

Abstract
Purpose
Growth hormone (GH) pituitary tumors are associated with significant morbidity and mortality. Current treatments, including surgery and medical therapy with somatostatin analogues (SSA), dopamine agonists and/or a GH receptor antagonist, result in disease remission in approximately half of patients. Predictors of GH tumor response to different therapies have been incompletely defined based on histologic subtype, particularly densely (DG) versus sparsely (SG) granulated adenomas. The aim of this study was to examine our own institutional experience with GH adenomas and correlate how subtype related to clinical parameters as well as response to surgery and medical therapies.
Methods
A retrospective chart review of 101 acromegalic patients operated by a single neurosurgeon was performed. Clinical data were correlated with histologic subtype and disease control, as defined by IGF-1 levels, and random growth hormone levels in response to surgery and/or medical therapies.
Results
SG tumors, compared to DG, occurred in younger patients (p=0.0010), were 3-fold larger (p=0.0030), but showed no differences in tumor-invasion characteristics (p=0.12). DG tumors had a higher rate of remission in response to surgery compared to SG, 65.7% vs. 14.3% (p<0.0001), as well as to medical therapy with SSAs (68.8% for DG vs. 28.6% for SG tumors; p=0.028). SG tumors not controlled with SSAs consistently responded to a switch to, or addition of, a GH receptor antagonist.
Conclusions
Histological GH tumor subtyping implicates a different clinical phenotype and biologic behavior, and provides prognostic significance for surgical success and response to medical therapies.
Keywords: Acromegaly, dense GH tumors, sparse GH tumors, somatostatin analogue, pegvisomant
Introduction
Acromegaly, a disorder of growth hormone over-secretion, is almost always due to a pituitary growth hormone (GH) adenoma. GH tumors represent up to 15-20% of all pituitary tumors, and more than 65% are macroadenomas, with associated compressive clinical signs of headaches, visual impairment and hypopituitarism [1,2]. Long-term persistent elevation of GH levels are associated with cardiovascular, pulmonary, neoplastic and metabolic complications, leading to significant morbidity, and a 10-year reduction in life span [2]. The management of patients with GH tumors is focused on normalization of GH levels and its downstream target, insulin-like growth factor 1 (IGF-1) levels adjusted for age and gender [3,4].
First-line therapy for most GH tumors is surgical resection [3]. However, more than 50% of acromegalic patients have persistent disease after surgery, even in experienced pituitary centers [5,6]. When biochemical remission in not achieved by a surgical approach, single or combined medical therapies with somatostatin analogues (SSA), growth hormone receptor agonist and/or a dopamine agonist are indicated [4]. Although early studies reported remission rates of 50-60% with SSAs in pre-selected patients, recent literature suggests remission rates are actually less than 30% in unselected patients [7-9]. Pegvisomant (PEG), a GH receptor antagonist, achieved IGF-1 normalization in 60-70% of patients, although some of the non-response might be attributable to inadequate dose titration [10,11]. The dopamine agonist, cabergoline, achieved GH and IGF-1 normalization in approximately 10-30% of cases, and has a role in acromegaly treatment limited to those with mild IGF-1elevations [3,12-14]. Radiotherapy, used in patients with persistent uncontrolled disease, has a prolonged onset of action and carries risk of long term untoward side-effects, such as hypopituitarism and possible increased stroke risk [3] .
The variable and inconsistent response of GH tumors to primary surgical resection, as well as post- operative medical therapies, has increased interest in identifying clinical, radiological, histological, and genetic markers to better predict GH tumor behavior. Clinical factors such as age, gender, and tumor size have been previously examined, albeit with inconsistent results [5,15]. GH tumor sub-typing has been also evaluated as it relates to clinical outcomes.
Using electron microscopy (EM), GH tumors have been subtyped into densely granulated (DG) adenomas that contain abundant secretory granules and sparsely granulated (SG) adenomas with sparse granules [16,17]. Yamada et al. showed that immunohistochemistry for cytokeratin (CAM5.2 keratin stain), to identify circular intracytoplasmic collections of keratin filaments known as fibrous bodies (FBs), can be used as a surrogate to EM to distinguish between DG and SG growth hormone tumor sub-types [18]. All studies agree that large numbers of diffusely distributed FBs are indicative of the SG growth hormone adenoma type [18-22]. Some GH adenomas otherwise indistinguishable from DG adenomas on light microscopy do contain small numbers of FBs; Obari and coworkers coined the term “intermediate variant” for these adenomas and noted that these tumors behaved similarly to DG growth hormone adenomas [20].
Although cytokeratin is known to have important roles in cellular structure and function , the mechanism underlying variable cytokeratin patterns in GH tumors remains unknown [23]. Prognosis based on specific histologic cytokeratin pattern has been suggested, although the literature is conflicting [24-26]. Prior reports found that SG tumors are more common in young patients (<50 yrs), often are larger, and may have lower GH/IGF-1 levels than DG tumors [20,24,27-30]. SG tumors are thought to be less responsive to SSAs than DG tumors, although the mechanisms underlying the differences in tumor behavior are incompletely defined [25,29-32]. Importantly, although GH tumor cytokeratin histologic subtyping has been suggested as a potential marker of response to therapy and overall prognosis [28-32], it is not used routinely by clinicians in the clinical management of acromegalic patients. The most recent guidelines, for the first time, suggested that SG tumors might be associated with disease persistence, although this was not fully agreed on by consensus participants [33].
Therefore, we reviewed our own experience with GH pituitary tumors to assess whether GH adenoma subtypes correlated with baseline clinical, laboratory or radiologic characteristics, and/or predicted a differential response to surgical and medical therapies.
Material and Methods
Patients
A retrospective chart review of 111 consecutive patients with a diagnosis of acromegaly was completed with Colorado Multiple Institutional Review Board (COMIRB) approval. The diagnosis of acromegaly was made based on clinical findings and biochemical elevation of GH and IGF-1 levels, and confirmed on pathological examination by the neuropathologist on the study (B.K.D.). All of the patients underwent transsphenoidal pituitary surgery, for a GH-secreting adenoma by a single neurosurgeon (K.O.L.), between January 1997 and September 2012. Patients who did not achieve disease remission, defined as normalization of IGF-1 at 3-months post-operative visit and had an inappropriately elevated random GH level, were treated with medical therapy with an SSA, with titration every 3-4 months until IGF-1 normalization or maximum dose was achieved for ≥3 months. Patients whose IGF did not normalize with maximal dose of SSA were either switched to the GH receptor agonist, pegvisomant (PEG) as a single agent, or received combination therapy (SSA/PEG). No patient received prolonged treatment with a dopamine agonist. Importantly, if the patient received pituitary radiation therapy, their inclusion in the study was terminated at that point.
