

Abstract
Background
The development of robust methods for chemical named entity recognition, a challenging natural language processing task, was previously hindered by the lack of publicly available, large-scale, gold standard corpora. The recent public release of a large chemical entity-annotated corpus as a resource for the CHEMDNER track of the Fourth BioCreative Challenge Evaluation (BioCreative IV) workshop greatly alleviated this problem and allowed us to develop a conditional random fields-based chemical entity recogniser. In order to optimise its performance, we introduced customisations in various aspects of our solution. These include the selection of specialised pre-processing analytics, the incorporation of chemistry knowledge-rich features in the training and application of the statistical model, and the addition of post-processing rules.
Results
Our evaluation shows that optimal performance is obtained when our customisations are integrated into the chemical entity recogniser. When its performance is compared with that of state-of-the-art methods, under comparable experimental settings, our solution achieves competitive advantage. We also show that our recogniser that uses a model trained on the CHEMDNER corpus is suitable for recognising names in a wide range of corpora, consistently outperforming two popular chemical NER tools.
Conclusion
The contributions resulting from this work are two-fold. Firstly, we present the details of a chemical entity recognition methodology that has demonstrated performance at a competitive, if not superior, level as that of state-of-the-art methods. Secondly, the developed suite of solutions has been made publicly available as a configurable workflow in the interoperable text mining workbench Argo. This allows interested users to conveniently apply and evaluate our solutions in the context of other chemical text mining tasks.
Keywords: Chemical named entity recognition, Text mining, Sequence labelling, Conditional random fields, Feature engineering, Configurable workflows, Workflow optimisation
Background
In carrying out scientific work, most researchers rely on published information in order to keep abreast of recent developments in the field, to avoid repetition of work and to guide the direction of current studies. This is especially true in the field of chemistry where endeavours such as drug discovery and development are largely driven by information screened from the copious amounts of data available. Whilst databases storing structured chemical information have proliferated in the last few years, published scientific articles, technical reports, patent documents and other forms of unstructured data remain to be the richest source of the most current information.
Text mining facilitates the efficient distillation of information from the plethora of scientific literature. Whilst most of the scientific text mining efforts in the last decade have focussed on the identification of biomedical entities such as genes, their products and the interactions between them, the community has recently begun to appreciate the need for automatically extracting chemical information from text. Applications in chemoinformatics, drug discovery and systems biology such as automatic database curation [1], compound screening [2], detection of adverse drug reactions [3], drug repurposing [4] and metabolic pathway curation [5] are facilitated and informed by the outcomes of chemical text mining, a fundamental task of which is the recognition of chemical named entities.
Chemical named entity recognition (NER), the automatic demarcation of expressions pertaining to chemical entities within text, is considered a challenging task for a number of reasons. First, chemical names may appear in various forms, ranging from the popular and human-readable trivial and brand names to the more obscure abbreviations, molecular formulas and database identifiers, to long nomenclature-conforming expressions, e.g., International Union of Pure and Applied Chemistry (IUPAC) names and Simplified Molecular-Input Line-Entry System (SMILES) strings [6-8]. Moreover, researchers working on lead compound identification and discovery sometimes tend to report their results using their own arbitrarily assigned abbreviations, further aggravating the proliferation of chemical names. Also considered a barrier to the development of chemical named entity recognisers is the relatively small number of available supporting corpora, compared to those developed for biological, such as gene and protein, name recognition [9]. Whilst a few notable data sets containing chemical named entity annotations have been developed, there was a lack of publicly available, wide-coverage, large-scale gold standard corpora of scientific publications. Although the SciBorg corpus [10,11] contains a substantial number of manually annotated chemical names in its 42 full-text articles, it had not been publicly available until very recently. In contrast, the large-scale CALBC corpus [12] is publicly available, but is considered "silver standard" as it contains annotations resulting from the harmonisation of the outputs of five different automatic tools, rather than manual annotations. The similarly publicly available SCAI pilot corpus [13,14] contains gold standard annotations for various types of chemical names but is relatively small with only 100 MEDLINE abstracts.
This limited number of resources has influenced the means by which the state-of-the-art chemical named entity recognisers have been developed and evaluated. Built as a pipeline of several Markov model-based classifiers, the publicly available OSCAR tool [15] was tuned to recognise the annotation types defined in the SciBorg corpus. The system was evaluated by means of three-fold cross validation on this corpus as well as on a bespoke data set of 500 annotated MEDLINE abstracts. ChemSpot [16], another publicly available chemical named entity recogniser, is a hybrid between methods for dictionary matching and machine learning. For capturing brand names, this tool uses a lexicon-based approach for matching expressions against the Joint Chemical Dictionary [17]. For recognising nomenclature-based expressions, however, it employs a conditional random fields (CRF) [18] model trained on the SCAI corpus subset that contains annotations for only IUPAC names. The developers carried out a comparative evaluation of ChemSpot and OSCAR on the SCAI pilot corpus, in which the former was reported to have outperformed the latter by a margin of 10.8 percentage points. It is worth noting, however, that both of these tools have not been comparatively evaluated nor benchmarked against any large-scale, gold standard corpora.
