

Abstract
The study of protein interactions in the context of living cells can generate critical information about localization, dynamics, and interacting partners. This information is particularly valuable in the context of host-pathogen interactions. Many pathogen proteins function within host cells in a variety of way such as, enabling evasion of the host immune system and survival within the intracellular environment. To study these pathogen-protein host-cell interactions, several approaches are commonly used, including: in vivo infection with a strain expressing a tagged or mutant protein, or introduction of pathogen genes via transfection or transduction. Each of these approaches has advantages and disadvantages. We sought a means to directly introduce exogenous proteins into cells. Electroporation is commonly used to introduce nucleic acids into cells, but has been more rarely applied to proteins although the biophysical basis is exactly the same. A standard electroporator was used to introduce affinity-tagged bacterial effectors into mammalian cells. Human epithelial and mouse macrophage cells were cultured by traditional methods, detached, and placed in 0.4 cm gap electroporation cuvettes with an exogenous bacterial pathogen protein of interest (e.g. Salmonella Typhimurium GtgE). After electroporation (0.3 kV) and a short (4 hr) recovery period, intracellular protein was verified by fluorescently labeling the protein via its affinity tag and examining spatial and temporal distribution by confocal microscopy. The electroporated protein was also shown to be functional inside the cell and capable of correct subcellular trafficking and protein-protein interaction. While the exogenous proteins tended to accumulate on the surface of the cells, the electroporated samples had large increases in intracellular effector concentration relative to incubation alone. The protocol is simple and fast enough to be done in a parallel fashion, allowing for high-throughput characterization of pathogen proteins in host cells including subcellular targeting and function of virulence proteins.
Keywords: Immunology, Issue 95, electroporation, protein, transfection, expression, localization, confocal microscopy, Salmonella, effector
Introduction
Many Gram-negative bacteria employ specialized secretion systems to inject virulence-related proteins (referred to as effectors) directly into host cells 1-5. These effectors have a wide range of biological functions including: suppression of host immunity, cytoskeletal changes, modification of intracellular trafficking and signaling, transcriptional changes, and host proteasome alterations 6-9. The functions of some effectors are known, however the host targets and biochemical action(s) of many others remains to be determined. While comparing wild-type and recombinant bacterial infections is a valid approach to study intracellular effector virulence mechanisms, it is often advantageous to introduce an individual effector into the host cell. Thus, simple methods for introducing and characterizing bacterial effector proteins in the context of host cells is highly desirable.
Simplifying the experimental analysis with a single effector is critical as other effectors may have opposing or redundant functions. To accomplish this simplification, researchers have previously introduced macromolecules into cells by many different methods, including viral transduction 10, microinjection 11, scrape loading 12,13, cell fusion with chemically induced microinjection 14, proprietary protein “transfection” reagents 15, calcium phosphate precipitations 16, and electroporation 17-20. The introduced molecules range from nucleic acids including DNA, RNA, & RNAi species to proteins, cell-impermeable dyes, and antibodies for intracellular targets 21,22. Some methods have limitations including the type of macromolecule that can be introduced, and particular downstream analyses may be limited due to high cellular toxicity, damaging mechanisms of action, low efficacy, or introduction efficiency. Transfection, an often-used method for expressing bacterial genes in mammalian cells, also suffers the limitation that some relevant host cell types, such as macrophages and primary cells, are particularly resistant towards transfection. Beyond this, it is difficult to control the levels of bacterial protein produced upon introduction of foreign DNA.
Much work has established electroporation of nucleic acids into both bacterial and mammalian cells as a common laboratory technique; however, there is ongoing research into the best methods for delivering proteins into cells under physiological conditions. Reports on protein transfection are promising, but require expensive reagents and optimization. The desire to introduce potentially toxic bacterial effectors into a wide variety of cell targets with minimal cost led us to consider electroporation as a method for studying these proteins in vivo.
Protein electroporation 23-25 is a method to introduce proteins into living cells via electropermeabilization, also known as electro-transfection or electro-injection 26. This technique uses high-intensity electrical pulses to create pores in cell membranes. These reversible pores allow macromolecules that are normally excluded from intracellular space to enter the cell. Upon removal of the external electric field, the membrane can reseal, allowing the cell to retain molecules that passed through the pores 27,28.
A standard electroporator was used in this study to consistently introduce bacterial effectors into both mouse macrophage-like cells and human epithelial cells. The method is quick, efficient, and inexpensive, with no appreciable decrease in cellular viability. The introduced proteins can be visualized via immunofluorescence microscopy or used for functional assays. This has been demonstrated using green fluorescent protein (GFP) as a non-toxic standard, as well as two Salmonella effector proteins, SspH1 and GtgE. We propose protein electroporation as an additional tool in the repertoire for the study of bacterial virulence proteins and their functions in eukaryotic host cells.
Protocol
1. Prepare in Advance
Warm sterile phosphate buffered saline (PBS) to 37 °C.
Warm Dulbecco’s Modification of Eagle’s Medium (DMEM) and Minimal Essential Medium (MEM) supplemented with 10% fetal bovine serum (FBS), 100 I.U./ml penicillin, and 100 µg/ml streptomycin to 37 °C. Note: These represent Normal Growth Media (NGM) for RAW and HeLa cells respectively.
2. Preparation of Cells
- Grow RAW 264.7 cells to 70-90% confluency in NGM.
- Maintain cells at 37 °C in a humidified 95% air/5% CO2 atmosphere.
- Grow HeLa cells to 70-90% confluency in NGM.
- Maintain cells at 37 °C in a humidified 95% air/5% CO2 atmosphere.
Before collection, wash cell monolayer once with sterile PBS.
- Collect pre-confluent cells in a sterile conical tube.
- Gently scrape RAW cells with a rubber policeman in PBS, with repeated pipetting to disperse cell aggregations.
- Detach HeLa cells with 0.25% trypsin solution until visual examination shows dissociation from culture surface. For example, use 2 to 3 ml for a T-75 flask. Adjust volume accordingly to ensure trypsin solution covers entire growth surface.
- Quench dissociation reaction with NGM containing 10% FBS.
- Use at least twice the volume of NGM to trypsin, with repeated pipetting to disperse cell aggregations.
