the fusion-competent state. In liposome-based flotation experiments, the whole input antibody co-floated with CpreTM-primed vesicles (Fig. 4A). In contrast, the antibody did not co-float with the vesicles loaded with the Cala peptide, which bears the 4E10 key epitope residues Trp-Phe, critical for binding, mutated to Ala-Ala (Fig. 4B). This peptide otherwise partitions into POPC:Chol vesicles almost as efficiently as CpreTM (partitioning constants, Kx, of 3×106 and 5×106, respectively), and induces lipid-mixing [32]. In the Cala sample the presence of a faint protein band in the floating fractions evidenced the existence of some residual antibody-membrane binding. Similarly, the bulk of membrane binding was absent in the vesicles devoid of peptide (Fig. 4C). Floating antibody was not detected either when incubated in solution in the absence of vesicles (Fig. 4D). These observations underpin MAb4E10 binding to CpreTM-primed vesicles, and the dependence of the process on specific epitope recognition.
Figure 4. 4E10 antibody association to fusion-committed vesicles in flotation experiments.

A) MAb4E10 (45 μg mL−1) was incubated for 15′ with CpreTM-containing POPC:Chol (1:1) vesicles before centrifugation. Consistent with antibody binding to CpreTM-primed vesicles, MAb4E10 was recovered from the floating fractions. B) MAb4E10 was mainly recovered from pellets when vesicles were loaded with Cala peptide. C) MAb4E10 incubated with POPC:Chol (1:1) vesicles in the absence of CpreTM was also detected mainly in the pellets. D) Antibody incubated in the absence of vesicles was not present in floating fractions. The presence of the antibody in the different fractions was probed with anti-human IgG by Western Blot. Rhodamine emission denoted the presence of the vesicles in the fractions (spectra displayed in the panels corresponding to vesicle-containing samples).
Figure 5. Lipid-mixing inhibition by 4E10 antibody.
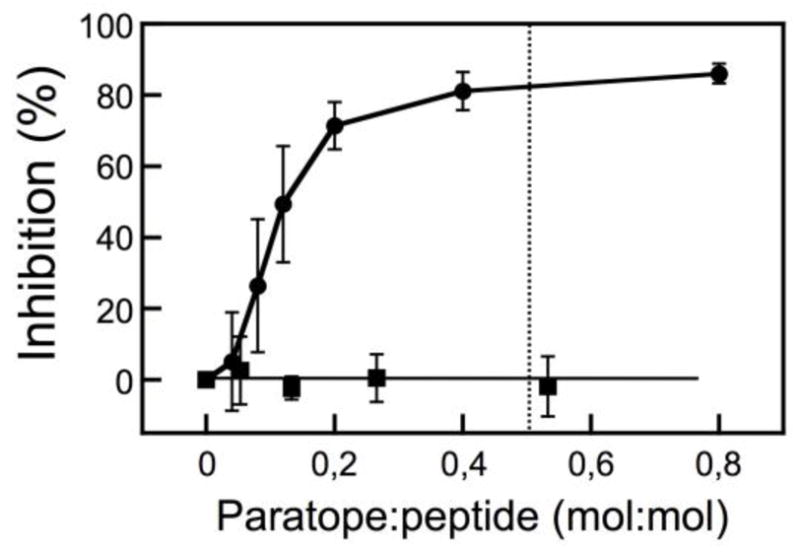
CpreTM-primed vesicles were incubated with increasing amounts of MAb4E10 prior to the addition of target vesicles (circles). Cala-primed vesicles were used as negative control to probe the specificity of the process (squares). The dotted line begins at the paratope-to-peptide ratio of 0.5:1. Plotted values are means±SD of three experiments.
As shown by the dotted trace in the previous Fig. 1B-right, antibody-treated vesicles underwent ameliorated lipid-mixing. Antibody titrations shown in Fig. 5 further indicate that 4E10 inhibited the lipid-mixing process in a dose and epitope recognition-dependent way. Moreover the inhibition curve saturated at approximately the 0.5 value (paratope:peptide mole ratio) consistent with an apparent 1:2 stoichiometry for the paratope-peptide association reaction.