In aiming to alleviate these issues, the Critical Assessment of Information Extraction in Biology (BioCreative) initiative organised a track in the Fourth BioCreative Challenge Evaluation workshop to encourage the text mining community to develop methods for chemical named entity recognition, and enable the benchmarking of these methods against substantial gold standard data [19]. Known as CHEMDNER, this track publicly released a large corpus of documents containing manually annotated chemical named entities. The 10,000 MEDLINE abstracts in the CHEMDNER corpus [20], which were grouped into disparate sets for training (3,500), development (3,500) and testing (3,000), came from various chemical subdomains including pharmacology, medicinal chemistry, pharmacy, toxicology and organic chemistry. Each annotated chemical name was labelled with one of the following mention types: systematic, trivial, family, abbreviation, formula, identifier, coordination and a catch-all category. The corpus served as the primary resource for the two CHEMDNER subtasks, namely, chemical entity mention recogniton (CEM) and chemical document indexing (CDI). Whilst the former required participating systems to return the locations of all chemical mention instances found within a given document, the latter expects a ranked listing of unique mentions without any location information.
Having participated in the CHEMDNER challenge, we have developed our own chemical named entity recogniser that obtained top-ranking performance in both the CDI (1st) and CEM (3rd) tasks. Extending that work, we describe in this paper the details of our proposed methods for optimising chemical NER performance. In the next section, we compare the performance of our methods with the state of the art and present results of our evaluation on several corpora. Furthermore, we share details on how our contributions, publicly available as a service, can be accessed and utilised by the community. The Experiments section contains a detailed discussion of our proposed methods and the experiments we have performed in order to identify the optimal solution on each of the data sets considered. We summarise the results of our work in the Conclusions section. Lastly, we provide some technical background on the techniques and evaluation metrics we have used in this study in the Methods section.
Results and discussion
We developed a conditional random fields (CRF)-based method for chemical named entity recognition whose performance was optimised by (a) the selection of best-suited pre-processing components, (b) the incorporation of CRF features capturing chemistry-specific information, and (c) the application of post-processing heuristics.
We begin with describing the results from the evaluation of our method under the settings of the CHEMDNER challenge. Next, we demonstrate that our method obtains competitive performance compared to the state of the art. We then show that a statistical model trained on a large-scale, gold standard corpus such as CHEMDNER is suitable for recognising chemical names in a wider range of corpora, on which it consistently outperforms two known chemical NER tools. Finally, we describe the availability of our approach as a configurable workflow in the interoperable text mining platform Argo [21]. Hereafter, we refer to our suite of solutions collectively as Chemical Entity Recogniser, or ChER.
Performance evaluation under the CHEMDNER challenge settings
The first set of experiments was performed based on the specifications of the BioCreative IV CHEMDNER track [19], which our research team participated in. The micro-averaged results on the CHEMDNER test set obtained by our solutions using specialised pre-processing analytics (i.e., Cafetiere Sentence Splitter and OSCAR4 Tokeniser) are presented in Table 1. These closely approximate the results which were reported for our submissions during the official BioCreative challenge evaluation [22], in which the variant employing knowledge-rich features and abbreviation recognition achieved the best performance in both the CEM and CDI subtasks.
Table 1.
Performance of ChER under the BioCreative IV CHEMDNER track setting.
| Custom Features | Post-processing | CEM | CDI | |||||
|---|---|---|---|---|---|---|---|---|
| Abbr. | Comp. | P | R | F1 | P | R | F1 | |
| ✓ | ✗ | ✗ | 92.76 | 81.02 | 86.49 | 91.39 | 85.29 | 88.23 |
| ✓ | ✓ | ✗ | 92.76 | 81.30 | 86.65 | 91.37 | 85.45 | 88.31 |
| ✓ | ✗ | ✓ | 92.14 | 81.41 | 86.44 | 90.55 | 85.72 | 88.07 |
| ✓ | ✓ | ✓ | 92.14 | 81.69 | 86.60 | 90.53 | 85.88 | 88.14 |
Key: Abbr. = Abbreviation recognition, Comp. = Chemical composition-based token relabelling
Performance comparison against state-of-the-art methods
We conducted a performance-wise comparison of our solution, ChER, against previously reported machine learning-based chemical NER methods, namely, that of Corbett et al. [15], Rocktäschel et al. [16] and Nobata et al. [23]. To facilitate a fair comparison, we performed a series of benchmarking tasks under the same experimental settings used in their previously reported work.
Following Corbett et al. [15], we performed three-fold cross validation on the SciBorg corpus, taking into consideration only annotations for mentions of chemical molecules. As summarised in Table 2, the F1 score obtained by our methods (79.66%) is slightly lower than that reported by Corbett et al. (81.20%). We cannot remark on precision and recall, however, as the authors did not report them. It is worth noting that their work became the foundation of what is now known as the OSCAR chemical NER tool. Although the software is freely available [24], we have not been able to replicate their reported results on the SciBorg corpus as the models bundled with the downloadable release were trained on documents from the same data set.
Table 2.