Lightly pellet cells by centrifugation at 900 x g for 4 min.
Resuspend in same volume as trypsin/quench solution using sterile PBS.
Count cells using hemocytometer or particle counter.
Lightly pellet cells by centrifugation at 900 x g for 4 min.
Resuspend in adequate volume of PBS for 5.5 x 106 to 6.0 x 106 cells/ml. Note: For example, one T-75 flask will yield approximately 7.5 x 106 cells 29.
Keep cell suspension on ice until electroporation.
3. Preparation for Electroporation
Pre-chill electroporation cuvettes (0.4 cm gap) on ice.
Turn on electroporation apparatus and set the voltage to 0.3 kV. NOTE: Capacitance and resistance were not adjustable settings on our electroporator (set at 10 μF and 600 Ω by the manufacturer).
Fill recovery plates with NGM and equilibrate in humidified 95% air/5% CO2 atmosphere at 37° C.
4. Electroporation
Place 400 µl of cell suspension in pre-chilled cuvette and add 20 µg of selected protein to cuvette (50 µg/ml).
Flick cuvette gently ~10 times to mix without damaging cells. Note: The cuvette may also be inverted several times to mix thoroughly, but do not pipet up and down or vortex to avoid damaging cells.
Dry outside of cuvette with paper towel or other wiper to avoid electrical arcing in the electroporator.
Electroporate sample at 0.3 kV for 1.5-1.7 msec. Note: This was typical for this study.
Immediately after electroporation flick cuvette gently ~10 times to mix thoroughly.
5. Plating of Cells
Store cuvette with electroporated cells on ice until ready to place in recovery plates.
- For most downstream analyses, wash cells 1x with pre-warmed NGM to remove extraneous effector protein.
- Remove cells for analysis and suspend in 3-5 ml of NGM.
- Pellet cells by centrifugation at 900 x g for 4 min.
- Resuspend in adequate volume of NGM for desired plate size (e.g. 2 ml for 35 mm dish).
- Remove appropriate amount of cells for downstream analysis.
- Example 1: plate into glass bottom dishes for microscopy analysis.
- Example 2: plate into cell culture plastic for protein analysis such as affinity purifications.
Allow cells to recover in equilibrated plates in humidified 95% air/5% CO2 atmosphere at 37° C for at least 4 hr.
6. Microscopy Analysis
6.1) Fixation/Immunofluorescence Staining
Wash cells 1x with sterile PBS after 4 hr recovery period.
Fix cells in 100% methanol for 2 min at room temperature. Use enough methanol to completely cover cells (e.g. 2 ml for 35 mm dish).
Wash 3x with sterile PBS.
- Permeabilize cells with 0.4% Triton X-100 in PBS for 15 min. For example, use 1 ml for a 35 mm diameter plate.
- Adjust length of permeabilization and strength of Triton X-100 according to epitope and location of target protein. Note: Best results will need to be empirically determined for each target, however the above conditions should be adequate for most cytosolic targets.
Block with 5% Bovine Serum Albumin (BSA) in PBS for 1 hr at RT. For example, use 1 ml for a 35 mm diameter plate.
Wash 3x with PBS.
- Incubate primary antibody in antibody binding solution (0.1% Triton X-100 and 1% BSA in PBS) overnight at 4 °C with gentle rocking. For example, use 0.5 ml for a 35 mm diameter plate.
- Follow manufacturer’s recommendations for antibody dilution. Note: For example, the streptavidin-binding peptide tag (SBP-tag) antibody was used at 1:1,000 dilution, while the PKN1 antibody was used at 1:200 dilution.
Wash 4x with PBS.
- Incubate with appropriate fluorescently conjugated secondary antibody in antibody binding solution for 1 hr at room temperature, protected from light.
- For example, use Alexa 488 or Alexa 647 for 1 hr at room temperature.
- Follow manufacturer’s recommendations for antibody dilution. (e.g. about 1:1,000 for this study).
- Add other stains as needed, for example, 5 μM Wheat germ agglutinnin (WGA) conjugated to Alexa 647 or DAPI according to manufacturer’s recommendation, for 1 hr at room temperature.
Wash 5x with PBS and store at 4 °C, protected from light until ready to image.
6.2) Confocal Microscopy and Image Analysis
- Image samples on an inverted confocal microscope using a 63x oil immersion objective.
- Image green channel using the 488nm line of an argon laser, with bandwidth emission between 492-542 nm.
- Image red channel using a 633 nm diode laser, with bandwidth emission between 640-718 nm.
- Image blue channel using a 405 nm diode laser, with bandwidth emission between 407-453 nm.
- Ensure the multiple channel z-stacks cover the entirety of the cellular volume.
Process images with appropriate image processing software.
7. Affinity Purification
Wash electroporated cells twice with 4 °C PBS after 4 hr recovery period.
Lyse with ~1.0 ml of lysis buffer (1% Triton X-100 with protease inhibitor cocktail and phosphatase inhibitor in PBS) on ice. Use inhibitors according to manufacturer instructions. Note: For example, both the phosphatase and protease inhibitors used in this study were supplied at 100x, but other formulations should work equally well.
Promptly scrape cells with rubber policeman and collect into conical tubes.
- Lyse by vigorous vortexing and sonication. Note: Other lysis methods may work equally as well, but efficiency will need to be empirically determined as to the suitability of assay requirements.
- Sonicate 3 x 30 sec with intermittent vortexing.
Centrifuge at 10,000 x g for 10 min at 4 °C to collect cellular debris and insoluble aggregates; save the supernatant.
Combine equal volumes (~1.0 ml) of electroporated cell lysate with 50 µl of Streptavidin agarose resin suspension at 4 °C overnight with end-over-end rotation. Note: This resin captures the electroporated protein (and associated complexes) via its streptavidin-binding peptide affinity tag.
Centrifuge at 2,500 x g for 2 min and discard supernatant.
Wash twice with 40 bed volumes (~1 ml) PBS.
Add 30 µl 4x LDS loading buffer (141 mM Tris base, 2% LDS, 10% glycerol, 0.51 mM EDTA, 0.22 mM SERVA Blue G, 0.175 mM phenol red, pH 8.5), 20 µl diH2O, and 1 µl of 0.5 M tris(2-carboxyethyl)phosphine (TCEP), allowing for some volume to remain in beads.