3.3 Heterotypic vesicle fusion dependence on the Chol content of membranes
The lipid-mixing assay based on the use of fusion-committed vesicles allowed performing heterotypic vesicle fusion assays. Given the relevance of Chol for the HIV fusion process [36, 37] (see also discussion in [14]), we first assessed the effects of this compound as a component of either primed or target vesicles (Fig. 6). The kinetic traces displayed in Fig. 6A illustrate the effect of reducing Chol from 50 mole % (POPC:Chol (1:1)) to 33 mole % (POPC:Chol (2:1)) in either CpreTM-primed or target vesicles. Priming POPC:Chol (1:1) vesicles with CpreTM at peptide-to-lipid ratios ranging between 1:1000 and 1:100 resulted in efficient and dose-dependent lipid-mixing with target vesicles of the same composition (left panel). Reduction of the Chol content in the CpreTM-primed vesicles severely interfered with the process (center panel). In contrast, the same Chol reduction in the target vesicle composition had little effect on the measured lipid-mixing (right panel).
Figure 6. Lipid-mixing dependence on the Chol content of vesicles.

A) Kinetics of lipid-mixing were measured for different combinations of POPC:Chol mole ratios in Primed or Target vesicles, as indicted on top of the panels. Unlabeled vesicles were primed by adding CpreTM at the peptide-to-lipid ratios indicated in the traces. The t = 0 s corresponded to the addition time of labeled target-vesicles. The final lipid concentration was 100 μM. B) Top panels: Final extents of lipid mixing for different compositions in Primed or Target vesicles. Symbols: (●), POPC:Chol (1:1); (■), POPC:Chol (2:1); (▲), POPC:Chol (4:1); (▼), POPC. The percentage of lipid-mixing measured after a 10 min incubation was plotted as a function of the CpreTM concentration used to prime unlabeled vesicles. Bottom panels: Levels of lipid-mixing measured after 10 min were plotted vs. the Chol mole fraction. Unlabeled vesicles were primed by adding CpreTM at a final peptide-to-lipid mole ratio of 1:100 in this case. Means±SD values of three experiments are shown.
Plots of the extents of lipid-mixing after 10 min vs. CpreTM concentration disclosed more clearly the differential effects of Chol on either CpreTM-primed or target vesicles (Fig. 6B, left and right panels, respectively). Effective promotion of lipid-mixing was above all observed for the primed 1:1 vesicles and for all target vesicles that contained Chol (top panels). The titration curves exhibited a sigmoidal shape consistent with the involvement of cooperative effects in peptide-induced fusion (Fig. 5 and reference [32]). Plotting the extents of fusion as a function of the Chol mole fraction further revealed a sharp enhancement of primed vesicle fusion at the highest Chol concentration (bottom-left). In contrast, the target vesicles showed a more gradual increase with values close to its maximum already attained with 33 mole % of Chol (bottom-right). In conclusion, in the heterotypic vesicle fusion assay, CpreTM-induced lipid mixing showed more stringent dependency on the Chol content of primed membranes as compared to target ones.
3.4 Effect of target membrane lipid composition
The heterotypic vesicle fusion approach was finally applied to study the effect of other relevant lipids as components of the target membranes (Figs. 7 and 8). Phospholipids with larger cross-sectional area available for the acyl chain tails and a small area available for the headgroups, such as phosphatidylethanolamine, impart mean negative curvature on membrane monolayers and sustain membrane fusion phenomena by promoting the formation of non-lamellar lipid structures [38]. On the other hand, the outer layer of the plasma membrane is enriched in sphingolipids and sterols, which are tightly packed and resist mechanical stress [39, 40]. Thus, phosphatidylethanolamine in one hand, and sphingomyelin/Chol on the other, are predicted to contribute in disparate ways to lipid-mixing: the former by eliciting the process, the latter by interfering with it. To compare lipids of each kind, half of the POPC in the POPC:Chol (2:1) mixture was substituted by either POPE or SM to render POPC:POPE:Chol (1:1:1) or POPC:SM:Chol (1:1:1) target vesicles, respectively. As shown in Fig 7A-left, in experiments using POPC:Chol (1:1) vesicles primed with CpreTM at a 1:200 peptide-to-lipid ratio, POPC:POPE:Chol (1:1:1) and POPC:SM:Chol (1:1:1) vesicles underwent fusion following kinetics comparable to those measured for the POPC:Chol (2:1) reference vesicles. Interestingly however, upon lowering the CpreTM dose to 1:1000 peptide-to-lipid ratio, substantial lipid-mixing was only and consistently observed in the case of POPC:SM:Chol (1:1:1) target vesicles (Fig. 7A-right). Calculations of final extents and initial rates put forward that, besides the significant SM effect observed with low CpreTM concentrations, vesicles containing POPE or SM, while slightly more susceptible to fusion, did not differ substantially in fusogenicity from the reference vesicle population (Fig. 7B). These observations suggest that propagation of the CpreTM-induced perturbation into the target membrane follows a mechanism that is not dependent on its propensity to form non-lamellar structures. Moreover, vesicles reflecting the stability of the plasma membrane outer leaflet were also competent in exchanging lipids with the fusion-committed vesicles.