Comparative evaluation of ChER against state-of-the-art chemical name recognition methods.
| SciBorg (chemical molecules) | SCAI-100 (systematic names) | ||||||
|---|---|---|---|---|---|---|---|
| P | R | F1 | P | R | F1 | ||
| ChER | 85.96 | 74.22 | 79.66 | ChER | 86.70 | 67.50 | 75.90 |
| OSCAR | - | - | 81.20 | ChemSpot | 57.47 | 67.70 | 62.17 |
OSCAR's F1 score was taken from the paper of Corbett et al. [15].
Following the experimental setup employed by Rocktäschel et al. [16] in evaluating their ChemSpot tool, we trained a CRF model on the SCAI training corpus containing annotations for systematic names. Consequently, the version of ChER driven by this particular model can recognise only systematic names, and was thus evaluated only against the gold standard systematic name annotations in the SCAI pilot corpus of 100 abstracts (SCAI-100). The results shown in Table 2 indicate that whilst ChER and the CRF-based component of ChemSpot achieve similar recall (67.50% and 67.70%, respectively), the former obtains far more superior precision and F1 score (86.70% and 75.90%) over the latter (57.47% and 62.17%). We note that in conducting this comparison, we ran ChemSpot [25] on the SCAI-100 corpus ourselves, enabling its capability to recognise multiple chemical name subtypes, in order to segregate recognised systematic names.
Last in this series of evaluations is the performance-wise comparison of ChER with MetaboliNER [23], a tool based on a CRF model that utilised the Chemical Entities of Biological Interest (ChEBI) [26] and Human Metabolome (HMDB) [27] databases as dictionaries. The tools were evaluated on the NaCTeM Metabolites corpus [28] in a 10-fold cross validation manner [23]. The obtained results, presented in Table 3, indicate that MetaboliNER achieves higher precision (83.02% vs. 81.42%); however, it is outperformed by our method in terms of recall and F1 score (79.66% and 80.53% vs. 74.42% and 78.49%).
Table 3.
Comparative evaluation of ChER against a state-of-the-art metabolite name recognition method.
| NaCTeM Metabolites | |||
|---|---|---|---|
| P | R | F1 | |
| ChER | 81.42 | 79.66 | 80.53 |
| MetaboliNER | 83.02 | 74.42 | 78.49 |
We surmise that our solution's superior performance over the similarly CRF-based ChemSpot and MetaboliNER tools can be explained by the richer feature set we employed in developing ChER. As described in the Experiments section below, ChER utilises a comprehensive set of character and word n-grams as well as orthographic features, which were then augmented with ones which capture chemical knowledge, e.g., number of chemical basic segments, dictionary and chemical symbol matches. Meanwhile, ChemSpot employs only size-two affixes, a check for leading or trailing whitespace, a quite limited set of orthographic features and bag-of-words [16]. MetaboliNER uses a similar feature set, with the addition of word shape, part-of-speech tags and dictionary features [23]. Based on the evaluation presented, ChER's rich feature set proved to be more informative and powerful over that of ChemSpot and MetaboliNER.
Performance evaluation on a variety of chemical corpora
As stipulated earlier, one of the barriers to the development of chemical named entity recognisers was the lack of publicly available, wide-coverage, large-scale gold standard corpora. The public release of the CHEMDNER corpus directly alleviates this issue, allowing us to train our CRF model on a massive number and variety of learning examples. We argue that a model trained on the CHEMDNER corpus produces satisfactory NER performance even on documents of different types (e.g., patents, DrugBank descriptions) and from various specialised subject domains (e.g., pharmacology, metabolomics). In validating this, we utilised the CHEMDNER training and development sets to train CRF models under the various configurations detailed in the Experiments section. Taking the best performing variant, we compared its performance with that of OSCAR and ChemSpot by also running their latest versions (OSCAR4.1 and ChemSpot 2.0) on each corpus of interest. Across all five corpora we used, ChER consistently outperformed the other two NER tools, often with a noticeable margin.
Presented in Table 4 are results of this evaluation scheme on general chemical corpora. On the SCAI-100 corpus, with all chemical name types taken into consideration, ChER achieved a good balance between precision and recall, giving an F1 score of 78.27% which is almost four percentage points higher than that of the second-best performing ChemSpot. An even larger margin of about 12 percentage points (also in terms of F1 score) was obtained by ChER over ChemSpot on the Patents corpus [29,30]. The relatively low F1 score on this corpus (64.75%) can be explained by the difference in document types between the corpus for model training (i.e., scientific abstracts) and evaluation (i.e., patent applications). We note that an evaluation on a third chemical corpus, SciBorg, was not carried out under this scheme. Since OSCAR was trained on the SciBorg corpus, a comparative evaluation of ChER, OSCAR and ChemSpot on this data would not have given fair results.
Table 4.
Applicability of ChER with the CHEMDNER model to other chemical corpora.
| SCAI-100 (all names) | Patents | |||||
|---|---|---|---|---|---|---|
| P | R | F1 | P | R | F1 | |
| ChER | 77.85 | 78.69 | 78.27 | 73.43 | 57.91 | 64.75 |
| ChemSpot | 76.35 | 72.55 | 74.41 | 67.79 | 41.97 | 51.84 |
| OSCAR4 | 50.88 | 81.34 | 62.60 | 49.90 | 60.73 | 54.79 |
The model trained on CHEMDNER data was proven suitable even for recognising mentions of drugs, which comprise a more specific chemical type (Table 5). When evaluated on each of the Drug-Drug Interaction (DDI) test [31,32] and Pharmacokinetics (PK) [33,34] corpora, more than satisfactory F1 scores (≈83%) were obtained. ChemSpot's F1 score on the DDI test corpus trails behind by only two percentage points, but is significantly lower than ChER's on the PK corpus with a margin of almost 10 percentage points. Applying the same model to the NaCTeM Metabolites corpus, however, did not yield results as satisfactory as those on the drug corpora, with the highest F1 score being 73.07% (Table 6). This, nevertheless, still indicates a significant advantage over ChemSpot, whose F1 score is 8 percentage points behind.
