

Abstract
CPT-11 is a camptothecin analog used for the clinical treatment of colorectal adenocarcinoma. CPT-11 is converted into the therapeutic anti-cancer agent SN-38 by liver enzymes and can be further metabolized to a non-toxic glucuronide SN-38G, resulting in low SN-38 but high SN-38G concentrations in the circulation. We previously demonstrated that adenoviral expression of membrane-anchored beta-glucuronidase could promote conversion of SN-38G to SN-38 in tumors and increase the anticancer activity of CPT-11. Here, we identified impediments to effective tumor therapy with E. coli that were engineered to constitutively express highly active E. coli beta-glucuronidase intracellularly to enhance the anticancer activity of CPT-11. The engineered bacteria, E. coli (lux/βG), could hydrolyze SN-38G to SN-38, increased the sensitivity of cultured tumor cells to SN-38G by about 100 fold and selectively accumulated in tumors. However, E. coli (lux/βG) did not more effectively increase CPT-11 anticancer activity in human tumor xenografts as compared to non-engineered E. coli. SN-38G conversion to SN-38 by E. coli (lux/βG) appeared to be limited by slow uptake into bacteria as well as by segregation of E. coli in necrotic regions of tumors that may be relatively inaccessible to systemically-administered drug molecules. Studies using a fluorescent glucuronide probe showed that significantly greater glucuronide hydrolysis could be achieved in mice pretreated with E. coli (lux/βG) by direct intratumoral injection of the glucuronide probe or by intratumoral lysis of bacteria to release intracellular beta-glucuronidase. Our study suggests that the distribution of beta-glucuronidase, and possibly other therapeutic proteins, in the tumor microenvironment might be an important barrier for effective bacterial-based tumor therapy. Expression of secreted therapeutic proteins or induction of therapeutic protein release from bacteria might therefore be a promising strategy to enhance anti-tumor activity.
Introduction
Specific delivery of therapeutic enzymes to cancer cells for subsequent activation of anticancer drugs in the tumor microenvironment is a promising approach to improve the selectivity of cancer chemotherapy [1,2]. Different vehicles including viruses [3,4], liposomes [5] and antibodies [6,7] have been evaluated to deliver therapeutic enzymes to cancer cells. Recently, bacteria directed enzyme prodrug therapy (BDEPT) has been investigated for cancer therapy [8]. In this approach, engineered bacteria are employed to deliver therapeutic enzymes to tumors. Although the mechanism of tumor localization is still unclear, many bacterial species have been found to selectively localize and proliferate in tumors after systemic administration, including Clostridium [9,10], Bifidobacterium [11,12], Salmonella [13,14] and E. coli [15,16]. Using bacteria as a delivery vehicle possesses potential advantages as compared to other delivery systems. Bacteria can proliferate in tumors for several weeks [17,18], thereby providing sustained expression of therapeutic enzymes to facilitate multiple rounds of drug therapy. Bacteria tumor targeting might also represent a universal treatment method since tumor colonization seems to be independent of the type of cancer being treated [15,19,20]. Bacteria colonization of tumors is highly specific and can achieve much greater tumor/normal tissue ratios than other targeted therapies such as antibodies [21–24] and nanoparticles [25–28]. This can help decrease off-target toxicity of anticancer drug therapy.
CPT-11 (Irinotecan) is currently used as a single agent or in combination with 5-FU/leucovorin for the chemotherapy of colorectal adenocarcinoma [29]. CPT-11 is hydrolyzed by carboxylesterases into SN-38, which is believed to be the active form of the drug responsible for the anticancer activity of CPT-11 [30] (Fig. 1A). SN-38 is a topoisomerase I poison that causes the formation of a stable complex between DNA, topoisomerase I and SN-38, resulting in stabilization of single-strand DNA breaks and cancer cell apoptosis [31,32]. However, SN-38 is rapidly metabolized to the non-toxic and largely inactive glucuronide conjugate SN-38G by UDP glucuronosyltransferase (UDPGT) in the liver [33]. The concentration of SN-38G is up to 10-fold higher than SN-38 in the plasma of patients receiving CPT-11 [34,35], and therefore represents a potential target for enzyme-mediated prodrug therapy. Indeed, we previously showed that expression of a membrane-anchored form of murine beta-glucuronidase in tumors can enhance CPT-11 anti-tumor efficacy by conversion of SN-38G into SN-38 in the tumor microenvironment [4,36,37].
Fig 1. Beta-glucuronidase expression and activity of transformed bacteria.

(A) CPT-11 metabolism. CPT-11 can be converted to SN-38 by carboxylesterase (CE). SN-38 can be further metabolized to SN-38G by UDP-glucuronosyltransferase (UDPGT). SN-38G can be reconverted by beta-glucuronidase (βG) to SN-38. (B) Lysates prepared from 4 x 106 c.f.u. E. coli were immunoblotted with mouse anti-E. coli beta-glucuronidase (1E8) monoclonal antibody and goat anti—mouse IgG-HRP. The chemiluminescence signal was detected by X-ray film. (C) Defined amounts of recombinant E. coli beta-glucuronidase or lysates prepared from 2 x 107 c.f.u. E. coli (lux/βG) were immunoblotted as above. The chemiluminescence signal was detected by Fuji LAS-3000. (D) 500 μM 4-MUG or (E) 2 μM SN-38G was incubated with 25 ng purified recombinant. E. coli beta-glucuronidase, 2 x 106 c.f.u. E. coli (lux), 2 x 106 c.f.u. E. coli (lux/βG) or lysates prepared form the same number of bacteria. The formation of 4-MU and SN-38 at each time point was measured as described in materials and methods (n = 3). 2 x 106 c.f.u. E. coli were mixed with (F) 500 μM 4-MUG or (G) 2 μM SN-38G at 37°C for 30 min and then centrifuged to separate the supernatant and pellet. Fluorescence was detected and analyzed (n = 3).
Here, we investigated if the anticancer activity of CPT-11 could be improved by systematic administration of E. coli that was engineered to express E. coli beta-glucuronidase. E. coli were selected for tumor colonization because they proliferate less in normal tissues compared to other strains of bacteria [18,19,38]. We anticipated that tumor-located expression of E. coli beta-glucuronidase would more effectively convert SN-38G to SN-38 as compared to viral-mediated expression of murine beta-glucuronidase in tumors [4] since the bacterial enzyme displays about one hundred fold greater catalytic activity as compared to murine beta-glucuronidase [36]. We found that E. coli engineered to over express beta-glucuronidase can convert SN-38G to SN-38 and enhance the anticancer activity of SN-38G to cultured cancer cells by about 100 fold. In vivo biodistrubution studies demonstrated that E. coli specifically colonized tumors in both immunodeficient and immune competent mice. However, in contrast to adenovirus-mediated expression of murine beta-glucuronidase in tumors, microbially-delivered E. coli beta-glucuronidase did not significantly enhance the antitumor activity of CPT-11. Slow uptake of glucuronides into E. coli coupled with the preferential accumulation of E. coli in necrotic regions of tumors appears to hinder efficient activation of systemically-administered glucuronide drugs, leading to poor in vivo antitumor activity. Our results suggest that secreted enzymes might be more effective for BDEPT approaches to cancer therapy.
Materials and Methods
Bacteria and plasmids
E. coli DH5α (F- φ80lacZΔM15 Δ(lacZYA-argF) U169 recA1 endA1 hsdR17 (rk-, mk+) phoA supE44 λ- thi-1 gyrA96 relA1) was from Bioman Scientific (Taipei, Taiwan). The plasmid constructs have been described [15]. Briefly, to constitutively express beta-glucuronidase or luciferase, we first replaced the T7 promoter in the pRSETB vector (Invitrogen, Grand Island, NY) with the fen promoter from pGHL6 [39] to produce pRSETB-fen. The E. coli beta-glucuronidase gene was PCR amplified from genomic DNA isolated from E. coli and cloned into pRESTB-fen to generate pRESTB-fen-βG. The luxCDABE gene cluster (generously provided by Dr. EA Meighen, Department of Biochemistry, McGill University, Montreal, Quebec, Canada) [39] was cloned into pRSETB-fen to create pRSETB-fen-lux. The fen-βG fragment was also cloned into pRSETB-fen-lux to form pRSETB-lux/βG. These three plasmids were individually transformed into E. coli DH5alpha to constitutively produce E. coli beta-glucuronidase, luciferase or both gene products, respectively.
Reagents
RPMI-1640, MEM-α, and fluorescein di-β-D-glucuronide (FDGlcU) were from Invitrogen (Grand Island, NY). Bovine calf serum (BCS) was from HyClone (Logan, Utah). 4-methylumbelliferyl-β-D-glucuronide (4-MUG), CPT-11, lysozyme, and DNase I were from Sigma-Aldrich (Saint Louis, MO). SN-38G was purified by HPLC from urine of CPT-11-treated mice [40]. SN-38 was from Sinopharm (Beijing, China). 3H-thymidine was from PerkinElmer, Inc. (Boston, MA).
Animals and cells
6 to 8-week-old NOD.CB17-Prkdcscid/IcrCrlBltw mice were purchased from BioLASCO Taiwan Co., Ltd (Taipei, Taiwan). Animal experiments were performed under specific pathogen free conditions in accordance with institute guidelines. Protocols were approved by Academia Sinica Institutional Animal Care & Utilization Committee (Permit Number: 12–12–451). HCT116 (ATCC No. CCL-247) and LS174T (ATCC No. CL-188) human colorectal carcinoma cells were from the American Type Culture Collection (Manassas, VA). CL1–5 human lung adenocarcinoma cells were a gift from Dr. Pan-Chyr Yang (National Taiwan University, Taipei, Taiwan) [41]. HCT116 cells and CL1–5 cells were cultured in RPMI-1640 medium containing 2.98 mg/ml HEPES, 1 mg/ml sodium bicarbonate, and 10% BCS. LS174T cells were cultured in MEM-α medium containing 1 mg/ml sodium bicarbonate and 10% BCS. All cells were cultured in a humidified atmosphere of 5% CO2 in air at 37°C.
