

Abstract
Autism spectrum disorder (ASD) is increasingly thought to result from low-level deficits in synaptic development and neural circuit formation that cascade into more complex cognitive symptoms. However, the link between synaptic dysfunction and behavior is not well understood. By comparing the effects of abnormal circuit formation and behavioral outcomes across different species, it should be possible to pinpoint the conserved fundamental processes that result in disease. Here we use a novel model for neurodevelopmental disorders in which we expose Xenopus laevis tadpoles to valproic acid (VPA) during a critical time point in brain development at which neurogenesis and neural circuit formation required for sensory processing are occurring. VPA is a commonly prescribed antiepileptic drug with known teratogenic effects. In utero exposure to VPA in humans or rodents results in a higher incidence of ASD or ASD-like behavior later in life. We find that tadpoles exposed to VPA have abnormal sensorimotor and schooling behavior that is accompanied by hyperconnected neural networks in the optic tectum, increased excitatory and inhibitory synaptic drive, elevated levels of spontaneous synaptic activity, and decreased neuronal intrinsic excitability. Consistent with these findings, VPA-treated tadpoles also have increased seizure susceptibility and decreased acoustic startle habituation. These findings indicate that the effects of VPA are remarkably conserved across vertebrate species and that changes in neural circuitry resulting from abnormal developmental pruning can cascade into higher-level behavioral deficits.
Keywords: autism, developmental, neural circuit, valproic acid, Xenopus
Introduction
Autism spectrum disorder (ASD) is characterized by highly recognizable symptoms, including deficits in social interaction, language, sensory integration, and increased seizure susceptibility, yet across the population, ASD can have very different underlying etiologies. For example, >40 different mutations and various environmental risk factors have been associated with ASD (Abrahams and Geschwind, 2008; Lichtenstein et al., 2010; Neale et al., 2012). Despite this great diversity of potential causes and complex cognitive symptoms, there is reason to believe that these symptoms originate from a few fundamental abnormalities in nervous system development and function (Pratt and Khakhalin, 2013). These include abnormal synaptic maturation and plasticity, defects in microcircuitry organization, disruption of the excitation to inhibition balance, and local overconnectivity and long-range underconnectivity throughout the brain (Krey and Dolmetsch, 2007; Abrahams and Geschwind, 2008; Markram et al., 2008; Rinaldi et al., 2008; Markram and Markram, 2010; Bhakar et al., 2012; Marín, 2012). Thus, ASD could be a manifestation unique to humans of low-level neurodevelopmental abnormalities, but links between synaptic dysfunction, neural circuit development, and ASD-related behaviors still remain to be fully explored. These links could be better studied in simpler animal models even if they are not necessarily capable of expressing higher-level cognitive symptoms of ASD, as long as links between neural circuit function and behavior can be drawn. Furthermore, finding conserved behavioral effects resulting from abnormalities in synaptic development across species is a powerful method for identifying fundamental biological processes underlying complex human disorders.
One known cause of ASD conserved between humans and animal models is prenatal exposure to valproic acid (VPA). VPA is one of the most commonly prescribed antiepileptic drugs (Gerstner et al., 2008), but it has well known teratogenic effects. If a developing fetus is exposed to VPA, this exposure greatly increases the risk of ASD later in life (Bath and Scharfman, 2013; Bromley et al., 2013). In rodents, in utero exposure to VPA results in autistic-like behaviors in offspring, namely, decreased socialization, increased repetitive behaviors, and hypersensitivity to sensory stimuli (Schneider and Przewlocki, 2005; Markram et al., 2008). These behavioral deficits are accompanied by corresponding deficits in brain physiology, including increased local recurrent connectivity, synaptic activity, and decreased intrinsic neuronal excitability (Rinaldi et al., 2008; Walcott et al., 2011; Martin and Manzoni, 2014). Although studies such as these provide a host of phenomenological results, whether these results are reproducible in other model organisms and thus whether they represent a fundamental evolutionary conserved mechanism underlying neurodevelopmental disorders remain unknown.
The development of the Xenopus tadpole visual system has served as a useful model for understanding basic processes mediating neural circuit development, as well as a model for a variety of neurological and neurodevelopmental disorders (Pratt and Khakhalin, 2013). Here, we test electrophysiological, behavioral, and neuroanatomical effects of exposure to VPA during a key period in Xenopus visual system development, in which synapses and circuits in the optic tectum undergo a large amount of maturation and refinement that correlate with robust changes in visual function and behavior.
Materials and Methods
All animal experiments were performed in accordance with and approved by Brown University Institutional Animal Care and Use Committee standards.
Experimental animals.
Tadpoles were raised in Steinberg's rearing media on a 12 h light/dark cycle at 18–21°C for 7–8 d, until they reached developmental stage 42 (Nieuwkoop and Faber, 1956). They were then transferred to either control rearing media or 1 mm solution of VPA in Steinberg's solution and raised at temperatures ranging from 18°C to 24°C until they reached developmental stages of either 47 or 49, depending on the protocol (see below). Developmental stages of tadpoles were determined according to Nieuwkoop and Faber (1956). The rearing medium was renewed every 3 d. Immediately before behavioral experiments, tadpoles were transferred to Steinberg's media and left for 1 h to recover from acute action of the drug. Tadpoles that were used for acoustic startle, schooling, or seizure protocols were not used again for other experiments, whereas after visual avoidance experiments, tadpoles were in some cases reused in startle and schooling experiments after having 24 h of rest in respective rearing solution. At least two different clutches of tadpoles from different husbandry were used for every set of behavioral experiments. Animals of either sex were used because, at these developmental stages, tadpoles of either sex are phenotypically indistinguishable.
Behavioral experiments.
Previous research using Xenopus tadpoles has demonstrated that exposing developing tadpoles to concentrations of 2 mm VPA caused teratogenic effects and developmental delays (Terbach et al., 2011). In vivo and in vitro studies in various organisms have shown that concentrations of VPA from 0.75 to 3.5 mm can serve as an effective dose for modeling teratogenic effects of VPA (Rinaldi et al., 2008; Terbach et al., 2011; Wang et al., 2012). For the present study, we performed dose–response assays and found that exposing developing tadpoles to 1 mm of VPA beginning at stage 41/42 results in no statistically significant changes in mortality rate compared with controls and resulted in no visible physically deforming teratogenic effects. Thus, this concentration was selected for all experiments presented in this study.
Seizures.
For seizure experiments, stage 47 tadpoles were transferred into individual wells in a six-well plate (Corning), each filled with 7 ml of 5 mm pentylenetetrazol (PTZ) solution in Steinberg's media. The plate was diffusely illuminated from below and imaged from above with a SCB 2001 color camera (Samsung) at 30 frames/s. Tadpole positions were tracked in Noldus EthoVision XT (Noldus Information Technology) and processed offline in a custom MATLAB program (MathWorks). Onset of regular seizures happened on average 3.9 ± 1.3 min into the recording; seizure events were defined as periods of rapid and irregular movement, interrupted by periods of immobility (Bell et al., 2011), and were detected automatically using swimming speed thresholding at a level of half of the maximal swimming speed. Frequency of seizures and length of seizure events were measured across 5 min intervals of a 20-min-long recording.
Startle response habituation.
Stage 49 tadpoles were placed into individual wells in a six-well plate, which was fixed between two audio speakers (SPA2210/27; Philips), and mechanically connected to diaphragms of these speakers by inflexible plastic struts. Acoustic stimuli (one period of a 200 Hz sine wave, 5 ms long) were delivered every 5 s. These stimuli evoked a reliable startle response. Because of the nature of the stimulus, the startle response likely reflects a combination of inner ear-mediated and lateral line-mediated inputs, and, for the purposes of this study, we did not attempt to differentiate between the two. The amplitude of stimuli for habituation experiments was set at two times above the startle threshold. Each train of stimuli lasted for 2 min; trains 1 to 5 were separated by 5 min gaps, whereas train 6 was separated from train 5 by a 15 min gap. The location of VPA-raised tadpoles and matched controls within the six-well plate was alternated across experiments to compensate for possible variation in stimulus delivery across the wells. Videos were acquired and tracked in EthoVision and processed offline in a custom MATLAB script.
Peak speed of each startle response was measured across a 2 s interval after stimulus delivery. To quantify habituation of startle responses at different timescales, we adopted the nomenclature from previous studies (Eddins et al., 2010; Roberts et al., 2011) and averaged startle speeds across 1-min-long periods of acoustic stimulation, comparing these averages across periods. For rapid habituation, we compared responses during minutes 1 and 2 of the first 2-min train of stimuli. For short-term habituation (acting at timescales of 5–20 min and measured after a short 5 min period of rest), we compared average response during minute 1 of train 1 with that in minute 1 of train 4, whereas for long-term habituation (acting at timescales of ∼1 h and measured after a longer 15 min period of rest), a similar comparison was performed between trains 1 and 6 (see Fig. 7D).
Figure 7.

VPA-reared tadpoles exhibit increased seizure susceptibility and decreased acoustic startle habituation. A, A typical C-shaped bend exhibited by tadpoles during pharmacologically induced seizures. B, A sample trace of swimming speed evolution during a seizure protocol; onset of regular seizures is marked with an arrow. C, Comparison of seizures frequency during the first 5 min of testing for control (blue) and VPA-reared (red) tadpoles. All data, medians, and 20–80% quantiles are shown. D, Speed of acoustic startle responses in the acoustic habituation protocol; the plot shows all data points and averages across all tested animals in each group. Brackets on the top illustrate segments of data that were averaged and compared for each tadpole to produce estimations of rapid, short-term, and long-term habituation. E, Comparison of short-term habituation amount, defined as the ratio of average startle response amplitudes at different stages of habituation protocol (see Results), for control (blue) and VPA-reared (red) tadpoles. F, Comparison of long-term habituation amount for control and VPA-reared tadpoles. *p < 0.05.