Hormone evaluation
Serum growth hormone (GH) and/or insulin-like growth factor 1 (IGF-1) levels were measured at baseline pre-operatively, then post-operatively at 2-weeks and 3-months, followed by annual visits. Plasma GH was measured using Quantitative Chemiluminescent Immunoassay (Immulite Immunoassay Systems; Siemens Medical Solutions Diagnostics) with lower limit of detection at 0.01ng/mL. This assay is standardized to the Recombinant Second International Standard, National Institute for Biological Standards and Control (NIBSC) 98/574. IGF-1 level was determined using Quantitative Chemiluminescent Immunoassay (Immunodiagnositic Systems; Isys technologies) with detection level of 10 ng/mL. IGF-1 is standardized to World Health Organization (WHO) international standard 02\254. IGF-1 levels were calculated and reported as fold-change from upper limit of normal (ULN), matched for age and sex. Remission was defined as IGF-1 level within normal limits for age and gender. Random GH levels and OGTT were assessed where available, with oral glucose tolerance test (OGTT) GH cut off at < 1 ng/mL.
Neuroimaging evaluation
Standard pre-operative and post-surgical magnetic resonance images (MRI) of pituitary, with and without gadolinium contrast, were retrospectively evaluated for the purposes of this study by the neuroradiologist (M.T.B.) on a PACS (Picture archiving communication system) (McKesson). Tumor length, width and depth were recorded and tumor volume was calculated using formula: 0.5 × width × length × height (mm3) [34]. Cavernous sinus invasion was also evaluated and defined as tumor circumferentially around an internal carotid artery and lateral tumor extension beyond medial wall of cavernous sinus. Optic chiasm compression was documented as perceived physical contact between tumor and optic chiasm structure as assessed by the neuroradiologist.
Histopathological evaluation
Tumor tissues resected by surgery were submitted in toto and were fixed overnight in 10% buffered formalin. Sections from the paraffin blocks were cut at 4 microns and stained with hematoxylin and eosin, Gomori reticulin, and periodic acid Schiff-orange G histochemical stains. Immunohistochemical staining (IHC) staining for anterior pituitary hormones was performed using the peroxidase-antiperoxidase complex technique, with light hematoxylin counterstaining. Antisera used in IHC studies included synaptophysin (monoclonal, pre-diluted; Ventana, Tucson, AZ), adrenocorticotropin hormone, growth hormone (both polyclonal, pre-diluted; Cell Marque, Rocklin, CA), alpha-subunit (monoclonal, 1:400, Biogenex, San Ramon, CA), follicle-stimulating hormone (1:50), luteinizing hormone (1:50), thyroid-stimulating hormone (1:100) (all monoclonal, Dako, Carpinteria, CA), prolactin (polyclonal, pre-diluted; Signet). Growth hormone tumor subtyping using CAM5.2 (monoclonal, pre-diluted; Becton Dickinson) was assessed based on the distribution pattern of keratin filaments.
Positivity for CAM5.2, staining for cytokeratin, shows several patterns in GH tumors as shown in Figure 1: densely-granulated (DG) growth hormone adenomas with cytokeratin filaments throughout the cytoplasm and an absence of rounded CAM5.2 IHC-positive fibrous bodies (FBs) (panel A); sparsely-granulated (SG) growth hormone adenomas with numerous, diffusely-distributed FBs (panel B.), and intermediate (DG/SG) granulated tumors, resembling DG growth hormone adenomas by light microscopy but containing scattered FBs (panels C and D). DG subtype was assigned when perinuclear cytokeratin immunostaining without FB formation was present in over 70% of cells, SG type was categorized when FBs were present in over 70% of cells, and “intermediate pattern” was assigned when more than 30% of cells deviated from the dominant CAM5.2 pattern, as previously described [20]. Record was made as to the presence or absence of co-secretion of prolactin and alpha subunit; other hormone types were not usually found in these GH adenomas.
Figure 1.

Cam5.2 staining of growth hormone adenoma. A. Densely granulated tumor (60X) shows keratins diffusely distributed throughout cytoplasm and perinuclear distribution pattern. B. Sparsely granulated adenoma (60X) with characteristics dot-like keratin immunoreactivity representing fibrous bodies C. Intermediate tumors (40X) typically comprised of two keratin pattern populations with dot-like pattern and perinuclear pattern interspersed D. Intermediate tumors (60X) with DG and SG pattern
Statistical analysis
Patient and tumor characteristics were compared between the two tumor subtypes SG and DG (containing both DG and intermediate histology) using two-sample t-tests or Chi-square tests as appropriate. Tumor volume and the hormone measurements were skewed. Thus, analyses were completed after log (base-e) transformation. Results are presented as geometric means and 95% confidence intervals (CI). Differences in response to surgical and medical treatment by tumor subtype were assessed using Fisher's exact test given the small samples. The association between tumor subtype and remission from primary surgery was further assessed using logistic regression and the final model adjusted for age and log tumor volume. Given the small samples, a similar analysis on remission from SSA or PEG could not be performed. All analyses were performed using SAS v9.3 (Cary, NC). P-values < 0.05 were considered statistically significant.
Results
Patients
A retrospective chart review identified 111 patients with a diagnosis of acromegaly that had undergone transsphenoidal resection at the University of Colorado Hospital by K.O.L. between January 1997 and September 2012 (Figure 2). Importantly, all the patients were medication-naïve prior to surgery. Histologic subtyping could not be performed for 10 tumors, due to absence of archival paraffin tissue blocks on the oldest cases in the series, and these cases were excluded from the analysis. Thus, 101 patient samples were included in the analyses.
Figure 2.

Flow diagram of patient population. N represents the number of records with follow up information for specific treatment modality.
