

Abstract
Folding of transmembrane and secretory proteins occurs in the lumen of the endoplasmic reticulum (ER) before transportation to the cell surface and is monitored by the unfolded protein response (UPR) signaling pathway. The accumulation of unfolded proteins in the ER activates the UPR that restores ER homeostasis by regulating gene expression that leads to an increase in the protein-folding capacity of the ER and a decrease in the ER protein-folding load. However, prolonged UPR activity has been associated with cell death in multiple pathological conditions, including neurodegeneration. Here, we report a spontaneous recessive mouse mutation that causes progressive cerebellar granule cell death and peripheral motor axon degeneration. By positional cloning, we identify the mutation in this strain as a retrotransposon insertion in the Clcc1 gene, which encodes a putative chloride channel localized to the ER. Furthermore, we demonstrate that the C3H/HeSnJ inbred strain has late onset cerebellar degeneration due to this mutation. Interestingly, acute knockdown of Clcc1 expression in cultured cells increases sensitivity to ER stress. In agreement, GRP78, the major HSP70 family chaperone in the ER, is upregulated in Clcc1-deficient granule cells in vivo, and ubiquitinated proteins accumulate in these neurons before their degeneration. These data suggest that disruption of chloride homeostasis in the ER disrupts the protein-folding capacity of the ER, leading to eventual neuron death.
Keywords: ataxia, cerebellum, degeneration, endoplasmic reticulum, ER stress, granule cell
Introduction
The endoplasmic reticulum (ER) generates and folds secreted and membrane-bound proteins that together account for more than one-third of newly synthesized proteins in the cell. Disruption of the protein-folding capacity of the ER, or accumulation of misfolded proteins in the ER lumen, results in ER stress. To restore ER homeostasis under conditions of ER stress, cells activate the unfolded protein response (UPR) signal transduction cascade, which acts to limit new protein synthesis, extend the protein-folding capacity of the ER, and accelerate the degradation of unfolded or misfolded proteins (Kaufman, 1999; Xu et al., 2005; Lindholm et al., 2006). However, excessive exposure to ER stressors can lead to prolonged ER stress resulting in the activation of signals leading to cell death (Xu et al., 2005; Zhao and Ackerman, 2006).
Many neurodegenerative diseases, including Alzheimer's disease, Parkinson's disease, Huntington's disease, and amyotrophic lateral sclerosis are associated with the accumulation of specific misfolded proteins (Xu et al., 2005; Lindholm et al., 2006; Zhao and Ackerman, 2006). Previous evidence suggests that these aberrant proteins disrupt the ER quality control pathways, leading to ER stress and neuron death (Saxena et al., 2009; Doyle et al., 2011). A few mutations in genes that regulate protein folding or other aspects of the ER microenvironment have been shown to induce neuron death. For example, loss of function of Sil1, a cochaperone of BiP (Grp78), the major HSP70 chaperone of the ER lumen and a UPR signaling protein, leads to ER stress in cerebellar Purkinje cells and the accumulation of ubiquitinated proteins in these neurons (Zhao et al., 2005). Similarly, disruption of the ER type 1 inositol 1,4,5-trisphosphate receptor (IP3R1) Ca2+ release channel that regulates release of calcium ions from the ER store has been associated with increased vulnerability to ER stress and neurodegeneration (Higo et al., 2010).
Here we describe our identification of a loss-of-function mutation in the chloride channel CLIC-like 1 (Clcc1) gene in a mouse model of late-onset ataxia and peripheral neuropathy. CLCC1 is an ER-localized transmembrane protein with high permeability to anions, in particular, chloride (Nagasawa et al., 2001). Notably, downregulation of this gene sensitizes cultured cells to chemically induced ER stress. Furthermore, loss of Clcc1 function in vivo results in upregulated expression of UPR target genes and the accumulation of ubiquitin-positive inclusions in neurons before their degeneration, suggesting that disruption of chloride/anion concentrations in the ER leads to loss of ER homeostasis and eventual neuron death.
Materials and Methods
Mice and genetic mapping.
The nm2453 mutant strain arose spontaneously in a colony of mice on a segregating C3H/HeSnJ, C57BL/6J, and C57BL/10J background. The original mutant strain was backcrossed to C57BL/6J for 10 generations to generate congenic B6.nm2453 mice (B6.Cgnm2453).
To map the nm2453 mutation, B6.Cgnm2453−/− mice were crossed with BALB/cByJ mice, and F1 mice were intercrossed to generate F2 mice. Initially, genomic DNA from 15 affected and 15 unaffected F2 mice was used for genome scans with polymorphic microsatellite markers. The critical interval was fine mapped in 517 F2 mice from an intercross of B6.Cgnm2453−/− and CAST/EiJ mice, using microsatellite markers and single nucleotide polymorphisms (SNPs). After the identification of the nm2453 mutation, mice were genotyped by PCR using the primer pairs Clcc1ex2F (5′ggctgctcctttgtgaatgt3′) and IAP forward (5′ggctcatgcgcagattattt3′) and Clcc1ex2F and Clcc1ex3R (5′ctgacagttcctcggcttct3′) that amplify the mutant and wild-type alleles of Clcc1, respectively.
To generate bacterial artificial chromosome (BAC) transgenic mice, the C57BL/6J-derived RP24-306B18 BAC was injected into the pronuclei of C57BL/6J zygotes. Founder mice were identified by PCR using the primer pairs T7306B18 (5′taatacgactcactataggg3′) and R1306B18 (5′ctcagggctgtcctttcttg3′) and SP6306B18 (5′gttttttgcgatctgccgtttc3′) and R2306B18 (5′cagacaagttgccagcacat3′), and then bred to C57BL/6J mice. All animal protocols were approved by The Jackson Laboratory Animal Care and Use Committee.
Histological and immunohistological analysis.