Collision avoidance.
Stage 49 tadpoles were placed in a clear plastic Petri dish (8.5 cm in diameter) filled to an approximate depth of 1 cm with Steinberg's solution at 18°C. The dish was put on top of a CRT monitor screen (maximum luminance, 57 cd/m2 and minimum luminance, 0.3 cd/m2; Dell Ultrascan 1600 SH Series; Dell Computer Company) and screened from all sides with an opaque black cloth. Stimuli were generated by a custom-written MATLAB program using the Psychophysics Toolbox (Brainard, 1997). A black circle of a radius of either 0.28 or 0.56 cm was projected in the center of the dish. Every 30 s, this circle was sent toward the tadpole at a speed of either 1.4 or 2.9 cm/s. Only collisions in which the animal was swimming within 1 s before the encounter with the circle were included in the dataset. Experiments were performed in the morning (from 9:00 A.M. to 1:00 P.M.), because animals seemed to be less responsive in the afternoon; each testing session lasted for 5 min. Videos were acquired in EthoVision; both the tadpole and the stimulus were manually tracked offline, and trajectories were exported for additional automated analysis in MATLAB. Avoidance response initiation points were identified as points of peak acceleration immediately after an encounter with a visual stimulus; escape speed was averaged over a 17 ms window (five frames) around the swimming velocity peak.
Schooling.
Fifteen to 20 tadpoles at developmental stage 49 were transferred to a glass bowl 17 cm in diameter (for each batch, control tadpoles matched VPA-raised tadpoles in number). A still image of tadpole distribution in the bowl was made every 5 min using Yawcam software (Magnus Lundvall, Yawcam) for 1 h (13 images per experiment). A strong acoustic stimulus was delivered 2.5 min after each photo was taken to elicit a startle response and force tadpoles to redistribute (Katz et al., 1981). Coordinates of tadpole heads and tails were tracked manually in NIH ImageJ and exported for additional processing in MATLAB. We defined neighboring tadpoles through point set triangulation and used a Kolmogorov–Smirnov test to compare distributions of inter-tadpole distances between VPA-raised tadpoles and matched controls. For all pairs of “neighboring tadpoles” that were located closer than 5.7 cm to each other (two-thirds of the bowl radius), we also estimated the angle between their orientations in the bowl (Wassersug and Hessler, 1971).
Statistics and data presentation.
For behavioral data, averages and SDs are given in the text, whereas median values and 20–80% percentiles are shown in the figures. When the Mann–Whitney test was used to compare values between the groups, significance values were reported as PMW, whereas for Kolmogorov–Smirnov test, p values are reported as PKS. Sample sizes are reported as n = x, N = y, where lowercase n stands for the number of measurements and capital N stands for the number of animals.
Electrophysiology experiments.
For whole-brain recordings, tadpole brains were prepared as described by Wu et al. (1996) and Aizenman et al. (2003). In brief, tadpoles were anesthetized in 0.02% tricainemethane sulfonate (MS-222). To access the ventral surface of the tectum, brains were filleted along the dorsal midline and dissected in HEPES-buffered extracellular saline [in mm: 115 NaCl, 2 KCl, 3 Cacl2, 3 MgCl2, 5 HEPES, 10 glucose, and 0.1 picrotoxin, pH 7.2 (osmolarity, 255 mOsm)]. Brains were then pinned to a submerged block of Sylgard in a recording chamber and maintained at room temperature (24°C). To access tectal cells, the ventricular membrane surrounding the tectum was carefully removed using a broken glass pipette. For evoked synaptic response experiments, a bipolar stimulating electrode (FHC) was placed on the optic chiasm to activate RGC axons.
Whole-cell voltage-clamp and current-clamp recordings were performed using glass micropipettes (8–12 MΩ) filled with K-gluconate intracellular saline [in mm: 100 K-gluconate, 8 KCl, 5 NaCl, 1.5 MgCl2, 20 HEPES, 10 EGTA, 2 ATP, and 0.3 GTP, pH 7.2 (osmolarity, 255 mOsm)]. Recordings were restricted consistently to retinorecipient neurons in the middle one-third of the tectum, thus avoiding any developmental variability existing along the rostrocaudal axis (Wu et al., 1996; Khakhalin and Aizenman, 2012; Hamodi and Pratt, 2014). Electrical signals were measured with an Axopatch 200B or a Multiclamp 700B amplifier (Molecular Devices), digitized at 10 kHz using a Digidata 1322A analog-to-digital board, and acquired using pClamp 10 software. Leak subtraction was done in real time using the acquisition software. Membrane potential in the figures was not adjusted to compensate for a predicted 12 mV liquid junction potential. Data were analyzed using AxographX software. The GABAA antagonist picrotoxin (100 μm) was added to the external saline in a subset of experiments. Spontaneous synaptic events were collected and quantified using a variable amplitude template (Clements and Bekkers, 1997). Spontaneous EPSCs (sEPSCs) were recorded at −60 mV in the presence of picrotoxin, whereas sIPSCs were collected in control media at 5 mV (the reversal for glutamatergic currents). For each cell, 60 s of spontaneous activity was recorded. For evoked synaptic response experiments, a bipolar stimulating electrode (FHC) was placed on the optic chiasm to activate RGC axons. Synaptic stimulation experiments were conducted by collecting EPSCs evoked by stimulating the optic chiasm at a stimulus intensity that consistently evoked maximal amplitude EPSCs. Evoked responses at −45 mV (excitation) and 5 mV (inhibition) were used to calculate the excitation/inhibition (E/I) ratio. Excitation and inhibition were calculated as a measure of area under the curve for a 250 ms time window beginning at the onset of the synaptic response. Evoked monosynaptic events (at a stimulus intensity that does not evoke polysynaptic activity, typically 30–60% of the maximum) were used to collect AMPA/NMDA ratios. Peak current amplitude at −65 mV (1 ms window at peak; 10–15 trials per cell) was used to calculate AMPAR-mediated currents, and average current amplitude collected at 55mV (10 ms window 20 ms after peak AMPA; 5–15 trials per cell) was used to calculate NMDAR-mediated currents. NMDAR current decay rates were collected from evoked monosynaptic currents collected at 55 mV in the presence of the AMPAR blocker NBQX (20 μm). The rate of decay (Tau) was calculated from the peak of the NMDA current fit to a single exponential (10–20 trials per cell). Polysynaptic stimulation experiments were performed by collecting EPSCs evoked by stimulating the optic chiasm at a stimulus intensity that evoked the maximal amplitude EPSC. Quantification of polysynaptic activity was calculated by measuring the total change in current over 100 ms time bins beginning at the onset of the evoked response. A spontaneous barrage was defined as a change in holding current of 10 or 20 pA intervals for a period of >200 ms. To quantify intrinsic cell excitability, cells were presented with a series of depolarizing steps (20 pA intervals) in current clamp, starting from −65 mV. The number of spikes elicited by current injection was quantified using the following criteria: to qualify as a spike, the height of the spike had to be at least half the height of its preceding spike and no wider than three times the width of the first original spike (Pratt and Aizenman, 2007). Voltage-gated Na+ and K+ current–voltage (I–V) curves were calculated as in the study by Aizenman et al. (2003), by measuring the early Na+ peak current and the steady-state K+ current. All statistics used nonparametric Mann–Whitney U tests. Graphs show median and interquartile ranges (IQRs) as error bars, and data in the text show averages and SDs.
Neuronal morphology.
Tadpoles were reared in either VPA or control media as above. To label single tectal cells, after 3–4 d in rearing media (stage 44–45), tadpoles were temporarily removed, and whole-brain electroporation (Haas et al., 2002) was used to transfect tectal neurons: tadpoles were anesthetized with 0.02% MS-222, and plasmid DNA (4–5 μg/μl in water, colored with fast green) was pressure injected into the middle ventricle of stage 44–46 tadpoles using a glass micropipette. Then, two platinum electrodes (1–2 mm) were placed on the skin overlying both sides of the midbrain to electroporate the DNA into the tectal cells. One to two electrical pulses at 50 V with an exponential decay of 70 ms were delivered. We used the pCALNL–GFP plus pCAG–Cre plasmids (courtesy Ed Ruthazer, McGill University, Montreal, Quebec, Canada; Ruthazer et al., 2013) at a ratio of 5000:1 to limit coexpression to a very small subset of neurons. Immediately after electroporation, the tadpoles were transferred back into bowls containing their respective rearing media. Tadpoles were allowed to develop until stage 49 and were then anesthetized for imaging. Tadpoles were selected for those in which a single neuron was expressing GFP and could be imaged clearly. A Zeiss LSM-510 Meta confocal microscope was used to image tadpoles containing labeled neurons. Optical sections at 1 μm intervals were acquired from the confocal microscope with a 20× or 63× objective. The confocal images were then used to reconstruct the three-dimensional morphology of tectal cells using Neuromantic software (http://www.reading.ac.uk/neuromantic/). Three-dimensional reconstructions of dendritic arbors were created by Neuromantic software and were used to calculate the total dendritic branch length, branch numbers, and tip numbers. L-Measure (http://cng.gmu.edu:8080/Lm/) was used for three-dimensional Sholl analysis. The number of dendritic branch tips at each sphere segment was summed in each 10 μm bin for plotting and statistical analysis. Results were nearly the same if number of branches intersecting each sphere was used as a metric. All morphometric data except Sholl analysis were tested for significance by using Mann–Whitney U tests. Sholl data were analyzed by comparing branch-tip distributions from cells across experimental groups. A two-way ANOVA with multiple comparisons was performed.