Western blotting and quantification of beta-glucuronidase expression
0.5 ml (Fig. 1B) or 2 ml (Fig. 1C) E. coli (lux/βG) (OD600 = 0.5, ~8 x 107 c.f.u./ml) and defined amounts of purified recombinant E. coli beta-glucuronidase [42] were separated on a 10% SDS-PAGE gel and transferred to nitrocellulose paper. The membranes were incubated with 0.6 μg/ml mouse anti-E. coli beta-glucuronidase antibody 1E8 [43] followed by goat anti-mouse IgG Fc-HRP (Organon Teknika Corporation, Durham, NY). Antibody binding was detected by SuperSignal West Pico Chemiluminescence Substrate (Thermo Scientific, Rockford, IL). The luminescence was visualized on an X-ray film (Fig. 1B) or in a Fuji LAS-3000 imager (Fig. 1C) to quantify the protein amounts using Metamorph software (Molecular Devices, Sunnyvale, CA).
E. coli beta-glucuronidase activity
1 ml of E. coli (OD600 = 0.5) was washed with PBS and suspended in 1 ml reaction buffer (50 mM bis-Tris, 50 mM triethanolamine, 100 mM acetic acid, 0.1% bovine serum albumin, pH 7). To break the cell wall, 1 ml of E. coli (OD600 = 0.5) was washed with reaction buffer and then suspended in reaction buffer containing 0.2 mg/ml lysozyme and DNase I at 37°C for 15 min followed by sonication for 15 min on ice. 50 μl live E. coli or E. coli lysate was incubated with 50 μl 4-MUG (500 μM) or SN-38G (4 μM) in 96-well black microtiter plates at 37°C. Relative fluorescence was measured with a fluorescence microplate reader (Molecular Devices) and the 4-MU formation was calculated from a standard curve. The excitation and emission wavelengths of 4-MU, SN-38 and SN-38G were 355/460, 375/470, and 375/560, respectively. Fluorescence was detected every 2 minutes for 20 minutes. The SN-38 concentration was calculated by:
where A is the absorbance, ε is the absorption coefficient and C is the concentration of each molecule.
To determine whether the hydrolyzed substrates were released from the bacteria, 50 μl live E. coli were incubated with 50 μl 4-MUG (500 μM) or SN-38G (4 μM) at 37°C for 5 minutes and then the bacteria were pelleted at 5000xg for 5 minutes. The fluorescence of the cell pellet and supernatant were measured and converted into 4-MU and SN-38 concentrations. To detect beta-glucuronidase activity of E. coli in tumor tissues, tumors were isolated from mice and then homogenized in PBS on ice. 50 μl tissue lysates were reacted with 50 μl 4-MUG (500 μM) at 37°C for 30 minutes and the 4-MU formation was calculated.
Recombinant E. coli and mouse beta-glucuronidase specific activity
Recombinant E. coli and mouse beta-glucuronidase were produced as described [44]. 1 ng E. coli beta-glucuronidase and 10 ng mouse beta-glucuronidase were incubated with 2 μM SN-38G at 37°C. The SN-38 formation at pH 7 was measured at 0, 5, 10, 15, 20 and 30 minutes (n = 3). One unit beta-glucuronidase activity corresponds to the hydrolysis of 1 pmole SN-38G per h at 37°C.
Luciferase activity
E. coli (50 μl, OD600 = 0.5) suspended in PBS were added into 96-well white plates and luminescence was detected on a Top Count Luminescence Counter (Perkin-Elmer Life Sciences, Waltham, MA).
3H-thymidine incorporation assay
5000 HCT116 or CL1–5 cells per well were seeded in 96-well cell culture plates for 8 h. 1.5 x 107 c.f.u. E. coli (lux) or E. coli (lux/βG) were mixed with 1.5 ml 2 μM or 50 nM SN-38G at 37°C for 2 h and then the bacteria were removed by filtration through a 0.2 μm filter. SN-38, SN-38G or E. coli treated SN-38G were 3 fold serially-diluted in culture medium and then added to HCT116 or CL1–5 cells for 48 h. The culture medium was removed and fresh culture medium containing 3H-thymidine (1 μCi/well) was added for another 16 h. Radiation incorporated into cellular DNA was then measured on a Top Count scintillation counter. The results are expressed as 3H-thymidine incorporation (% control) = (c.p.m.(S) x 100)/c.p.m.(C) where c.p.m. represents counts per minute of sample (S) or untreated controls (C).
Adenovirus preparation
The pAd-CMV plasmids containing membrane-tethered mouse beta-glucuronidase or anti-dansyl single-chain antibody transgenes [45] were co-transfected with pJM17, which carries the entire Ad5 genome, lacking E1 and E3 functions, in E1-complementing 293N cells to produce adenoviruses expressing membrane tethered mouse beta-glucuronidase (Ad/mβG) or anti-dansyl single-chain antibody (Ad/DNS). Virus production, purification and titer determination were as previously described [4].
In vivo imaging
NOD/SCID mice were s.c. inoculated with 107 CT26 mouse colon cancer cells, EJ human bladder cancer cells, HCT116 human colon cancer cells, or 2 x 106 LS174T human colon cancer cells on the right flank. BALB/c mice were s.c. inoculated with 107 CT26 mouse colon cancer cells. 4 x 107 c.f.u. E. coli were i.v. injected in each mouse when the tumor volumes reached ~150 mm3. The relative body weight was measured every 2 or 3 days. The localization of E. coli in mice was detected by luminescence emission. Under gas anesthesia, the luminescence in tumors was detected daily on an IVIS Spectrum (Caliper Lifesciences, Hopkinton, MA). The region of interest (ROI) was analyzed with Living Image Software (Caliper lifesciences). To measure the in vivo enzyme activity, mice were either intratumorally-injected on two consecutive days with 109 p.f.u. Ad/mβG or Ad/DNS when tumor volumes reached ~100 mm3 or i.v. injected with 4 x 107 c.f.u. E. coli (lux) or E. coli (lux/βG) when the tumor volume reached ~150 mm3. On the fourth day after administration of E. coli or adenovirus, mice received 500 μg FDGlcU by intravenous injection or 5 μg FDGlcU by direct intratumor injection, respectively. The fluorescence in tumors was detected on an IVIS Spectrum 30 minutes later. To investigate the effect of releasing beta-glucuronidase from E. coli, E. coli (lux/βG) treated mice were i.v. injected with 500 μg FDGlcU to determine baseline beta-glucuronidase imaging. Two days later, each mouse was treated with 50 μl 4 mg/ml lysozyme and DNase I via intratumor injection to lyse the bacteria and then i.v. immediately injected with 500 μg FDGlcU. After 30 minutes, both control and treated mice were imaged on an IVIS Spectrum for luminescence and fluorescence analysis.
Ex vivo E. coli counting, imaging and tissue section staining
To examine the biodistribution of E. coli in mice, tumors and other normal organs were removed and imaged on an IVIS Spectrum 8 days after i.v. administration of 4 x 107 c.f.u. E. coli (lux) or E. coli (lux/βG). Samples were homogenized and spread on carbenicillin-containing LB plates to count bacterial colonies. Spleen weights from each group were measured. For immunofluorescence histology, tumor samples were fixed with 4% paraformaldehyde in PBS and embedded with paraffin. Sections were stained with goat anti-E. coli polyclonal antibody (Abcam, Cambridge, MA) and biotinylated 1E8 (anti-E. coli beta-glucuronidase) followed by rhodamine-conjugated rabbit anti-goat IgG (Organon Teknika Corporation, West Chester, PA) and Alexa Fluor 488 conjugated streptavidin (Invitrogen), respectively. Sections were incubated with 1 μg/ml DAPI to visualize nuclei.
In vivo anti-tumor activity
6 to 8 week-old NOD/SCID mice were s.c. injected on the right flank with 107 HCT116 or LS174T colon cancer cells. The mice were divided into 5 groups (n = 6) and treated with PBS, CPT-11, E. coli (lux) and CPT-11, E. coli (lux/βG) and PBS, or E. coli (lux/βG) and CPT-11. We injected 4 x 107 c.f.u. E. coli (lux) or E. coli (lux/βG) by a single i.v. injection when tumors reached a mean size of 150 mm3. Four days later, mice were i.v. injected with 10 mg/kg CPT-11 or vehicle for two consecutive days. Tumor volumes were calculated according to the formula: length x width x height x 0.5.
Statistical significance
Statistical significance of differences between mean values was estimated with Excel (Microsoft, Redmond, WA, USA) using the independent t-test for unequal variances. For in vivo enzyme releasing image, the ROI value differences were analyzed by GraphPad Prism Version 5 paired t-test. P-values of < 0.05 were considered statistically significant.