Initially GH tumors were subdivided into three separate groups (SG, intermediate and DG) and we concluded, as others previously [20,29,30], that intermediate and DG tumors were similar. DG and intermediate tumors were thus combined into a single DG group. The tumors included 70.3% (N=71/101) DG and 29.7% (N=30/101) SG subtype. As expected, tumors showed immunostaining for GH only (N=86), with small amounts of PRL immunostaining in a minority (N=15). Similar to the study by Obari et al.[20], we found that alpha subunit was detected more often in DG (40%, N=29/71) compared to SG tumors (6%, N=2/30) (p=0.0015). Mean follow-up was similar for the 2 groups (mean±SD): 2.4±2.5 years for DG and 3.2±2.3 years for SG (p=0.19).
Post-operatively, data on normalization of IGF-1 level were available in 28 SG and 67 DG tumors, and absolute IGF-1 values with reference IGF-1 levels were reported in 22 SG and 42 DG tumors, respectively. Patients who did not achieve remission with surgery (49.5%, N=47/95) were started on SSA (octreotide LAR or lanreotide autogel) at 3-12 months post-op, and follow-up was available for 30 patients (16 DG and 14 SG subtypes). Patients not controlled with a maximum SSA dose (monthly octreotide LAR 30-40mg or lantreotide autogel 120mg for ≥3 months), were switched to pegvisomant (10-40 mg/d) or pegvisomant was added to SSA therapy (data available on 10 patients (3 DG and 7 SG)). In our cohort, treatment with dopamine agonist was given only short-term in 5 patients, and thus was not included in the analysis.
Clinical characteristics
Clinical patient characteristics are outlined in Table 1. Consistent with prior literature, age differed by histologic subtype (p=0.0010). Patients with SG tumors were younger than those with DG, 40.4 ± 13.7 vs 50.0 ±12.8 years, respectively (Mean+/−SD). There was no difference in gender across GH tumor subtypes (p=0.62).
Table 1.
Clinical baseline characteristics
| Variable | Sparse GH | Dense GH | P value |
|---|---|---|---|
| Age (Mean ± SD) | 40.4 ± 13.7 | 50.0 ± 12.8 | 0.0010 |
| Gender (N: % Female) | 16 (53.3%) | 34 (47.9%) | 0.62 |
| Mean follow up (Yrs ± SD) | 3.2 ± 2.3 | 2.4 ± 2.5 | 0.19 |
| Tumor volume (mm3)* Geo. Mean (95% CI) | 2392, N=23 (1274-4537) | 665, N=60 (428-1043) | 0.0030 |
| Cavernous sinus invasion N/N total (%) | 12/23 (54.6%) | 21/59 (35.6%) | 0.12 |
| Optic chiasm compression N/N total (%) | 5/18 (21.7%) | 8/59 (13.6%) | 0.50 |
| Baseline IGF-1 FC ULN Geo. Mean (95% CI) | 2.80 (2.34-3.39) | 2.88 (2.60-3.19) | 0.80 |
| Baseline GH level (ng/mL) Geo. Mean (95% CI) | 9.77 (4.39-21.76) | 11.59 (8.08-16.61) | 0.54 |
| GH index (GH baseline/tumor volume) Geo. Mean (95% CI) | 0.0035 (0.0012-0.011) | 0.019 (0.012-0.030) | 0.013 |
| Alpha subunit IHC expression N/N total (%) | 2/30 (6%) | 19/71 (40%) | 0.0015 |
| 3-Month IGF-1 × FC ULN Geo. Mean (95% CI) | 1.49 (1.15-1.95) | 0.83 (0.70-0.98) | 0.0007 |
| 3-Month GH level (ng/mL) Geo. Mean (95% CI) | 3.42 (1.26-9.12) | 0.64 (0.40-1.00) | 0.0061 |
| Ratio GH/IFG Geo Mean 95% CI | 0.32 (0.11-0.90) | 0.19 (0.098-0.36) | 0.36 |
Tumor volume = 0.5 × (height × width × depth); FC ULN-fold change from upper limit of normal. Geo. (geometric) mean is the mean of the log values back transformed to the observed scale.
Neuroimaging results
The results of the analysis of tumor size and invasion assessed by MRI are summarized in Table 1. MRI was available in 83/101 of the patients. Approximately 75% of tumors were classified as macroadenomas (≥1cm), but the frequency differed by histologic subtype: 65.0% (N=39/60) of DG and 100% (N=23/23) of SG group. In addition, the volume of SG (N=23) tumors was significantly larger than DG (N=60) (p=0.0030), with SG tumors being, on average, 3-fold larger at clinical presentation. Despite the size differential however, rates of cavernous sinus invasion did not differ between the subtypes (p=0.12; 54.6% SG and 35.6% DG). Rates of optic chiasm compression also did not differ significantly between the tumor types (p=0.50; SG 21.7%, compared to 13.6% of DG). Evidence of residual tumor postoperatively on 3-month follow-up MRI was higher in SG 45.4% (N=5/11) compared to 18.2% DG (N=6/27) tumors, although these differences did not reach statistical significance (p=0.11).
Hormonal results
As shown in Table 1, despite a difference in the size of tumors based on histological subtyping, the pre-operative IGF-1 level fold-change above the upper limit of normal (ULN), and random GH level did not differ between GH tumor subtypes, p=0.80 and p=0.54, respectively. GH index, defined as ratio of baseline GH and tumor volume, was higher in the DG subtype (0.019 (95% CI: 0.012-0.030) compared to SG (0.0035 (95% CI: 0.0012-0.011); p=0.013). At 3 months after surgery, IGF-1 fold change from ULN was significantly higher in SG (N=21) than DG tumors (N=42) (p=0.0007). Random GH levels, were also higher in SG (N=13) than DG (N=37) (p=0.0061). Overall 67.3% (N=68/101) of patients had postoperative GH levels measured with IGF-1 to determine whether their disease was in remission. Only three patients displayed discordant tests with high GH and normal IGF-1. However, each had normal OGTT, suggesting that the random GH level was a result of normal GH pulsatility.