Results
Collision avoidance
The retinotectal circuit and local circuits within the optic tectum, the primary visual area in the frog brain, are known to refine during development in an activity-dependent manner and mediate robust visually guided behaviors that are very sensitive to abnormal circuit development (Dong et al., 2009; Lee et al., 2010; Khakhalin et al., 2014), providing a strong link between neural circuit physiology and behavior. We first examined whether VPA exposure had any behavioral effects, particularly in tasks that required sensorimotor integration, such as visually guided behavior and schooling. Visual avoidance behavior relies on normal development of the retinotectal circuitry and can be used to detect abnormal wiring in this brain region (Lee et al., 2010; Spawn and Aizenman, 2012). Tadpoles were either reared in control media or exposed to VPA between developmental stages 42 and 49, a critical time period for tectal circuit establishment and maturation (Akerman and Cline, 2006; Pratt and Aizenman, 2007). To compare processing of visual information in VPA-treated and control tadpoles, we assessed their ability to implement visually guided avoidance maneuvers and prevent collisions with objects that approach them (Fig. 1A). It was shown previously that, depending on the collision dynamics, tadpoles can perform two types of adaptive maneuvers: (1) slow “avoidance” that is used to dodge large slowly moving visual stimuli; and (2) a fast startle-like “escape” maneuver that is typically evoked by smaller and faster visual stimuli (Khakhalin et al., 2014). We tested VPA-raised and control tadpoles in both “fast” and “slow” types of collision-avoidance protocols. When fast escape maneuvers were elicited, VPA-raised animals escaped from the stimulus less effectively than control tadpoles: only 39 ± 28% of collisions were avoided by VPA-raised tadpoles compared with 71 ± 26% for control animals (PMW = 0.001; N = 21 and 19 respectively). Threshold distance at which startle responses were initiated was lower in VPA-raised animals (1.1 ± 0.8 cm) than for control ones (1.6 ± 1.2; PMW = 6e-6; n = 111 and 161; N = 22 and 19 respectively; Fig. 1B). Escape maneuver speed did not differ significantly across conditions (7.5 ± 3.7 cm/s for VPA; 6.9 ± 2.8 cm/s for control; PMW = 0.1; n = 111 and 161; N = 22 and 19; Fig. 1D).
Figure 1.

VPA-reared tadpoles show impaired visual avoidance and schooling behaviors. A, Three sample frames representing dynamics of a typical fast collision-avoidance maneuver (see Results). B, Reaction distance during collision avoidance from a fast-moving object; all points, median, and 20–80% quantiles are shown. Blue, Control tadpoles; red, VPA-reared tadpoles. C, Peak speed during avoidance maneuver from a fast-moving object. D, Reaction distance during avoidance from a slowly moving object. E, Peak speed during avoidance from a slowly moving object. F, Distribution of inter-tadpole distances during schooling for both control (blue) and VPA-reared (red) animals; control animals have higher representation of short and long distances but fewer middle distances compared with VPA-reared tadpoles. G, Typical distributions and orientations of control and VPA-reared animals in the bowl during schooling; control tadpoles (blue) tend to form clusters, whereas VPA-reared animals (red) are distributed evenly. H, Distribution of angles between orientations of neighboring tadpoles; VPA-reared animals are markedly less co-oriented. K-S, Kolmogorov–Smirnov test. *p < 0.05 (Mann–Whitney).
In experiments designed to measure slow avoidance of large visual stimuli, VPA-treated tadpoles also initiated an avoidance maneuver at a slightly lower distance from the moving circle center than control animals (1.6 ± 0.8 cm in VPA vs 1.8 ± 0.7 cm in control, PMW = 0.005, n = 154 and 155, N = 19 and 20 respectively; Fig. 1C). Unlike in experiments measuring fast escape behavior, for slow avoidance, the speed of avoidance maneuvers was very different between treated and control tadpoles (Fig. 1E): VPA-raised animals escaped with speeds characteristic of fast escape responses (7.9 ± 7.7 cm/s), whereas control tadpoles maneuvered slowly (3.2 ± 4.5 cm/s; PMW = 9e-6; n = 153 and 155; N = 19 and 20, respectively). Although in this study we did not attempt to explicitly classify responses into fast and slow subtypes, it likely that some VPA-reared animals performed “inappropriate” fast escapes instead of slow avoidance maneuvers, because, for control tadpoles, the distribution of avoidance speeds was unimodal [Hartigan's dip test (Hartigan and Hartigan, 1985), PH = 0.98], whereas in VPA-treated tadpoles, respective distribution was markedly bimodal (PH = 1e-5).
Based on the pattern of background swimming occurring between visually evoked avoidance trials, VPA-raised tadpoles were slightly less active overall than their control counterparts. VPA-raised and control tadpoles spent a similar share of time actively swimming (69 ± 27% for VPA vs 77 ± 27% in control; PMW = 0.2; N = 21 and 19), but VPA-raised animals had a slightly lower average swimming speed (1.4 ± 0.4 cm/s in VPA; 1.8 ± 0.5 cm/s in control; PMW = 0.005; N = 21 and 19) and exhibited less frequent spontaneous accelerations (1.9 ± 2.3 starts/min in VPA vs 3.4 ± 2.8 starts/min in control; PMW = 0.02; N = 21 and 19).
Schooling
Tadpoles normally demonstrate schooling behavior when put in a large arena (Katz et al., 1981). This social behavior is likely a result of integration of multiple sensory and social cues (Wassersug and Hessler, 1971; Lum et al., 1982; Villinger and Waldman, 2012). We compared schooling in control and VPA-raised tadpoles: a total of 1861 inter-tadpole distances were measured for VPA-raised tadpoles and 1859 distances for matched controls. Empirical distributions of inter-tadpole distances differed significantly between groups (PKS = 0.01; N = 60 and 59 for VPA and control respectively; Fig. 1F). Compared with VPA-reared animals, control tadpoles had a higher share of short and long inter-tadpole distances and fewer medium distances, because they aggregated socially and schooled around the bowl in small clusters of five to six animals (Fig. 1G). In contrast, VPA-raised tadpoles did not aggregate but distributed evenly across the bowl, resulting in a higher share of medium inter-tadpole distances and fewer short and long distances.
When body-axis orientation of neighboring tadpoles was considered, the distribution of inter-tadpole angles also differed between VPA-raised and control groups (PKS = 7e-8; n = 1861 and 1859; N = 60 and 59), because neighboring VPA-raised animals did not co-orient as well as control tadpoles (Fig. 1H). Together, these data indicate that VPA-reared tadpoles not only do not tend to aggregate but also do not swim in the same direction as their immediate neighbors, indicating an abnormal schooling behavior.
Deficits in both avoidance and schooling behaviors suggest that VPA-reared tadpoles may have abnormally developed circuitry for sensory processing and sensorimotor integration, which is known to take place in the optic tectum (Dong et al., 2009). Thus, we looked at both the dendritic anatomy and network connectivity of tectal neurons in control and VPA-treated animals, especially because it is believed that neurodevelopmental disorders are marked by abnormalities in synaptic activity (Markram et al., 2008; Rinaldi et al., 2008; Markram and Markram, 2010; Walcott et al., 2011; Martin and Manzoni, 2014).
Dendritic structure of tectal neurons
We used electroporation to express GFP in single tectal neurons from control and VPA-treated groups (Fig. 2A, ref). We then reconstructed the dendritic arbors and quantified various aspects of dendritic morphology, including total dendritic branch length (control, 518 ± 242 μm, n = 11; VPA, 385 ± 154 μm, n = 12; p = 0.08), number of branch tips (control, 18.7 ± 13.5, n = 11; VPA, 11.8 ± 5.8, n = 12; p = 0.34), and number of bifurcations (control, 17.5 ± 13.7, n = 11; VPA, 10.8 ± 5.8, n = 12; p = 0.38). We found no significant differences in these parameters between control and VPA tadpoles, although across measures, the VPA neurons tended to be smaller and less branched. We next examined the branching pattern by performing a three-dimensional Sholl analysis (see Materials and Methods; Fig. 2B) and found significant differences in the branch distribution between control and VPA-treated tectal neurons. Control neurons had most branches concentrated midway along the dendritic arbor, whereas tectal neurons from VPA-raised tadpoles tended to have branches throughout the apical dendrite and a lot more branching closer to the soma (two-way ANOVA, p < 0.0001). This indicates that tectal neuron dendrites develop abnormally in VPA-reared tadpoles, possibly as a result of abnormal patterns of synaptic activity during development. However, such subtle neuroanatomical changes cannot necessarily explain the large-scale behavioral deficits. Thus, we performed a functional assessment of synaptic connectivity using electrophysiology.
Figure 2.