Results
Characterization of beta-glucuronidase expression and activity of transformed E. coli
E. coli that constitutively display luminescence and/or express E. coli beta-glucuronidase were generated as previously described [15]. Beta-glucuronidase expression in E. coli is inducible and normally low unless the bacteria are cultured in the presence of high concentrations of glucuronide carbon sources [46]. Western blot analysis of transformed bacteria showed that beta-glucuronidase was detected in E. coli (βG) and E. coli (lux/βG) but not in wild-type E. coli or E. coli (lux) (Fig. 1B). By comparing the levels of beta-glucuronidase in E. coli (lux/βG) with purified E. coli beta-glucuronidase protein, we estimated that E. coli (lux/βG) expressed about 60 ng beta-glucuronidase/107 c.f.u. (Fig. 1C). We assessed if E. coli (lux/βG) can hydrolyze glucuronides by adding a synthetic glucuronide substrate (4-MUG) to live bacteria and then measuring the fluorescence of the hydrolyzed product (4-MU). E. coli (lux/βG) converted 4-MUG to 4-MU whereas E. coli (lux) did not produce measurable 4-MU (Fig. 1D), indicating that ectopically-produced beta-glucuronidase in E. coli was active. However, 4-MUG was hydrolyzed much faster by a similar amount of recombinant beta-glucuronidase (25 ng), suggesting that 4-MUG hydrolysis in E. coli (lux/βG) may be limited by transport of 4-MUG into E. coli. In support of this hypothesis, the rate of 4-MUG hydrolysis in lysates prepared from the same number of E. coli (lux/βG) was similar to the rate of 4-MUG hydrolysis by recombinant beta-glucuronidase (Fig. 1D). By contrast, lysates prepared from E. coli (lux) did not hydrolyze 4-MUG, verifying that overexpression of beta-glucuronidase was required for glucuronide hydrolysis. Similar results were found for hydrolysis of SN-38G by E. coli (lux/βG) (Fig. 1E). Since the substrates were hydrolyzed inside E. coli, we examined if 4-MU and SN-38 were released from the bacteria by mixing 4-MUG or SN-38G with E. coli and then measuring 4-MU and SN-38 in the medium or cell pellet. 4-MU (Fig. 1F) and SN-38 (Fig. 1G) were mostly found in the supernatant of E. coli (lux/βG), indicating that the reaction products were efficiently released from E. coli. We conclude that E. coli (lux/βG) can uptake 4-MUG and SN-38G and convert and subsequently release 4-MU and SN-38 into the culture medium. However, uptake of the glucuronides into E. coli appears to be a rate-limiting step in the conversion process.
E. coli (lux/βG) can increase the cytotoxicity of SN-38G
We investigated if cytotoxic concentrations of SN-38 could be generated from SN-38G by E. coli (lux/βG). SN-38 was about 355 and 150 fold more toxic than SN-38G to HCT116 human colon cancer cells (Fig. 2A) and CL1–5 human lung cancer cells (Fig. 2B), respectively. Preincubation of SN-38G with E. coli (lux) slightly increased the inhibition of HCT116 and CL1–5 cell growth by 1.08 and 1.15 fold, respectively (Table 1). By contrast, incubation of SN-38G with E. coli (lux/βG) increased inhibition of HCT116 and CL1–5 cell growth by 135 and 90 fold, respectively. We conclude that E. coli (lux/βG) can generate cytotoxic concentrations of SN-38 from SN-38G.
Fig 2. Cytotoxicity of bacterially-activated SN-38G.
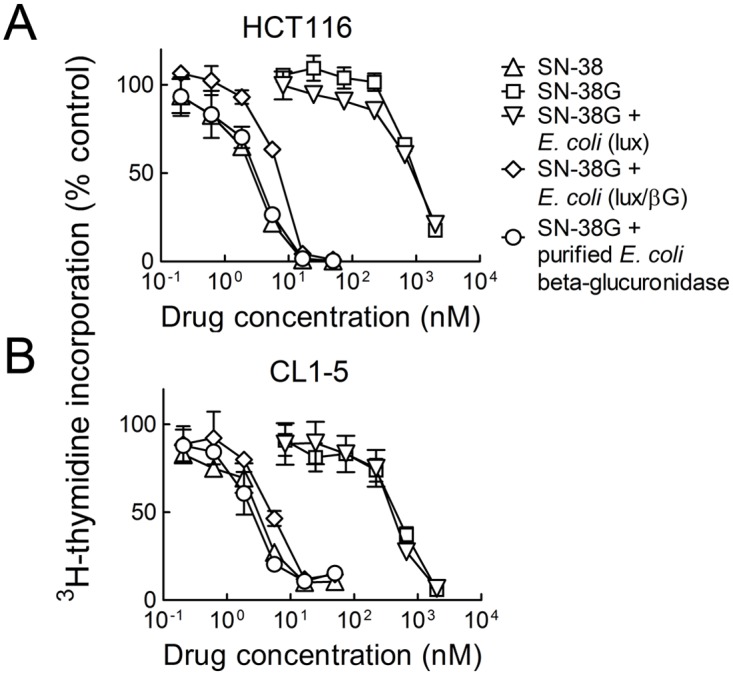
Defined concentrations of SN-38G were reacted with 1 μg recombinant E. coli beta-glucuronidase or 2 x 106 c.f.u. E. coli (lux) or E. coli (lux/βG) at 37°C for 2 h. The bacteria were removed by filtration and serial dilutions were then incubated with HCT116 human colorectal carcinoma cells (A) or CL1–5 human lung adenocarcinoma cells (B) for 48 h. HCT116 and CL1–5 cells were also treated with serial dilutions of SN-38 and SN-38G as controls. The cells were incubated with fresh medium for an additional 24 h before incorporation of 3H-thymidine into cellular DNA was measured. n = 3. Bars, s.d.
Table 1. E. coli (lux/βG) sensitization of cancer cells to SN-38G.
| Cells | SN-38 | SN-38G | SN-38G + 1 μg E. coli βG | SN-38G + E. coli (lux) | SN-38G + E. coli (lux/βG) |
|---|---|---|---|---|---|
| HCT116 | 2.7 ± 0.1 | 960 ± 9.1 | 3.0 ± 0.3 | 890 ± 21.1 | 7.1 ± 0.3 |
| CL1–5 | 3.0 ± 0.3 | 455 ± 36.2 | 2.4 ± 0.6 | 390 ± 57.4 | 5.0 ± 0.6 |
Results show the mean IC50 values in nM of triplicate determinations ± standard deviation.
E. coli (lux/βG) can specifically accumulate in tumors
Both E. coli (lux) and E. coli (lux/βG) can emit luminescence in vitro (Fig. 3A) and in vivo (Fig. 3B) to facilitate tracing their locations in mice. Imaging of tumor-bearing mice after i.v. injection of E. coli (lux/βG) demonstrated specific localization of E. coli in tumors that was relatively independent of the tumor type and immune status of the mice (Fig. 3B and 3C). Systemic administration of E. coli caused about 12% weight loss to mice on day 1 (Fig. 3D). Mice body weight partially rebounded but remained less than untreated mice. Although most experiments were performed in immune deficient mice to allow investigation of therapy in human xenograft models, we also examined the toxicity of systemic E. coli administration to immune competent BALB/c mice. E. coli in immune-competent mice retained the ability to colonize tumors (Fig. 3B and 3C). Interestingly, body weight loss was not significantly different in immune deficient and immune competent mice injected with E. coli (Fig. 3E), suggesting that E. coli (DH5±) do not cause excessive immune-related toxicity.
Fig 3. Localization of engineered E. coli in tumors.

(A) The luminescence of 10 fold serial dilutions of the indicated wild-type or engineered E. coli was detected on a Top Count scintillation counter (n = 3). (B) NOD/SCID mice bearing established CT26 mouse colon, EJ human bladder, HCT116 human colon or LS174T human colon tumors and BALB/c mice bearing CT26 colon tumors were i.v. injected with 4 x 107 c.f.u. E. coli (lux/βG). Mice were imaged on an IVIS Spectrum before E. coli injection and on day 4 to trace the bacteria location. (C) Mean luminescence values of tumors is shown. The Y-axis indicates the luminescence intensity. N = 5. Bars, s.d. (D) Relative body weights of mice are indicated (n = 6). Bars, s.d. * p < 0.05, ** p < 0.005. (E) Comparison of relative body weight loss of BALB/c and NOD/SCID mice bearing established CT26 mouse colon tumors after i.v. injection of 4 x 107 c.f.u. E. coli (lux/βG). N = 5. Bars, s.d.
The time course of E. coli accumulation in ~150 mm3 HCT116 tumors in NOD/SCID mice was examined after i.v. injection of 4x107 c.f.u. E. coli. Luminescence could be detected in tumors within 24 hour [4/6 for E. coli (lux), 3/6 for E. coli (lux/βG)] (Fig. 4A). The luminescence of E. coli (lux) and E. coli (lux/βG) in tumors reached a plateau from days 3 to 7 (Fig. 4B), demonstrating that E. coli localized and proliferated in tumors. Imaging of isolated tumors and normal tissues taken from mice on day 8 showed strong luminescence in tumors but not in normal tissues (Fig. 4C). Determination of the numbers of E. coli in different organs also showed that tumors accumulated about 6,500 and 23,000 fold higher levels of E. coli than did the liver and spleen, respectively (Fig. 4D). Mice injected with E. coli (lux) or E. coli (lux/βG) displayed splenomegaly (Fig. 4E), suggesting that the systemically-administered E. coli induced an immune response. This immune response may cause the transient body weight loss (9.2%) on day 3 observed after systematic administration of E. coli (Fig. 4F).
Fig 4. In vivo localization of E. coli in HCT116 tumors.