Response to surgery
Rates of remission, defined as IGF-1 normalization are shown in Figure 3A. The rate of remission after primary surgical resection of GH tumors was 50.5% (N=48/95). With respect to GH subtypes, significantly fewer patients with SG tumors 14.3%, (N=4/28) were in remission at 3 months and during the follow-up period compared to other tumor subtypes, DG 65.7%, (N=44/67) (p<0.0001). This finding was attenuated but consistent, after adjusting for age at surgery and log tumor size (p=0.0012). Prior to adjustment, the odds of remission in the DG subtype was 11.5 times (95% CI:3.6-37.0) those with SG tumors (p<0.0001). After adjusting for log tumor volume and age, the odds of remission in the DG subtype was 7.4 times (95% CI: 2.0-32.5) those with SG tumors (p=0.0036). Increased log tumor volume was associated with reduced odds of remission (p=0.047; OR=0.70; 95% CI 0.50-0.99). Age was not associated with the odds of remission (p=0.60; OR=1.01; 95% CI 0.97-1.06).
Figue 3.
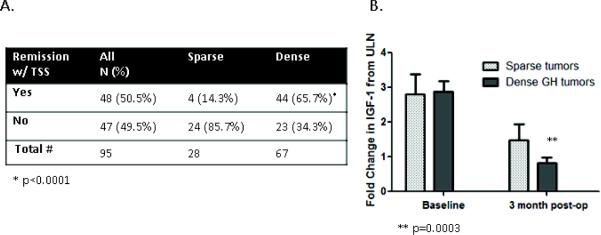
Postoperative remission rates. A. There is a significant difference in the rate of remission between sparse and dense tumor subtypes (*p<0.0001) B. The graph shows the baseline and post operative IGF-1 change from ULN. SG tumors had significantly less IGF-1 reduction than DG (**p=0.0003). IGF-1 change from ULN decreases 44% (95% CI: 24-58%) between baseline and 3-month follow up compared to DG decrease at 72 % (95% CI: 66-78%).
At 3-months post-operatively compared to baseline, the difference in the drop in the IGF-1 fold change from ULN, was significantly lower in SG (44% reduction; N=19) compared to DG (72% reduction; N=37; p=0.0003), as shown in Figure 3B. Similarly, the difference in the drop in random GH between baseline and 3-months post-operatively was lower in SG (80% reduction; N=12) compared to DG (95% reduction; N=27; p=0.021).
Response to medical therapy
When surgical resection was not curative, the overall response rate (defined as age and gender normalization of IGF1 levels) after treatment with somatostatin analogues was 50% (N=15/30). The cohort of patients treated with SSAs had mean follow up for 3.54±2.02 years. As shown in Figure 4A, there was a significant difference in SSA response between the GH tumor subtypes. Only 28.6% of SG (N=4/14) responded to treatment with SSA, in contrast to 68.8% of DG (N=11/16) with normalization of IGF-1 levels, (p=0.028). Only one patient (with a SG tumor) who was SSA responder, by IGF-1 criteria, had an abnormal GH level without OGTT as its utility has been questionable in assessing response to SSA and is generally not recommended [35].
Figure 4.
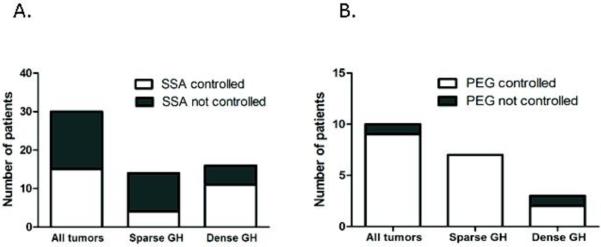
A. GH tumor response to somatostatin analogues (SSA). SG tumors were significantly less likely to be controlled with SSA compared to DG (p=0.028) B. GH adenoma subtypes and response to GH receptor antagonist, pegvisomant (PEG). The remission rates in response to PEG do not depend on tumor type (p=0.30)
Data were available for 10 patients who were not controlled with SSA and were placed on PEG. Of these, 4 SG and 2 DG were switched to PEG, whereas 3 SG and 1 DG were on combination SSA and PEG treatment. As shown in Figure 4B, the response to PEG was similar for the SG and DG subtypes (p=0.30). In contrast to a low response rate of SG tumors to SSA therapy, 100% (N=7/7) of SG patients showed normalization of IGF-1 while on PEG treatment.
Discussion
In this large cohort of consecutive GH adenomas examined from a single institution, with maximal surgical resection performed by a single neurosurgeon (K.O.L.) and retrospective neuroimaging review by a single neuroradiologist (M.T.B.) for residual disease assessment, we show that clinical characteristics and response to surgical and medical therapies correlate with GH adenoma subtype. This is a useful finding for both pathologists and clinicians who wish to use CAM5.2 on GH adenomas for diagnosis, clinical management, or medication stratification purposes.
The distribution of GH tumors in our series (54.9% SG, 30.4% DG and 14.7% intermediate) was within the range of prior literature, which actually has been quite variable, with DG representing 39-79%, SG 13-35% and intermediate 5-57% of GH subtypes (see Table 2) [18,30,27,31,28,20,29,36,37]. These wide ranges may be attributed to variable subtyping classification systems used, as well as the grouping of DG and intermediate subtypes together, as preferred by some authors [29]. In our analysis, we combined DG and intermediate groups and confirmed that SG tumors more often occur in younger patients compared to DG [27,20,28,30,37]. Several studies reported that SG tumors are more frequently found in female patients [18,29,37,32]; however, in our population, and in agreement with four previous reports, there was no correlation between the gender and GH tumor subtype [28,30,20,31,29].
Table 2.