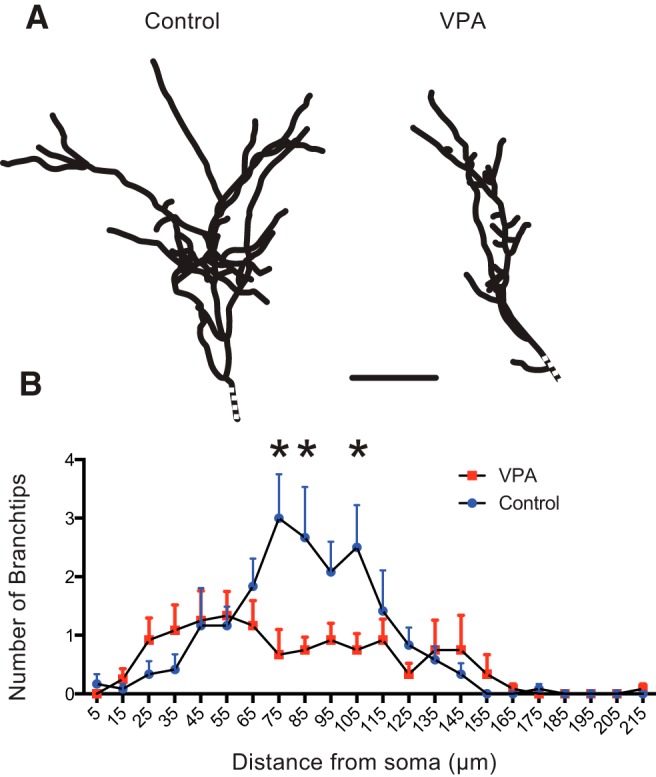
Abnormal dendritic morphology in VPA-reared tadpoles. A, Reconstructed dendritic arbors from representative tectal neurons from control and VPA-exposed tadpoles show different branching structure. Dotted line indicates the position of the soma. B, Three-dimensional Sholl analysis indicating the number of branch tips at different distances from the soma. Notice the differences in branch-tip distribution. Each group represents an average of 12 (control) and 13 (VPA) neurons, and error bars are SD. *p < 0.05.
Synaptic properties
To assess the effects of VPA treatment on the synaptic connectivity in the developing tectum, we performed whole-cell patch-clamp recordings using an isolated whole-brain preparation (Wu et al., 1996). We observed a significant increase in the frequency of sEPSCs and sIPSCs in VPA-treated tadpoles compared with controls [sEPSC frequency (events/s): control, 2.28 ± 2.17, n = 29; VPA, 3.78 ± 2.97, n = 33; PMW = 0.03 (Fig. 3A,B); sIPSC frequency (events/s): control, 1.79 ± 1.07, n = 10; VPA, 3.35 ± 2.11, n = 12; PMW = 0.04 (Fig. 3A,B)]. Consistent with the literature (Walcott et al., 2011), there were no significant differences in sEPSC or sIPSC amplitude (sEPSC amplitude: control, 9.4 ± 9.1 pA, n = 10; VPA, 10.2 ± 4.8 n = 12; PMW = 0.57; sIPSC amplitude: control, 7.8 ± 2.1 pA, n = 10; VPA, 8.2 ± 2.9, n = 12; PMW = 0.93; Fig. 3C). We further assessed changes in excitation and inhibition by looking at evoked synaptic responses by directly shocking the optic chiasm with a bipolar stimulating electrode. We found that both maximal excitatory and inhibitory responses were significantly increased in VPA-treated animals (excitation: n = 8; control peak amplitude, −737 ± 373 pA; VPA peak, −1486 ± 819 pA; PMW = 0.049; inhibition: n = 8; control, 866 ± 824 pA; VPA, 3156 ± 1968 pA; PMW = 0.0144; Fig. 3D). However, the E/I ratio in tectum remained consistent between the control and VPA-treated populations (E/I ratio: control, 1.85 ± 3.1, n = 6; VPA, 0.78 ± 0.77, n = 8; PMW = 0.74; Fig. 3D).
Figure 3.

VPA exposure enhances spontaneous and evoked excitation and inhibition in the tectum. A, Example sEPSCs and sIPSCs recorded at 5 and −60 mV, respectively, from control and VPA-treated tadpoles. B, Frequency of sEPSCs and sIPSCs are significantly enhanced in the VPA group. C, Plot of sEPSC and sIPSC amplitudes from both groups show no difference. D, Total charge from evoked synaptic responses over a 100 ms window for both EPSCs and IPSCs for both experimental groups (left) and E/I ratios. Although evoked currents are larger in VPA tadpoles, the E/I ratio is unchanged. The inset shows sample EPSC and IPSC traces. Error bars are IQRs and medians. *p < 0.05.
The AMPA/NMDA current ratio is a useful metric for assessing synaptic maturation, because glutamatergic synapses incorporate greater levels of AMPARs as they mature (Wu et al., 1996; Isaac, 2003). We found no significant change in the AMPA/NMDA current ratio between control and VPA-reared tadpoles (control, 2.12 ± 1.28, n = 13; VPA, 1.63 ± 1.63, n = 11; PMW = 0.24; Fig. 4A,B), suggesting that, on average, VPA-treated tectal cells do not contain a higher proportion of immature synapses. Another indicator of synaptic maturation is a shift between NMDAR NR2B to NR2A (Tovar and Westbrook, 1999, Ewald et al., 2008). Because NR2A subunits have a faster decay kinetics, this shift can be detected as a speeding up of the decay rate of NMDAR-mediated synaptic responses (Flint et al., 1997). The increased levels of NR2B earlier in development are thought to promote synaptic plasticity required for synapse formation (Quinlan et al., 1999). We found the decay of NMDAR-mediated currents was slower in VPA-treated tadpoles (control, 287 ± 143 ms, n = 15; VPA, 409 ± 166 ms, n = 12; PMW = 0.03; Fig. 4C,D), suggesting that, although VPA treatment does not impair insertion of AMPAR, overall synapses in treated tadpoles do show some characteristics of a state that would promote more synaptic plasticity (Rinaldi et al., 2007). Together, these results indicate that VPA-treated tadpoles have abnormally increased levels of excitatory and inhibitory synaptic transmission, as well as enhanced NMDAR-mediated transmission.
Figure 4.
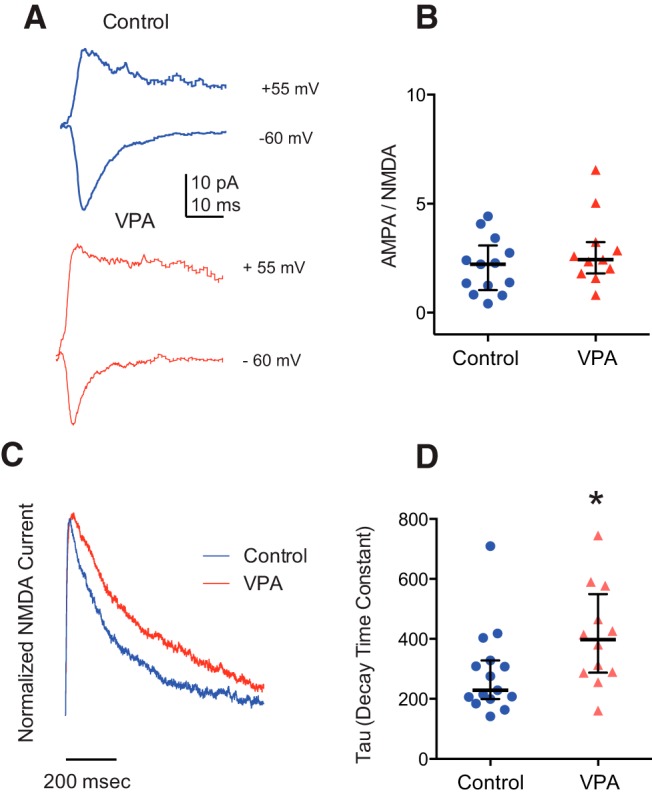
Effects of VPA rearing on synaptic maturation. A, Example traces of evoked AMPA-mediated (−65 mV) and AMPA/NMDA-mediated (55 mV) currents. B, Ratio of AMPA current peak/NMDA current amplitude. Control, n = 13; VPA, n = 11. Error bars indicate the IQR. C, Examples traces depicting isolated NMDAR-mediated currents from control and VPA-treated tadpoles. D, Scatter plot showing decay time constants of the NMDAR-mediated response. PMW = 0.03; control, n = 15; VPA, n = 12. Error bars are IQR. *p < 0.05.
Increased network connectivity and excitability
One possibility is that the observed increase in synaptic transmission reflects an abnormal developmental process in which synaptic pruning of both visual and local synaptic connections that occurs between developmental stages 45 and 49 (Pratt et al., 2008) is impaired. This would not only result in increased synaptic drive from visual synapses but also increased hyperconnectivity from local tectal synapses (Lee et al., 2010). In fact, various theories of autism and other neurodevelopmental disorders suggest that these disorders are marked by hyperconnected and excitable networks (Markram et al., 2007; Markram and Markram, 2010; Pescosolido et al., 2012). In the developing Xenopus tectum, evoked polysynaptic responses and spontaneous barrages of recurrent activity have been used to quantify intra-tectal connectivity and network excitability (Pratt and Aizenman, 2007; Pratt et al., 2008). To determine whether chronic exposure to VPA resulted in increased network excitability, we measured evoked and spontaneous polysynaptic activity in the tectum. We found that VPA-treated tadpoles had significantly greater amounts of polysynaptic activity as calculated during the 100 ms after the stimulus onset, as measured by total charge (n = 6; control, 529 ± 441 pA/s; VPA, 1425 ± 719 pA/s; PMW = 0.02; Fig. 5A,B). We also observed increased frequency of barrages of spontaneous recurrent activity (barrage frequency: control, 1.14 ± 1.26 barrages/min, n = 28; VPA, 2.58 ± 2.47 barrages/min, n = 26; PMW = 0.038; Fig. 5C,D). This suggests that VPA-reared tadpoles have a greater degree of recurrent connectivity within the optic tectum, consistent with abnormal developmental refinement and pruning of these circuits.