Table 5.
Applicability of ChER with the CHEMDNER model to drug corpora.
| DDI test | PK | |||||
|---|---|---|---|---|---|---|
| P | R | F1 | P | R | F1 | |
| ChER | 75.88 | 92.05 | 83.18 | 79.83 | 88.34 | 83.87 |
| ChemSpot | 73.09 | 89.49 | 80.46 | 65.29 | 86.07 | 74.25 |
| OSCAR4 | 60.20 | 85.51 | 70.66 | 42.65 | 81.71 | 56.04 |
Table 6.
Applicability of ChER with the CHEMDNER model to the NaCTeM Metabolites corpus.
| NaCTeM Metabolites | |||
|---|---|---|---|
| P | R | F1 | |
| ChER | 65.08 | 83.29 | 73.07 |
| ChemSpot | 58.02 | 73.99 | 65.04 |
| OSCAR4 | 35.37 | 84.18 | 49.81 |
Whilst the model obtained balanced precision and recall on the chemical corpus SCAI-100 (P = 77.85% vs. R = 78.69%), the suboptimal precision values on the DDI (P = 75.88% vs. R = 92.05%), PK (P = 79.83% vs. R = 88.34%) and Metabolites (P = 65.08% vs. R = 83.29%) corpora are noticeable. This drop in precision is to be expected, and can be explained by the differences between the annotation scopes of the training data, CHEMDNER, and of each of the latter three evaluation corpora. Both of the DDI and PK corpora contain only drug name annotations, whilst only metabolite mentions were annotated in the Metabolites corpus. Whereas the model was trained to recognise all chemical mentions, each of the DDI, PK and Metabolites corpora considers only a subset of them as correct, leading to an increase in the number of false positives.
Configurable chemical entity recognition workflows in Argo
In order to facilitate the reproduction of results and further experimentation, we have made the presented named entity recognition methods available in our publicly accessible, Web-based, text mining workbench Argo [35]. The workbench aims to bring text mining to non-technical audiences by providing a graphical user interface for building and running custom text-processing applications. Applications are built in Argo visually as block diagrams forming processing pipelines, or more generally, workflows. Individual blocks in a diagram correspond to elementary processing components that are selected by users from the available, ever-growing library of analytics. The components in the library range from simple data (de)serialisers to syntactic and semantic analytics to user-interactive components.
The proposed recogniser is available as a single component and exposes multiple configurations to choose from. Users may select one of chemical, drug or metabolite, as the model that will be used for the recognition. Additional options include the disabling of post-processing steps discussed in the next section.
Figure 1 shows how the chemical entity recogniser component can be used in Argo workflows. Both workflows shown in the figure contain components that proved to yield the best performance on the CHEMDNER corpus. The left-hand-side workflow is set up to process PubMed articles (supplied by specifying abstract identifiers in the reader's configuration) and save the result of processing (recognised chemical names) in an RDF file. The right-hand-side workflow is a sample set up for experimenting with components available in Argo. The ultimate component in this workflow, Reference Evaluator, reports evaluation metrics based on two inputs: the reference input, which in this workflow comes directly from the CHEMDNER corpus reader and contains golden annotations, and the other branch in the workflow that attempts to reproduce the annotations in the input corpus. Users may experiment with this workflow by replacing the components (specifically the preprocessing components) with other, similar-purpose analytics available in Argo.
Figure 1.
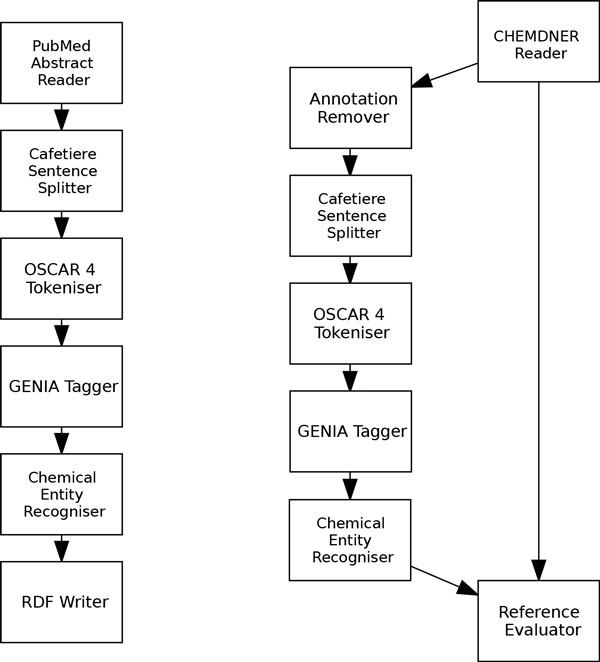
The chemical entity recogniser in Argo. The proposed chemical entity recogniser is available as a processing component in the Web-based, text mining workbench Argo. The component is shown here as part of two individual workflows. The left-hand-side workflow produces an RDF file containing annotated chemicals in user-specified PubMed abstracts. The right-hand-side workflow reports effectiveness metrics for the CHEMDNER corpus.