Studies evaluating clinical characteristics of histological growth hormone tumor subtypes
| Study | Number of patients | Gender (M/F) | Age mean (years) | Tumor Size/ Invasive | Hormone levels basal |
|---|---|---|---|---|---|
| Mayr et al [36] | N=28 DG=21, SG=7 |
DG=11/10 SG=2/5 |
Overall – 46.9±10.1 | DG – 5/21 micro SG – 2/7 micro |
Not reported |
| Larkin et al [32] | N=52 DG=23, SG=19 INTERM=10 |
DG= 14/9* SG= 5/14 INTERM = 2/8 |
DG - 52±14** SG - 41±15 INTERM - 54±10 |
DG – 11.8±6.6 mm*** SG – 21.3±10.9 mm INTERM – 13.2±5.7mm Significant cavernous sinus invasion and suprasellar extension in SG vs DG |
DG – GH: 30.98±53.88 μg/L IGF1 SDS: 35.1±53.88 SG – GH: 18.97±17.83 μg/L IGF SDS: 40.4±28.2 INTERM – GH: 10.45±5.44 μg/L IGF SDS: 36.4±13.7 |
| Mori et al [37] | N =108 DG=78, SG=26, INTERM=4 |
DG=42/38* SG=7/19 INTERM=3/1 |
DG=49.8±13** SG=41.3±7.80 INTERM=46.2±15.4 |
DG – 1.28±2.29 cm3*** SG – 3.02±3.43 cm3 INTERM – 1.69±1.11 cm3 Significant cavernous sinus invasion in SG vs DG |
DG – GH: 17.6±24.9 ng/mL IGF1: 650±269 U/mL SG – GH: 17.0±14.7 ng/mL IGF1: 631±275 U/mL INTERM – GH: 25.0±21.1 ng/mL IGF1: 665±332 U/mL |
| Brzana et al [29] | N=59 DG=22, SG=14 GH/PRL=23 |
DG=10/12* SG=3/11 GH/PRL=5/18 |
DG=52.0±16 SG=41.6±13.3 GH/PRL==48.6±15.6 |
DG – 19.2±10.6 mm*** SG – 21.6±8.1 mm GH/PRL – 14.9±7.8mm No significant difference in invasiveness |
DG –IGF1 × ULN: 2.98±1.28 SG – IGF1 × ULN: 2.81±1.34 GH/PRL - IGF1 × ULN: 2.79±1.51 |
| Fougner et al [31] | N=78 DG=31, SG=10 INTERM= 37 |
DG - 3/7 SG -13/18 |
DG – 56 (46-60) SG - 46 (40-52) INTERM – 47 (39-54) |
DG – 0.99 (0.43-2.19) cm3 SG – 2.03 (1.09-5.60) cm3 INTERM – 0.85 (0.50-2.25) cm3 No significant difference in invasiveness |
DG - GH: 27.1 (10.5-47.5) mU/L IGF: 89 (72.2-109) nmol/L SG - GH: 33.1 (10.1-96) mU/L IGF: 81.8 (47.5-109) nmol/L INTERM - GH: 25.1 (17.3-51.3) mU/L IGF: 89.6 (76.5-121.2) nmol/L |
| Bakhtiar et al [30] | N=141 DG=83, SG=30 INTERM=28 |
DG – 40/43 SG – 11/19 INTERM – 13/15 |
DG - 50.1±12.6 SG - 45.0±16.1 INTERM – 48.7±12.9 |
DG – 5.8±11.04 cm3*** SG – 11.7±18.4 cm3 INTERM – 4.6±10.5 cm3 No significant difference in invasiveness |
DG - GH: 46.2±130.5 ng/mL SG - GH: 115.7±326.9 ng/mL INTERM - GH: 51.5±65.8 ng/mL |
| Obari et al [20] | N=104 DG=47, SG=31 INTERM = 26 |
DG – 26/21 SG – 15/16 INTERM – 12/14 |
DG - 49.6±13.8** SG - 43.6±11.1 INTERM - 49.2±11.1 |
DG – 14/33 micro*** SG – 3/21 micro INTERM – 9/23 micro Significant cavernous sinus invasion in SG vs DG |
DG - GH: 59.4 ± 121.3 ng/mL SG - GH: 40.6 ± 43.9 ng/mL INTERM - GH: 59.5 ± 82.9 ng/mL |
| Mazal et al [28] | N=76 DG=47, SG=29 |
DG - 20/27 SG - 9/20 |
DG - 46.4 (14-70) SG - 38.7 (18-65) |
Significant cavernous sinus invasion and suprasellar extension in SG vs DG | Not reported |
| Yamada et al [18] | N=31 DG=23, SG=8 |
DG - 18/5* SG - 1/7 |
DG - 47.4 (26-66)** SG - 36.6 (22-46) |
Significant cavernous sinus invasion in SG vs DG | DG – GH: 44 ± 38 ng/mL IGF-I: 3.3±1.0 U/mL SG - GH: 71 ± 52 ng/mL IGF-I: 3.7±0.7 U/mL |
| Bando et al [27] | N=21 DG=13, SG=7 INTERM=1 |
DG - 4/9 SG - 2/5 INTERM – 0/1 |
DG - 47.4 ±14.6 SG - 39.5 ± 4.2 INTERM - 70 |
Significant cavernous sinus invasion in SG vs DG | DG - GH: 33.3 ng/mL (mean) SG - GH: 59.9 ng/mL (mean) |
DG- densely granulated SG – sparsely granulated INTERM – intermediate granulated growth hormone tumors
Significant female predominance in SG group compared to DG
Patients with SG tumors are significantly younger compared to DG
SG tumors have larger diameter/tumor volume compared to DG tumors
The literature has also been inconsistent regarding correlation of GH tumor subtypes and tumor volume and invasive characteristics, partially due to a lack of a widely-accepted pituitary tumor staging criteria and small sample sizes. Similar to prior publications of Bakhtiar et al., and Brzana et al., we documented that SG tumors were significantly larger, compared to other tumor subtypes [29,30]. Although cavernous sinus invasion characteristics and optic chiasm compression did not statistically differ, we noted a trend towards higher rate of invasive characteristics in SG tumors. Other groups have used the Wilson modification of Hardy's criteria and/or Knosp criteria and suggested that SG tumors are both larger and have more suprasellar extension or diffuse sellar invasion [27,18,20,28,38,37]. In contrast, a recent study from Fougner et al., reported no difference in tumor volume or tumor invasiveness between GH tumor subtypes, but small numbers of SG tumors likely limited the statistical power in that study [31]. Collectively, the data suggest that SG tumors more often occur in a younger people, are larger, and likely have more invasive tumor characteristics.
Baseline IGF-1 levels, corrected for age and gender, and GH levels have been shown to correlate with GH tumor subtype, although random GH levels need to be interpreted with caution due to the normal physiologic pulsatility of GH [20,30,27,18,31]. In our study, similar to previous reports, baseline IGF-1 and random GH levels were similar across GH tumor subtypes [27,18,29,31,30,20]. Some authors have suggested that DG tumors have higher GH or IGF-1-to-tumor volume ratios [30,31], while others suggested the ratio is higher in SG tumors [25]. In our study, GH-to-tumor volume ratios were higher in DG tumor subtypes.