For histological analysis of brains, anesthetized mice were transcardially perfused with Bouin's fixative. Brains were postfixed in the same fixative overnight and embedded in paraffin for sectioning. For immunoflorescence analysis, mice were perfused with 4% PFA, and brains were postfixed in the same fixative before paraffin embedding. Antigen retrieval was performed in 0.01 m citrate buffer, pH 6, and sections were incubated at 4°C overnight with the following primary antibodies: rabbit anti-GRP78 (BiP; Cell Signaling Technology, Abcam), mouse anti-NeuN (Millipore Bioscience Research Reagents), mouse anti-ubiquitin (Cell Signaling Technology), and rat anti-GRP94 (Neomarkers). Fluorescent detection was performed with Alexa Fluor-conjugated secondary antibodies (Invitrogen), and images were collected by confocal microscopy. For neuron counts, granule cells, identified by their distinct nuclei, were counted in a 50 × 100 μm area of lobule II from hematoxylin and eosin midline sections. Cells were counted from three sections spaced 10 μm apart, and counts were averaged. The number of granule cells in lobule II was extrapolated using the area of the external granule cell layer as determined for each section using an image-processing package (Fuji). All histological analyses were performed on at least three mice from each genotype and time point using mice from both sexes.
Nerve dissection and analysis.
Dissection of femoral and sciatic nerves was performed as described previously (Seburn et al., 2006; Burgess et al., 2010). Briefly, for L5 ventral root analysis, mice were transcardially perfused with fixative (2% glutaraldehyde/2% paraformaldehyde in 0.1 m cacodylate buffer), and the ventral root was postfixed overnight. For femoral nerve analysis, the nerve was exposed in killed mice and briefly fixed (2% glutaraldehyde/2% paraformaldehyde in 0.1 m cacodylate buffer) before dissection and overnight postfixation in the same fixative. Dissected motor and sensory branches of femoral nerves were processed for plastic embedding and transmission electron microscopy by standard procedures. Nerve cross sections (0.5 μm) were stained with toluidine blue and examined by light microscopy. Myelinated axons were counted in nerves from three mice per genotype for each time point. To calculate the g-ratio (inner axon diameter/outer axon diameter), the inner diameter (axon only) and outer diameter (axon and myelin sheath) were determined in nerves of 10-month-old mice (three mice per genotype).
RT-PCR.
Total RNA from brain tissues was extracted with TRIzol Reagent (Life Technologies) and treated with DNase I (Ambion). RT-PCR was performed on random-primed cDNA (Invitrogen) using the primer pair Clcc1ex2F and reverse primer Clcc1ex7 (5′TGCTGAGCAAAAGCCATCT3′). Quantitative RT-PCR was performed in triplicate using Sybr Green Supermix (Bio-Rad) and the primer pair Clcc1ex5 (5′agaagcatggaagtggcagt3′) and Clcc1ex7 (5′atgctgagcaaaagccatct3′). GAPDH PCR with the primer pair GapdhF (5′atcactgccacccagaagac3′) and GapdhR (5′cacattgggggtaggaacac3′) was used as an internal control. Relative gene expression was calculated as described previously using the 2ΔΔ method (Livak, 2001).
Western blot analysis.
Tissues or cultured cells were lysed in RIPA buffer (25 mm Tris-HCl, pH 8.0, 100 mm NaCl, 1 mm EDTA, 0.5% sodium deoxycholate, 0.1% SDS, 1% NP-40, supplemented with complete protease inhibitor mixture; Roche). Protein concentration was determined using the DC Protein Assay (Bio-Rad). Lysates were separated on SDS-PAGE gels and transferred to polyvinylidene difluoride membranes (GE Healthcare) using standard techniques. Blots were incubated with the anti-CLCC1 antibodies (Sigma) overnight at 4°C. Incubation with antibodies to GAPDH (Cell Signaling Technology) was used as a loading control. Blots were incubated with HRP-conjugated secondary antibodies, and signals were detected by ECL (GE Healthcare).
Cell culture, transfection, and luciferase assays.
HEK293T cells were grown in DMEM with 10% FBS. For immunostaining, cells were plated on gelatin-coated (0.2% in PBS) coverslips. For transient transfection studies, 3 × 105 cells per well were seeded in a 12-well plate, and DNA plasmids were delivered into cells with Lipofectamine 2000 (Invitrogen) 24 h after plating. For siRNA transfection, cells were transfected with siRNA against human Clcc1 (5′agagatgagaugagauaagacuugu3′ and 5′acaagucuuaucucaucucaucucuuu3′) or negative control nontarget siRNAs (5′cguuaaucgcguauaauacgcguau3′ and 5′auacgcguauuauacgcgauuaacgac3′) after seeding using Lipofectamine RNAi MAX (Invitrogen). Luciferase activities were measured as per the manufacturer's instructions (Promega) in a multilabel counter.
Statistical analysis.
All data are presented as means ± SEM. Data were analyzed by one-way or two-way ANOVA (SPSS). p < 0.05 was considered statistically significant.
Results
The nm2453 mutation causes progressive cerebellar granule neuron degeneration and peripheral neuropathy
The nm2453 mutation arose spontaneously in a colony of mice segregating C3H/HeSnJ, C57BL/6J, and C57BL/10J alleles. Mice that were homozygous for this mutation developed mild truncal ataxia and muscle wasting in their hindquarters by 1 year of age. Before detailed analysis, the nm2453 mutation was introgressed onto the C57BL6/J background for 10 generations.
Pyknotic granule cells were present in low numbers in the inner granule cell layer (IGL) of the mutant, but not the wild-type, cerebellum beginning at 2 months of age. At 11 and 17 months of age, the IGL of the rostral cerebellum was obviously depleted of granule cells, with a loss of over 50% of these neurons (Fig. 1A–I). Granule cells in the caudal cerebellum also degenerated, although the onset of cell death was later than in the rostral cerebellum, and the extent of degeneration was more variable than in rostral neurons (data not shown). In addition, we observed vacuoles in the IGL of mutant mice, and the size and number of vacuoles increased in older mice. However, Purkinje cells or neurons in the cortex and hippocampus did not degenerate even in aged mutant animals, nor was granule cell death observed in heterozygous mice (Fig. 1J,K; data not shown).
Figure 1.

Progressive cerebellar granule neuron degeneration in nm2453 mutant mice. A–H, Sagittal sections of cerebella from 2-month-old (A, C), 11-month-old (B, D), and 17-month-old (E, G) nm2453−/− and 26-month-old wild-type (WT; F, H) mice were stained with hematoxylin and eosin. Higher-magnification images of lobule II are shown (C, D, G, H). Note the presence of vacuoles in the 11- and 17-month-old mutant cerebella. I,The number of granule cells in cerebellar lobule II from the indicated ages. Results are shown as mean ± SEM (n = 3). *p < 0.001 (statistically significant differences between similarly aged mutant and WT mice and aged and 2-month-old mutant mice). J, K, Immunohistochemistry with antibodies to calbindin D-28 on cerebellar sections from 17-month-old nm2453−/− (J) and 18-month-old WT mice. Note the intact Purkinje cell layer in the mutant cerebellum. Cerebellar lobules are indicated by Roman numerals (F). Scale bars: F, 500 μm; H, 50 μm; K, 1 mm.