Figure 7. Effect of the target vesicle composition on fusion.

A) Kinetics of lipid mixing. Unlabeled POPC:Chol (1:1) vesicles were primed by adding CpreTM at the peptide-to-lipid ratio of 1:200 (left) or 1:1000 (right). Labeled target-vesicles composed of POPC:Chol (2:1), POPC:POPE:Chol (1:1:1) or POPC:SPM:Chol (1:1:1) were subsequently added at t = 0 s and ensuing lipid-mixing scored over time (continuous, dotted and dashed traces, respectively). The final lipid concentration was 100 μM. B) Final extents and initial rates. Percentages of lipid-mixing measured after 10 min (left) or 10 s (right) have been plotted as a function of the CpreTM concentration used to prime unlabeled vesicles. Symbols: (●), POPC:Chol (2:1); (■), POPC:POPE:Chol (1:1:1); (○), POPC:SM:Chol (1:1:1). Means±SD values of three experiments are shown.
Figure 8. Effects of Chol and SM on target membrane susceptibility to undergo fusion at low CpreTM doses.

A) Kinetics of lipid mixing. Unlabeled POPC:Chol (1:1) vesicles were primed by adding CpreTM at the peptide-to-lipid ratio of 1:500. Conditions otherwise as in the previous figure. Dashed traces correspond to controls without CpreTM. B) Significant enhancement of lipid-mixing levels upon inclusion of SM. Lipid-mixing extents were measured after 10 min incubation of target and primed-blank vesicles. The latter were pre-incubated with CpreTM at 1:500 or 1:1000 peptide-to-lipid ratios. The t-probability values were *p< 0.05 for unpaired set of data.
The results displayed in Fig. 8 illustrate the effect of approximating the plasma membrane external leaflet composition on the susceptibility of target membranes to undergo fusion with blank vesicles, which were primed with low CpreTM doses (i.e., 1:500 peptide-to-lipid mole ratio). Inclusion of Chol and then SM increased the rate and extent of the lipid mixing (Fig. 8A). Of note, even if included at low doses, the lipid exchange was in all cases dependent on the presence of CpreTM. Moreover, data shown in Fig. 8B confirm that the extent of the fusion process measured under those conditions was significantly higher upon inclusion of SM. These results suggest that target vesicles including lipids that are predominantly located at the external side of the plasma membrane, not only were competent to undergo fusion, but also seemed to reflect an optimal condition for the process to evolve.
4-Discussion
Analyses of the HIV envelope lipidome are consistent with viral assembly taking place within Chol-rich domains of the host-cell plasma membrane [41, 42]. Thus, to initiate a new replicative cycle the rigid envelope must be forced to merge with the less ordered target-cell membrane (see [14] for a discussion). Our recent findings indicate that the fusogenic activity of CpreTM, a peptide representing the MPER-TMD connection, is elicited at the Chol concentrations existing at the viral envelope [32]. MDS studies further revealed the capacity of this sequence to perturb the membrane interface, by promoting exposure of acyl-chains to solvent, or even by extracting complete phospholipid molecules under those conditions [32]. The observation of CpreTM-induced fusion would be in accordance with the finding that peptides containing the LWYIK CRAC motif lowered the bending modulus (kc) in Chol-containing membranes [43]. A similar reduction of the kc parameter was described for the inclusion in model membranes of the N-terminal FP [44, 45]. Thus, reduction of kc supports that both sequences, FP and CpreTM, might in a concerted manner assist bending of the membranes into highly curved fusion intermediates during viral fusion.