Heat at 95 °C for 10 min and cool on ice.
Spin >10,000 x g at 4 °C for 5 min.
Collect supernatant.
Perform a western blot using appropriate antibodies Note: Here, anti-PKN1 primary antibody is used.
Representative Results
As an initial proof of concept, purified green fluorescent protein was successfully introduced into mammalian cells using electroporation. GFP, an approximate 27 kD a molecular weight protein is commonly introduced into mammalian cells (normally expressed from plasmid DNA) as a molecular biology tool without significant cellular toxicity. HeLa cells were incubated (Figure 1A) or electroporated (Figure 1B) with 25 µg/ml GFP, followed by immunofluorescence confocal microscopy to check for fluorescent GFP signal. To demonstrate that the GFP was inside the cellular cytosol, the cells were also stained with wheat germ agglutinin (WGA) to delineate the cytosolic boundary (red) as well as a nucleic acid stain, DAPI (blue) to indicate the nucleus. Intracellular GFP signal was observed only upon electroporation, while no intracellular protein was observed in cells incubated with GFP. Similar results were observed with RAW264.7 mouse macrophage-like cells (Figure 1C and 1D). Note that WGA is a lectin that preferentially binds to residues in the plasma membrane and thus can exhibit punctate staining based on the length of incubation. Compare Figure 1A and Figure 2A, where Figure 2A was incubated for a longer duration than Figure 1A.
Electroporation was then extended to a tagged, purified Salmonella effector, GtgE. GtgE, a known virulence factor 30, was discovered by our group to be secreted into host cells 31, and was recently shown to be a cysteine protease 32. HeLa cells were incubated or electroporated with 50 µg/ml GtgE. For immunofluorescence analysis, the cells were stained with an antibody against the streptavidin-binding peptide affinity tag on GtgE. The cells were also stained with WGA and DAPI. In incubated HeLa cells (Figure 2A), there is no intracellular GtgE as visualized by a lack of green fluorescent foci. In contrast, electroporated HeLa cells (Figure 2B) show significant fluorescent intracellular signal indicating effector protein had entered the cells due to the electroporation process. RAW cells showed a slightly increased propensity to accumulate protein on the cellular surface during incubation (Figure 2C), but intracellular signal was seen only upon electroporation (Figure 2D).
Determining the limits of detection for visualizing sub-cellular localization via confocal microscopy was important to ensure enough protein entered the cell, while avoiding overloading the cell with potentially toxic bacterial effectors. In Figure 3, GtgE was electroporated into RAW macrophage-like cells at 2.5, 25, and 50 μg/ml. The cells were then fixed and stained with an antibody against the tag on GtgE. Only a very small number of foci were observed at 2.5 μg/ml GtgE (Figure 3A), with increased numbers of foci apparent at 25 μg/ml, (Figure 3B), but the greatest number of distinct foci were seen at the maximum protein concentration tested, 50 μg/ml (Figure 3C). Similar staining patterns were observed between the samples indicating at these protein concentrations intracellular protein could be easily verified by confocal microscopy. Protein concentrations above 50 μg/ml were not tested because as the concentration of protein increased, so did the propensity for the target protein to become adsorbed to the surface of the cells. To avoid these aggregates of membrane associated protein, it became necessary to pool two electroporation cuvettes to obtain enough host interacting protein to visualize on a western blot (Figure 6, far right lane).
To further demonstrate that the introduced protein was not simply adsorbed to the surface of the cell, consecutive optical sections were imaged at focal planes every 0.35 to 0.43 micrometers (Z-stacks) using confocal microscopy. RAW cells were electroporated with 50 µg/ml GtgE and stained as described above (Figure 4, A and B). The Z-stacks showed that GtgE was indeed intracellular and the foci extend from the bottom (Figure 4A, plane 6 of 36) to the top of the cell (Figure 4B, plane 26 of 36). Similar results were obtained for all electroporated proteins. The intrinsic fluorescent profile of control cell populations with and without effector protein or electroporation was examined by fluorescent confocal microscopy, and was found to be negligible.
Additionally, the electroporated protein was examined to see if it was targeted for degradation via endocytic pathways. Cells were stained for Ras-related protein 5A (Rab5), a marker for early endosomes (Figure 4C), or Lysosome-associated membrane protein 1 (Lamp1), a marker for late endosomes and lysosomes (Figure 4D). Co-localization between the electroporated protein and these cellular markers would indicate a close physical interaction between them (i.e. that the electroporated protein was inside the endosomes/lysosomes). Confocal microscopy showed that the electroporated protein (GFP or GtgE) did not co-localize with LAMP1 or Rab5. It was inferred from this that introduced protein was not directly endocytosed into the cells or targeted to the endocytotic pathway within four hours of treatment.
Exogenous protein introduction can have cellular effects over the course of several hours or days, and thus it is often beneficial to examine the persistence of introduced proteins. To determine how long an electroporated protein would persist without degradation after electroporation. Based on visualization of attachment and cell spreading, 4 hr was determined to be the approximate minimum recovery time for electroporated cells. To temporally observe protein persistence a time-course experiment was performed, staining the cells 4, 24, or 96 hr after electroporation. After 4 hr (Figure 5A), when cell morphology suggested recovery, there was an appreciable amount of intracellular protein. After 24 hr (Figure 5B), there was still substantial electroporated protein inside the cells. Even when the time after electroporation was extended to 96 hr before fixation and staining (Figure 5C) there were observable foci albeit at a reduced apparent abundance, demonstrating protein persistence days after initial treatment.
Two important caveats were noted during the course of these experiments. RAW cells exhibited a greater tendency than HeLa cells to accumulate exogenous proteins on the cellular surface irrespective of incubation (Figure 6A) or electroporation (Figure 6B). Despite this, all electroporated samples had increased internal protein (Figures 1, 2, 5B). Additionally, the membrane-associated protein disappeared over time. A second caveat was a small population of cells exhibiting a phenotype consisting of smaller, rounded cells loaded with high amounts of exogenous proteins in both control (incubated) and treated (electroporated) cells (Figure 6 C and D, incubation samples shown). The nuclei of these cells showed condensed chromatin based on DAPI staining, and filled the entirety of the cellular volume, suggestive of apoptosis. It is therefore possible that apoptotic cells (arising as a normal part of the cell cycle or due to harvest/treatment) have a propensity to absorb protein from the buffer.