Overt signs of muscle wasting were also observed in the hindquarters of aged nm2453 mutant mice. Indeed, atrophied muscle fibers, as identified by their decrease in fiber diameter, were observed upon histological analysis of the gastrocnemius muscle in 1-year-old mutant mice (Fig. 2A,B). Although normal fibers were still present, the number of atrophied muscle fibers increased in 2-year-old mutant mice, and many regenerating fibers, identified by their centrally located nuclei, were also observed (Fig. 2C,D), suggesting that mutant mice have neurogenic muscular atrophy.
Figure 2.
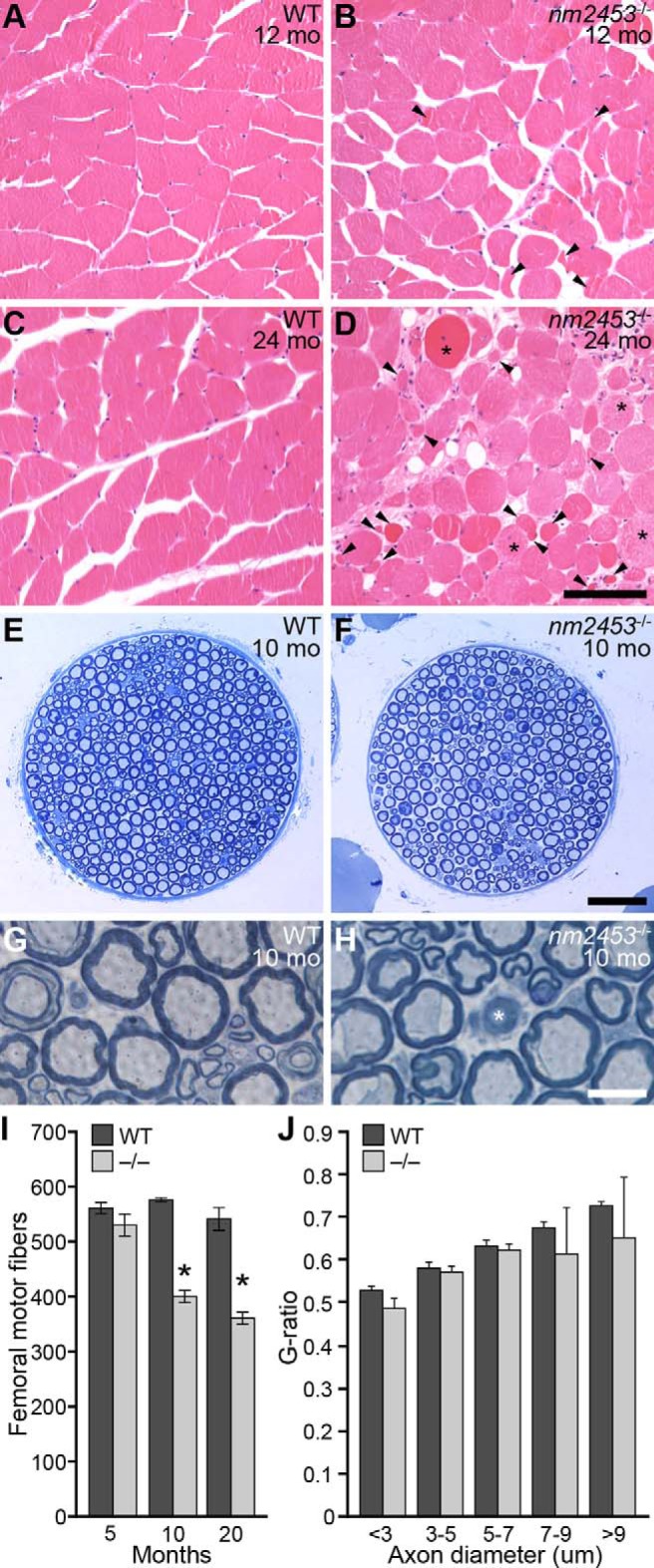
Muscle atrophy and peripheral neuropathy in nm2453 mutant mice. A–D, Hematoxylin- and eosin-stained cross sections of gastrocnemius muscles from wild-type (WT) and nm2453−/− mice. Arrowheads and asterisks indicate atrophied muscle fibers and fibers with central nuclei, respectively (D). E–H, Toluidine blue-stained cross sections of femoral nerve motor branches from 10-month-old WT and mutant mice. Axon degeneration (asterisk), but no typical signs of demyelination, was observed as shown in higher-magnification images (G, H). I, Quantitation of axon numbers in the motor branch of the WT and mutant femoral nerve from 5-, 10-, and 20-month-old mice. J, The g-ratio of axons in the motor branch of the femoral nerve from 12-month-old WT and nm2453−/− mice. No significant changes were observed between WT and mutant axons. Results in I and J are presented as mean ± SEM (n = 3). *p < 0.01 (statistically significant differences between similarly aged mutant and WT mice and aged and 2-month-old mutant mice). Scale bars: D, 100 μm; F, 50 μm; H, 10 μm.
To look for possible loss of motor innervation by spinal motor neurons, we analyzed the ventral (motor) root as a surrogate for motor neuron loss (Maddatu et al., 2004; Burgess et al., 2010; Gorbatyuk et al., 2012). No changes in axon numbers between 20-month-old nm2453 mutant and wild-type mice were observed (1085.9 ± 47 and 1052.7 ± 43, respectively). However, examination of the more distal peripheral motor branch of the femoral nerve revealed progressive axon degeneration in NMF2453 mutant mice between 5 and 10 months of age. Nerve fibers of all sizes were significantly reduced in mutant nerves from 10- and 20-month-old mutant mice relative to those in wild-type nerves from similarly aged mice (Fig. 2E–I; data not shown). Although degenerating nerve fibers were seen at high magnification, signs of demyelination, including ‘onion bulb’ pathology or thinly myelinated axons, were not observed (Fig. 2G,H). In agreement, no difference was observed in the g-ratio, the ratio of the diameter of the outer axon (including the myelin sheath) to the diameter of the inner axon (the nerve fiber without myelin), between wild-type and mutant axons from 10-month-old mice (Fig. 2J). Unlike motor axons, no difference in axon number was observed in the sensory branches of the femoral nerves from 2-year-old wild-type and mutant mice (849.3 ± 41 and 816.0 ± 34, respectively). These data suggest that muscular atrophy seen in nm2453−/− mice is associated with the degeneration of distal peripheral motor axons independently of demyelination.