Surgical response based upon GH tumor subtype has been evaluated by several groups [29,28,30]. Mazal et al., reported that compared to DG tumors, patients with SG tumors more often have incomplete resections leading to re-operation[28]. Bakhtiar et al. reported surgical cure in 42.3% of SG (N=30) tumors compared to 60.4 % (N=111) in non-SG tumors (p=0.076) [30]. In contrast, a recent study reported similar rates of surgical cure across different subtypes between 50-55% [29], which might be attributed to relatively short-term mean follow up of approximately 1 year. In our patient cohort, there was a marked difference in post-operative remission with approximately 65% of non-SG tumors achieving cure after surgery, compared to 14% of SG tumors, independent of patients’ age and tumor size. Consistent with this finding, differences in the drop in post-surgical IGF-1 compared to baseline fold change from ULN between different GH tumor subtypes was observed. We confirmed that DG tumor subtype demonstrate a better response to medical therapy with SSAs than SG tumors [38,31,29,25].
We also evaluated the correlation of GH tumor subtype to response to GH receptor antagonist, pegvisomant (PEG). Patients with SG tumors showed a uniform rate of remission to PEG compared to their inconsistent response to SSAs. To our knowledge, this is the first report in the literature correlating GH tumor histologic subtypes to responses to medical therapy with PEG. We acknowledge that residual adenoma volume after surgical resection may affect response to anti-tumor therapies, but have eliminated variable surgical expertise and approaches as a confounding variable since a single neurosurgeon performed all procedures and with the intent of a gross-total resection whenever possible.
Although this, and previous reports, have suggested a differential clinical behavior between GH tumor histological subtypes, the underlying mechanisms for these responses are not clear. Several groups have reported downregulation of E-cadherin and epithelial splicing regulatory protein 1 (ESRP1) in SG tumors, as part of epithelial-to-mesenchymal transition pathway, but the functional significance remains unknown [20,30,31,39]. A recent study explored the roles of calcium, cAMP and zinc-finger protein (ZAC1)/ (pleomorphic adenoma gene-like 1) PLAGL1 pathways, but concluded none were driving the differences in GH tumor subtype behavior [36]. Several groups have suggested that decreased somatostatin receptor subtype 2a (SSTR2a), expression in SG compared to DG tumors predicted responsiveness to SSA [29,36]. Alternatively, SSA resistance in patients with high SSTR2 expression might be explained by aryl hydrocarbon receptor-interacting protein (AIP) expression, although differential AIP expression has not been reported based upon histologic subtype to date [40]. Several groups have explored SSTR5 expression in SG tumors but the findings have been inconsistent [36,38]. Currently, none of these markers have been validated for routine diagnostic assessment of GH adenomas. Immunohistochemistry for CAM5.2, on the other hand, is widely available and its use has been recommended for subtyping of GH adenomas in the recent College of American Pathologists guidelines [41].
Limitations of this study, and in conjunction with the relative rarity of the condition include: 1) the retrospective study design, 2) tumor size and invasion assessment limited to MRI imaging, 3) no data on the erosion of sellar floor, 3) the limited number of patients treated with pegvisomant, and 4) the lack of post- operative oral glucose tolerance tests in addition to normalization of IGF-1 levels to confirm remission or cure. Although the IGF-1 quantitative chemiluminescent immunoassay has been used recently, various assays have been used in the past. To circumvent the variability in the assays we used the fold change from the upper normal range of IGF-1 level. Strengths of the study include: 1) a relatively large sample size for a single institution, 2) lack of any medical pretreatment before surgery, 3) uniformity of the surgical approach, and radiographic and histological evaluations by a single, experienced neurosurgeon, radiologist and neuropathologist ,respectively, and 4) a relatively long follow-up period.
In conclusion, our data support the assertion that the GH tumor histologic subtyping is a useful tool to predict tumor behavior and associated response to surgical and medical therapies. Our study confirms prior literature that SG tumors are less likely to be cured by surgery independent of patient age and tumor size. The patients with residual SG adenomas are less likely to achieve remission with SSA, but consistently showed an excellent response to PEG. Although this is an expected response, based on the PEG mechanism of GH receptor antagonism, and not GH tumor-directed therapy, this is still an important clinical finding suggesting consideration of the potential need for a different prioritization of medical therapy based upon histologic subtyping. Further research is needed to identify additional molecular biomarkers to better predict treatment response for patients with acromegaly. Nevertheless, we have demonstrated that GH subtyping yields important clinical prognostic and therapeutic information, and suggest that it should be considered in the standard pathological evaluation of all GH-secreting pituitary adenomas.
Acknowledgments
The authors would like to thanks Molly Levitt for help in constructing initial patient database and participation in data collection.