Experiments
The following is a detailed description of our proposed methods and the experiments carried out to facilitate the identification of the most optimal chemical NER solutions.
Selection of pre-processing analytics
Coming from a specialised domain, chemical literature exhibits unique properties, e.g., unusually long names, which are not typically encountered in documents from other subject domains. Whilst pre-processing steps to text mining have not been given much attention, we argue that the selection of suitable analytics for preprocessing chemical documents brings about a significant impact on NER performance, inspired by the findings of a prior exploratory work [36]. This is especially relevant in our case where features employed in training our CRF models were extracted at the basic level of tokens. In this work, we focus on the two pre-processing tasks of sentence boundary detection and tokenisation. For each of these, specialised and non-specialised implementations were explored.
Sentence splitters
In segmenting documents into sentences, two heuristics-based tools, i.e., the LingPipe Indo-European sentence model [37] and NaCTeM's Cafetiere sentence splitter [38], were individually employed in our experiments. Whilst the former was tuned for documents written in general language, the latter was designed specifically for scientific text, having been enriched with specialised rules that, for instance, account for the possibility of sentences beginning with lower-case characters, as with protein names, e.g., p53.
Tokenisers
For the decomposition of each sentence into tokens, we explored each of the tokenisers built into the GENIA tagger [39] and the OSCAR4 NER tool [40]. The former employs a statistical model trained on biomedical documents, whilst the latter applies segmentation rules specifically tuned for chemical texts. The OSCAR 4 tokeniser, for example, is capable of keeping intact long chemical names (e.g., 4,9-Diazadodecane-1,12-diamine).
Model training using a chemical knowledge-rich feature set
In building a model, we employed NERsuite, a combination of tools that include a CRF implementation [41] and utilities for embedding custom dictionary features. The following sections describe the features we used with this tool. They include basic, weakly chemical-indicative features and chemical-specific features.
Weakly chemical-indicative features
By default, NERsuite extracts the character and word n-gram features presented in Table 7. To exemplify, we provide the tokenised sentence in Table 8 as sample input, with GSK214a as the active token, i.e., the token currently under consideration. We note that the extraction of the word n-grams was done within a distance of two from the active token. Aside from these features, a token's symbol-level composition is also captured by means of the orthographic features listed in Table 9. We have augmented this set with the following:
Table 7.
Character and word n-gram features extracted by NERsuite by default.
| Feature | Brief description | Sample features (bigrams) |
|---|---|---|
| Character n-grams | the set of all possible combinations of a token's consecutive characters, taken n at a time (n = 2, 3, 4) | {GS}, {SK}, {K2}, {21}, {14}, {4a} |
| Token n-grams | unigrams and bigrams of surface forms; unigrams and bigrams of normalised surface forms where numbers numbers are replaced with '0's, the consecutive instances of which are compressed | {It, attenuated}, {attenuated, GSK214a}; {Aa, aaaaaaaaaa}, {aaaaaaaaaa, AAA000a} |
| Lemma n-grams | unigrams and bigrams of lemmatised surface forms | {It, attenuate}, {attenuate, GSK214a} |
| POS tag n-grams | unigrams and bigrams of part-of-speech (POS) tags | {PRP, VBD}, {VBD, NN}, |
| Lemma & POS tag n-grams |
unigrams and bigrams of lemmatised forms combined with POS tags | {It:PRP, attenuate:VBD}, {attenuate:VBD, GSK214a:NN} |
| Chunk information | chunk tag of current token; surface form of the enclosing chunk's | {B-NP}; {gestation} |
Table 8.
Example of a sentence tokenised and labelled with part-of-speech and chunk tags.
| Surface form | Lemma | Part-of-speech tag | Chunk tag |
|---|---|---|---|
| It | It | PRP | B-NP |
| attenuated | attenuate | VBD | B-VP |
| GSK214a | GSK214a | NN | B-NP |
| -induced | -induced | JJ | I-NP |
| gestation | gestation | NN | I-NP |
| in | in | IN | B-PP |
| rats | rat | NN | B-NP |
| . | . | . | O |
Table 9.