The nm2453 mutation disrupts the Clcc1 gene
The nm2453 mutation was localized to Chromosome 3 by genome scans with polymorphic markers on affected F2 mice from an nm2453−/− and BALB/cByJ cross. Fine mapping of the mutation in F2 offspring from an nm2453−/− and CAST/Ei cross determined that the nm2453 mutation resided in a 0.44 Mb (0.29 centimorgans; three recombinants/1034 meioses), C3H/HeSnJ-derived interval between two SNP markers (rs29603979 and rs29651778; Fig. 3A).
Figure 3.

The nm2453 mutation disrupts the Clcc1 gene. A, The nm2453 mutation was mapped to Chromosome 3 between rs29603979 and rs29651778. The IAP insertion in intron 2 of Clcc1 is shown and primer locations for subsequent analyses indicated. Filled boxes represent coding exons (EX); open boxes represent UTR regions. B, RT-PCR of cerebellar mRNA from wild-type (WT; (+/+) and nm2453−/− (−/−) tissues using primers in Clcc1 exons 2 and 7. Note the presence of an additional band and the reduced intensity of the normal amplicon in reactions with mutant cDNA. GAPDH served as a control. C, Normally spliced Clcc1 transcripts were dramatically reduced in the cerebellum (Cer), hippocampus (Hippo), cortex, and spinal cord (SC) of 2-month-old nm2453−/− mice. Real-time PCR was performed with primers in Clcc1 exons 2 and 3, and results normalized to GAPDH expression levels. D, Western blot analysis of CLCC1 in tissues from 1-month-old WT mice. SM, Skeleton muscle. E, Western blot analysis of CLCC1 in 1-month-old WT and mutant tissues. GAPDH was used as a loading control in D and E.
Analysis of genes in the nm2453 interval demonstrated that abnormal transcripts were generated from the Clcc1 gene in the mutant cerebellum. RT-PCR using wild-type cDNA and primers corresponding to exon 2, the first coding exon of Clcc1, and exon 7 generated a single band of the predicted size (Fig. 3B). However, analysis of the mutant cDNA gave two amplicons: a band corresponding to exons 2–7 of Clcc1 that was greatly reduced in intensity relative to that generated in reactions using wild-type cDNA, and a higher molecular weight band that was not present in wild-type reactions. Sequence analysis of the aberrant band revealed that it contained exon 2 of Clcc1 followed by 529 nucleotides of an intracisternal A-particle (IAP) retrotransposon (Maksakova et al., 2006). Exons 3–7 of Clcc1 were present downstream of the IAP sequence, indicating that splicing occurs both into and out of the IAP.
Although normally spliced Clcc1 transcripts are still present in the nm2453−/− cerebellum, quantitative RT-PCR revealed that the mutant cerebellum, hippocampus, cerebral cortex, and spinal cord showed a >10-fold decrease in normally spliced Clcc1 transcripts (Fig. 3C). Similar to a previous study of the CLCC1 expression profile on rat tissues (Nagasawa et al., 2001), Western blot analysis of wild-type tissues demonstrated that CLCC1 is widely expressed (Fig. 3D). In agreement with lower levels of normally spliced transcripts in nm2453−/− mice and the multiple in-frame stop codons in the IAP insertion, protein levels were dramatically reduced in the nm2453 mutant tissues, including cerebellum and spinal cord (Fig. 3E; data not shown).
To confirm that the nm2453 mutant phenotype is caused by mutation of the Clcc1 gene, we performed an in vivo complementation assay. Transgenic mice were generated carrying a BAC (RP24-306B18) spanning the distal boundary of the nm2453 critical interval (Fig. 4A). The wild-type Clcc1, Gpsm2, and Aknad1 genes were present on the BAC; however, no polymorphisms were observed in the Gpsm2 coding sequence, nor was expression of this gene altered in the mutant cerebellum (data not shown). The Aknad1 gene was not within the critical interval for the nm2453 mutation, excluding this gene as a candidate for the nm2453-associated phenotype (Figs. 3A, 4A).
Figure 4.
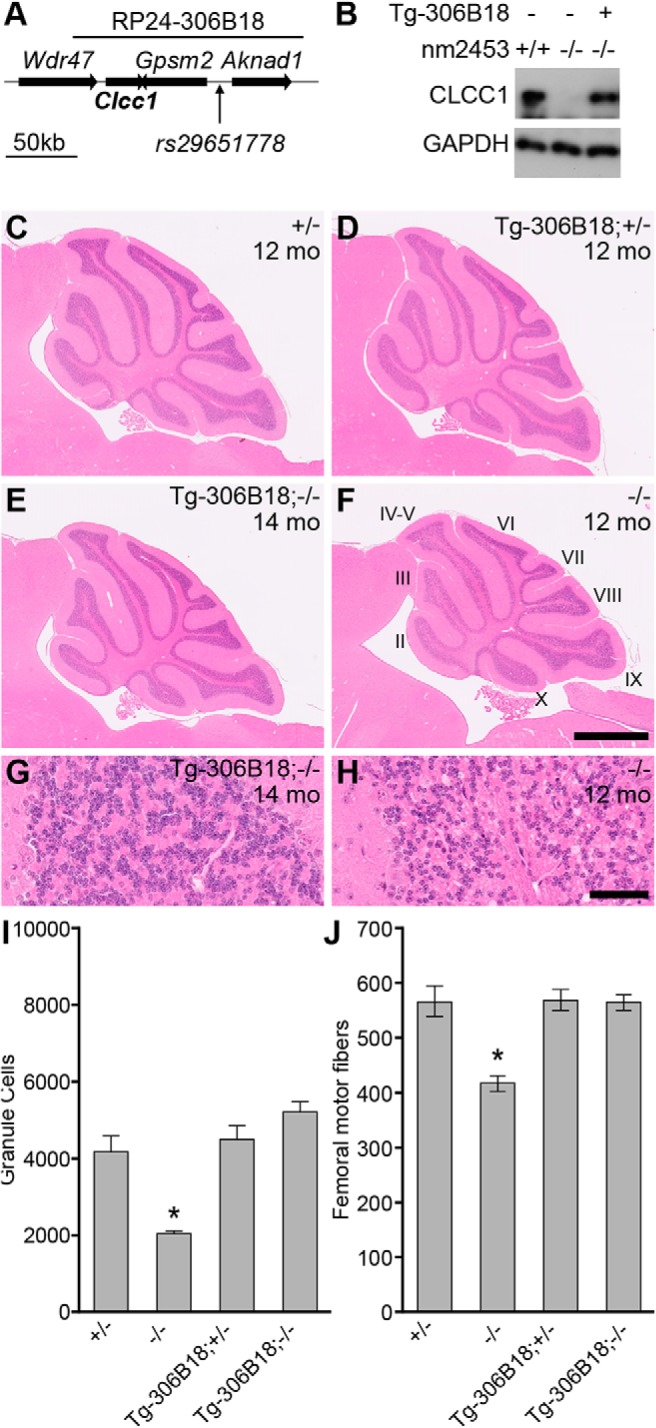