PBS or 4 x 107 c.f.u. E. coli (lux) or E. coli (lux/βG) were i.v. injected into mice bearing 150 mm3 HCT116 tumors. (A) Luminescence images were captured on an IVIS Spectrum on days 0, 1, 4 and 7 (n = 6). (B) The region of the interesting (ROI) of mice treated as in panel A on different days were analyzed (n = 6). The Y-axis indicates the luminescence intensity. Bars, s.d. (C) The luminescence of organs (clockwise order: heart, liver, spleen, lung, kidney, intestine, colon, and stomach) and tumors isolated from mice treated with PBS or 4 x 107 c.f.u. E. coli (lux/βG) were imaged on an IVIS spectrum on day 8. Bars, s.d. ** P < 0.005. (D) E. coli (lux/βG)-injected mice were sacrificed on day 8. Liver, spleen and tumor tissues were homogenized and spread on LB agar plates to count the bacterial colonies (n = 6). Bars, s.d. ** P < 0.005. (E) The spleen weight on day 8 and (F) relative body weight of mice during the experiment were measured (n = 6). Bars, s.d. ** P < 0.005.
E. coli accumulates in necrotic areas of tumors
The distribution of bacteria in HCT116 tumors was examined in more detail by immunohistochemical analysis of E. coli and beta-glucuronidase in tumor sections from E. coli (lux/βG), E. coli (lux), or PBS-treated mice. DAPI (blue) stained DNA in both live tumor cells and E. coli. DAPI-stained E. coli were observed as tiny spots that formed a dense cluster under UV excitation (Fig. 5A). Antibody staining for E. coli further confirmed the location of the bacteria. Anti-E. coli beta-glucuronidase antibody staining showed that E. coli (lux/βG) but not E. coli (lux) expressed beta-glucuronidase in the tumor microenvironment. 72.4% ±13.9% of E. coli (lux) and 84.9% ± 9.2% of E. coli (lux/βG) were located in necrotic areas of tumors as distinguished by DAPI staining. E. coli (lux) and E. coli (lux/βG) didn’t show significant differences in localization to tumor necrotic areas (Fig. 5B). The bacteria distribution indicates that E. coli primarily accumulated at the border between viable and necrotic cells.
Fig 5. The distribution of E. coli in tumors.

(A)Tumor sections obtained on day 8 from mice injected with PBS, E. coli (lux/βG) or E. coli (lux) were stained with DAPI (blue), mouse anti-E. coli beta-glucuronidase antibody (green) and goat anti-E. coli antibody (red). Arrows indicate the location of E. coli. Dotted lines indicate the approximate border between necrotic areas and live cells. N, necrotic area. Bars, 50 μm. (B) The percentage of E. coli in the necrotic area of tumor tissue sections. n = 4. n.s., no significant difference.
Anti-tumor activity of E. coli (lux/βG) and CPT-11 in vivo
To determine whether E. coli (lux/βG) could enhance the anti-tumor activity of CPT-11, NOD/SCID mice bearing established HCT116 human colon tumors were i.v. injected with PBS or E. coli. Four days later, the mice were i.v. injected with 10 mg/kg CPT-11 or vehicle on two consecutive days. Treatment of mice with either CPT-11 or E. coli (lux/βG) alone significantly suppressed tumor growth as compared to treatment with vehicle alone (Fig. 6A). Combination treatment with CPT-11 and E. coli (lux/βG) produced significantly greater antitumor activity that either individual treatment. However, this effect did not depend on bacterial expression of beta-glucuronidase because treatment of mice with CPT-11 and E. coli (lux) suppressed tumor growth to a similar extent (Fig. 6A). Similar results were found for LS174T human colorectal tumors in NOD/SCID mice; treatment with CPT-11 and either E. coli (lux/βG) or E. coli (lux) produced significant but similar suppression of tumor growth (Fig. 6B), indicating that therapeutic outcome was not affected by expression of beta-glucuronidase in E. coli (lux/βG).
Fig 6. Anti-tumor activity of CPT-11 and E. coli (lux/βG).

(A) 4 x 107 c.f.u. E. coli (lux) or E. coli (lux/βG) were i.v. injected into NOD/SCID mice bearing ~150 mm3 HCT116 tumors. After 4 days, 10 mg/kg CPT-11 or vehicle (PBS) were i.v. injected on two consecutive days. Tumor sizes were measured 21 days after treatment. Results show mean values of 7 mice. Bars, s.d. (B) NOD/SCID mice bearing established HCT116 (n = 7) or LS174T tumors (n = 6) were i.v. injected with PBS, or 4 x 107 c.f.u. E. coli (lux) or E. coli (lux/βG). All the mice were treated with 10 mg/kg CPT-11 on days 4 and 5. Tumor sizes were measured on day 10. Bars, s.d. * P < 0.05; ** P < 0.005; n.s. no significant difference.
In vivo beta-glucuronidase activity
To investigate if the similar anticancer activity observed for treatment with CPT-11 and either E. coli (lux/βG) or E. coli (lux) could be caused by in vivo induction of endogenous beta-glucuronidase expression in E. coli (lux) or by in vivo loss of beta-glucuronidase expression in E. coli (lux/βG), we i.v. injected E. coli (lux/βG) or E. coli (lux) into mice bearing established HCT116 tumors and then i.v. injected a fluorescence glucuronide probe, FDGlcU, to measure in vivo beta-glucuronidase activity (Fig. 7A). FDGlcU does not emit fluorescence unless the glucuronide group is enzymatically removed. E. coli (lux/βG) displayed similar luminescence emission but higher fluorescence emission than E. coli (lux) (Fig. 7B), indicating that similar numbers of bacteria were in tumors but that E. coli (lux/βG) expressed significantly more beta-glucuronidase activity in tumors than did E. coli (lux). We conclude that E. coli (lux/βG) and E. coli (lux) displayed the expected high and low beta-glucuronidase activity in tumors.
Fig 7. In vivo beta-glucuronidase activity of tumors treated with E. coli (lux/βG).
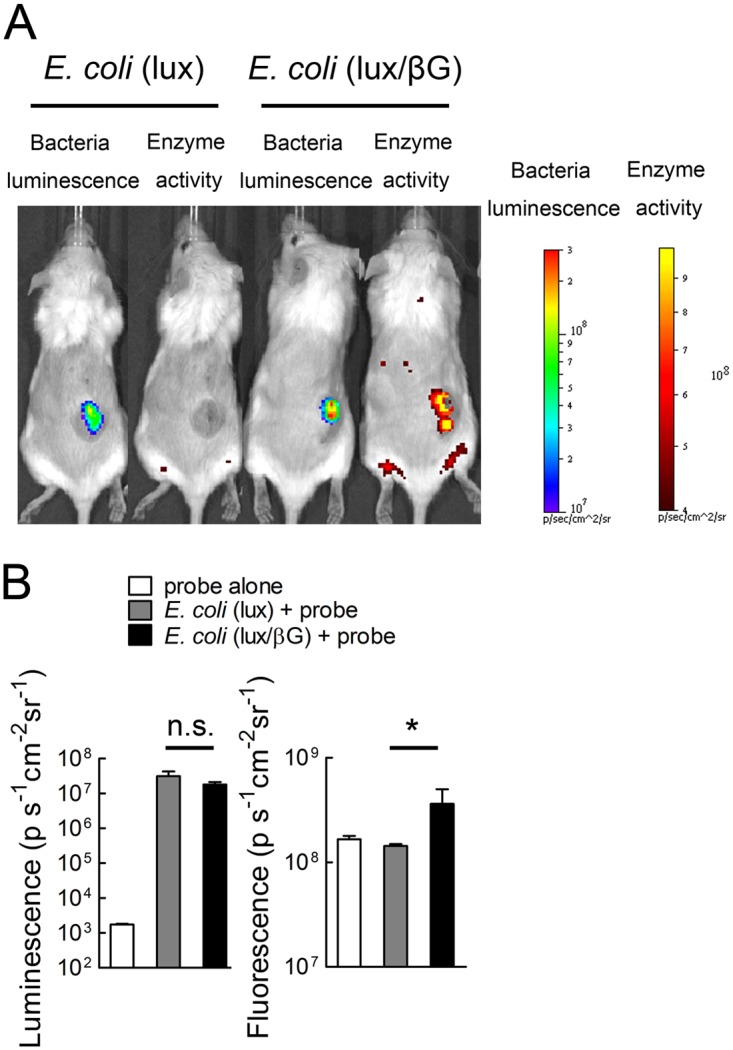
(A) NOD/SCID mice bearing HCT116 tumors were i.v injected with 4 x 107 c.f.u. E. coli (lux) or E. coli (lux/βG). After 4 days, 500 μg FDGlcU was i.v. injected into the mice. After 30 min, the luminescence (representing bacterial numbers) and fluorescence (representing beta-glucuronidase activity) in tumors were detected on an IVIS Spectrum (n = 3). (B) The mean luminescence and fluorescence intensities in tumors are shown. Unit is photons sec-1cm-2sr-1. Bars, s.d. * P < 0.05.