Orthographic features extracted by NERsuite by default.
| Feature | Example |
|---|---|
| Initial letter is in uppercase | Boc-L-leucine |
| Contains only digits | 206553 |
| Contains digits | 5-HTP |
| Contains only alphanumeric characters | HClO4 |
| Contains only uppercase letters and digits | AFB1 |
| Contains only uppercase letters | NO |
| Does not contain any lowercase letters | SKF81297 |
| Contains non-initial uppercase letters | PbS |
| Contains two consecutive uppercase letters | PAHs |
| Has a Greek letter name as a substring | alpha-ketoacid |
| Contains a comma | 3,14-dibromo |
| Contains a full stop | In(0.2)Ga(0.8)As |
| Contains a hyphen | HP-β-CD |
| Contains a forward slash | (E/Z)-Goniothalamin |
| Contains an opening square bracket | [(14)C]pazopanib |
| Contains a closing square bracket | pyrido[3,2-d]pyrimidines |
| Contains an opening parenthesis | I3 (-) |
| Contains a closing parenthesis | Fe(C10 H15)2 |
| Contains a semi-colon | R = Me, Et; X = O, S; |
| Contains a percentage symbol | 85% |
| Contains an apostrophe | 5-methyl-2'-deoxycytidine |
Occurrence of Greek characters. This feature reflects an observation that several chemical names contain Greek characters, e.g., (S)-α,ε-diaminohexonoic acid.
Word shape. The active token is transformed to a representation in which numericals are converted to the '0' characters, uppercase letters to the 'A' characters, lowercase letters to the 'a' characters and everything else to the '_' characters. Full and brief word shape variants were extracted for each token. In the former, each character in the resulting representation is retained, whereas consecutive similar character types are collapsed into one in the latter. For example, the name 10-amino-20(S)-camptothecin would have 00_aaaaa_00_A_ _aaaaaaaaaaaa and 0_a_0_A_a as its full and brief word shapes, respectively.
Chemical dictionary matches
Recognising that the occurrence of a token in an expert-curated dictionary indicates a high likelihood of it being a chemical name constituent, we utilised matches between token surface forms in text and entries in well-known chemical resources. Five dictionaries were compiled based on the chemical names and synonyms available in the Chemical Entities of Biological Interest (ChEBI) database [26], DrugBank [42], the Comparative Toxicogenomics Database (CTD) [43], PubChem Compound [44] and the Joint Chemical Dictionary (Jochem) [17]. The dictionary tools available in the NERsuite package were employed in the compilation and subsequent application of these dictionaries. We configured the compiler utility to generate a compiled dictionary whose entries were normalised by the conversion of alphabetic characters to their lower-case equivalents, numericals to the '0' characters and special characters/punctuation to the '_' characters. In the matching phase, the dictionary tagging tool performs the same conversion step on input text and then captures longest possible matches between the normalised token sequences and dictionary entries. The dictionary tagging results, exemplified in Table 10, were encoded in the begin-inside-outside (BIO) format. For an active token, unigrams and bigrams formed based on the BIO labels (within a distance of 2), as well as their combination with the corresponding surface forms, were generated as features. The token starch, for instance, would have the following as some of its CTD dictionary features: {on:O, hydroxyethyl:B}, {hydroxyethyl:B, starch:I} as surface form and dictionary label bigrams, and {O, B}, {B, I} as dictionary label bigrams.
Table 10.
Example of a token sequence tagged with matches against chemical dictionaries.
| Token | Normal form | ChEBI | DrugBank | CTD | PubChem | Jochem |
|---|---|---|---|---|---|---|
| For | for | O | O | O | O | O |
| the | the | O | O | O | O | O |
| preparation | preparation | O | O | O | O | O |
| of | of | O | O | O | O | O |
| hydrogel | hydrogel | O | O | B | O | B |
| microspheres | microsphere | O | O | O | O | O |
| based | base | O | O | O | O | O |
| on | on | O | O | O | O | O |
| hydroxyethyl | hydroxyethyl | O | O | B | O | B |
| starch | starch | B | O | I | O | I |
| - | _ | B | O | O | O | O |
| hydroxyethyl | hydroxyethyl | I | O | B | O | B |
| methacrylate | methacrylate | I | O | I | B | I |
| ( | _ | O | O | O | O | O |
| HES-HEMA | hes_hema | O | O | O | O | O |
| ) | _ | O | O | O | O | O |
Chemical affix matches
Many of the chemical names, especially nomenclature-based ones, contain chemical affixes (i.e., prefixes and suffixes). We attempt to capture this property by matching tokens in text against lists of commonly used chemical prefixes and suffixes whose lengths range from two to four. Shown in Table 11 is a sequence of tokens matched against our compiled affix lists, which are provided in an additional file (see Additional file 1). The feature set is augmented with the resulting affix matches.
Table 11.
Example of a token sequence tagged with matches against our affix lists.
| Prefixes | Suffixes | |||||
|---|---|---|---|---|---|---|
| Token | size 2 | size 3 | size 4 | size 2 | size 3 | size 4 |
| Incubation | O | O | O | O | O | O |
| with | O | O | O | O | O | O |
| diisopropyl | di | O | O | yl | O | O |
| fluorophosphate | O | O | fluo | O | ate | O |
| and | O | O | O | O | O | O |
| bis-(4-nitrophenyl) | O | O | O | O | O | O |
| phosphate | O | O | O | O | ate | O |
Number of chemical basic segments
Nomenclature-based chemical expressions, e.g., systematic and semi-systematic names, are formed from combinations of chemical segments. These segments are documented in the American Chemical Society's Registry File Basic Name Segment Dictionary, which contains a total of 3,307 entries as well as a description of the procedure for decomposing a name into its basic segments [45]. Following this algorithm, we process the surface form of each token to determine the number of constituent chemical basic segments. Table 12 lists the basic chemical segments found within the given expressions. We note that the number of basic segments also includes fragments which remain unmatched against the segment dictionary. For instance, only the fragments methyl, ergo and novi in the name methylergonovine can be found by the procedure; however, the remaining fragment ne was also counted as a basic segment.