Transgenic expression of Clcc1 rescues nm2453−/− phenotypes. A, Schematic of the RP24–306B18 BAC used to produce transgenic mice. B, The transgene (Tg-306B18) restores CLCC1 expression in nm2453−/− mice as shown by Western blot of cerebellar lysates from 1-month-old wild-type (+/+) or nm2453−/− (−/−) mice with (+) or without (−) the BAC transgene. GAPDH was used as a loading control. C–H, Transgenic expression of Clcc1 rescues cerebellar granule cell degeneration in nm2453−/− mice. Representative images of hematoxylin- and eosin-stained sagittal sections of cerebella from nm2453−/− and nm2453+/− (+/−) mice with or without the BAC transgene (Tg-306B18) at the indicated ages. Cerebellar lobules are indicated by Roman numerals. Higher-magnification images from lobule II are shown in G and H. Scale bars: F, 500 μm; H, 50 μm. I, Granule cells in lobule II of the cerebella from the indicated genotypes demonstrating that transgenic expression of Clcc1 reduces cell death in the nm2453−/− cerebellum. J, Axon degeneration was also rescued in nm2453−/− mice by transgenic expression of Clcc1, as shown by counts of nerve fibers in the femoral motor branch of mice with the same genotypes as in C–H. Values in I and J are means ± SEM (n = 3). *p < 0.01.
Transgenic mice were bred with nm2453 mutant mice, and offspring were intercrossed to generate homozygous mutant mice with the BAC transgene with wild-type levels of CLCC1 in the cerebellum (Fig. 4B). Unlike 12-month-old nm2453−/− mice, no granule cell loss was observed in cerebellum from Tg-306B18; nm2453−/− mice relative to controls (Fig. 4C–I). Similarly, the BAC transgene rescued femoral motor axon degeneration as observed by axon counts in 12-month-old mice (Fig. 4J). Together, these data confirm that a loss of Clcc1 function leads to cerebellar granule cell and peripheral axon degeneration.
Clcc1 is mutated in the C3H/HeSnJ inbred strain
Our genetic mapping of the nm2453 mutation demonstrated that the IAP insertion in Clcc1 occurred in a genomic region derived from the C3H/HeSnJ inbred strain. Many retroviral-induced spontaneous mutations have been reported on the various C3H-derived substrains (Maksakova et al., 2006; Sun et al., 2008). Therefore, we investigated whether this mutation occurred on the segregating background where it was ascertained or if it originated on the C3H background.
PCR using primers corresponding to Clcc1 exons flanking the Clcc1 intron 2 generated a single band in reactions with C57BL6/J genomic DNA, but failed to amplify products in reactions using NMF2453−/− DNA as expected. When these primers were used in reactions containing genomic DNA from five closely related C3H substrains (C3H/HeOuJ, C3HeB/FeJ, C3H/HeJ, C3H/HeSnJ, and C3H/HeN), amplicons were generated in all reactions except those with C3H/HeSnJ DNA (Fig. 5A). The presence of the IAP insertion in intron 2 of Clcc1 in the C3H/HeSnJ strain was confirmed by reactions using primers corresponding to exons 2 or 3 of Clcc1 and the 5′ or 3′ end of the nm2453-associated IAP, respectively (Fig. 5A). These results demonstrate that the nm2453 mutation was derived from an IAP insertion in the C3H/HeSnJ inbred strain and that the retrotransposition of this IAP is a recent event occurring during, or after, the establishment of the C3H/HeSnJ substrain.
Figure 5.

The IAP insertion in Clcc1 induces granule cell degeneration in the C3H/HeSnJ inbred strain. A, The nm2453-associated IAP insertion is present in the C3H/HeSnJ genome. PCR was performed with the indicated primers in Clcc1 (Fig. 3A) using genomic DNA from the indicated C3H substrains. B–K, Hematoxylin- and eosin-stained sagittal sections of cerebella from the indicated C3H substrains. Higher-magnification images from lobule IX are shown (D, E, I, J, K). Note that, like the aged NM2354−/− mutant cerebellum (Fig. 1D,G), numerous vacuoles are observed in the 22-month-old C3H/HeSnJ cerebellum, but not in that of other aged C3H substrains. Scale bars: H, 500 μm; K, 50 μm.
To determine whether neurodegeneration occurs in C3H/HeSnJ mice, we examined the cerebella of C3H/HeSnJ mice (Fig. 5B–E). Although cell death progressed more slowly than in the B6J.nm2453−/− cerebellum, pyknotic granule cells were observed in cerebellum from 2-month old C3H/HeSnJ mice (data not shown). By 22 months of age, the C3H/HeSnJ cerebellum was clearly hypoplastic, with numerous vacuoles. Granule cell death was not observed in aged mice from the C3HeB/FeJ, C3H/HeJ, and C3H/HeOuJ substrains that lack the IAP insertion (Fig. 5F–K). Similar to the slower progression of granule cell death in C3H/HeSnJ mice relative to that observed in the B6J.nm2453−/− cerebellum, loss of axons in the motor branch of the femoral nerve was not observed between 2- and 12-month-old C3H/HeSnJ mice (2 months, 550.3 ± 17.9; 12 months, 549.7 ± 4.7). These data further confirm that the Clcc1 IAP insertion causes neurodegeneration and suggest that the C57BL6/J, relative to C3H/HeSnJ, genetic background enhances the Clcc1-deficient phenotype.