Here, we describe two characteristics of CpreTM-induced perturbation that underline its biological relevance. On the one hand, a neutralizing antibody could associate to the fusion-committed vesicles and revert their state (Figs. 4 and 5). On the other, prominently at low CpreTM doses, requirements of the target vesicle composition reflected conditions of the plasma membrane leaflet that first exchanges lipids with the viral envelope (Figs. 6–8). We surmise that the lipid-mixing of fusion-committed vesicles described in this work (Fig. 1) might comprise a bona-fide reflection of MPER-TMD region activity within pre-hairpin structures [2, 3].
Even though perturbations triggered by CpreTM may result in phospholipid extraction [32], the experimental results displayed in Figs. 2 and 3 demonstrate that the peptide remains associated to vesicles and that its interaction does not cause their micellization. Indeed, incubation with CpreTM led to the formation of bigger vesicles with sizes ranging between 300 and 700 nm. Thus, our data are consistent with the generation of a fusion-competent state that lasts at the surface of the larger vesicles, and with the subsequent transmission of the perturbations into the supplement of target vesicles devoid of peptide. Importantly, the CpreTM peptide primed the vesicles for fusion at membrane doses ranging between 1:1000 and 1:100 peptide-to-lipid mole ratios (Fig. 6A). These doses are ca. 1 order of magnitude lower than those commonly used to assay synthetic peptides as fusogens [46], which further underlines the specificity of the measured process.
Evidencing the biological relevance of this phenomenon, the 4E10 antibody bound to fusion-committed vesicles and blocked the lasting fusion state (Figs. 4 and 5). This antibody targets the subset of pre-hairpin intermediates to block fusion [47]. Interestingly, the dose-inhibition curve suggests that MAb4E10 blocks fusion by targeting CpreTM dimers (Fig. 5). Consistent with the possibility that CpreTM dimers represent the membrane-active form of the peptide, SDS-resistant dimers were the predominant species observed after gel-electrophoresis [28], and MDS disclosed dimers as the active forms inducing acyl-chain splay and phospholipid protrusion [32]. Recently solved crystals of trimeric, pre-fusion Env display the C-termini of the CHR domains hanging free from each protomer, consistent with the absence of inter-protomer interactions at the level of the MPER D664 residue [48]. Thus, although MPER constitutes a part of the trimeric gp41 in the native glycoprotein, its sequence is predicted to insert into the viral membrane in monomeric form. We speculate that MPER-TMD dimerization might occur upon fusion activation, possibly when 4E10 binding is boosted [47, 49].
In addition, the flotation assays displayed a small fraction of antibody bound to membranes devoid of peptide (Fig. 5A). A similar finding has been recently reported and discussed as indicative of MAb4E10 capacity for directly interacting with the viral membrane [49]. Since the 4E10 epitope would be only fully accessible upon fusion activation, it has been argued that pre-concentrating the antibody at the viral membrane surface is a required step for the neutralization mechanism to evolve [49]. Alternatively, cross-reactivity with lipids has led to the proposal that the 4E10 epitope may comprise a proteo-lipid complex [15, 25, 27]. The observations that generation of the fusion-committed state may involve exposure of phospholipid moieties by CpreTM [32], and that the reversion of this condition may be attained by incubation with antibody (Fig. 5) would support this latter idea.
The fusion-competent vesicles were further used to assess heterotypic vesicle fusion (Figs. 6–8). This approach allowed getting insights into the Chol requirements in either primed or target vesicles (Fig. 6). Our experiments revealed that a Chol concentration comparable to that existing within virions [41, 42], was required in the vesicles to effectively generate the fusion-committed state. In contrast to the critical dependence observed for the primed vesicles, lipid-mixing increased gradually upon increasing the Chol content of the target vesicles. Thus, target vesicles containing Chol concentrations in the range found in the plasma membrane of virus-producing cells (ca. 30 mol %, [41, 42]), efficiently mixed their lipids with the fusion-committed vesicles. The fact that MPER-derived sequences are dependent on Chol for fusing vesicles was noted in an earlier study [14]. Our results add to this notion that the critical Chol-dependency specifically pertains to the vesicles acting as surrogates of the viral membrane. Interestingly, HIV membrane-like complex lipid mixtures that contain 45 mol % Chol, display low kc values allowing highly curved intermediates to form [50]. This suggests that the viral membrane composition and the CpreTM sequence might have co-evolved to sustain efficient fusion.