To demonstrate that electroporated proteins were functional and could localize correctly inside the host cell, co-localization and protein-protein interaction between the Salmonella effector protein SspH1 and its known host target, protein kinase N1 (PKN1) 33 was shown. After electroporation, the physical interaction between SspH1 and PKN1 was verified by an affinity based immunoprecipitation followed by western blot (Figure 7A). Co-localization was also observed by confocal microscopy, which indicates the fluorophores are close enough spatially to overlap 34. After electroporation with 50 µg/ml SspH1, HeLa cells (Figure 7B) were fixed and stained for SspH1 (green) and PKN1 (red). At low laser power distinct foci (yellow) were observed in the nucleus indicating SspH1 and PKN1 were physically close enough to interact. This interaction has been established in the literature; however this study is the first to show co-localization in the nucleus.
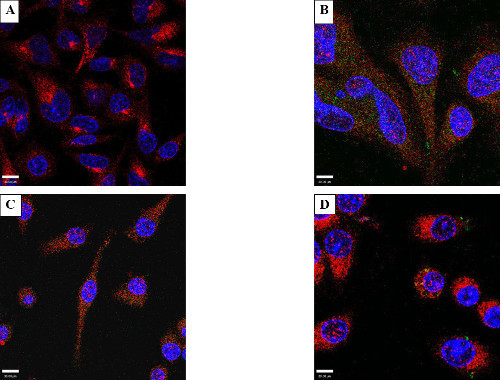 Figure 1: Electroporation of GFP - HeLa or RAW 264.7 cells. Cells were incubated or electroporated with 25 µg/ml of purified green fluorescent protein (GFP) and stained with anti-GFP antibody (Green), DAPI, a nuclear specific probe (Blue), and WGA (Red) to delineate the cytosolic boundary. Incubated (A) HeLa cells show an absence of green fluorescence indicating a lack of internalization of GFP. (B) shows a representative photomicrograph of electroporated GFP. Note the appearance of intracellular foci indicating GFP had entered the cells. (C) and (D) are representative images of RAW cells incubated (C), or electroporated (D) with 25 µg/ml of purified green fluorescent protein.
Figure 1: Electroporation of GFP - HeLa or RAW 264.7 cells. Cells were incubated or electroporated with 25 µg/ml of purified green fluorescent protein (GFP) and stained with anti-GFP antibody (Green), DAPI, a nuclear specific probe (Blue), and WGA (Red) to delineate the cytosolic boundary. Incubated (A) HeLa cells show an absence of green fluorescence indicating a lack of internalization of GFP. (B) shows a representative photomicrograph of electroporated GFP. Note the appearance of intracellular foci indicating GFP had entered the cells. (C) and (D) are representative images of RAW cells incubated (C), or electroporated (D) with 25 µg/ml of purified green fluorescent protein.
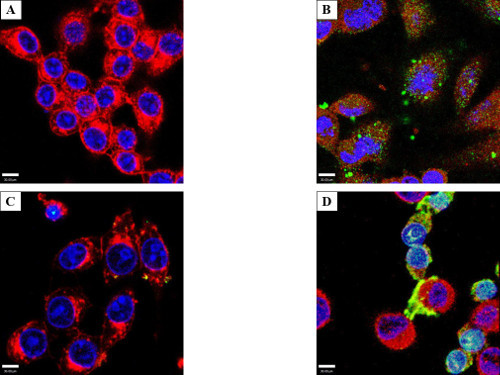 Figure 2: Electroporation of Salmonella effector GtgE - HeLa cells. Cells were incubated (A) or electroporated (B) with 50 µg/ml of an affinity tagged Salmonella effector, GtgE, and stained with an anti-effector tag antibody (Green), DAPI, a nuclear mask (Blue), and WGA (Red) to delineate the cytosolic boundary. In (A), incubated HeLa cells show an absence of green fluorescence indicating a lack of internalization of GtgE. (B) shows a representative photomicrograph of electroporated GtgE, showing intracellular electroporated effector protein. (C) and (D) are representative images of RAW cells that were either incubated or electroporated, respectively, in the same fashion with 50 µg/ml of Salmonella GtgE. Light blue is the combination, or overlay, of red fluorescence from the WGA and green fluorescence from the secondary antibody.
Figure 2: Electroporation of Salmonella effector GtgE - HeLa cells. Cells were incubated (A) or electroporated (B) with 50 µg/ml of an affinity tagged Salmonella effector, GtgE, and stained with an anti-effector tag antibody (Green), DAPI, a nuclear mask (Blue), and WGA (Red) to delineate the cytosolic boundary. In (A), incubated HeLa cells show an absence of green fluorescence indicating a lack of internalization of GtgE. (B) shows a representative photomicrograph of electroporated GtgE, showing intracellular electroporated effector protein. (C) and (D) are representative images of RAW cells that were either incubated or electroporated, respectively, in the same fashion with 50 µg/ml of Salmonella GtgE. Light blue is the combination, or overlay, of red fluorescence from the WGA and green fluorescence from the secondary antibody.
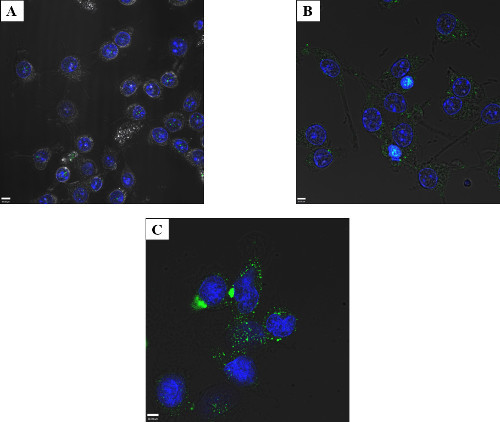 Figure 3: Titration of electroporated protein – RAW macrophage-like cells. Cells were electroporated with 2.5 μg/ml (A), 25 μg/ml (B), or 50 μg/ml (C) Salmonella effector GtgE and allowed to recover for 4 hr. The cells were fixed and stained with an anti-SBP-tag antibody (green) and the nuclei mask, DAPI (blue). It is possible to visualize fluorescent foci, indicative of intracellular GtgE at 2.5 μg/ml via confocal microcopy, although better results were obtained when higher starting amount of protein were used. Satisfactory results (including intracellular protein easily observed by confocal microscopy without excessive membrane associated aggregates) were obtained at 50 μg/ml for GtgE and thus no greater concentrations were tested.