Clcc1 knockdown sensitizes cells to ER stress
Previous studies of overexpression of CLCC1 in CHO cells suggested a predominant endoplasmic reticular localization of this protein (Nagasawa et al., 2001). In agreement, ER localization was observed by transfection of HEK293T cells with a plasmid encoding epitope-tagged human CLCC1 (data not shown). Immunostaining of nontransfected cells with antibodies to CLCC1 also gave a reticular distribution suggestive of ER localization, a finding that was confirmed by coimmunostaining with antibodies to GRP94 that is largely localized to the ER lumen (Reddy et al., 1999; Fig. 6A–F). A strong signal was also observed surrounding the nucleus, as expected from the ER membrane localization of CLCC1.
Figure 6.

Knockdown of the ER-localized CLCC1 protein increases cell sensitivity to chemically induced ER stress. A–F, Endogenous CLCC1 colocalizes with GRP94, an ER marker. Immunofluorescence was performed on HEK293T cells with CLCC1 (A, D) and GRP94 (B, E) primary antibodies. Merged images are shown in C and F. Note the fine reticular ER localization of endogenous CLCC1. Scale bars: C, 10 μm; F, 5 μm. G, The experiment paradigm of ATF6-dependent in vitro reporter assays in control and Clcc1 knockdown HEK293T cells. H, Western blot analysis of CLCC1 levels 48 h after mock transfection or transfection of siRNAs. I, ATF6-dependent reporter activities before treatment (Ctrl) or after drug treatment. Transfection efficiency was normalized with pCMV-Renilla luciferase, as reflected in the graph. Note the increased reporter activity in Clcc1 knockdown cells after drug treatment. Statistically significant changes in reporter activity are shown by asterisks: *p < 0. 05; **p < 0. 01. n = 3.
Disruption of ER microenvironment can lead to ER stress and UPR induction (Kaufman, 1999; Xu et al., 2005), and prolonged ER stress has been implicated in the etiology of many neurodegenerative diseases (Lindholm et al., 2006; Zhao and Ackerman, 2006). Thus, we asked whether loss of Clcc1 function could sensitize the cellular response to ER stress-inducing agents. Human Clcc1 was knocked down by siRNAs in HEK293T cells, which consistently resulted in ∼90% reduction in protein levels (Fig. 6G,H). For controls, cells were mock transfected or transfected with nontarget siRNA. Twenty-four hours after siRNA treatment, cells were transfected with an ATF6 firefly luciferase reporter construct (p5XATF6), the expression of which is controlled by the ER-stress-regulated transcription factor ATF6 (Wang et al., 2000; Samali et al., 2010). A constitutively active Renilla luciferase reporter was cotransfected with p5XATF6 to monitor transfection efficiency. Cells were subsequently treated with either brefeldin A (BFA), an agent that perturbs anterograde transport of proteins from the ER to the Golgi apparatus; thapsigargin (TG), an ER calcium-ATPase inhibitor that disturbs ER calcium homeostasis; or tunicamycin (TU), a glycosylation inhibitor that causes accumulation of misfolded proteins in the ER, using the lowest dose that induced upregulation of the ATF6-dependent reporter but resulted in the minimal level of cell death in nontransfected cells (data not shown). ATF6 reporter activity was induced in cells treated with BFA, TG, or TU, as expected (Fig. 6I). Interestingly, reporter activity was significantly higher in Clcc1 knockdown cells than in control cells, suggesting that loss of Clcc1 function causes increased sensitivity to chemically induced ER stress.
Loss of Clcc1 function causes ER stress in the nm2453 mutant mouse
Given that Clcc1 knockdown increased the UPR response of cultured cells, we asked whether ER stress is associated with neurodegeneration in nm2453−/− mice. Coimmunofluorescence with antibodies to BiP (GRP78), the major HSP70 family member in the ER and a central regulator of ER homeostasis, and NeuN revealed that BiP was strikingly upregulated in granule cells in the 3-month-old nm2453 mutant cerebellum (Fig. 7A,D,G; data not shown). Notably, at this stage, very few granule cells undergo cell death, indicating that ER stress occurs before neurodegeneration in mutant cerebella.
Figure 7.

Upregulation of GRP78 and accumulation of misfolded proteins in the nm2453−/− cerebellar granule cells. A–I, Immunofluorescence with antibodies to BiP (GRP78; A, D, G) and ubiquitin (B, E, H) on cerebellar sections from 3-month-old wild-type (WT; A–C) and nm2453−/− (D–I) mice. Merged images are shown in C, F, and I. Higher-magnification images of D–F are shown in G–I. Note the upregulation of BiP in mutant granule cells that is in part coincident with the accumulation of misfolded proteins, as indicted by the punctate staining in E and H. Scale bars: C, F, 5 μm; I, 10 μm.
Previous studies indicated that prolonged ER stress leads to accumulation of ubiquitin-positive misfolded proteins in degenerating neurons (Zhao et al., 2005; Saxena et al., 2009). To determine whether misfolded proteins accumulate in nm2453 mutant granule cells, we immunostained cerebellar sections from homozygous mutant mice and wild-type littermate controls with an antibody to ubiquitin. A punctate staining pattern, typical for ubiquitin-positive protein aggregates, was observed in the cytoplasm of mutant, but not wild-type, granule cells (Fig. 7D–I). These puncta were commonly observed in granule neurons with upregulated expression of BiP or two other ER-resident chaperones, ERp72 and GRP94 (Fig. 7G–I; data not shown). In addition to the cerebellum, we also examined other brain areas, including the hippocampus, cortex, and striatum. No cell death was observed in these regions, nor was upregulation of BiP expression or Ub-positive inclusions observed (data not shown). Together, these data suggests that loss of Clcc1 function specifically results in disruption of ER homeostasis in the cerebellum, leading to neurodegeneration.
Discussion
Although expression of specific, misfolded proteins has been shown previously to cause neuron degeneration through ER stress, few ER resident molecules have been identified that when defective cause ER stress and neuron death. Here, we identified a novel mouse mutant with late-onset cerebellar granule cell degeneration and peripheral axon degeneration. By positional cloning, we identified the molecular defect in this mutant strain as an IAP insertion in the Clcc1 gene that causes dramatic downregulation of this gene. Interestingly, this mutation originally arose on the inbred C3H/HeSnJ strain, but the neurodegeneration associated with the Clcc1 mutation is likely enhanced by C57BL6/J alleles. Although very few pyknotic granule cells are observed in the mutant cerebellum at 3 months of age, upregulation of the ER chaperone BiP and ubiquitin-positive inclusions were widely observed in these neurons. These findings suggest that CLCC1 plays an important role in maintaining ER homeostasis even in the young adult cerebellum, and that levels of ER stress may progressively increase with neuron death occurring when a critical threshold is reached.