The heterotypic fusion measurements allowed further analysis of the target-membrane composition requirements (Figs 7 and 8). In the plasma membrane PE is predominantly exposed on the cytosolic leaflet, whereas PC and SM are predominantly located on the outer leaflet [39, 40]. Thus, the cell membrane (cis-)monolayer that makes first contact with the viral envelope is presumptively devoid of non-lamellar lipids. On the contrary, this monolayer is expected to be enriched in lipids conferring high packing density and, hence, resistance to bending. Thus, the observations that PE did not appreciably stimulate lipid-mixing, or that the inclusion of SM did not interfere with the process (Fig. 7), would not be at odds with the putative requirements for the initial lipid exchange with virions occurring at the surface of the infected cell [2, 9]. Moreover, at low CpreTM doses (i.e. 1:1000 or 1:500 peptide-to-lipid ratio) a significant enhancement of lipid mixing was observed upon inclusion of SM into the target membrane (Fig. 8). We note that the low CpreTM dose condition might be relevant for the HIV fusion process, since the low number spikes existing at the envelope (10–20 peplomers per virion [51]) implies an MPER density close to ca. 1:5000 (MPER-to-lipid mole ratio). Thus, perturbations generated within the viral membrane surrogate by low CpreTM doses seem to suffice for induction of lipid exchange with target membranes resembling the outer leaflet of the plasma membrane.
In conclusion, our recent MDS studies and the heterotypic vesicle fusion assays described herein have rendered relevant information for the understanding of the initial steps of the HIV fusion reaction triggered by fusogenic pre-hairpins. They point to the MPER-TMD connection as a pivotal component of the HIV fusion machinery, which would function by facilitating lipid exchange between the virus and plasma cell membranes. In this regard, this region should be considered as internal HIV-1 fusion peptide [52].
Highlights.
CpreTM peptide generates a fusion-competent state in Cholesterol-containing vesicles.
Neutralizing 4E10 antbody binds to fusion-competent vesicles and precludes fusion.
High cholesterol concentrations are required to prime vesicles.
Primed vesicles can fuse with sphingolipid/cholesterol-containing target membranes.
Acknowledgments
We acknowledge the financial support of Basque Government (Grant IT838-13) and the National Institutes of Health and the Canadian Institutes for Health Research (combined 1R01AI097051-01 and MOP-114941 grants).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Melikyan GB. Common principles and intermediates of viral protein-mediated fusion: the HIV-1 paradigm. Retrovirology. 2008;5:111. doi: 10.1186/1742-4690-5-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Melikyan GB. Membrane fusion mediated by human immunodeficiency virus envelope glycoprotein. Current topics in membranes. 2011;68:81–106. doi: 10.1016/B978-0-12-385891-7.00004-0. [DOI] [PubMed] [Google Scholar]
- 3.Blumenthal R, Durell S, Viard M. HIV entry and envelope glycoprotein-mediated fusion. J Biol Chem. 2012;287:40841–40849. doi: 10.1074/jbc.R112.406272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eckert DM, Kim PS. Mechanisms of viral membrane fusion and its inhibition. Annu Rev Biochem. 2001;70:777–810. doi: 10.1146/annurev.biochem.70.1.777. [DOI] [PubMed] [Google Scholar]
- 5.Weissenhorn W, Dessen A, Harrison SC, Skehel JJ, Wiley DC. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387:426–430. doi: 10.1038/387426a0. [DOI] [PubMed] [Google Scholar]