Figure 5.
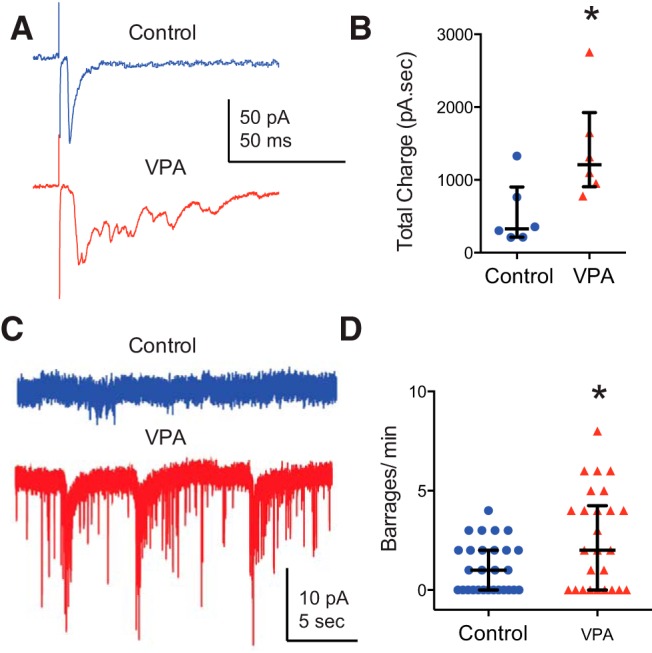
Exposure to VPA enhances recurrent connectivity in the optic tectum. A, Example traces of maximally evoked synaptic responses from control and VPA-treated tadpoles at −65 mV. Notice the enhanced polysynaptic activity in the VPA response. B, Total charge calculated over 100 ms from maximally evoked synaptic responses reflect increased polysynaptic activity in the VPA group. Mann–Whitney U test, p = 0.026, N = 6. Error bars are IQRs and medians. C, Example traces of spontaneous synaptic activity from control and VPA tectal cells (recorded at −65 mV with PTX in the external recording solution). D, The VPA group shows increased frequency of barrages. PMW = 0.038; control, n = 28; VPA, n = 26. Error bars are IQRs. *p < 0.05.
Changes in intrinsic excitability of tectal neurons
One possibility is that increased network activity is driven by an increase in the intrinsic neuronal excitability of tectal neurons (Aizenman et al., 2003), but previous research has indicated that VPA exposure can result in a reduction in neuronal excitability (Rinaldi et al., 2008; Walcott et al., 2011). To test whether either of these possibilities occurred, we assessed intrinsic properties of tectal cells in VPA-reared tadpoles. We performed current-clamp recordings and measured spike output in response to a series of current injections of increasing amplitude. We found that intrinsic cell excitability was slightly, but significantly, reduced in tectal neurons after VPA exposure (maximum spike output: n = 19; control, 2.53 ± 0.42 spikes; VPA, 1.77 ± 0.35; PMW = 0.0001; Fig. 6A,B). VPA-exposed cells also had a small but significant reduction in spike threshold, time to spike peak, and an increase in voltage-gated Na+ current amplitude (Fig. 6C; Table 1). Thus, the reduction in excitability cannot be explained by changes in Na+ currents, because these occur in the direction opposite of what would be expected. Furthermore, there were no changes in membrane resistance and membrane capacitance between groups (Table 1). One possibility is that the decrease in excitability was attributable to changes in Na+ channel inactivation properties, such as faster inactivation or slower recovery of inactivation. The fact that action potentials in the VPA group have a faster time-to-peak and faster repolarization are both consistent with this hypothesis (Fig. 6A, inset). However, because of the small change in excitability and the cell-to-cell variability, it was not practical to test inactivation directly.
Figure 6.
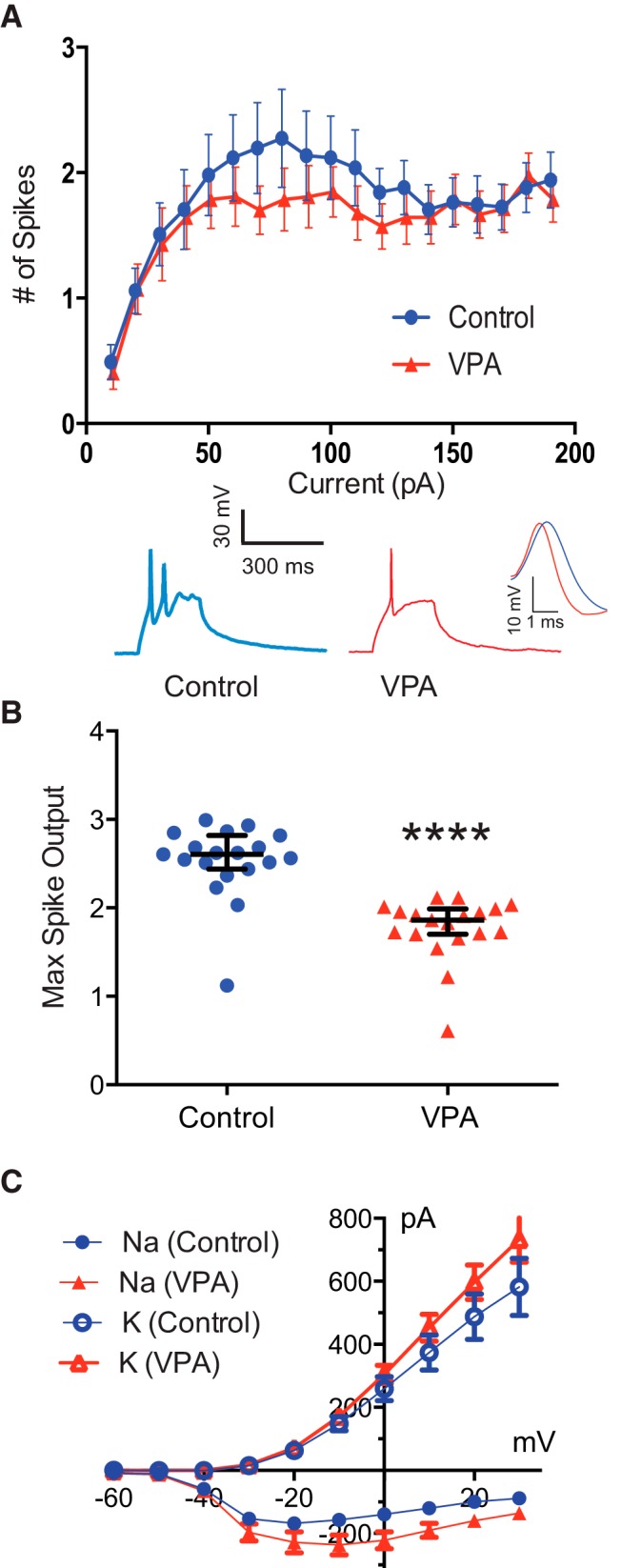
Exposure to VPA decreases intrinsic excitability in tectal neurons. A, Reduced intrinsic cell excitability. Line graph depicting number of evoked spikes as a function of injected current steps. Control, n = 17; VPA, n = 14. The inset shows sample responses to a depolarizing current injection. B, Scatter plot depicting the maximum spike output for each cell. p = 0.0001, N = 19. Error bars are IQRs. C, I–V plot depicting Na+ and K+ currents. Control, n = 21; VPA, n = 21. *p < 0.001.
Table 1.
Active and passive intrinsic neuronal properties
| Median |
pa | ||
|---|---|---|---|
| Control (n) | VPA (n) | ||
| Action potential threshold (mV) | −25.63 (28) | −31.12 (18) | 0.03 |
| Time-to-peak (ms) | 1.631 (28) | 1.267 (18) | 0.02 |
| 50% of peak (ms) | 3.462 (28) | 2.988 (18) | 0.06 |
| Rise time (20–80%; ms) | 0.9220 (28) | 0.7797 (18) | 0.06 |
| Membrane capacitance (pf) | 11.43 (40) | 12.07 (46) | 0.65 |
| Input resistance (GΩ) | 1.200 (70) | 1.200 (63) | 0.43 |
| Na+ peak amplitude (pA) | 193.5 (21) | 269.8 (21) | 0.01 |
| K+ peak amplitude (pA) | 412.8 (21) | 829.3 (21) | 0.06 |
aMann-Whitney U test.
Seizure susceptibility
Together, the data so far suggest that increases in network excitability resulting from VPA treatment are a result of increased connectivity within the tectum and possibly other brain regions. One prediction from this interpretation is that behaviors that reflect increased connectivity, such as seizure susceptibility and habituation to startle stimuli, should be altered significantly in VPA-reared tadpoles.