Financial support: This work was supported by Investigator Initiated grant from Pfizer, Inc and VA Merit Review to MEW
Footnotes
Disclosures: The authors have nothing to disclose
References
- 1.Asa SL, Ezzat S. The pathogenesis of pituitary tumors. Annu Rev Pathol. 2009;4:97–126. doi: 10.1146/annurev.pathol.4.110807.092259. [DOI] [PubMed] [Google Scholar]
- 2.Melmed S. Acromegaly pathogenesis and treatment. J Clin Invest. 2009;119(11):3189–3202. doi: 10.1172/JCI39375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Melmed S, Colao A, Barkan A, Molitch M, Grossman AB, Kleinberg D, Clemmons D, Chanson P, Laws E, Schlechte J, Vance ML, Ho K, Giustina A. Guidelines for acromegaly management: an update. J Clin Endocrinol Metab. 2009;94(5):1509–1517. doi: 10.1210/jc.2008-2421. [DOI] [PubMed] [Google Scholar]
- 4.Clemmons DR, Chihara K, Freda PU, Ho KK, Klibanski A, Melmed S, Shalet SM, Strasburger CJ, Trainer PJ, Thorner MO. Optimizing control of acromegaly: integrating a growth hormone receptor antagonist into the treatment algorithm. J Clin Endocrinol Metab. 2003;88(10):4759–4767. doi: 10.1210/jc.2003-030518. [DOI] [PubMed] [Google Scholar]
- 5.Shimon I, Cohen ZR, Ram Z, Hadani M. Transsphenoidal surgery for acromegaly: endocrinological follow-up of 98 patients. Neurosurgery. 2001;48(6):1239–1243. doi: 10.1097/00006123-200106000-00008. discussion 1244-1235. [DOI] [PubMed] [Google Scholar]
- 6.Donangelo I, Melmed S. Treatment of acromegaly: future. Endocrine. 2005;28(1):123–128. doi: 10.1385/ENDO:28:1:123. [DOI] [PubMed] [Google Scholar]
- 7.Freda PU. Somatostatin analogs in acromegaly. J Clin Endocrinol Metab. 2002;87(7):3013–3018. doi: 10.1210/jcem.87.7.8665. [DOI] [PubMed] [Google Scholar]
- 8.Mercado M, Borges F, Bouterfa H, Chang TC, Chervin A, Farrall AJ, Patocs A, Petersenn S, Podoba J, Safari M, Wardlaw J. A prospective, multicentre study to investigate the efficacy, safety and tolerability of octreotide LAR (long-acting repeatable octreotide) in the primary therapy of patients with acromegaly. Clin Endocrinol (Oxf) 2007;66(6):859–868. doi: 10.1111/j.1365-2265.2007.02825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Melmed S, Cook D, Schopohl J, Goth MI, Lam KS, Marek J. Rapid and sustained reduction of serum growth hormone and insulin-like growth factor-1 in patients with acromegaly receiving lanreotide Autogel therapy: a randomized, placebo-controlled, multicenter study with a 52 week open extension. Pituitary. 2010;13(1):18–28. doi: 10.1007/s11102-009-0191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buchfelder M, Schlaffer S, Droste M, Mann K, Saller B, Brubach K, Stalla GK, Strasburger CJ. The German ACROSTUDY: past and present. Eur J Endocrinol. 2009;161(Suppl 1):S3–S10. doi: 10.1530/EJE-09-0350. [DOI] [PubMed] [Google Scholar]
- 11.van der Lely AJ, Biller BM, Brue T, Buchfelder M, Ghigo E, Gomez R, Hey-Hadavi J, Lundgren F, Rajicic N, Strasburger CJ, Webb SM, Koltowska-Haggstrom M. Long-term safety of pegvisomant in patients with acromegaly: comprehensive review of 1288 subjects in ACROSTUDY. J Clin Endocrinol Metab. 2012;97(5):1589–1597. doi: 10.1210/jc.2011-2508. [DOI] [PubMed] [Google Scholar]
- 12.Sandret L, Maison P, Chanson P. Place of cabergoline in acromegaly: a meta-analysis. J Clin Endocrinol Metab. 2011;96(5):1327–1335. doi: 10.1210/jc.2010-2443. [DOI] [PubMed] [Google Scholar]
- 13.Marazuela M, Ramos-Levi A, Sampedro-Nunez M, Bernabeu I. Cabergoline treatment in acromegaly: pros. Endocrine. 2014 doi: 10.1007/s12020-014-0206-1. [DOI] [PubMed] [Google Scholar]
- 14.Kasuki L, Vieira Neto L, Gadelha MR. Cabergoline treatment in acromegaly: cons. Endocrine. 2014 doi: 10.1007/s12020-014-0183-4. [DOI] [PubMed] [Google Scholar]
- 15.Nomikos P, Buchfelder M, Fahlbusch R. The outcome of surgery in 668 patients with acromegaly using current criteria of biochemical ‘cure’. Eur J Endocrinol. 2005;152(3):379–387. doi: 10.1530/eje.1.01863. [DOI] [PubMed] [Google Scholar]
- 16.Kovacs K, Horvath E, Corenblum B, Sirek AM, Penz G, Ezrin C. Pituitary chromophobe adenomas consisting of prolactin cells: a histologic, immunocytological and electron microscopic study. Virchows Arch A Pathol Anat Histol. 1975;366(2):113–123. doi: 10.1007/BF00433585. [DOI] [PubMed] [Google Scholar]
- 17.Horvath E, Kovacs K. Ultrastructural classification of pituitary adenomas. Can J Neurol Sci. 1976;3(1):9–21. doi: 10.1017/s0317167100025944. [DOI] [PubMed] [Google Scholar]
- 18.Yamada S, Aiba T, Sano T, Kovacs K, Shishiba Y, Sawano S, Takada K. Growth hormone-producing pituitary adenomas: correlations between clinical characteristics and morphology. Neurosurgery. 1993;33(1):20–27. doi: 10.1227/00006123-199307000-00003. [DOI] [PubMed] [Google Scholar]
- 19.Sano T, Ohshima T, Yamada S. Expression of glycoprotein hormones and intracytoplasmic distribution of cytokeratin in growth hormone-producing pituitary adenomas. Pathol Res Pract. 1991;187(5):530–533. doi: 10.1016/S0344-0338(11)80135-4. [DOI] [PubMed] [Google Scholar]
- 20.Obari A, Sano T, Ohyama K, Kudo E, Qian ZR, Yoneda A, Rayhan N, Mustafizur Rahman M, Yamada S. Clinicopathological features of growth hormone-producing pituitary adenomas: difference among various types defined by cytokeratin distribution pattern including a transitional form. Endocr Pathol. 2008;19(2):82–91. doi: 10.1007/s12022-008-9029-z. [DOI] [PubMed] [Google Scholar]
- 21.Neumann PE, Goldman JE, Horoupian DS, Hess MA. Fibrous bodies in growth hormone-secreting adenomas contain cytokeratin filaments. Archives of Pathology and Laboratory Medicine. 1985;109(6):505–508. [PubMed] [Google Scholar]