- 6.Markosyan RM, Cohen FS, Melikyan GB. HIV-1 envelope proteins complete their folding into six-helix bundles immediately after fusion pore formation. Molecular biology of the cell. 2003;14:926–938. doi: 10.1091/mbc.E02-09-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miyauchi K, Kim Y, Latinovic O, Morozov V, Melikyan GB. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell. 2009;137:433–444. doi: 10.1016/j.cell.2009.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de la Vega M, Marin M, Kondo N, Miyauchi K, Kim Y, Epand RF, Epand RM, Melikyan GB. Inhibition of HIV-1 endocytosis allows lipid mixing at the plasma membrane, but not complete fusion. Retrovirology. 2011;8:99. doi: 10.1186/1742-4690-8-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Padilla-Parra S, Marin M, Gahlaut N, Suter R, Kondo N, Melikyan GB. Fusion of mature HIV-1 particles leads to complete release of a gag-GFP-based content marker and raises the intraviral pH. PloS one. 2013;8:e71002. doi: 10.1371/journal.pone.0071002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Apellaniz B, Huarte N, Largo E, Nieva JL. The three lives of viral fusion peptides. Chem Phys Lipids. 2014;181:40–55. doi: 10.1016/j.chemphyslip.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salzwedel K, West JT, Hunter E. A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Env-mediated fusion and virus infectivity. J Virol. 1999;73:2469–2480. doi: 10.1128/jvi.73.3.2469-2480.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suarez T, Gallaher WR, Agirre A, Goni FM, Nieva JL. Membrane interface-interacting sequences within the ectodomain of the human immunodeficiency virus type 1 envelope glycoprotein: putative role during viral fusion. J Virol. 2000;74:8038–8047. doi: 10.1128/jvi.74.17.8038-8047.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saez-Cirion A, Arrondo JL, Gomara MJ, Lorizate M, Iloro I, Melikyan G, Nieva JL. Structural and functional roles of HIV-1 gp41 pretransmembrane sequence segmentation. Biophys J. 2003;85:3769–3780. doi: 10.1016/S0006-3495(03)74792-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shnaper S, Sackett K, Gallo SA, Blumenthal R, Shai Y. The C- and the N-terminal regions of glycoprotein 41 ectodomain fuse membranes enriched and not enriched with cholesterol, respectively. J Biol Chem. 2004;279:18526–18534. doi: 10.1074/jbc.M304950200. [DOI] [PubMed] [Google Scholar]
- 15.Huarte N, Lorizate M, Maeso R, Kunert R, Arranz R, Valpuesta JM, Nieva JL. The broadly neutralizing anti-human immunodeficiency virus type 1 4E10 monoclonal antibody is better adapted to membrane-bound epitope recognition and blocking than 2F5. J Virol. 2008;82:8986–8996. doi: 10.1128/JVI.00846-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Apellaniz B, Ivankin A, Nir S, Gidalevitz D, Nieva JL. Membrane-proximal external HIV-1 gp41 motif adapted for destabilizing the highly rigid viral envelope. Biophys J. 2011;101:2426–2435. doi: 10.1016/j.bpj.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lorizate M, Huarte N, Saez-Cirion A, Nieva JL. Interfacial pre-transmembrane domains in viral proteins promoting membrane fusion and fission. Biochim Biophys Acta. 2008;1778:1624–1639. doi: 10.1016/j.bbamem.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Munoz-Barroso I, Salzwedel K, Hunter E, Blumenthal R. Role of the membrane-proximal domain in the initial stages of human immunodeficiency virus type 1 envelope glycoprotein-mediated membrane fusion. J Virol. 1999;73:6089–6092. doi: 10.1128/jvi.73.7.6089-6092.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vishwanathan SA, Hunter E. Importance of the membrane-perturbing properties of the membrane-proximal external region of human immunodeficiency virus type 1 gp41 to viral fusion. J Virol. 2008;82:5118–5126. doi: 10.1128/JVI.00305-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Epand RF, Thomas A, Brasseur R, Vishwanathan SA, Hunter E, Epand RM. Juxtamembrane protein segments that contribute to recruitment of cholesterol into domains. Biochemistry. 2006;45:6105–6114. doi: 10.1021/bi060245+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vishwanathan SA, Thomas A, Brasseur R, Epand RF, Hunter E, Epand RM. Hydrophobic substitutions in the first residue of the CRAC segment of the gp41 protein of HIV. Biochemistry. 2008;47:124–130. doi: 10.1021/bi7018892. [DOI] [PubMed] [Google Scholar]