Efficient glucuronide hydrolysis by adenoviral expression of beta-glucuronidase
We previously showed that intratumoral injection of Ad/mβG, which allows expression of murine beta-glucuronidase as a membrane-anchored form on the surface of infected cells, can enhance the anti-tumor activity of CPT-11 [47]. Therefore, we used adenovirus therapy as a control to identify possible mechanisms that led to E. coli therapy failure. To investigate differences between bacterial and adenoviral beta-glucuronidase therapy, we first compared the enzymatic activities of E. coli beta-glucuronidase (produced in E. coli (lux/βG) and murine beta-glucuronidase (produced by Ad/mβG). E. coli beta-glucuronidase hydrolyzed the glucuronidase substrate 4-Nitrophenyl β-D-glucopyranoside (pNPG) faster than did murine beta-glucuronidase at pH 7 [36]. Similarly, we found that E. coli beta-glucuronidase displayed about 250 fold greater enzymatic activity for the hydrolysis of SN-38G compared to murine beta-glucuronidase at pH 7 (Fig. 8A). Next, we measured the relative beta-glucuronidase activity in tumor homogenates prepared from mice treated by i.v. injection of E. coli (lux/βG) or intratumoral injection of Ad/mβG. Again, the total beta-glucuronidase activity in tumors was significantly greater in mice treated with E. coli (lux/βG) than those treated with Ad/mβG (Fig. 8B). These results show that systemic administration of E. coli (lux/βG) can deliver more beta-glucuronidase activity to tumors as compared to direct injection of tumors with Ad/mβG.
Fig 8. Comparison of in vivo beta-glucuronidase activity of tumors treated with Ad/mβG or E. coli (lux/βG).

(A) The specific activities of E. coli and mouse beta-glucuronidase for hydrolysis of SN-38G at pH 7 were determined (n = 3). Bars, s.d. (B) Mice bearing HCT116 tumor were treated with 109 p.f.u. Ad/mβG intratumorally on two consecutive days or 4 x 107 c.f.u. E. coli (lux) or E. coli (lux/βG) via i.v. injection. After 4 days, 50 μl homogenized tumors lysates were incubated with 50 μl 500 μM 4-MUG to measure beta-glucuronidase activity. Results represent mean values from three mice. Bars, s.d. (C) Mice bearing HCT116 tumors were treated intratumorally with Ad/mβG or i.v. injected with E. coli (lux/βG). On day 4, 500 μg FDGlcU was i.v. injected into each mouse (n = 3). The fluorescence signal represents the in vivo beta-glucuronidase activity. (D) The mean fluorescence in tumors after i.v injection of FDGlcU in mice previously treated i.v. with E. coli (lux/βG) or intratumorally with Ad/mβG was quantified. (n = 3). (E) The relative in vivo FDGlcU conversion efficiency was calculated by dividing the in vivo FDGlcU fluorescence in tumors by the beta-glucuronidase activity in tumor lysates (n = 3). * P < 0.05; ** P < 0.005; n.s. no significant difference.
To assess the functional accessibility of beta-glucuronidase to systemically administered drugs, we i.v. injected the fluorescent glucuronide probe FDGlcU and then imaging the fluorescence intensity in tumors of mice treated i.v. with E. coli (lux/βG) or i.t. with Ad/mβG. Fluorescence signals were apparent in the tumors of mice receiving either treatment (Fig. 8C), but mice treated with Ad/mβG exhibited significantly more fluorescence in tumors as compared to the mice treated with E. coli (lux/βG) (Fig. 8D). A relative in vivo FDGlcU conversion efficiency was calculated by dividing the FDGlcU tumor fluorescence (from Fig. 8D) by the beta-glucuronidase activity in tumor lysates (from Fig. 8B). Tumors injected with Ad/mβG displayed about 22 fold greater FDGlcU conversion efficiency than tumors treated with E. coli (lux/βG) (Fig. 8E). We conclude that although treatment of mice with E. coli (lux/βG) produced more beta-glucuronidase activity in tumors, the beta-glucuronidase activity generated by Ad/mβG can more effectively hydrolyze a systemically administered glucuronide probe in the tumors.
Beta-glucuronidase distribution in tumors may affect hydrolysis of a systemically administered glucuronide probe
The relatively ineffective hydrolysis of FDGlcU in tumors treated with E. coli (lux/βG) suggested that the chemical probe delivered via the tumor blood supply may not effectively contact beta-glucuronidase in the tumor. To test this idea, we compared the fluorescence in tumors after systemic (i.v.) or direct intratumoral (i.t.) injection of FDGlcU to mice previously treated with E. coli (lux/βG) or Ad/mβG. The dose of FDGlcU was decreased by 100 fold for intratumoral injection groups to prevent saturation of beta-glucuronidase in the tumors. While mice treated with Ad/mβG displayed similar fluorescence after either i.v. or i.t. injection of FDGlcU, the mice treated with E. coli (lux/βG) exhibited more tumor fluorescence after i.t. injection of FDGlcU (Fig. 9A). Mean tumor fluorescence from groups of three mice treated with E. coli (lux/βG) was significantly greater when FDGlcU was directly injected into tumors as opposed to after i.v, administration (Fig. 9B), suggesting that systemically administered FDGlcU was less able to come into contact with beta-glucuronidase in E. coli as opposed to beta-glucuronidase expressed in cancer cells after administration of Ad/mβG.
Fig 9. Intratumoral injection of FDGlcU in mice treated with E. coli (lux/βG) enhances glucuronide hydrolysis.
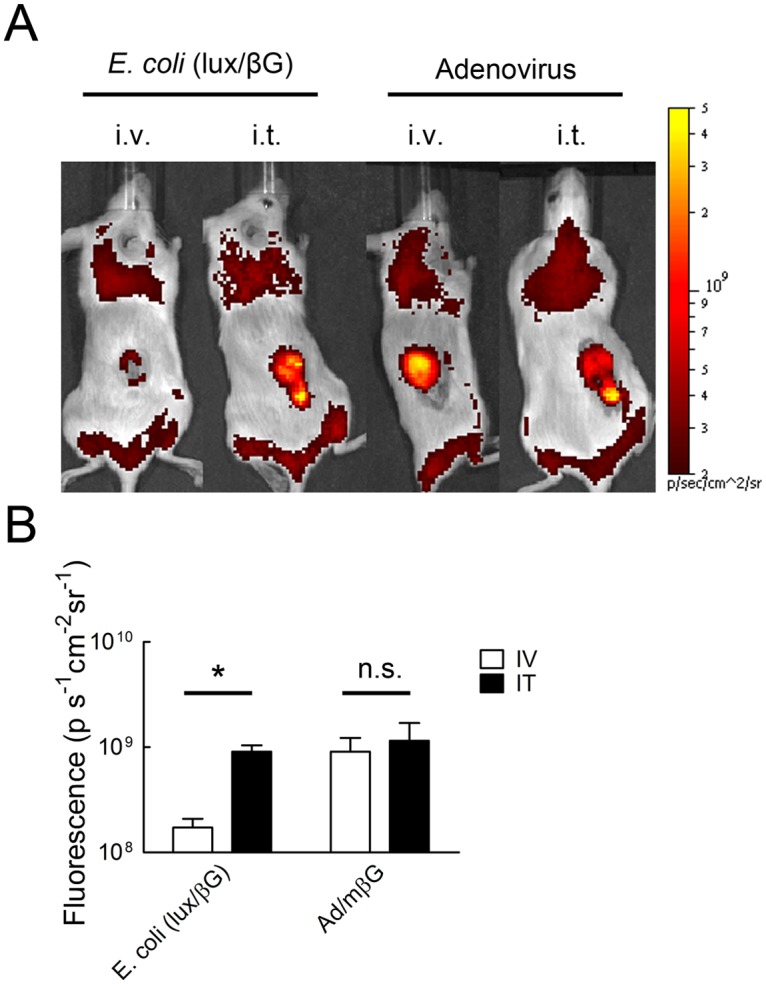
(A) NOD/SCID mice bearing HCT116 tumors were i.v. injected with 4 x 107 c.f.u. E. coli (lux/βG) or intratumorally-injected with 109 p.f.u. Ad/mβG on two consecutive days. On day 4, 500 μg FDGlcU was i.v. injected into mice or 5 μg FDGlcU was directly injected into tumors. After 30 min, the fluorescence of the tumors were detected on an IVIS Spectrum (n = 3). (B) The region of the interesting (ROI) of mice treated as in panel A were analyzed (n = 3). The Y-axis indicates the mean FDGlcU fluorescence intensity (n = 3). Bars, s.d. * P < 0.05; n.s. no significant difference.
Release of beta-glucuronidase from bacteria increases hydrolysis of a systemically administered glucuronide probe
To investigate if the limited probe hydrolysis by poor contact between beta-glucuronidase in E. coli and FDGlcU could be reversed by generating soluble beta-glucuronidase in tumors, we i.v. injected E. coli (lux/βG) to allow tumor colonization and then directly injected a mixture of lysozyme/DNase I into tumors to break the bacteria cell wall. This approach was used to release beta-glucuronidase from the bacteria because the enzyme is too large (~280 kDa) to be efficiently secreted (results not shown). Mice were first i.v. injected with FDGlcU on day 4 to measure the fluorescence generated by hydrolysis of FDGlcU by intact bacteria in the tumors. After 48 h, the lysozyme/DNase I mixture was injected into tumors and then FDGlcU was again i.v. injected and tumor fluorescence was imaged. Imaging of bacterial luminescence showed the number of bacteria in tumors on days 4 and 6 were not significantly different (Fig. 10A and B). By contrast, i.t. injection of lysis solution resulted in stronger tumor fluorescence in images (Fig. 10A right) and significantly increased (p = 0.01) tumor fluorescence from FDGlcU hydrolysis (Fig. 10B). We conclude that release of beta-glucuronidase from E. coli in tumors can enhance the effective hydrolysis of a systemically-administered glucuronide compound.
Fig 10. Lysing E. coli in tumors can increase glucuronide hydrolysis.