Figure 3: Titration of electroporated protein – RAW macrophage-like cells. Cells were electroporated with 2.5 μg/ml (A), 25 μg/ml (B), or 50 μg/ml (C) Salmonella effector GtgE and allowed to recover for 4 hr. The cells were fixed and stained with an anti-SBP-tag antibody (green) and the nuclei mask, DAPI (blue). It is possible to visualize fluorescent foci, indicative of intracellular GtgE at 2.5 μg/ml via confocal microcopy, although better results were obtained when higher starting amount of protein were used. Satisfactory results (including intracellular protein easily observed by confocal microscopy without excessive membrane associated aggregates) were obtained at 50 μg/ml for GtgE and thus no greater concentrations were tested.
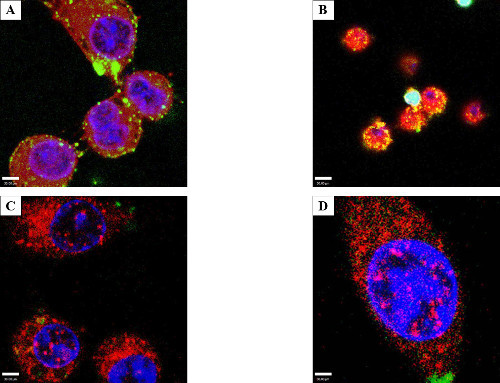 Figure 4: Protein internalization and lack of co-localization with endosome markers. Consecutive optical slices (z-stacks) obtained by confocal microscopy to visualize if the electroporated protein was internal. RAW cells were electroporated with 50 µg/ml GtgE. (A) shows optical slice 6 of 36, the z-stack (2.1 micrometers) above the bottom of the dish. (B) shows the same field of view but at slice 26 of 36. The intracellular foci extend throughout the cell indicating that the protein is truly intracellular and not aggregated on the surface of the cell. Light blue is the combination of red WGA, green secondary antibody, and blue DAPI. Co-localization assay with markers of endosomes/lysosomes, Rab5, (C) or a lysosome marker, LAMP1 (D). The effector protein has been stained green, the markers (Rab5 or LAMP1) stained red, and the nuclei delineated with DAPI, blue. There is no evidence of co-localization with either LAMP1 or Rab5 indicating that the protein was not phagocytized.
Figure 4: Protein internalization and lack of co-localization with endosome markers. Consecutive optical slices (z-stacks) obtained by confocal microscopy to visualize if the electroporated protein was internal. RAW cells were electroporated with 50 µg/ml GtgE. (A) shows optical slice 6 of 36, the z-stack (2.1 micrometers) above the bottom of the dish. (B) shows the same field of view but at slice 26 of 36. The intracellular foci extend throughout the cell indicating that the protein is truly intracellular and not aggregated on the surface of the cell. Light blue is the combination of red WGA, green secondary antibody, and blue DAPI. Co-localization assay with markers of endosomes/lysosomes, Rab5, (C) or a lysosome marker, LAMP1 (D). The effector protein has been stained green, the markers (Rab5 or LAMP1) stained red, and the nuclei delineated with DAPI, blue. There is no evidence of co-localization with either LAMP1 or Rab5 indicating that the protein was not phagocytized.
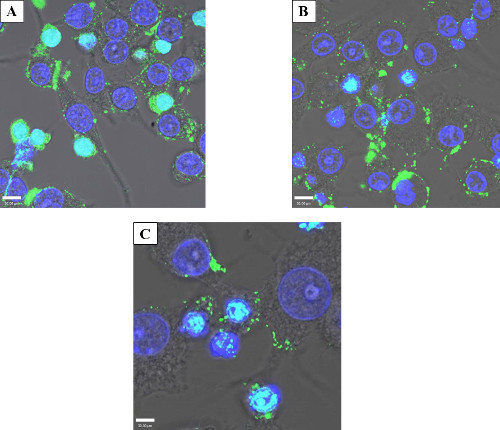 Figure 5: Protein persistence after electroporation. Confocal micrograph showing the persistence of electroporated GtgE over time. 50 µg/ml GtgE was electroporated into HeLa cells. The cells were then fixed and stained with an anti-SBP-tag antibody (green) and the nuclei mask, DAPI (blue). 4 hr (A) was determined to be the minimum amount of time to allow cells to recover and as expected shows maximum intracellular protein. After 24 hr (B) and 96 hr (C), green foci, both intracellular and on the cellular surface, show effector persistence indicating that the protein is not degraded days after the initial treatment. The light blue color in this image is the overlay of blue DAPI and the green antibody staining.
Figure 5: Protein persistence after electroporation. Confocal micrograph showing the persistence of electroporated GtgE over time. 50 µg/ml GtgE was electroporated into HeLa cells. The cells were then fixed and stained with an anti-SBP-tag antibody (green) and the nuclei mask, DAPI (blue). 4 hr (A) was determined to be the minimum amount of time to allow cells to recover and as expected shows maximum intracellular protein. After 24 hr (B) and 96 hr (C), green foci, both intracellular and on the cellular surface, show effector persistence indicating that the protein is not degraded days after the initial treatment. The light blue color in this image is the overlay of blue DAPI and the green antibody staining.