When incorporated into lipid bilayers, CLCC1 functions as an anion channel with the highest permeability to Cl− (Nagasawa et al., 2001), suggesting that disruption of Cl−/anion homeostasis in the ER leads to protein misfolding and neurodegeneration. Chloride channels/transporters are evolutionarily very diverse and are involved in a wide range of biological functions, including stabilization of membrane potential, regulation of synaptic activity, acidification of vesicles and organelles, and cell volume control (Duran et al., 2010). Studies provide evidence that dysfunction of intracellular organelle-localized chloride channels/transporters is associated with neurodegeneration (Jentsch et al., 2005). Disruption of the endosome and synaptic vesicle-localized chloride channel CLC3 impairs synaptic vesicle acidification, resulting in neuron loss in retina and hippocampus (Stobrawa et al., 2001). Mice with mutations in the endosomal and lysosomal chloride channel/transporter CLC7 also have neuron loss in multiple regions of the CNS in addition to osteopetrosis (Kornak et al., 2001; Kasper et al., 2005). In agreement with the subcellular localization of CLC7, degenerating neurons displayed typical features of neuronal ceroid lipofuscinosis, a human lysosomal storage disease (Kasper et al., 2005).
Previous studies suggest that chloride channels are present in the ER; however, the identity and the function of such channels are unknown. Our results suggest that CLCC1 is necessary for ER homeostasis and neuron survival. The ER is a major intracellular store for calcium, and disruption of Ca2+ concentrations in the ER can alter chaperone function, ultimately leading to protein misfolding and ER stress (Merksamer et al., 2008). Both ER Ca2+ uptake and release are electrogenic processes requiring the corresponding movement of counterions to maintain the electroneutrality of the ER membrane (al-Awqati, 1995; Edwards and Kahl, 2010). Although biochemical and physiological studies suggest the existence of chloride channels in ER fractions (Schmid et al., 1988; Morier and Sauvé, 1994), to the best of our knowledge, no chloride/anion channel has been identified as a counterion channel for ER Ca2+ movement; in fact, it has been suggested that the counterion current might be conducted by the ryanodine and IP3 receptors themselves (Gillespie and Fill, 2008). However, it is plausible that loss of CLCC1 function indirectly leads to disturbance of ER Ca2+ homeostasis, which in turn leads to the reduction of ER folding capability and accumulation of misfolded proteins.
Alternatively, CLCC1 may play a role in other ER functions. The phenotypic features of the nm2453 mutant mice, including peripheral neuropathy, ataxia, and spasticity in the lower limbs, occur later in adulthood and are reminiscent of clinical symptoms of some types of hereditary spastic paraplegia (HSP) in which spastic paraplegia is associated with ataxia (Dürr, 2008; Fink, 2014). However, no obvious axonopathy of the corticospinal track, which is typical for HSP, was observed in the aged mutant mouse (Y. Jia and S. L. Ackerman, unpublished observations). HSPs are heterogeneous diseases, and >70 genetic loci have been associated with these disorders (Dürr, 2008; Fink, 2014). Despite the genetic complexity of the HSPs, over half of HSP mutations are in one of three genes: atlastin-1 (SPG3A, also known as ATL1), spastin (SPG4, also known as SPAST), or receptor expression enhancing protein 1 (REEP1, also known as SPG31). The atlastin-1 GTPase and spastin AAA protein interact with REEP1 and other reticulon family members in the ER membrane to maintain its morphology (Park et al., 2010). The ER-localization and neurological phenotypes in Clcc1-deficient mice suggest that CLCC1 may be a component of protein complexes that regulate ER shape. Additional studies will be instructive in understanding the role of this protein.
Footnotes
This work was supported by the Mouse Mutant Resource Grant (NIHOD010972, to S.C.) and services used in this study were supported by CORE Grant CA34196. S.L.A. is an investigator of the Howard Hughes Medical Institute. The authors would like to thank Linda Washburn for her initial identification of the nm2453 mouse mutant, Dr. Kevin Seburn and Vivek Philip for assistance with statistics, Drs. Robert Burgess and Greg Cox for comments on this manuscript, Drs. Wayne Frankel and Cathleen Lutz for providing the aged C3H substrain mice, and The Jackson Laboratory sequencing, histology, microinjection, imaging, and multimedia services for their contributions.
The authors declare no competing financial interests.