(A) NOD/SCID mice bearing HCT116 tumors were i.v. treated with 4x 107 c.f.u. E. coli (lux/βG). 500 μg FDGlcU was i.v. injected into mice and imaged on day 4 (before lysis reagent treatment). Two days later, the same mice were treated with 50 μl 4 mg/ml lysozyme and DNase I by intratumor injection to break the bacteria and release the enzyme into tumor tissue. After treatment, 500 μg FDGlcU was immediately given by i.v. injection. Mice were imaged (after lysis reagent treatment) 30 min later and (B) the ROI were analyzed (n = 4). * P < 0.05; n.s. no significant.
Discussion
Methods to enhance the selectivity and efficacy of cancer chemotherapy are needed to improve patient outcome. Bacteria that have been modified to express enzymes for tumor-selective activation of proactive anticancer agents have been investigated as a promising approach to achieve more selective cancer therapy [8,48]. Here we investigated if the anticancer activity of the clinically used anticancer drug CPT-11 could be enhanced by systematic administration of E. coli that were engineered to express E. coli beta-glucuronidase. The rationale for this approach is that high levels of SN-38G, a non-toxic glucuronide metabolite, are present in the blood of patients that receive CPT-11 [34,35]. Conversion of SN-38G to the highly potent topoisomerase I poison SN38 in tumors might enhance the activity of CPT-11 therapy. Indeed, we previously demonstrated that expression of murine beta-glucuronidase on cancer cells can significantly enhance the anticancer activity of CPT-11 in mice bearing human tumor xenografts [4,36,37]. These results are encouraging because SN-38G/SN-38 ratios in human serum are greater than those found in mice receiving CPT-11, suggesting that intratumoral conversion of SN-38G to SN-38 may produce superior antitumor activity in humans. We predicted that E. coli engineered to constitutively express E. coli beta-glucuronidase would more effectively enhance CPT-11 anticancer activity because E. coli beta-glucuronidase is about one hundred times more active than murine beta-glucuronidase. Indeed, systemically-administration of engineered E. coli generated significantly more beta-glucuronidase activity in tumors as compared to direct tumor injection of an adenoviral vector that expressed murine beta-glucuronidase on transfected cells. However, we found that microbially-delivered E. coli beta-glucuronidase did not significantly enhance the antitumor activity of CPT-11. We identified two impediments that appear to reduce the effectiveness of BDEPT using beta-glucuronidase: 1) slow uptake of glucuronide compounds into E. coli may reduce the rate of prodrug hydrolysis and 2) the preferential accumulation of E. coli in necrotic regions of tumors may hamper efficient contact of systematically administered drugs with beta-glucuronidase. Our study suggests that both the cellular and spatial distribution of beta-glucuronidase, and possibly other therapeutic enzymes, might be an important barrier for effective bacteria-based prodrug cancer therapy.
E. coli was selected in our study to deliver beta-glucuronidase to tumors. There are two major advantages of using E. coli as a delivery vehicle for prodrug activation. First, E. coli can selectively colonize solid tumors, providing high tumor/normal tissue colonization ratios of around 10,000 fold [17,18,38]. In agreement with these studies, we observed tumor/liver and tumor/spleen bacterial ratios of 9,700 and 23,000 at eight days after i.v. injection of E. coli to mice. By contrast, Salmonella provides tumor/liver and tumor/spleen bacterial ratios of 100~1000 [13,14]. Likewise, tumor selective antibodies typically achieve tumor/liver ratios of 1.4~7 in mice [21–24]. The high selectivity of E. coli for tumors may help reduce prodrug activation in normal tissues. Second, unlike liposomes, antibodies or replication deficient viruses, bacteria can proliferate in tumors [49–51]. The long-term colonization of tumors may allow multiple rounds of prodrug administration after a single injection of E. coli. These advantages have been recognized as indicated by the increasing number of papers investigating E. coli for tumor imaging and tumor therapy [15,17,19,20,38,52]. Of note, the strain of E. coli used in our study (DH5α) does not express enterotoxins or lipopolysaccharides and is considered to be non-pathogenic [53]. We used immune-deficient mice in most studies to investigate tumor therapy in human xenografts, but E. coli were also able to accumulate in murine tumors in immune-competent mice with similar toxicity as observed in immune deficient mice, suggesting that E. coli (DH5α) may be a suitable delivery vehicle for clinical applications.
Beta-glucuronidase is normally expressed at low levels in E. coli [46]. Thus, beta-glucuronidase levels were undetectable in wild-type E. coli. We constitutively expressed E. coli beta-glucuronidase in E. coli (lux/βG). These bacteria expressed about 60 ng beta-glucuronidase per 107 c.f.u. of bacteria and could convert cytotoxic concentrations of SN-38 from SN-38G. Because E. coli beta-glucuronidase can hydrolyze SN-38G to SN-38 about 250-fold faster than murine beta-glucuronidase (Fig. 8A), we anticipated that bacterial-mediated delivery of E. coli beta-glucuronidase to tumors would greatly enhance the antitumor activity of CPT-11 based on our previous studies in which we showed that ectopic expression or viral-mediated expression of murine beta-glucuronidase on tumor cells resulted in intratumoral conversion of SN-38G to SN-38, thereby increasing the antitumor activity of CPT-11 in mouse models of human cancer [4,36,37]. Indeed, significantly more beta-glucuronidase activity accumulated in tumors of mice treated with E. coli (lux/βG) as compared to mice treated with Ad/mβG (Fig. 8B). However, microbally-expressed E. coli beta-glucuronidase did not increase CPT-11 antitumor activity beyond that achievable by treating tumors with wild-type E. coli (Fig. 6). This correlates with our observation that a systemically-administered glucuronide fluorescent probe was more effectively hydrolyzed in tumors treated with Ad/mβG than in tumors colonized by E. coli (lux/βG) (Fig. 8E).
Ad/mβG and E. coli (lux/βG) therapy differ in several important facets as indicated in Table 2. First, the enzyme distribution in the tumor microenvironment is different. Adenoviruses only infect live cells whereas E. coli preferred to colonize the margins between necrotic regions and live cells (Fig. 5A) [4]. Ad/mβG infected cells may therefore more easily interact than E. coli (lux/βG) with systemically-administered drugs. Second, the enzyme location in the cells differs. Ad/mβG was designed to express beta-glucuronidase on the cell membrane of infected cells whereas E. coli (lux/βG) expresses beta-glucuronidase in the periplasmic space of the bacteria. Glucuronide drugs can directly interact with membrane-anchored beta-glucuronidase but require receptor-mediated transport into E. coli to interact with beta-glucuronidase [54]. Third, the enzymes are derived from different sources; Ad/mβG expresses murine beta-glucuronidase whereas E. coli (lux/βG) expresses E. coli beta-glucuronidase. The enzymatic activity of E. coli beta-glucuronidase is much greater than mouse beta-glucuronidase (Fig. 8A).
Table 2. Differences between Ad/mβG and E. coli (lux/βG).
| Therapy | Ad/mβG | E. coli (lux/βG) |
|---|---|---|
| Tumor distribution | Live cells | Border between live cells and necrotic areas |
| Enzyme location | Cancer cell surface | E. coli periplasmic space |
| Enzyme source | Mouse beta-glucuronidase | E. coli beta-glucuronidase |
We identified two impediments that may account for the relatively poor performance of E. coli (lux/βG) as compared to Ad/mβG. First, beta-glucuronidase is expressed intracellularly in E. coli (lux/βG) but is present on the surface of cells as a membrane anchored form after infection with Ad/mβG. Thus, glucuronide substrates must enter bacteria, presumably via a glucuronide transporter complex present on E. coli [54], to contact beta-glucuronidase present inside the bacteria. This step appeared to be rate-limiting as shown by the approximately 10.1 fold and 7.7 fold slower hydrolysis of 4-MUG and SN-38G, respectively, by the same amount of beta-glucuronidase in intact bacteria compared to bacterial lysates (Fig. 1D and E). No such barrier exists for membrane-anchored beta-glucuronidase, which may allow efficient hydrolysis of glucuronide substrates [55]. The second factor contributing to the poor performance of microbially-delivered beta-glucuronidase appears to be related to localization of bacteria in necrotic areas of the tumor, which may hinder contact of systemically administered drugs and bacteria. In agreement with Westphal et al [38], we observed that E. coli preferentially accumulated at the border between viable cancer cells and necrotic regions (Fig. 5). Salmonella and Shigella have also been observed to colonize in the necrotic areas of tumors [38], suggesting that this may be a general phenomenon. The necrotic areas in tumors are distant from blood vessels and display high interstitial fluid pressure [56–58] which may constitute a physiological barrier for drug delivery in solid tumors [59,60]. The high interstitial fluid pressure might impede drug diffusion to areas of bacterial tumor colonization. On the other hand, adenoviruses infect live cells which are located in the outer region of tumor tissue. Systemically-administered drugs might therefore more easily interact with beta-glucuronidase on Ad/mβG infected cancer cells. In agreement with these ideas, direct intratumoral injection of a glucuronide probe to tumors previously colonized with E. coli (lux/βG) significantly increased probe hydrolysis (Fig. 9B), consistent with drug distribution as a limiting factor in conversion of the glucuronide probe to fluorescent product in the tumors. Likewise, lysing E. coli (lux/βG) in the tumor by intratumoral injection of lysozyme and DNase I to release beta-glucuronidase also significantly increased hydrolysis of the glucuronide probe (Fig. 10B). Taken together, these results support the idea that bacterial distribution and cellular enzyme location are important factors for BDEPT of glucuronide prodrugs. Future studies that measure activated drug concentrations in tumors and serum will be important to confirm these results.