Table 12.
Examples of chemical names with corresponding basic segments.
| Token | Basic segments | No. of basic segments |
|---|---|---|
| 10-acetoxyactinidine | 10, acet, oxy, actin, idine | 5 |
| methylergonovine | methyl, ergo, novi, ne | 4 |
| interleukin-2 | interleukin, 2 | 2 |
Chemical symbol matches
In order to account for chemical element symbols, which are not always covered by our five chosen dictionaries, we matched tokens in text against a list of symbols which occur in the periodic table of elements. This list has been provided in an additional file (see Additional file 2).
Heuristics-based post-processing
Two CRF models were initially learned from the CHEMDNER training corpus: one with only the default features and another with our engineered ones. For each input sentence, each of the models automatically generates a label sequence in BIO format, together with the confidence values with which the labels were assigned. Upon individually applying the models on the CHEMDNER development set, we obtained the results presented in Table 13. Whilst the performance boost brought about by our customised features was encouraging, the suboptimal recall prompted us to introduce post-processing steps for reducing the number of false negatives.
Table 13.
Performance of models learned from the CHEMDNER training set when evaluated on the development set.
| Macro | Micro | |||||
|---|---|---|---|---|---|---|
| P | R | F1 | P | R | F1 | |
| Default features | 86.66 | 79.01 | 80.89 | 88.55 | 76.82 | 82.27 |
| Enriched features | 88.26 | 81.11 | 82.86 | 89.87 | 78.99 | 84.07 |
| Margin | +1.6 | +2.1 | +1.97 | +1.32 | +2.17 | +1.8 |
By inspecting the distribution of false negatives according to chemical mention types (provided in Table 14), we identified the most prevalent problematic cases which we addressed with two rule-based post-processing steps.
Table 14.
Distribution (according to chemical subtype) of the instances incorrectly rejected by the model trained with enriched features.
| Subtype | Frequency | Percentage |
|---|---|---|
| Abbreviation | 1,882 | 30.32% |
| Formula | 1,291 | 20.80% |
| Family | 979 | 15.77% |
| Trivial | 926 | 14.92% |
| Systematic | 693 | 11.16% |
| Identifier | 293 | 4.72% |
| Multiple | 118 | 1.90% |
| No class | 25 | 0.40% |
Abbreviation recognition
To alleviate the problem of missed abbreviations which account for about 30% of the false negatives, we introduced an abbreviation recognition step which performs the following checks given the last token ti of a named entity e recognised by the CRF model:
• ti+1 is the opening parenthesis '(',
• ti+3 is the closing parenthesis ')', and
• ti+2 was recognised as a non-chemical token by the CRF model.
Token ti+2 becomes a candidate abbreviation for e if all three conditions hold true. As a verification step, a procedure [46] for checking the sequential occurrence of each character in ti+2 within the entity e is performed. Upon successful verification, all instances of token ti+2 within the document are relabelled as chemical tokens. In this manner, for instance, the chemical abbreviation STMP missed by the CRF model will be captured from the phrase, "... was phosphorylated with sodium trimetaphosphate (STMP) at ambient temperature..." assuming that sodium trimetaphosphate was recognised by the model as a chemical entity.
Chemical composition-based token relabelling
About 42% of the false negatives correspond to missed family, trivial and systematic names, all of which typically contain chemical segments. In attempting to increase recall for these mention types, we developed a procedure that analyses tokens which were labelled by the CRF model as non-chemical with confidence values lower than a chosen threshold t1. These tokens are of interest as the relatively low confidence values attached to them indicate their likelihood of being chemical name constituents. This likelihood was further verified by the computation of a token's chemical segment composition, given by the ratio of the number of characters comprising segments matched against the chemical basic segment dictionary to the total number of characters in the token. Sample tokens and the ratios calculated for them are provided in Table 15. The procedure relabels a token of interest as chemical if its chemical segment composition is greater than a chosen threshold t2.
Table 15.
Sample tokens and their chemical segment composition.
| Token initially recognised as non-chemical | Chemical basic segments | Ratio |
|---|---|---|
| polycalcium | poly, calcium | 1.0 |
| 2-methoxyestradiol | meth, oxy, estra, di, ol | 0.89 |
| palytoxin | toxin | 0.56 |
Different combinations of the thresholds t1 and t2 were investigated to establish the most optimal values. After a few exploratory runs, we decided to restrict our search space to the range [0.91, 0.99] for t1 and to [0.5, 0.9] for t2. These were exhaustively probed in increments of 0.01 and 0.1 for t1 and t2, respectively, by means of evaluation on the CHEMDNER development data set. Results showed that recall is optimal with t1 = 0.96 and t2 = 0.5, and that optimal precision and F1 scores are obtained with t1 = 0.93 and t2 = 0.9. In the rest of the experiments presented in this paper, we used the latter threshold settings.