To assess seizure susceptibility, tadpoles were exposed to 5 mm PTZ, a GABA agonist, over a 20 min period, during which seizure activity was measured (Fig. 7A,B; see Materials and Methods; Bell et al., 2011). Pharmacologically induced seizures were more frequent in VPA-reared tadpoles than in matched controls (frequency of events per minute, 0.90 ± 0.38 for VPA vs 0.51 ± 0.40 for control; N = 18 and 17 respectively; Fig. 7C). Although our behavioral protocol did not allow us to assess the severity of seizures, individual seizure events in VPA-reared tadpoles were significantly shorter than in control (20 ± 16 s for VPA vs 31 ± 16 s for control; N = 18 and 17), potentially indicating that, in VPA-reared tadpoles, synaptic transmission was exhausted and depressed sooner than in control animals.
Startle habituation
Habituation to startle stimuli is known to be impaired in individuals with autism (Madsen et al., 2014) and in various animal models (de Vrij et al., 2008). Habituation of the acoustic startle response was assessed by subjecting tadpoles to six 2-minute-long bouts of periodical acoustic stimuli, in which individual stimuli within each bout were delivered every 5 s (see Materials and Methods). Both control and VPA-reared tadpoles demonstrated startle response habituation at various timescales (Eddins et al., 2010; Roberts et al., 2011), including the following: (1) rapid habituation during the train of stimuli; (2) short-term habituation, measured after a 5 min gap in stimulation; and (2) long-term habituation, measured after several bouts of intermittent stimulation and a longer 15 min gap in the stimulation protocol (Fig. 7D). Average speed of startle responses of naive animals, measured over minute 1 of stimulation, did not differ significantly between the VPA and control groups (9 ± 4 cm/s for VPA-reared tadpoles and 11 ± 4 cm/s for control; N = 21 and 21; PMW = 0.07). The amount of rapid habituation, measured as the ratio between average speed of responses over minutes 1 and 2 of stimulation, also did not differ across groups (0.9 ± 0.4 for VPA-reared tadpoles and 0.8 ± 0.3 for control; N = 21 and 21; PMW = 0.7). However, the short-term habituation was almost absent in VPA-reared tadpoles but was present in control animals, with the ratio of average response speeds of 1.1 ± 0.6 in VPA-raised tadpoles and 0.7 ± 0.5 in control (N = 21 and 21; PMW = 0.01; Fig. 7E). Similarly, unlike control animals, VPA-reared tadpoles did not demonstrate long-term habituation (ratio of average response speeds was equal to 1.3 ± 1.0 for VPA-reared tadpoles compared with 0.8 ± 0.6 in control animals; N = 21 and 21; PMW = 0.03; Fig. 7F).
Both increased seizure susceptibility and decreased short- and long-term habituation to acoustic startle stimuli are consistent with our interpretation that chronic VPA treatment results in abnormally hyperconnected neural circuits and thus an increase in network excitability.
Discussion
Our data show that exposure to VPA between developmental stages 42 and 49 has widespread effects in neural circuit development. We found that VPA-exposed tadpoles had abnormal visual avoidance and schooling behaviors. These changes were accompanied by altered tectal neuron dendritic morphology, as well as increased sEPSC and sIPSC frequency and enhanced visually evoked excitatory and inhibitory synaptic drive. Visual synapses did not show alterations in the AMPA/NMDA ratio but did show immature NMDAR-mediated currents. VPA-treated tadpoles also had immature-like recurrent activity within the optic tectum as measured by large amounts of evoked polysynaptic activity and spontaneous barrages of synaptic activity. The enhanced network activity within the optic tectum was not attributable to increased neuronal excitability, because neurons from VPA-treated tadpoles fired fewer action potentials when depolarized. Finally, chronic VPA treatment resulted in increased seizure susceptibility and reduced acoustic startle habituation.
These data are consistent with a model in which VPA exposure, during a critical period in tectal circuit establishment and remodeling, results in abnormal refinement and pruning of retinotectal synapses, as well as of local intra-tectal circuits. This results in hyperconnected tectal circuitry and leads to deficits in sensory integration, as evidenced by abnormal visual avoidance and schooling behaviors. Furthermore, this abnormally developed tectal circuitry also leads to network hyperexcitability that correlates with increased susceptibility to seizures and decreased startle habituation. Thus, a simple deficit in developmental synaptic remodeling can result in more complex behavioral phenotypes, strengthening the view that complex syndromes can result from simple low-level deficits.
Our findings indicate that VPA exposure results in both an increase in synaptic connectivity and a decrease in intrinsic excitability, consistent with early developmental findings in rodents (Rinaldi et al., 2008; Walcott et al., 2011). Although an increase in synaptic conductivity will result in increased network excitability, a decrease in intrinsic neuronal excitability should have the opposite effect. So why do we find that there is an overall increase in network excitability? Previous work from our laboratory has shown that there is an inverse relationship between synaptic transmission and neuronal excitability (Aizenman et al., 2003; Pratt and Aizenman, 2007). When excitatory synaptic transmission is elevated, tectal neurons show decreased intrinsic excitability. In contrast, if synaptic transmission is low, tectal neurons will be more excitable. This homeostatic relationship serves to normalize the input–output function of the cell. We also found that intrinsic excitability responds to changes in synaptic transmission but not the other way around. Thus, in the current study, we believe that the decrease in excitability is a compensatory response to increased synaptic connectivity resulting from abnormal developmental pruning of synapses. However, this decrease is not sufficient to counteract the increase in synaptic connectivity, and therefore, we see a pathological condition, with an overall increase internetwork excitability.
How does VPA exert its effect on the developing nervous system? VPA is known primarily as an antiepileptic drug because of its effect on GABA transmission. VPA blocks GABA transaminase, an enzyme that breaks down GABA (Löscher, 1999, Johannessen, 2000), resulting in elevated GABA levels in the CNS. Long-term alterations in GABA signaling during development are known to cause abnormalities in neural circuit formation and in developmental plasticity during the critical period in both mammals and Xenopus (Iwai et al., 2003; Shen et al., 2011), and these could account for some of the effects of VPA in our system. VPA is also known to reduce neuronal excitability through effects on voltage-gated Na+ and Ca2+ channels (Löscher, 1999), and, in Xenopus tadpole tectum, long-term reductions in spike firing result in increased synaptic transmission, consistent with the findings in this study (Pratt and Aizenman, 2007). However, the effects of VPA on excitability occur at much higher concentrations than those used in this study and so are not likely to be a factor here (Johannessen, 2000). Finally, VPA is a potent inhibitor of type I and II histone deacetylases (HDAC) and can cause hyperacetylation of gene promoters and, thus, altered gene expression (Fukuchi et al., 2009; Wang et al., 2012). In a separate ongoing study in our laboratory, using gene microarrays, we have shown that exposing tadpoles to VPA between stages 42 and 49 results in large changes in gene expression (C. M. Ciarleglio and C.D.A., unpublished observations) and that rearing tadpoles during this period in the presence of a different HDAC inhibitor (suberanilohydroxamic acid; Whittle and Singewald, 2014) also results in electrophysiological effects in local connectivity, consistent with those caused by VPA (E.J.J. and C.D.A., unpublished observations). Thus, it is possible that at least some of the effects of VPA may be attributable to alterations in gene expression by HDAC inhibition.
How does the timing of VPA exposure in our experimental model compare with the known effects of VPA in rodent models? In mammals, prenatal exposure to VPA has its strongest effect if it occurs during the time of neural tube closure, when there is a large amount of cell proliferation in the CNS, and it is logical that a large disruption in gene expression during neurogenesis could have profound effects on developing neural circuitry (Bath and Scharfman, 2013). VPA also has effects if exposure occurs in early postnatal life when neural circuitry is being established and pruned (Bath and Scharfman, 2013). In Xenopus tadpoles, neural circuits mediating visual processing and motor output go online very early during brain development, at a time when most placental mammals would be in either late embryonic stages or early postnatal stages (Ruthazer and Aizenman, 2010). Although it is difficult to draw one-to-one correlations between Xenopus and rodent development, the time period when tadpoles were exposed to VPA (stages 42–49) corresponds to a period when the retinotectal circuit is established and undergoes growth and activity-dependent refinement, in conjunction with large amounts of neuronal proliferation and differentiation. During this time period, tectal neurons undergo large amounts of dendritic growth, retinotectal projections and local intratectal inputs are refined, excitatory synapses undergo increases in the AMPA/NMDA ratio, and the amount of inhibitory input increases (Akerman and Cline, 2006; Pratt and Aizenman, 2007; Pratt et al., 2008). In addition, new neurons from the proliferative zone of the tectum are being incorporated continuously into the tectal circuitry (Sharma and Cline, 2010). Thus, in our experiments, tadpoles were exposed to VPA during a critical developmental window when there is both a large amount of ongoing neurogenesis and neural circuit formation in the optic tectum, both processes known to be susceptible to abnormal patterns of gene expression (Bestman and Cline, 2008; Schwartz et al., 2009; Sharma and Cline, 2010) and therefore would also be most vulnerable to VPA exposure.