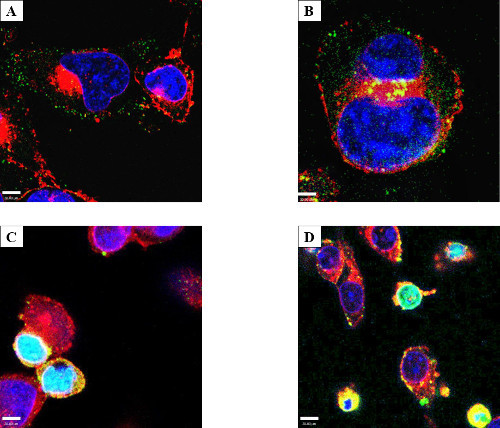 Figure 6: Cell surface protein aggregation and an electroporation phenotype. RAW macrophage-like cells were either incubated (A) or electroporated (B) with 50 µg/ml GtgE and stained with anti-SBP-tag antibody (Green), WGA (Red), and with DAPI (Blue). Both RAW 264.7 and to a lesser degree HeLa cells, tended to aggregate effector on the surface of the cell; all electroporated cells were shown to have increased internal protein (HeLa not shown). Yellow is the overlay of red from WGA and green from target protein. RAW (C) and HeLa (D) cells showing an altered phenotype after incubation with GtgE (shown). Several experiments showed a phenotype consisting of smaller cells with differential DAPI staining and a loss of cytoplasm. Light blue is the overlay from the red WGA, green secondary antibody, and blue DAPI. Confocal microscopy was used to show the target protein accumulating in the nucleus. Since this phenotype of exogenous protein-laden cells appeared after both incubation and electroporation, one could hypothesize this phenotype to be suggestive of apoptosis.
Figure 6: Cell surface protein aggregation and an electroporation phenotype. RAW macrophage-like cells were either incubated (A) or electroporated (B) with 50 µg/ml GtgE and stained with anti-SBP-tag antibody (Green), WGA (Red), and with DAPI (Blue). Both RAW 264.7 and to a lesser degree HeLa cells, tended to aggregate effector on the surface of the cell; all electroporated cells were shown to have increased internal protein (HeLa not shown). Yellow is the overlay of red from WGA and green from target protein. RAW (C) and HeLa (D) cells showing an altered phenotype after incubation with GtgE (shown). Several experiments showed a phenotype consisting of smaller cells with differential DAPI staining and a loss of cytoplasm. Light blue is the overlay from the red WGA, green secondary antibody, and blue DAPI. Confocal microscopy was used to show the target protein accumulating in the nucleus. Since this phenotype of exogenous protein-laden cells appeared after both incubation and electroporation, one could hypothesize this phenotype to be suggestive of apoptosis.
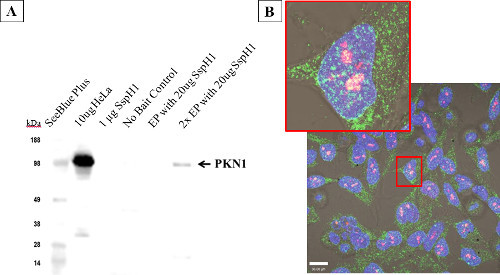 Figure 7: Host protein interactions with electroporated Salmonella SspH1 - HeLa cells. Cells were electroporated at the listed concentrations with SspH1, allowed to recover for 4 hr before performing a modified affinity purification followed by western blot (A) to enrich for and detect the known eukaryotic interacting partner: serine/threonine protein kinase N1 (PKN1). Note that the far right lane (2x EP) includes 2 pooled cuvettes, as the signal from one cuvette blended into background, yet PKN1 was still enriched above the lack of binding seen in the no-bait control. The western blot was run with native HeLa lysate, purified SspH1, and the affinity purification samples before being probed with a monoclonal antibody to PKN1. Target detection was obtained via a horseradish peroxidase conjugated secondary antibody with reactivity against the isotype of the primary antibody. HeLa cells (B) showing co-localization (yellow) between PKN1 (red) and SspH1 (green) via by confocal microscopy after electroporation with 50 µg/ml SspH1. This optical overlap indicates that the effector and the host protein are close enough to physically interact. The fact that the interaction was shown in the nucleus for the first time, offers additional support that the protein was trafficked correctly by the host.
Figure 7: Host protein interactions with electroporated Salmonella SspH1 - HeLa cells. Cells were electroporated at the listed concentrations with SspH1, allowed to recover for 4 hr before performing a modified affinity purification followed by western blot (A) to enrich for and detect the known eukaryotic interacting partner: serine/threonine protein kinase N1 (PKN1). Note that the far right lane (2x EP) includes 2 pooled cuvettes, as the signal from one cuvette blended into background, yet PKN1 was still enriched above the lack of binding seen in the no-bait control. The western blot was run with native HeLa lysate, purified SspH1, and the affinity purification samples before being probed with a monoclonal antibody to PKN1. Target detection was obtained via a horseradish peroxidase conjugated secondary antibody with reactivity against the isotype of the primary antibody. HeLa cells (B) showing co-localization (yellow) between PKN1 (red) and SspH1 (green) via by confocal microscopy after electroporation with 50 µg/ml SspH1. This optical overlap indicates that the effector and the host protein are close enough to physically interact. The fact that the interaction was shown in the nucleus for the first time, offers additional support that the protein was trafficked correctly by the host.
Discussion
Secreted effectors from pathogenic bacteria have evolved to function within the host cell environment and thus it is helpful to study them in situ within the host. Introduction of specific effectors of interest into host cells allows the relevant pathogen-host interactions to be studied in isolation without interference from other bacterial proteins. The goal was to explore electroporation as a means to introduce bacterial effector proteins into eukaryotic host cells, thereby avoiding some of the challenges associated with transfection or transduction. Green fluorescent protein was used as a control and Salmonella effector proteins were tested. Because of interest in bacterial pathogens that target host epithelial cells as well as immune cells, a human epithelial-like cell line (HeLa) and mouse macrophage-like cells (RAW 264.7) were used. Electroporation can deliver exogenous proteins into host cells with no appreciable decrease in cell viability, and the delivered proteins were detectable in cells for up to four days.
To isolate the effects of electroporated protein, pure protein is required. In this study, proteins were expressed in E. coli strain BL21 with N-terminal polyhistidine and streptavidin binding-peptide tags. Proteins were purified with Ni-NTA resin, assayed for concentration (generally between 5-25 mg/ml) and purity, and stored at -80 °C until electroporation. Commercially produced proteins would also be acceptable for electroporation, as they tend to be of high purity and well characterized.