- 22.Kiseljak-Vassiliades K, Shafi S, Kerr JM, Phang TL, Kleinschmidt-DeMasters BK, Wierman ME. Clinical implications of growth hormone-secreting tumor subtypes. Endocrine. 2012;42(1):18–28. doi: 10.1007/s12020-012-9660-9. [DOI] [PubMed] [Google Scholar]
- 23.Karantza V. Keratins in health and cancer: more than mere epithelial cell markers. Oncogene. 2011;30(2):127–138. doi: 10.1038/onc.2010.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Besser GM, Burman P, Daly AF. Predictors and rates of treatment-resistant tumor growth in acromegaly. Eur J Endocrinol. 2005;153(2):187–193. doi: 10.1530/eje.1.01968. [DOI] [PubMed] [Google Scholar]
- 25.Bhayana S, Booth GL, Asa SL, Kovacs K, Ezzat S. The implication of somatotroph adenoma phenotype to somatostatin analog responsiveness in acromegaly. J Clin Endocrinol Metab. 2005;90(11):6290–6295. doi: 10.1210/jc.2005-0998. [DOI] [PubMed] [Google Scholar]
- 26.Ezzat S, Kontogeorgos G, Redelmeier DA, Horvath E, Harris AG, Kovacs K. In vivo responsiveness of morphological variants of growth hormone-producing pituitary adenomas to octreotide. Eur J Endocrinol. 1995;133(6):686–690. doi: 10.1530/eje.0.1330686. [DOI] [PubMed] [Google Scholar]
- 27.Bando H, Sano T, Ohshima T, Zhang CY, Yamasaki R, Matsumoto K, Saito S. Differences in pathological findings and growth hormone responses in patients with growth hormone-producing pituitary adenoma. Endocrinol Jpn. 1992;39(4):355–363. doi: 10.1507/endocrj1954.39.355. [DOI] [PubMed] [Google Scholar]
- 28.Mazal PR, Czech T, Sedivy R, Aichholzer M, Wanschitz J, Klupp N, Budka H. Prognostic relevance of intracytoplasmic cytokeratin pattern, hormone expression profile, and cell proliferation in pituitary adenomas of akromegalic patients. Clin Neuropathol. 2001;20(4):163–171. [PubMed] [Google Scholar]
- 29.Brzana J, Yedinak CG, Gultekin SH, Delashaw JB, Fleseriu M. Growth hormone granulation pattern and somatostatin receptor subtype 2A correlate with postoperative somatostatin receptor ligand response in acromegaly: a large single center experience. Pituitary. 2013;16(4):490–498. doi: 10.1007/s11102-012-0445-1. [DOI] [PubMed] [Google Scholar]
- 30.Bakhtiar Y, Hirano H, Arita K, Yunoue S, Fujio S, Tominaga A, Sakoguchi T, Sugiyama K, Kurisu K, Yasufuku-Takano J, Takano K. Relationship between cytokeratin staining patterns and clinico-pathological features in somatotropinomae. Eur J Endocrinol. 2010;163(4):531–539. doi: 10.1530/EJE-10-0586. [DOI] [PubMed] [Google Scholar]
- 31.Fougner SL, Casar-Borota O, Heck A, Berg JP, Bollerslev J. Adenoma granulation pattern correlates with clinical variables and effect of somatostatin analogue treatment in a large series of patients with acromegaly. Clin Endocrinol (Oxf) 2012;76(1):96–102. doi: 10.1111/j.1365-2265.2011.04163.x. [DOI] [PubMed] [Google Scholar]
- 32.Larkin S, Reddy R, Karavitaki N, Cudlip S, Wass J, Ansorge O. Granulation pattern, but not GSP or GHR mutation, is associated with clinical characteristics in somatostatin-naive patients with somatotroph adenomas. Eur J Endocrinol. 2013;168(4):491–499. doi: 10.1530/EJE-12-0864. [DOI] [PubMed] [Google Scholar]
- 33.Melmed S, Casanueva FF, Klibanski A, Bronstein MD, Chanson P, Lamberts SW, Strasburger CJ, Wass JA, Giustina A. A consensus on the diagnosis and treatment of acromegaly complications. Pituitary. 2013;16(3):294–302. doi: 10.1007/s11102-012-0420-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lundin P, Pedersen F. Volume of pituitary macroadenomas: assessment by MRI. J Comput Assist Tomogr. 1992;16(4):519–528. doi: 10.1097/00004728-199207000-00004. [DOI] [PubMed] [Google Scholar]
- 35.Carmichael JD, Bonert VS, Mirocha JM, Melmed S. The utility of oral glucose tolerance testing for diagnosis and assessment of treatment outcomes in 166 patients with acromegaly. J Clin Endocrinol Metab. 2009;94(2):523–527. doi: 10.1210/jc.2008-1371. [DOI] [PubMed] [Google Scholar]
- 36.Mayr BM, Buslei R, Theodoropoulou M, Stalla GK, Buchfelder M, Schoefl C. Molecular and functional properties of densely and sparsely granulated GH-producing pituitary adenomas. Eur J Endocrinol. 2013 doi: 10.1530/EJE-13-0134. [DOI] [PubMed] [Google Scholar]
- 37.Mori R, Inoshita N, Takahashi-Fujigasaki J, Joki T, Nishioka H, Abe T, Fujii T, Yamada S. Clinicopathological Features of Growth Hormone-Producing Pituitary Adenomas in 242 Acromegaly Patients: Classification according to Hormone Production and Cytokeratin Distribution. ISRN Endocrinol. 2013;2013:723432. doi: 10.1155/2013/723432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kato M, Inoshita N, Sugiyama T, Tani Y, Shichiri M, Sano T, Yamada S, Hirata Y. Differential expression of genes related to drug responsiveness between sparsely and densely granulated somatotroph adenomas. Endocr J. 2012;59(3):221–228. doi: 10.1507/endocrj.ej11-0177. [DOI] [PubMed] [Google Scholar]
- 39.Lekva T, Berg JP, Fougner SL, Olstad OK, Ueland T, Bollerslev J. Gene expression profiling identifies ESRP1 as a potential regulator of epithelial mesenchymal transition in somatotroph adenomas from a large cohort of patients with acromegaly. J Clin Endocrinol Metab. 2012;97(8):E1506–1514. doi: 10.1210/jc.2012-1760. [DOI] [PubMed] [Google Scholar]
- 40.Kasuki L, Vieira Neto L, Wildemberg LE, Colli LM, de Castro M, Takiya CM, Gadelha MR. AIP expression in sporadic somatotropinomas is a predictor of the response to octreotide LAR therapy independent of SSTR2 expression. Endocr Relat Cancer. 2012;19(3):L25–29. doi: 10.1530/ERC-12-0020. [DOI] [PubMed] [Google Scholar]
- 41.Nose V, Ezzat S, Horvath E, Kovacs K, Laws ER, Lloyd R, Lopes MB, Asa SL. Protocol for the examination of specimens from patients with primary pituitary tumors. Arch Pathol Lab Med. 2011;135(5):640–646. doi: 10.5858/2010-0470-SAR1.1. [DOI] [PubMed] [Google Scholar]