The findings in this study are in line with a growing body of evidence suggesting that ASD is associated with disruption in synaptic development. For example, several mutations and abnormalities that are associated with various neurodevelopmental syndromes, such as MeCP2 (methyl CpG binding protein 2, which causes Rett syndrome in humans; Samaco and Neul, 2011), neurexin and neuroligins (associated with ASD; Buxbaum, 2009), FMRP (fragile X mental retardation protein; Abrahams and Geschwind, 2008), and altered immune signaling (also found in ASD; Cohly and Panja, 2005) all result in abnormal synaptic development and/or neural circuit formation in rodents (Samaco and Neul, 2011; Bhakar et al., 2012; Dixon-Salazar et al., 2014). Similarly in Xenopus tadpoles, disruptions of MeCP2 and neurexin/neuroligin signaling also result in abnormal dendritic morphology and synaptic connectivity in optic tectal neurons (Chen et al., 2010; Marshak et al., 2012). Altered levels of proinflammatory cytokines also cause abnormal dendritic development and hyperconnectivity in the Xenopus tadpole tectum (Lee et al., 2010). This, in combination with the present findings, not only suggests that key transcription pathways and signaling mechanisms are conserved from Xenopus to humans but that their effects on circuit development remains the same. Although an amphibian species cannot display the complex cognitive symptoms associated with ASD, the deficits in circuit formation are manifested as various abnormal behavioral phenotypes, including abnormal sensory integration and schooling, increased seizure susceptibility, and hyperexcitability.
In conclusion, this study follows the effects of VPA exposure during early development at multiple levels of analysis—synaptic, cellular, circuits, and behavior—and we describe how a relatively low-level alteration in neural circuit function cascades into a behavioral phenotype. Remarkably, the effects of VPA in tadpoles remain consistent with what is observed in rodent models, suggesting that VPA exposure is affecting a fundamental developmental process that is evolutionarily conserved across species. By understanding the mechanisms by which VPA exerts its effect on developing neural circuits, we may also learn something about the basic cellular and circuit level changes that VPA-induced deficits may share with other known causes of ASD.
Footnotes
This work was supported by the National Eye Institute and National Science Foundation Integrative Organismal Systems. E.J.J. received funding from the National Science Foundation Graduate Research Fellowship Program. J.G., C.M.R.-V., and M.H. received summer funding from Brown University, and A.S.K. received funding from a Fox Postdoctoral Fellowship from Brown University. We thank members of the Aizenman Laboratory for helpful discussion.
The authors declare no competing financial interests.
References
- Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9:341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aizenman CD, Akerman CJ, Jensen KR, Cline HT. Visually driven regulation of intrinsic neuronal excitability improves stimulus detection in vivo. Neuron. 2003;39:831–842. doi: 10.1016/S0896-6273(03)00527-0. [DOI] [PubMed] [Google Scholar]
- Akerman CJ, Cline HT. Depolarizing GABAergic conductances regulate the balance of excitation to inhibition in the developing retinotectal circuit in vivo. J Neurosci. 2006;26:5117–5130. doi: 10.1523/JNEUROSCI.0319-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bath KG, Scharfman HE. Impact of early life exposure to antiepileptic drugs on neurobehavioral outcomes based on laboratory animal and clinical research. Epilepsy Behav. 2013;26:427–439. doi: 10.1016/j.yebeh.2012.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell MR, Belarde JA, Johnson HF, Aizenman CD. A neuroprotective role for polyamines in a Xenopus tadpole model of epilepsy. Nat Neurosci. 2011;14:505–512. doi: 10.1038/nn.2777. [DOI] [PubMed] [Google Scholar]
- Bestman JE, Cline HT. The RNA binding protein CPEB regulates dendrite morphogenesis and neuronal circuit assembly in vivo. Proc Natl Acad Sci U S A. 2008;105:20494–20499. doi: 10.1073/pnas.0806296105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakar AL, Dölen G, Bear MF. The pathophysiology of fragile X (and what it teaches us about synapses) Annu Rev Neurosci. 2012;35:417–443. doi: 10.1146/annurev-neuro-060909-153138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brainard DH. The psychophysics toolbox. Spat Vis. 1997;10:433–436. doi: 10.1163/156856897X00357. [DOI] [PubMed] [Google Scholar]
- Bromley RL, Mawer GE, Briggs M, Cheyne C, Clayton-Smith J, García-Fiñana M, Kneen R, Lucas SB, Shallcross R, Baker GA. The prevalence of neurodevelopmental disorders in children prenatally exposed to antiepileptic drugs. J Neurol Neurosurg Psychiatry. 2013;84:637–643. doi: 10.1136/jnnp-2012-304270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD. Multiple rare variants in the etiology of autism spectrum disorders. Dialogues Clin Neurosci. 2009;11:35–43. doi: 10.31887/DCNS.2009.11.1/jdbuxbaum. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SX, Tari PK, She K, Haas K. Neurexin-neuroligin cell adhesion complexes contribute to synaptotropic dendritogenesis via growth stabilization mechanisms in vivo. Neuron. 2010;67:967–983. doi: 10.1016/j.neuron.2010.08.016. [DOI] [PubMed] [Google Scholar]
- Clements JD, Bekkers JM. Detection of spontaneous synaptic events with an optimally scaled template. Biophys J. 1997;73:220–229. doi: 10.1016/S0006-3495(97)78062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohly HH, Panja A. Immunological findings in autism. Int Rev Neurobiol. 2005;71:317–341. doi: 10.1016/S0074-7742(05)71013-8. [DOI] [PubMed] [Google Scholar]
- de Vrij FM, Levenga J, van der Linde HC, Koekkoek SK, De Zeeuw CI, Nelson DL, Oostra BA, Willemsen R. Rescue of behavioral phenotype and neuronal protrusion morphology in Fmr1 KO mice. Neurobiol Dis. 2008;31:127–132. doi: 10.1016/j.nbd.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon-Salazar TJ, Fourgeaud L, Tyler CM, Poole JR, Park JJ, Boulanger LM. MHC class I limits hippocampal synapse density by inhibiting neuronal insulin receptor signaling. J Neurosci. 2014;34:11844–11856. doi: 10.1523/JNEUROSCI.4642-12.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong W, Lee RH, Xu H, Yang S, Pratt KG, Cao V, Song YK, Nurmikko A, Aizenman CD. Visual avoidance in Xenopus tadpoles is correlated with the maturation of visual responses in the optic tectum. J Neurophysiol. 2009;101:803–815. doi: 10.1152/jn.90848.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddins D, Cerutti D, Williams P, Linney E, Levin ED. Zebrafish provide a sensitive model of persisting neurobehavioral effects of developmental chlorpyrifos exposure: comparison with nicotine and pilocarpine effects and relationship to dopamine deficits. Neurotoxicol Teratol. 2010;32:99–108. doi: 10.1016/j.ntt.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald RC, Van Keuren-Jensen KR, Aizenman CD, Cline HT. Roles of NR2A and NR2B in the development of dendritic arbor morphology in vivo. J Neurosci. 2008;28:850–861. doi: 10.1523/JNEUROSCI.5078-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint AC, Maisch US, Weishaupt JH, Kriegstein AR, Monyer H. NR2A subunit expression shortens NMDA receptor synaptic currents in developing neocortex. J Neurosci. 1997;17:2469–2476. doi: 10.1523/JNEUROSCI.17-07-02469.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuchi M, Nii T, Ishimaru N, Minamino A, Hara D, Takasaki I, Tabuchi A, Tsuda M. Valproic acid induces up- or down-regulation of gene expression responsible for the neuronal excitation and inhibition in rat cortical neurons through its epigenetic actions. Neurosci Res. 2009;65:35–43. doi: 10.1016/j.neures.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Gerstner T, Bell N, König S. Oral valproic acid for epilepsy—long-term experience in therapy and side effects. Expert Opin Pharmacother. 2008;9:285–292. doi: 10.1517/14656566.9.2.285. [DOI] [PubMed] [Google Scholar]
- Haas K, Jensen K, Sin WC, Foa L, Cline HT. Targeted electroporation in Xenopus tadpoles in vivo—from single cells to the entire brain. Differentiation. 2002;70:148–154. doi: 10.1046/j.1432-0436.2002.700404.x. [DOI] [PubMed] [Google Scholar]
- Hamodi AS, Pratt KG. Region-specific regulation of voltage-gated intrinsic currents in the developing optic tectum of the Xenopus tadpole. J Neurophysiol. 2014;112:1644–1655. doi: 10.1152/jn.00068.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartigan JA, Hartigan PM. The dip test for unimodality. Ann Stat. 1985;13:70–84. doi: 10.1214/aos/1176346577. [DOI] [Google Scholar]
- Isaac JT. Postsynaptic silent synapses: evidence and mechanisms. Neuropharmacology. 2003;45:450–460. doi: 10.1016/S0028-3908(03)00229-6. [DOI] [PubMed] [Google Scholar]
- Iwai Y, Fagiolini M, Obata K, Hensch TK. Rapid critical period induction by tonic inhibition in visual cortex. J Neurosci. 2003;23:6695–6702. doi: 10.1523/JNEUROSCI.23-17-06695.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen CU. Mechanisms of action of valproate: a commentary. Neurochem Int. 2000;37:103–110. doi: 10.1016/S0197-0186(00)00013-9. [DOI] [PubMed] [Google Scholar]