References
- al-Awqati Q. Chloride channels of intracellular organelles. Curr Opin Cell Biol. 1995;7:504–508. doi: 10.1016/0955-0674(95)80006-9. [DOI] [PubMed] [Google Scholar]
- Burgess RW, Cox GA, Seburn KL. Neuromuscular disease models and analysis. Methods Mol Bio. 2010;602:347–393. doi: 10.1007/978-1-60761-058-8_20. [DOI] [PubMed] [Google Scholar]
- Doyle KM, Kennedy D, Gorman AM, Gupta S, Healy SJ, Samali A. Unfolded proteins and endoplasmic reticulum stress in neurodegenerative disorders. J Cell Mol Med. 2011;15:2025–2039. doi: 10.1111/j.1582-4934.2011.01374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran C, Thompson CH, Xiao Q, Hartzell HC. Chloride channels: often enigmatic, rarely predictable. Ann Rev Physiol. 2010;72:95–121. doi: 10.1146/annurev-physiol-021909-135811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dürr A. Genetic testing for the spastic paraplegias: drowning by numbers. Neurology. 2008;71:236–238. doi: 10.1212/01.wnl.0000320131.36091.a5. [DOI] [PubMed] [Google Scholar]
- Edwards JC, Kahl CR. Chloride channels of intracellular membranes. FEBS Lett. 2010;584:2102–2111. doi: 10.1016/j.febslet.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink JK. Hereditary spastic paraplegia: clinical principles and genetic advances. Semin Neurol. 2014;34:293–305. doi: 10.1055/s-0034-1386767. [DOI] [PubMed] [Google Scholar]
- Gillespie D, Fill M. Intracellular calcium release channels mediate their own countercurrent: the ryanodine receptor case study. Biophys J. 2008;95:3706–3714. doi: 10.1529/biophysj.108.131987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbatyuk MS, Shabashvili A, Chen W, Meyers C, Sullivan LF, Salganik M, Lin JH, Lewin AS, Muzyczka N, Gorbatyuk OS. Glucose regulated protein 78 diminishes alpha-synuclein neurotoxicity in a rat model of Parkinson disease. Mol Ther. 2012;20:1327–1337. doi: 10.1038/mt.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higo T, Hamada K, Hisatsune C, Nukina N, Hashikawa T, Hattori M, Nakamura T, Mikoshiba K. Mechanism of ER stress-induced brain damage by IP(3) receptor. Neuron. 2010;68:865–878. doi: 10.1016/j.neuron.2010.11.010. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ, Poet M, Fuhrmann JC, Zdebik AA. Physiological functions of CLC Cl- channels gleaned from human genetic disease and mouse models. Ann Rev Physiol. 2005;67:779–807. doi: 10.1146/annurev.physiol.67.032003.153245. [DOI] [PubMed] [Google Scholar]
- Kasper D, Planells-Cases R, Fuhrmann JC, Scheel O, Zeitz O, Ruether K, Schmitt A, Poët M, Steinfeld R, Schweizer M, Kornak U, Jentsch TJ. Loss of the chloride channel ClC-7 leads to lysosomal storage disease and neurodegeneration. EMBO J. 2005;24:1079–1091. doi: 10.1038/sj.emboj.7600576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- Kornak U, Kasper D, Bösl MR, Kaiser E, Schweizer M, Schulz A, Friedrich W, Delling G, Jentsch TJ. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell. 2001;104:205–215. doi: 10.1016/S0092-8674(01)00206-9. [DOI] [PubMed] [Google Scholar]
- Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006;13:385–392. doi: 10.1038/sj.cdd.4401778. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schimittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Maddatu TP, Garvey SM, Schroeder DG, Hampton TG, Cox GA. Transgenic rescue of neurogenic atrophy in the nmd mouse reveals a role for Ighmbp2 in dilated cardiomyopathy. Hum Mol Genet. 2004;13:1105–1115. doi: 10.1093/hmg/ddh129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksakova IA, Romanish MT, Gagnier L, Dunn CA, van de Lagemaat LN, Mager DL. Retroviral elements and their hosts: insertional mutagenesis in the mouse germ line. PLoS genetics. 2006;2:e2. doi: 10.1371/journal.pgen.0020002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merksamer PI, Trusina A, Papa FR. Real-time redox measurements during endoplasmic reticulum stress reveal interlinked protein folding functions. Cell. 2008;135:933–947. doi: 10.1016/j.cell.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morier N, Sauvé R. Analysis of a novel double-barreled anion channel from rat liver rough endoplasmic reticulum. Biophys J. 1994;67:590–602. doi: 10.1016/S0006-3495(94)80519-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasawa M, Kanzaki M, Iino Y, Morishita Y, Kojima I. Identification of a novel chloride channel expressed in the endoplasmic reticulum, Golgi apparatus, and nucleus. J Biol Chem. 2001;276:20413–20418. doi: 10.1074/jbc.M100366200. [DOI] [PubMed] [Google Scholar]
- Park SH, Zhu PP, Parker RL, Blackstone C. Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network. J Clin Invest. 2010;120:1097–1110. doi: 10.1172/JCI40979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy RK, Lu J, Lee AS. The endoplasmic reticulum chaperone glycoprotein GRP94 with Ca(2+)-binding and antiapoptotic properties is a novel proteolytic target of calpain during etoposide-induced apoptosis. J Biol Chem. 1999;274:28476–28483. doi: 10.1074/jbc.274.40.28476. [DOI] [PubMed] [Google Scholar]
- Samali A, Fitzgerald U, Deegan S, Gupta S. Methods for monitoring endoplasmic reticulum stress and the unfolded protein response. Int J Cell Biol. 2010;2010:830307. doi: 10.1155/2010/830307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci. 2009;12:627–636. doi: 10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- Schmid A, Gogelein H, Kemmer TP, Schulz I. Anion channels in giant liposomes made of endoplasmic reticulum vesicles from rat exocrine pancreas. J Membrane Biol. 1988;104:275–282. doi: 10.1007/BF01872329. [DOI] [PubMed] [Google Scholar]
- Seburn KL, Nangle LA, Cox GA, Schimmel P, Burgess RW. An active dominant mutation of glycyl-tRNA synthetase causes neuropathy in a Charcot-Marie-Tooth 2D mouse model. Neuron. 2006;51:715–726. doi: 10.1016/j.neuron.2006.08.027. [DOI] [PubMed] [Google Scholar]
- Stobrawa SM, Breiderhoff T, Takamori S, Engel D, Schweizer M, Zdebik AA, Bosl MR, Ruether K, Jahn H, Draguhn A, Bösl MR, Ruether K, Jahn H, Draguhn A, Jahn R, Jentsch TJ. Disruption of ClC-3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron. 2001;29:185–196. doi: 10.1016/S0896-6273(01)00189-1. [DOI] [PubMed] [Google Scholar]
- Sun XY, Chen ZY, Hayashi Y, Kanou Y, Takagishi Y, Oda S, Murata Y. Insertion of an intracisternal A particle retrotransposon element in plasma membrane calcium ATPase 2 gene attenuates its expression and produces an ataxic phenotype in joggle mutant mice. Gene. 2008;411:94–102. doi: 10.1016/j.gene.2008.01.013. [DOI] [PubMed] [Google Scholar]
- Wang Y, Shen J, Arenzana N, Tirasophon W, Kaufman RJ, Prywes R. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J Biol Chem. 2000;275:27013–27020. doi: 10.1074/jbc.M003322200. [DOI] [PubMed] [Google Scholar]
- Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clinical Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Ackerman SL. Endoplasmic reticulum stress in health and disease. Curr Opin Cell Biol. 2006;18:444–452. doi: 10.1016/j.ceb.2006.06.005. [DOI] [PubMed] [Google Scholar]
- Zhao L, Longo-Guess C, Harris BS, Lee JW, Ackerman SL. Protein accumulation and neurodegeneration in the woozy mutant mouse is caused by disruption of SIL1, a cochaperone of BiP. Nat Genet. 2005;37:974–979. doi: 10.1038/ng1620. [DOI] [PubMed] [Google Scholar]