- 22.Vishwanathan SA, Thomas A, Brasseur R, Epand RF, Hunter E, Epand RM. Large changes in the CRAC segment of gp41 of HIV do not destroy fusion activity if the segment interacts with cholesterol. Biochemistry. 2008;47:11869–11876. doi: 10.1021/bi8014828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lorizate M, Cruz A, Huarte N, Kunert R, Perez-Gil J, Nieva JL. Recognition and blocking of HIV-1 gp41 pre-transmembrane sequence by monoclonal 4E10 antibody in a Raft-like membrane environment. J Biol Chem. 2006;281:39598–39606. doi: 10.1074/jbc.M605998200. [DOI] [PubMed] [Google Scholar]
- 24.Montero M, van Houten NE, Wang X, Scott JK. The membrane-proximal external region of the human immunodeficiency virus type 1 envelope: dominant site of antibody neutralization and target for vaccine design. Microbiol Mol Biol Rev. 2008;72:54–84. doi: 10.1128/MMBR.00020-07. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun ZY, Oh KJ, Kim M, Yu J, Brusic V, Song L, Qiao Z, Wang JH, Wagner G, Reinherz EL. HIV-1 broadly neutralizing antibody extracts its epitope from a kinked gp41 ectodomain region on the viral membrane. Immunity. 2008;28:52–63. doi: 10.1016/j.immuni.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 26.Montero M, Gulzar N, Klaric KA, Donald JE, Lepik C, Wu S, Tsai S, Julien JP, Hessell AJ, Wang S, Lu S, Burton DR, Pai EF, Degrado WF, Scott JK. Neutralizing epitopes in the membrane-proximal external region of HIV-1 gp41 are influenced by the transmembrane domain and the plasma membrane. J Virol. 2012;86:2930–2941. doi: 10.1128/JVI.06349-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nieva JL, Apellaniz B, Huarte N, Lorizate M. A new paradigm in molecular recognition? Specific antibody binding to membrane-inserted HIV-1 epitopes. Journal of molecular recognition : JMR. 2011;24:642–646. doi: 10.1002/jmr.1092. [DOI] [PubMed] [Google Scholar]
- 28.Apellaniz B, Nir S, Nieva JL. Distinct mechanisms of lipid bilayer perturbation induced by peptides derived from the membrane-proximal external region of HIV-1 gp41. Biochemistry. 2009;48:5320–5331. doi: 10.1021/bi900504t. [DOI] [PubMed] [Google Scholar]
- 29.Apellaniz B, Nieva JL, Schwille P, Garcia-Saez AJ. All-or-none versus graded: single-vesicle analysis reveals lipid composition effects on membrane permeabilization. Biophys J. 2010;99:3619–3628. doi: 10.1016/j.bpj.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Epand RM, Thomas A, Brasseur R, Epand RF. Cholesterol interaction with proteins that partition into membrane domains: an overview. Subcell Biochem. 2010;51:253–278. doi: 10.1007/978-90-481-8622-8_9. [DOI] [PubMed] [Google Scholar]
- 31.Gangupomu VK, Abrams CF. All-atom models of the membrane-spanning domain of HIV-1 gp41 from metadynamics. Biophys J. 2010;99:3438–3444. doi: 10.1016/j.bpj.2010.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Apellaniz B, Rujas E, Carravilla P, Requejo-Isidro J, Huarte N, Domene C, Nieva JL. Cholesterol-Dependent Membrane Fusion Induced by the gp41 Membrane-Proximal External Region-Transmembrane Domain Connection Suggests a Mechanism for Broad HIV-1 Neutralization. J Virol. 2014;88:13367–13377. doi: 10.1128/JVI.02151-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Melnyk RA, Partridge AW, Yip J, Wu Y, Goto NK, Deber CM. Polar residue tagging of transmembrane peptides. Biopolymers. 2003;71:675–685. doi: 10.1002/bip.10595. [DOI] [PubMed] [Google Scholar]
- 34.Yethon JA, Epand RF, Leber B, Epand RM, Andrews DW. Interaction with a membrane surface triggers a reversible conformational change in Bax normally associated with induction of apoptosis. J Biol Chem. 2003;278:48935–48941. doi: 10.1074/jbc.M306289200. [DOI] [PubMed] [Google Scholar]
- 35.Struck DK, Hoekstra D, Pagano RE. Use of resonance energy transfer to monitor membrane fusion. Biochemistry. 1981;20:4093–4099. doi: 10.1021/bi00517a023. [DOI] [PubMed] [Google Scholar]