- Katz LC, Potel MJ, Wassersug RJ. Structure and mechanisms of schooling in tadpoles of the clawed frog, Xenopus laevis. Anim Behav. 1981;29:20–33. doi: 10.1016/S0003-3472(81)80148-0. [DOI] [Google Scholar]
- Khakhalin AS, Aizenman CD. GABAergic transmission and chloride equilibrium potential are not modulated by pyruvate in the developing optic tectum of Xenopus laevis tadpoles. PLoS One. 2012;7:e34446. doi: 10.1371/journal.pone.0034446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakhalin AS, Koren D, Gu J, Xu H, Aizenman CD. Excitation and inhibition in recurrent networks mediate collision avoidance in Xenopus tadpoles. Eur J Neurosci. 2014;40:2948–2962. doi: 10.1111/ejn.12664. [DOI] [PubMed] [Google Scholar]
- Krey JF, Dolmetsch RE. Molecular mechanisms of autism: a possible role for Ca2+ signaling. Curr Opin Neurobiol. 2007;17:112–119. doi: 10.1016/j.conb.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Lee RH, Mills EA, Schwartz N, Bell MR, Deeg KE, Ruthazer ES, Marsh-Armstrong N, Aizenman CD. Neurodevelopmental effects of chronic exposure to elevated levels of pro-inflammatory cytokines in a developing visual system. Neural Dev. 2010;5:2. doi: 10.1186/1749-8104-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenstein P, Carlström E, Råstam M, Gillberg C, Anckarsäter H. The genetics of autism spectrum disorders and related neuropsychiatric disorders in childhood. Am J Psychiatry. 2010;167:1357–1363. doi: 10.1176/appi.ajp.2010.10020223. [DOI] [PubMed] [Google Scholar]
- Löscher W. Valproate: a reappraisal of its pharmacodynamic properties and mechanisms of action. Prog Neurobiol. 1999;58:31–59. doi: 10.1016/S0301-0082(98)00075-6. [DOI] [PubMed] [Google Scholar]
- Lum AM, Wassersug RJ, Potel MJ, Lerner SA. Schooling behavior of tadpoles: a potential indicator of ototoxicity. Pharmacol Biochem Behav. 1982;17:363–366. doi: 10.1016/0091-3057(82)90093-4. [DOI] [PubMed] [Google Scholar]
- Madsen GF, Bilenberg N, Cantio C, Oranje B. Increased prepulse inhibition and sensitization of the startle reflex in autistic children. Autism Res. 2014;7:94–103. doi: 10.1002/aur.1337. [DOI] [PubMed] [Google Scholar]
- Marín O. Interneuron dysfunction in psychiatric disorders. Nat Rev Neurosci. 2012;13:107–120. doi: 10.1038/nrn3155. [DOI] [PubMed] [Google Scholar]
- Markram H, Rinaldi T, Markram K. The intense world syndrome–an alternative hypothesis for autism. Front Neurosci. 2007;1:77–96. doi: 10.3389/neuro.01.1.1.006.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram K, Markram H. The intense world theory—a unifying theory of the neurobiology of autism. Front Hum Neurosci. 2010;4:224. doi: 10.3389/fnhum.2010.00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram K, Rinaldi T, La Mendola D, Sandi C, Markram H. Abnormal fear conditioning and amygdala processing in an animal model of autism. Neuropsychopharmacology. 2008;33:901–912. doi: 10.1038/sj.npp.1301453. [DOI] [PubMed] [Google Scholar]
- Marshak S, Meynard MM, De Vries YA, Kidane AH, Cohen-Cory S. Cell-autonomous alterations in dendritic arbor morphology and connectivity induced by overexpression of MeCP2 in Xenopus central neurons in vivo. PLoS One. 2012;7:e33153. doi: 10.1371/journal.pone.0033153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin HGS, Manzoni OJ. Late onset deficits in synaptic plasticity in the valproic acid rat model of autism. Front Cell Neurosci. 2014;8:23. doi: 10.3389/fncel.2014.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, Polak P, Yoon S, Maguire J, Crawford EL, Campbell NG, Geller ET, Valladares O, Schafer C, Liu H, Zhao T, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwkoop PD, Faber J. New York: Garland Publishing; 1956. Normal table of Xenopus laevis (Daudin) [Google Scholar]
- Pescosolido MF, Yang U, Sabbagh M, Morrow EM. Lighting a path: genetic studies pinpoint neurodevelopmental mechanisms in autism and related disorders. Dialogues Clin Neurosci. 2012;14:239–252. [PMC free article] [PubMed] [Google Scholar]
- Pratt KG, Aizenman CD. Homeostatic regulation of intrinsic excitability and synaptic transmission in a developing visual circuit. J Neurosci. 2007;27:8268–8277. doi: 10.1523/JNEUROSCI.1738-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt KG, Khakhalin AS. Modeling human neurodevelopmental disorders in the Xenopus tadpole: from mechanisms to therapeutic targets. Dis Model Mech. 2013;6:1057–1065. doi: 10.1242/dmm.012138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt KG, Dong W, Aizenman CD. Development and spike timing-dependent plasticity of recurrent excitation in the Xenopus optic tectum. Nat Neurosci. 2008;11:467–475. doi: 10.1038/nn2076. [DOI] [PubMed] [Google Scholar]
- Quinlan EM, Olstein DH, Bear MF. Bidirectional, experience-dependent regulation of N-methyl-d-aspartate receptor subunit composition in the rat visual cortex during postnatal development. Proc Natl Acad Sci U S A. 1999;96:12876–12880. doi: 10.1073/pnas.96.22.12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi T, Kulangara K, Antoniello K, Markram H. Elevated NMDA receptor levels and enhanced postsynaptic long-term potentiation induced by prenatal exposure to valproic acid. Proc Natl Acad Sci U S A. 2007;104:13501–13506. doi: 10.1073/pnas.0704391104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi T, Silberberg G, Markram H. Hyperconnectivity of local neocortical microcircuitry induced by prenatal exposure to valproic acid. Cereb Cortex. 2008;18:763–770. doi: 10.1093/cercor/bhm117. [DOI] [PubMed] [Google Scholar]
- Roberts AC, Reichl J, Song MY, Dearinger AD, Moridzadeh N, Lu ED, Pearce K, Esdin J, Glanzman DL. Habituation of the C-start response in larval zebrafish exhibits several distinct phases and sensitivity to NMDA receptor blockade. PLoS One. 2011;6:e29132. doi: 10.1371/journal.pone.0029132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthazer ES, Aizenman CD. Learning to see: patterned visual activity and the development of visual function. Trends Neurosci. 2010;33:183–192. doi: 10.1016/j.tins.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthazer ES, Schohl A, Schwartz N, Tavakoli A, Tremblay M, Cline HT. Cold Spring Harbor protocols. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 2013. Bulk electroporation of retinal ganglion cells in live Xenopus tadpoles. [DOI] [PubMed] [Google Scholar]
- Samaco RC, Neul JL. Complexities of Rett syndrome and MeCP2. J Neurosci. 2011;31:7951–7959. doi: 10.1523/JNEUROSCI.0169-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider T, Przewlocki R. Behavioral alterations in rats prenatally exposed to valproic acid: animal model of autism. Neuropsychopharmacology. 2005;30:80–89. doi: 10.1038/sj.npp.1300518. [DOI] [PubMed] [Google Scholar]
- Schwartz N, Schohl A, Ruthazer ES. Neural activity regulates synaptic properties and dendritic structure in vivo through calcineurin/NFAT signaling. Neuron. 2009;62:655–669. doi: 10.1016/j.neuron.2009.05.007. [DOI] [PubMed] [Google Scholar]
- Sharma P, Cline HT. Visual activity regulates neural progenitor cells in developing Xenopus CNS through musashi1. Neuron. 2010;68:442–455. doi: 10.1016/j.neuron.2010.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, McKeown CR, Demas JA, Cline HT. Inhibition to excitation ratio regulates visual system responses and behavior in vivo. J Neurophysiol. 2011;106:2285–2302. doi: 10.1152/jn.00641.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spawn A, Aizenman CD. Abnormal visual processing and increased seizure susceptibility result from developmental exposure to the biocide methylisothiazolinone. Neuroscience. 2012;205:194–204. doi: 10.1016/j.neuroscience.2011.12.052. [DOI] [PubMed] [Google Scholar]
- Terbach N, Shah R, Kelemen R, Klein PS, Gordienko D, Brown NA, Wilkinson CJ, Williams RS. Identifying an uptake mechanism for the antiepileptic and bipolar disorder treatment valproic acid using the simple biomedical model Dictyostelium. J Cell Sci. 2011;124:2267–2276. doi: 10.1242/jcs.084285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villinger J, Waldman B. Social discrimination by quantitative assessment of immunogenetic similarity. Proc Biol Sci. 2012;279:4368–4374. doi: 10.1098/rspb.2012.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walcott EC, Higgins EA, Desai NS. Synaptic and intrinsic balancing during postnatal development in rat pups exposed to valproic acid in utero. J Neurosci. 2011;31:13097–13109. doi: 10.1523/JNEUROSCI.1341-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CC, Chen PS, Hsu CW, Wu SJ, Lin CT, Gean PW. Valproic acid mediates the synaptic excitatory/inhibitory balance through astrocytes—a preliminary study. Prog Neuropsychopharmacol Biol Psychiatry. 2012;37:111–120. doi: 10.1016/j.pnpbp.2012.01.017. [DOI] [PubMed] [Google Scholar]
- Wassersug R, Hessler CM. Tadpole behaviour: aggregation in larval Xenopus laevis. Anim Behav. 1971;19:386–389. doi: 10.1016/S0003-3472(71)80021-0. [DOI] [PubMed] [Google Scholar]
- Whittle N, Singewald N. HDAC inhibitors as cognitive enhancers in fear, anxiety and trauma therapy: where do we stand? Biochem Soc Trans. 2014;42:569–581. doi: 10.1042/BST20130233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Malinow R, Cline HT. Maturation of a central glutamatergic synapse. Science. 1996;274:972–976. doi: 10.1126/science.274.5289.972. [DOI] [PubMed] [Google Scholar]