Besides E. coli, other bacteria such as Salmonella and Shigella also prefer to colonize in the necrotic regions of tumors [38]. Thus, designing secreted forms of therapeutic proteins [61–67] or inducing bacteria lysis to release intracellular proteins from the bacteria [66] might represent general approaches to maximize cancer cell and drug contact by allowing diffusion of the proteins more evenly in tumor tissue as well as by removing bacterial barriers to allow unhindered contact between enzymes and drug molecules.
Acknowledgments
The authors appreciated the helps with tissue section preparation by the Pathology Core Lab, Institute of Biomedical Sciences, at Academia Sinica, Taipei, Taiwan.
Data Availability
All relevant data are within the paper.
Funding Statement
This work was supported by the research grant from the National Science Council, Taipei, Taiwan (NSC102-2320-B-001-013-MY3). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Sreeramoju P, Libutti SK (2010) Strategies for targeting tumors and tumor vasculature for cancer therapy. Adv Genet 69: 135–152. 10.1016/S0065-2660(10)69015-3 [DOI] [PubMed] [Google Scholar]
- 2. Xu G, McLeod HL (2001) Strategies for enzyme/prodrug cancer therapy. Clin Cancer Res 7: 3314–3324. [PubMed] [Google Scholar]
- 3. Chung-Faye G, Palmer D, Anderson D, Clark J, Downes M, et al. (2001) Virus-directed, enzyme prodrug therapy with nitroimidazole reductase: a phase I and pharmacokinetic study of its prodrug, CB1954. Clin Cancer Res 7: 2662–2668. [PubMed] [Google Scholar]
- 4. Huang PT, Chen KC, Prijovich ZM, Cheng TL, Leu YL, et al. (2011) Enhancement of CPT-11 antitumor activity by adenovirus-mediated expression of beta-glucuronidase in tumors. Cancer Gene Ther 18: 381–389. 10.1038/cgt.2011.3 [DOI] [PubMed] [Google Scholar]
- 5. Fonseca MJ, Jagtenberg JC, Haisma HJ, Storm G (2003) Liposome-mediated targeting of enzymes to cancer cells for site-specific activation of prodrugs: comparison with the corresponding antibody-enzyme conjugate. Pharm Res 20: 423–428. [DOI] [PubMed] [Google Scholar]
- 6. Chen KC, Wu SY, Leu YL, Prijovich ZM, Chen BM, et al. (2011) A humanized immunoenzyme with enhanced activity for glucuronide prodrug activation in the tumor microenvironment. Bioconjug Chem 22: 938–948. 10.1021/bc1005784 [DOI] [PubMed] [Google Scholar]
- 7. Springer CJ, Niculescu-Duvaz II (1997) Antibody-directed enzyme prodrug therapy (ADEPT): a review. Adv Drug Deliv Rev 26: 151–172. [DOI] [PubMed] [Google Scholar]
- 8. Forbes NS (2010) Engineering the perfect (bacterial) cancer therapy. Nat Rev Cancer 10: 785–794. 10.1038/nrc2934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Malmgren RA, Flanigan CC (1955) Localization of the vegetative form of Clostridium tetani in mouse tumors following intravenous spore administration. Cancer Res 15: 473–478. [PubMed] [Google Scholar]
- 10. Moese JR, Moese G (1964) Oncolysis by Clostridia. I. Activity of Clostridium Butyricum (M-55) and Other Nonpathogenic Clostridia against the Ehrlich Carcinoma. Cancer Res 24: 212–216. [PubMed] [Google Scholar]
- 11. Kimura NT, Taniguchi S, Aoki K, Baba T (1980) Selective localization and growth of Bifidobacterium bifidum in mouse tumors following intravenous administration. Cancer Res 40: 2061–2068. [PubMed] [Google Scholar]
- 12. Yazawa K, Fujimori M, Amano J, Kano Y, Taniguchi S (2000) Bifidobacterium longum as a delivery system for cancer gene therapy: selective localization and growth in hypoxic tumors. Cancer Gene Ther 7: 269–274. [DOI] [PubMed] [Google Scholar]
- 13. Pawelek JM, Low KB, Bermudes D (1997) Tumor-targeted Salmonella as a novel anticancer vector. Cancer Res 57: 4537–4544. [PubMed] [Google Scholar]
- 14. Clairmont C, Lee KC, Pike J, Ittensohn M, Low KB, et al. (2000) Biodistribution and genetic stability of the novel antitumor agent VNP20009, a genetically modified strain of Salmonella typhimurium. J Infect Dis 181: 1996–2002. [DOI] [PubMed] [Google Scholar]
- 15. Cheng CM, Lu YL, Chuang KH, Hung WC, Shiea J, et al. (2008) Tumor-targeting prodrug-activating bacteria for cancer therapy. Cancer Gene Ther 15: 393–401. 10.1038/cgt.2008.10 [DOI] [PubMed] [Google Scholar]
- 16. Yu YA, Shabahang S, Timiryasova TM, Zhang Q, Beltz R, et al. (2004) Visualization of tumors and metastases in live animals with bacteria and vaccinia virus encoding light-emitting proteins. Nat Biotechnol 22: 313–320. [DOI] [PubMed] [Google Scholar]
- 17. Zhang HY, Man JH, Liang B, Zhou T, Wang CH, et al. Tumor-targeted delivery of biologically active TRAIL protein. Cancer Gene Ther 17: 334–343. 10.1038/cgt.2009.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stritzker J, Weibel S, Hill PJ, Oelschlaeger TA, Goebel W, et al. (2007) Tumor-specific colonization, tissue distribution, and gene induction by probiotic Escherichia coli Nissle 1917 in live mice. Int J Med Microbiol 297: 151–162. [DOI] [PubMed] [Google Scholar]
- 19. Min JJ, Kim HJ, Park JH, Moon S, Jeong JH, et al. (2008) Noninvasive real-time imaging of tumors and metastases using tumor-targeting light-emitting Escherichia coli. Mol Imaging Biol 10: 54–61. [DOI] [PubMed] [Google Scholar]
- 20. Jiang SN, Phan TX, Nam TK, Nguyen VH, Kim HS, et al. Inhibition of tumor growth and metastasis by a combination of Escherichia coli-mediated cytolytic therapy and radiotherapy. Mol Ther 18: 635–642. 10.1038/mt.2009.295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Press OW, Eary JF, Appelbaum FR, Martin PJ, Badger CC, et al. (1993) Radiolabeled-antibody therapy of B-cell lymphoma with autologous bone marrow support. N Engl J Med 329: 1219–1224. [DOI] [PubMed] [Google Scholar]
- 22. Reddy S, Shaller CC, Doss M, Shchaveleva I, Marks JD, et al. (2011) Evaluation of the anti-HER2 C6.5 diabody as a PET radiotracer to monitor HER2 status and predict response to trastuzumab treatment. Clin Cancer Res 17: 1509–1520. 10.1158/1078-0432.CCR-10-1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jang JK, Khawli LA, Park R, Wu BW, Li Z, et al. (2013) Cytoreductive chemotherapy improves the biodistribution of antibodies directed against tumor necrosis in murine solid tumor models. Mol Cancer Ther 12: 2827–2836. 10.1158/1535-7163.MCT-13-0383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hess C, Neri D (2014) Tumor-targeting properties of novel immunocytokines based on murine IL1beta and IL6. Protein Eng Des Sel 27: 207–213. 10.1093/protein/gzu013 [DOI] [PubMed] [Google Scholar]
- 25. Chang DK, Li PC, Lu RM, Jane WN, Wu HC (2013) Peptide-mediated liposomal Doxorubicin enhances drug delivery efficiency and therapeutic efficacy in animal models. PLoS One 8: e83239 10.1371/journal.pone.0083239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu M, Li W, Larregieu CA, Cheng M, Yan B, et al. (2014) Development of Synthetic Peptide-Modified Liposomes with LDL Receptor Targeting Capacity and Improved Anticancer Activity. Mol Pharm 11: 2305–2312. 10.1021/mp400759d [DOI] [PubMed] [Google Scholar]
- 27. Qhattal HS, Hye T, Alali A, Liu X (2014) Hyaluronan polymer length, grafting density, and surface poly(ethylene glycol) coating influence in vivo circulation and tumor targeting of hyaluronan-grafted liposomes. ACS Nano 8: 5423–5440. 10.1021/nn405839n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang L, Zhou H, Belzile O, Thorpe P, Zhao D (2014) Phosphatidylserine-targeted bimodal liposomal nanoparticles for in vivo imaging of breast cancer in mice. J Control Release 183: 114–123. 10.1016/j.jconrel.2014.03.043 [DOI] [PubMed] [Google Scholar]
- 29. Basili S, Moro S (2009) Novel camptothecin derivatives as topoisomerase I inhibitors. Expert Opin Ther Pat 19: 555–574. 10.1517/13543770902773437 [DOI] [PubMed] [Google Scholar]
- 30. Rivory LP, Bowles MR, Robert J, Pond SM (1996) Conversion of irinotecan (CPT-11) to its active metabolite, 7-ethyl-10-hydroxycamptothecin (SN-38), by human liver carboxylesterase. Biochem Pharmacol 52: 1103–1111. [DOI] [PubMed] [Google Scholar]
- 31. Kawato Y, Aonuma M, Hirota Y, Kuga H, Sato K (1991) Intracellular roles of SN-38, a metabolite of the camptothecin derivative CPT-11, in the antitumor effect of CPT-11. Cancer Res 51: 4187–4191. [PubMed] [Google Scholar]