Key steps in this protocol included completely removing the cell culture medium by washing and suspending cells in protein-free buffer. This ensures that any downstream phenotypes observed are due to the exogenous protein of interest and not to proteins present in the culture medium. Better efficiency of electroporation was observed when cell density was higher (e.g. 6 x 106 vs. 1 x 106 cells), though the best cell number will vary with cell size and type. Another critical step in the experiment was to allow cells time to recover and reattach to dishes; this was necessary because adherent cells were used in this study. A recovery period should be less important for suspension cells, although it may be necessary to give proteins time for proper intracellular trafficking, protein-protein interactions, or enzymatic activity, depending on the nature of the intended study. Since delivered proteins were observed inside cells for up to 96 hr, there is much room for adjustment to the incubation period prior to microscopy visualization or cellular assay.
Several variables in this protocol may be optimized for different cell types, protein targets, or downstream analysis schemes. The amount of protein to be electroporated can be fine-tuned for the desired intracellular concentration. For visualization of protein localization, as little as 1 µg can be used. For functional studies, higher starting amounts of proteins may be needed. In addition, the amount of time for post-electroporation cell recovery can be shorted or extended. Labile protein targets or fast downstream processes would require a shorter incubation time. Also, the amount of cells plated can be adjusted beyond the suggestions here. Cells may be plated more sparsely if the introduced protein is to be monitored by microscopy, or plated more densely if the electroporated protein is to be recovered for applications such as western blots.
While electroporation is a simple and straightforward method for introducing proteins into living cells, there are some limitations to the technique. The method requires highly pure proteins in microgram amounts. As little as 1 µg was electroporated and visualized with confocal microscopy, but higher starting amounts of protein gave better visual results. While protein function was not assessed at all concentrations after electroporation, protein concentrations equal to or greater than 50 μg/ml were required for adequate signal for western blot. Also, due to the size constraints of the electroporation cuvettes, scaling up may require processing of multiple cuvettes of cells. However, considering that the electroporation process takes mere seconds once cells are prepared, parallel processing requires little extra time and effort.
If the introduced protein is to be visualized by western blot, microscopy, or used for affinity purification, the protein should have a tag 35 for affinity purification or an available antibody for detection. However, for unknown reasons, some interactions known from the literature or other biochemical methods (data not shown) were not able to be recapitulated after electroporation. It may be that the host recognizes some electroporated proteins as foreign and quickly marks them for proteasome degradation.
Electroporation holds several advantages over other existing methods for protein delivery. Compared to microinjection, electroporation is much faster and simpler, and a large number of cells can be treated at once with almost 100 percent viability for the tested conditions. Electroporation is also cheaper than protein transfection with commercially available reagents. DNA transfections are often used to study bacterial effector proteins in host cells; however, this causes the bacterial proteins to be synthesized non-ideally by the host. On the other hand, electroporation of bacterial proteins removes the reliance on mammalian protein expression machinery, allowing for a better representation of the infectious process where effector proteins are expressed by the bacteria.
Several applications could use protein electroporation as a method in the study of bacterial-host interactions. First, primary cells that are often difficult to transfect may be more amenable to electroporation for protein delivery. This will allow for the study of effector function in more biologically relevant cell types. Electroporation also gives the option of multiplexing proteins and/or treatments. For example, multiple proteins may be introduced simultaneously, or drugs and other small molecules can be delivered alongside proteins. Most important for obtaining a functional understanding of the effect of the introduced proteins, protein electroporation could be coupled with assays for protein function and interactions, such as affinity purification, western blots against known targets, whole-cell expression analysis, and microscopy to determine subcellular localization of the introduced protein. Hence, this method has great potential as a tool for elucidating bacterial pathogen biology alongside other cellular and molecular biology techniques.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by NIGMS, National Institutes of Health (GM094623). Significant portions of this work were performed in the Environmental Molecular Sciences Laboratory, a DOE/BER national scientific user facility located at the Pacific Northwest National Laboratory (PNNL). PNNL is operated for the DOE by Battelle under Contract DE-AC05-76RLO1830.
References
- Mota LJ, Cornelis GR. The bacterial injection kit: type III secretion systems. Annals of Medicine. 2005;37:234–249. doi: 10.1080/07853890510037329. [DOI] [PubMed] [Google Scholar]
- Galan JE, Collmer A. Type III secretion machines: bacterial devices for protein delivery into host cells. Science. 1999;284:1322–1328. doi: 10.1126/science.284.5418.1322. [DOI] [PubMed] [Google Scholar]
- Hueck CJ. Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiology and molecular biology reviews : MMBR. 1998;62:379–433. doi: 10.1128/mmbr.62.2.379-433.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelis GR, Van Gijsegem F. Assembly and function of type III secretory systems. Annual Review of Microbiology. 2000;54:735–774. doi: 10.1146/annurev.micro.54.1.735. [DOI] [PubMed] [Google Scholar]
- He SY. Type III protein secretion systems in plant and animal pathogenic bacteria. Annual Review of Phytopathology. 1998;36:363–392. doi: 10.1146/annurev.phyto.36.1.363. [DOI] [PubMed] [Google Scholar]
- Dean P. Functional domains and motifs of bacterial type III effector proteins and their roles in infection. FEMS Microbiology Reviews. 2011;35:1100–1125. doi: 10.1111/j.1574-6976.2011.00271.x. [DOI] [PubMed] [Google Scholar]
- Espinosa A, Alfano JR. Disabling surveillance: bacterial type III secretion system effectors that suppress innate immunity. Cellular Microbiology. 2004;6:1027–1040. doi: 10.1111/j.1462-5822.2004.00452.x. [DOI] [PubMed] [Google Scholar]