- 36.Graham DR, Chertova E, Hilburn JM, Arthur LO, Hildreth JE. Cholesterol depletion of human immunodeficiency virus type 1 and simian immunodeficiency virus with beta-cyclodextrin inactivates and permeabilizes the virions: evidence for virion-associated lipid rafts. J Virol. 2003;77:8237–8248. doi: 10.1128/JVI.77.15.8237-8248.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guyader M, Kiyokawa E, Abrami L, Turelli P, Trono D. Role for human immunodeficiency virus type 1 membrane cholesterol in viral internalization. J Virol. 2002;76:10356–10364. doi: 10.1128/JVI.76.20.10356-10364.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siegel DP, Epand RM. The mechanism of lamellar-to-inverted hexagonal phase transitions in phosphatidylethanolamine: implications for membrane fusion mechanisms. Biophys J. 1997;73:3089–3111. doi: 10.1016/S0006-3495(97)78336-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Devaux PF, Lopez-Montero I, Bryde S. Proteins involved in lipid translocation in eukaryotic cells. Chem Phys Lipids. 2006;141:119–132. doi: 10.1016/j.chemphyslip.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 40.Devaux PF, Morris R. Transmembrane asymmetry and lateral domains in biological membranes. Traffic. 2004;5:241–246. doi: 10.1111/j.1600-0854.2004.0170.x. [DOI] [PubMed] [Google Scholar]
- 41.Brugger B, Glass B, Haberkant P, Leibrecht I, Wieland FT, Krausslich HG. The HIV lipidome: a raft with an unusual composition. Proc Natl Acad Sci U S A. 2006;103:2641–2646. doi: 10.1073/pnas.0511136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chan R, Uchil PD, Jin J, Shui G, Ott DE, Mothes W, Wenk MR. Retroviruses human immunodeficiency virus and murine leukemia virus are enriched in phosphoinositides. J Virol. 2008;82:11228–11238. doi: 10.1128/JVI.00981-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Greenwood AI, Pan J, Mills TT, Nagle JF, Epand RM, Tristram-Nagle S. CRAC motif peptide of the HIV-1 gp41 protein thins SOPC membranes and interacts with cholesterol. Biochim Biophys Acta. 2008;1778:1120–1130. doi: 10.1016/j.bbamem.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tristram-Nagle S, Chan R, Kooijman E, Uppamoochikkal P, Qiang W, Weliky DP, Nagle JF. HIV fusion peptide penetrates, disorders, and softens T-cell membrane mimics. J Mol Biol. 2010;402:139–153. doi: 10.1016/j.jmb.2010.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tristram-Nagle S, Nagle JF. HIV-1 fusion peptide decreases bending energy and promotes curved fusion intermediates. Biophys J. 2007;93:2048–2055. doi: 10.1529/biophysj.107.109181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nieva JL, Agirre A. Are fusion peptides a good model to study viral cell fusion? Biochim Biophys Acta. 2003;1614:104–115. doi: 10.1016/s0005-2736(03)00168-8. [DOI] [PubMed] [Google Scholar]
- 47.Alam SM, Morelli M, Dennison SM, Liao HX, Zhang R, Xia SM, Rits-Volloch S, Sun L, Harrison SC, Haynes BF, Chen B. Role of HIV membrane in neutralization by two broadly neutralizing antibodies. Proc Natl Acad Sci U S A. 2009;106:20234–20239. doi: 10.1073/pnas.0908713106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Julien JP, Cupo A, Sok D, Stanfield RL, Lyumkis D, Deller MC, Klasse PJ, Burton DR, Sanders RW, Moore JP, Ward AB, Wilson IA. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science. 2013;342:1477–1483. doi: 10.1126/science.1245625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen J, Frey G, Peng H, Rits-Volloch S, Garrity J, Seaman MS, Chen B. Mechanism of HIV-1 neutralization by antibodies targeting a membrane-proximal region of gp41. J Virol. 2014;88:1249–1258. doi: 10.1128/JVI.02664-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boscia AL, Akabori K, Benamram Z, Michel JA, Jablin MS, Steckbeck JD, Montelaro RC, Nagle JF, Tristram-Nagle S. Membrane structure correlates to function of LLP2 on the cytoplasmic tail of HIV-1 gp41 protein. Biophys J. 2013;105:657–666. doi: 10.1016/j.bpj.2013.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roux KH, Taylor KA. AIDS virus envelope spike structure. Curr Opin Struct Biol. 2007;17:244–252. doi: 10.1016/j.sbi.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 52.Peisajovich SG, Shai Y. Viral fusion proteins: multiple regions contribute to membrane fusion. Biochim Biophys Acta. 2003;1614:122–129. doi: 10.1016/s0005-2736(03)00170-6. [DOI] [PubMed] [Google Scholar]