- 32. Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB Jr., et al. (2002) The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc Natl Acad Sci U S A 99: 15387–15392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Iyer L, King CD, Whitington PF, Green MD, Roy SK, et al. (1998) Genetic predisposition to the metabolism of irinotecan (CPT-11). Role of uridine diphosphate glucuronosyltransferase isoform 1A1 in the glucuronidation of its active metabolite (SN-38) in human liver microsomes. J Clin Invest 101: 847–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rivory LP, Haaz MC, Canal P, Lokiec F, Armand JP, et al. (1997) Pharmacokinetic interrelationships of irinotecan (CPT-11) and its three major plasma metabolites in patients enrolled in phase I/II trials. Clin Cancer Res 3: 1261–1266. [PubMed] [Google Scholar]
- 35. Mathijssen RH, van Alphen RJ, Verweij J, Loos WJ, Nooter K, et al. (2001) Clinical pharmacokinetics and metabolism of irinotecan (CPT-11). Clin Cancer Res 7: 2182–2194. [PubMed] [Google Scholar]
- 36. Chen KC, Cheng TL, Leu YL, Prijovich ZM, Chuang CH, et al. (2007) Membrane-localized activation of glucuronide prodrugs by beta-glucuronidase enzymes. Cancer Gene Ther 14: 187–200. [DOI] [PubMed] [Google Scholar]
- 37. Prijovich ZM, Chen KC, Roffler SR (2009) Local enzymatic hydrolysis of an endogenously generated metabolite can enhance CPT-11 anticancer efficacy. Mol Cancer Ther 8: 940–946. 10.1158/1535-7163.MCT-08-0812 [DOI] [PubMed] [Google Scholar]
- 38. Westphal K, Leschner S, Jablonska J, Loessner H, Weiss S (2008) Containment of tumor-colonizing bacteria by host neutrophils. Cancer Res 68: 2952–2960. 10.1158/0008-5472.CAN-07-2984 [DOI] [PubMed] [Google Scholar]
- 39. Lin TP, Chen CL, Chang LK, Tschen JS, Liu ST (1999) Functional and transcriptional analyses of a fengycin synthetase gene, fenC, from Bacillus subtilis. J Bacteriol 181: 5060–5067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen CP, Hsieh YT, Prijovich ZM, Chuang HY, Chen KC, et al. (2012) ECSTASY, an adjustable membrane-tethered/soluble protein expression system for the directed evolution of mammalian proteins. Protein Eng Des Sel 25: 367–375. 10.1093/protein/gzs033 [DOI] [PubMed] [Google Scholar]
- 41. Chu YW, Yang PC, Yang SC, Shyu YC, Hendrix MJ, et al. (1997) Selection of invasive and metastatic subpopulations from a human lung adenocarcinoma cell line. Am J Respir Cell Mol Biol 17: 353–360. [DOI] [PubMed] [Google Scholar]
- 42. Cheng TL, Chen BM, Chan LY, Wu PY, Chern JW, et al. (1997) Poly(ethylene glycol) modification of beta-glucuronidase-antibody conjugates for solid-tumor therapy by targeted activation of glucuronide prodrugs. Cancer Immunol Immunother 44: 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cheng TL, Wu PY, Wu MF, Chern JW, Roffler SR (1999) Accelerated clearance of polyethylene glycol-modified proteins by anti-polyethylene glycol IgM. Bioconjug Chem 10: 520–528. [DOI] [PubMed] [Google Scholar]
- 44. Wu CH, Balasubramanian WR, Ko YP, Hsu G, Chang SE, et al. (2004) A simple method for the production of recombinant proteins from mammalian cells. Biotechnol Appl Biochem 40: 167–172. [DOI] [PubMed] [Google Scholar]
- 45. Roffler SR, Wang HE, Yu HM, Chang WD, Cheng CM, et al. (2006) A membrane antibody receptor for noninvasive imaging of gene expression. Gene Ther 13: 412–420. [DOI] [PubMed] [Google Scholar]
- 46. Novel G, Didier-Fichet ML, Stoeber F (1974) Inducibility of beta-glucuronidase in wild-type and hexuronate-negative mutants of Escherichia coli K-12. J Bacteriol 120: 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Huang PT, Chen KC, Prijovich ZM, Cheng TL, Leu YL, et al. Enhancement of CPT-11 antitumor activity by adenovirus-mediated expression of beta-glucuronidase in tumors. Cancer Gene Ther 18: 381–389. 10.1038/cgt.2011.3 [DOI] [PubMed] [Google Scholar]
- 48. Lehouritis P, Springer C, Tangney M (2013) Bacterial-directed enzyme prodrug therapy. J Control Release 170: 120–131. 10.1016/j.jconrel.2013.05.005 [DOI] [PubMed] [Google Scholar]
- 49. Loessner H, Leschner S, Endmann A, Westphal K, Wolf K, et al. (2009) Drug-inducible remote control of gene expression by probiotic Escherichia coli Nissle 1917 in intestine, tumor and gall bladder of mice. Microbes Infect 11: 1097–1105. 10.1016/j.micinf.2009.08.002 [DOI] [PubMed] [Google Scholar]
- 50. Zhang HY, Man JH, Liang B, Zhou T, Wang CH, et al. (2010) Tumor-targeted delivery of biologically active TRAIL protein. Cancer Gene Ther 17: 334–343. 10.1038/cgt.2009.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cronin M, Le Boeuf F, Murphy C, Roy DG, Falls T, et al. (2014) Bacterial-mediated knockdown of tumor resistance to an oncolytic virus enhances therapy. Mol Ther 22: 1188–1197. 10.1038/mt.2014.23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Critchley RJ, Jezzard S, Radford KJ, Goussard S, Lemoine NR, et al. (2004) Potential therapeutic applications of recombinant, invasive E. coli. Gene Ther 11: 1224–1233. [DOI] [PubMed] [Google Scholar]
- 53. Chart H, Smith HR, La Ragione RM, Woodward MJ (2000) An investigation into the pathogenic properties of Escherichia coli strains BLR, BL21, DH5alpha and EQ1. J Appl Microbiol 89: 1048–1058. [DOI] [PubMed] [Google Scholar]
- 54. Liang WJ, Wilson KJ, Xie H, Knol J, Suzuki S, et al. (2005) The gusBC genes of Escherichia coli encode a glucuronide transport system. J Bacteriol 187: 2377–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cheng CM, Chen FM, Lu YL, Tzou SC, Wang JY, et al. (2013) Expression of beta-glucuronidase on the surface of bacteria enhances activation of glucuronide prodrugs. Cancer Gene Ther 20: 276–281. 10.1038/cgt.2013.17 [DOI] [PubMed] [Google Scholar]
- 56. Heldin CH, Rubin K, Pietras K, Ostman A (2004) High interstitial fluid pressure—an obstacle in cancer therapy. Nat Rev Cancer 4: 806–813. [DOI] [PubMed] [Google Scholar]
- 57. Simonsen TG, Gaustad JV, Leinaas MN, Rofstad EK (2012) High interstitial fluid pressure is associated with tumor-line specific vascular abnormalities in human melanoma xenografts. PLoS One 7: e40006 10.1371/journal.pone.0040006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Boucher Y, Baxter LT, Jain RK (1990) Interstitial pressure gradients in tissue-isolated and subcutaneous tumors: implications for therapy. Cancer Res 50: 4478–4484. [PubMed] [Google Scholar]
- 59. Heine M, Freund B, Nielsen P, Jung C, Reimer R, et al. (2012) High interstitial fluid pressure is associated with low tumour penetration of diagnostic monoclonal antibodies applied for molecular imaging purposes. PLoS One 7: e36258 10.1371/journal.pone.0036258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Minchinton AI, Tannock IF (2006) Drug penetration in solid tumours. Nat Rev Cancer 6: 583–592. [DOI] [PubMed] [Google Scholar]
- 61. Loeffler M, Le’Negrate G, Krajewska M, Reed JC (2007) Attenuated Salmonella engineered to produce human cytokine LIGHT inhibit tumor growth. Proc Natl Acad Sci U S A 104: 12879–12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu SC, Ahn GO, Kioi M, Dorie MJ, Patterson AV, et al. (2008) Optimized clostridium-directed enzyme prodrug therapy improves the antitumor activity of the novel DNA cross-linking agent PR-104. Cancer Res 68: 7995–8003. 10.1158/0008-5472.CAN-08-1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Loeffler M, Le’Negrate G, Krajewska M, Reed JC (2009) Salmonella typhimurium engineered to produce CCL21 inhibit tumor growth. Cancer Immunol Immunother 58: 769–775. 10.1007/s00262-008-0555-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jiang SN, Phan TX, Nam TK, Nguyen VH, Kim HS, et al. (2010) Inhibition of tumor growth and metastasis by a combination of Escherichia coli-mediated cytolytic therapy and radiotherapy. Mol Ther 18: 635–642. 10.1038/mt.2009.295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Jiang SN, Park SH, Lee HJ, Zheng JH, Kim HS, et al. (2013) Engineering of bacteria for the visualization of targeted delivery of a cytolytic anticancer agent. Mol Ther 21: 1985–1995. 10.1038/mt.2013.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jeong JH, Kim K, Lim D, Jeong K, Hong Y, et al. (2014) Anti-tumoral effect of the mitochondrial target domain of Noxa delivered by an engineered Salmonella typhimurium. PLoS One 9: e80050 10.1371/journal.pone.0080050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Friedlos F, Lehouritis P, Ogilvie L, Hedley D, Davies L, et al. (2008) Attenuated Salmonella targets prodrug activating enzyme carboxypeptidase G2 to mouse melanoma and human breast and colon carcinomas for effective suicide gene therapy. Clin Cancer Res 14: 4259–4266. 10.1158/1078-0432.CCR-07-4800 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.