- Orchard RC, Alto NM. Mimicking GEFs: a common theme for bacterial pathogens. Cellular Microbiology. 2012;14:10–18. doi: 10.1111/j.1462-5822.2011.01703.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan JE. Common themes in the design and function of bacterial effectors. Cell Host Microbe. 2009;5:571–579. doi: 10.1016/j.chom.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis BL, et al. A survey of ex vivo/in vitro transduction efficiency of mammalian primary cells and cell lines with Nine natural adeno-associated virus (AAV1-9) and one engineered adeno-associated virus serotype. Virology Journal. 2013;10:74. doi: 10.1186/1743-422X-10-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreelatha A, et al. Vibrio effector protein, VopQ, forms a lysosomal gated channel that disrupts host ion homeostasis and autophagic flux. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:11559–11564. doi: 10.1073/pnas.1307032110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil PL, Murphy RF, Lanni F, Taylor DL. A method for incorporating macromolecules into adherent cells. The Journal of Cell Biology. 1984;98:1556–1564. doi: 10.1083/jcb.98.4.1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafon M, Lafage M. Antiviral activity of monoclonal antibodies specific for the internal proteins N and NS of rabies virus. The Journal of General Virology. 1987;68(Pt. 12):3113–3123. doi: 10.1099/0022-1317-68-12-3113. [DOI] [PubMed] [Google Scholar]
- Kriegler MP, Livingston DM. Chemically facilitated microinjection of proteins into intact monolayers of tissue culture cells) Somatic Cell Genetics. 1977;3:603–610. doi: 10.1007/BF01539068. [DOI] [PubMed] [Google Scholar]
- Nossa CW, et al. Activation of the abundant nuclear factor poly(ADP-ribose) polymerase-1 by Helicobacter pylori. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:19998–20003. doi: 10.1073/pnas.0906753106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan M, Schallhorn A, Wurm FM. Transfecting mammalian cells: Optimization of critical parameters affecting calcium-phosphate precipitate formation. Nucleic Acids Research. 1996;24:596–601. doi: 10.1093/nar/24.4.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winegar RA, Phillips JW, Youngblom JH, Morgan WF. Cell Electroporation Is a Highly Efficient Method for Introducing Restriction Endonucleases into Cells. Mutation Research. 1989;225:49–53. doi: 10.1016/0165-7992(89)90032-8. [DOI] [PubMed] [Google Scholar]
- Cortes F, Ortiz T. Chromosome damage induced by restriction endonucleases recognizing thymine-rich DNA sequences in electroporated CHO cells. International Journal of Radiation Biology. 1992;61:323–328. doi: 10.1080/09553009214551001. [DOI] [PubMed] [Google Scholar]
- Cortes F, Ortiz T. Induction of chromosomal aberrations in the CHO mutant EM9 and its parental line AA8 by EcoRI restriction endonuclease: electroporation experiments. Mutation Research. 1991;246:221–226. doi: 10.1016/0027-5107(91)90125-8. [DOI] [PubMed] [Google Scholar]
- Baron S, Poast J, Rizzo D, McFarland E, Kieff E. Electroporation of antibodies, DNA, and other macromolecules into cells: a highly efficient method. Journal of Immunological Methods. 2000;242:115–126. doi: 10.1016/s0022-1759(00)00242-8. [DOI] [PubMed] [Google Scholar]
- Lewis R. Electroporation edges toward clinic for both gene therapy and drug delivery. Genetic Engineering and Biotechnology News. 1997;17 [Google Scholar]
- Kotzamanis G, et al. CFTR expression from a BAC carrying the complete human gene and associated regulatory elements. Journal of Cellular and Molecular Medicine. 2009;13:2938–2948. doi: 10.1111/j.1582-4934.2008.00433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti R, Wylie DE, Schuster SM. Transfer of monoclonal antibodies into mammalian cells by electroporation. The Journal of Biological Chemistry. 1989;264:15494–15500. [PubMed] [Google Scholar]
- Graziadei L, Burfeind P, Bar-Sagi D. Introduction of unlabeled proteins into living cells by electroporation and isolation of viable protein-loaded cells using dextran-fluorescein isothiocyanate as a marker for protein uptake. Analytical Biochemistry. 1991;194:198–203. doi: 10.1016/0003-2697(91)90168-s. [DOI] [PubMed] [Google Scholar]
- Wilson AK, Horwitz J, De Lanerolle P. Evaluation of the electroinjection method for introducing proteins into living cells. The American Journal of Physiology. 1991;260:C355–C363. doi: 10.1152/ajpcell.1991.260.2.C355. [DOI] [PubMed] [Google Scholar]
- Prasanna GL, Panda T. Electroporation: Basic principles, practical considerations and applications in molecular biology. Bioprocess Engineering. 1997;16:261–264. [Google Scholar]
- Weaver JC, Chizmadzhev YA. Theory of electroporation: A review. Bioelectrochemistry and Bioenergetics. 1996;41:135–160. [Google Scholar]
- Weaver JC. Electroporation theory. Concepts and mechanisms. Methods in Molecular Biology. 1995;55:3–28. doi: 10.1385/0-89603-328-7:3. [DOI] [PubMed] [Google Scholar]
- McAteer JA, Douglas WH. Monolayer culture techniques. Methods in Enzymology. 1979;58:132–140. doi: 10.1016/s0076-6879(79)58131-2. [DOI] [PubMed] [Google Scholar]
- Ho TD, et al. Identification of GtgE, a novel virulence factor encoded on the Gifsy-2 bacteriophage of Salmonella enterica serovar Typhimurium. Journal of Bacteriology. 2002;184:5234–5239. doi: 10.1128/JB.184.19.5234-5239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemann GS, et al. Discovery of novel secreted virulence factors from Salmonella enterica serovar Typhimurium by proteomic analysis of culture supernatants. Infection and Immunity. 2011;79:33–43. doi: 10.1128/IAI.00771-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spano S, Liu X, Galan JE. Proteolytic targeting of Rab29 by an effector protein distinguishes the intracellular compartments of human-adapted and broad-host Salmonella. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:18418–18423. doi: 10.1073/pnas.1111959108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraga A, Miller SI. A Salmonella type III secretion effector interacts with the mammalian serine/threonine protein kinase PKN1. Cellular Microbiology. 2006;8:837–846. doi: 10.1111/j.1462-5822.2005.00670.x. [DOI] [PubMed] [Google Scholar]
- Zinchuk V, Grossenbacher-Zinchuk O, et al. Quantitative colocalization analysis of confocal fluorescence microscopy images. Current Protocols in Cell Biology / Editorial Board, Juan S. Bonifacino ... [et al] 2011;4(Unit 4.19) doi: 10.1002/0471143030.cb0419s52. [DOI] [PubMed] [Google Scholar]
- Kimple ME, Sondek J, et al. Overview of affinity tags for protein purification. Current Protocols in Protein Science / Editorial Board, John E. Coligan ... [et al] 2004;9(Unit 9.9) doi: 10.1002/0471140864.ps0909s36. [DOI] [PubMed] [Google Scholar]