Evaluation
With the proposed extensions presented above, chemical NER can be optimised according to the following five dimensions:
1 Pre-processing: Sentence splitting (LingPipe Indo-European model or Cafetiere)
2 Pre-processing: Tokenisation (GENIA or OSCAR4)
3 Model training: Knowledge-rich features (include or exclude)
4 Post-processing: Abbreviation recognition (enable or disable)
5 Post-processing: Chemical composition-based token relabelling (enable or disable)
Out of the possible combinations from these five dimensions, we selected 20 for each of our experiments, enabling abbreviation recognition and chemical composition-based token relabelling only when knowledge-rich features were employed in model training. This comprehensive evaluation was carried out with the utilisation of the CHEMDNER, SCAI and SciBorg corpora, as well as the following document collections:
Patents [30]. This corpus is the outcome of the collaborative effort of curators from the European Patent Office and the ChEBI project who annotated all mentions of chemical entities in 40 patent application documents [29].
Drug-Drug Interaction (DDI) [32]. Consisting of 233 MEDLINE abstracts and 792 textual descriptions from the DrugBank database, this corpus contains annotated drug mentions pertaining to generic names, brands, groups (e.g., antibiotic) and non-human applications (e.g., pesticide) [31]. Released as a resource for the SemEval 2013 DDI Extraction task [47], the corpus is divided into subsets for training and testing.
Pharmacokinetics (PK) [34]. Also containing drug name annotations, this corpus is comprised of a selection of 541 MEDLINE abstracts on the topics of clinical pharmacokinetics and phamacogenetics as well as in vitro and in vivo drug-drug interactions [33].
NaCTeM Metabolites [28]. This document collection contains 296 MEDLINE abstracts with annotations for metabolite and enzyme names [23]. For our evaluation, only metabolite name annotations were taken into consideration.
Table 16 summarises the results of the best performing ChER combination in each of the experiments we conducted. For the purpose of comparison, we have also provided results obtained by our baseline, i.e., the variant of the named entity recogniser that employs non-specialised pre-processing analytics (i.e., the LingPipe Indo-European sentence model and the GENIA tokeniser) and none of the knowledge-rich features and post-processing heuristics. It can be observed that in majority of the nine sets of experiments in this table, the optimal combination for ChER incorporates the use of specialised pre-processing tools, feature set enrichment and abbreviation recognition. The lack of a unique combination yielding optimal results across all evaluation data sets can be explained by the differences of the corpora in terms of the guidelines which were adhered to during their annotation. Enabling chemical composition-based token relabelling brought about improved F1 scores on the SciBorg, Patents, Metabolites and DDI corpora (owing to increased recall), but resulted in lower values of F1 on the CHEMDNER, SCAI and PK corpora (due to decreased precision). This post-processing step, for example, captured mentions of anions which were considered as chemical names in the SciBorg and Metabolites corpora (e.g., silicate, glutamate, succinate) but were counted as false positives by the SCAI corpus. Similarly, some chemical umbrella terms, such as esters and nucleotides, captured by this step were treated as true positives under evaluation against the Patents corpus, but stand for false positives in the CHEMDNER, SCAI and PK corpora. Another source of discrepancy are chemical named entities which have ambiguous meanings, that this rule-based step is oblivious to. Iron as a metallic element, for example, was not annotated in CHEMDNER and SCAI, but is considered a drug (i.e., a vitamin) in the DDI corpus. Meanwhile, abbreviation recognition boosted ChER's performance on all corpora except for the DDI corpus, where no impact was observed due to it not having been annotated with abbreviation information.
Table 16.
Summary of ChER's performance under the CHEMDNER track setting (set 1), under similar experimental settings as state-of-the-art methods (sets 2-4), and when applied to various corpora (sets 5-9).
| Data | Pre-processing | Cust. | Post-processing | Micro-averages | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Training | Test | Splitter | Tokeniser | Feats. | Abbr. | Comp. | P | R | F1 | |
| 1 | CHEMDNER | CHEMDNER | LingPipe | GENIA | ✗ | ✗ | ✗ | 88.87 | 70.95 | 78.91 |
| training & dev. | test | Cafetiere | OSCAR4 | ✓ | ✓ | ✓ | 92.76 | 81.30 | 86.65 | |
| 2 | SciBorg (CM):3-fold CV | LingPipe | GENIA | ✗ | ✗ | ✗ | 80.44 | 55.16 | 65.45 | |
| Cafetiere | OSCAR4 | ✓ | ✓ | ✓ | 85.96 | 74.22 | 79.66 | |||
| 3 | SCAI-IUPAC | SCAI-100 | LingPipe | GENIA | ✗ | ✗ | ✗ | 84.78 | 66.87 | 74.77 |
| training | (IUPAC) | Cafetiere | GENIA | ✓ | ✓ | ✓ | 86.70 | 67.50 | 75.90 | |
| 4 | NaCTeM Metabolites:10-fold CV | LingPipe | GENIA | ✗ | ✗ | ✗ | 81.72 | 64.49 | 72.09 | |
| Cafetiere | OSCAR4 | ✓ | ✓ | ✓ | 81.42 | 79.66 | 80.53 | |||
| 5 | CHEMDNER | SCAI-100 | LingPipe | GENIA | ✗ | ✗ | ✗ | 72.56 | 66.00 | 69.13 |
| training & dev. | (All) | Cafetiere | OSCAR4 | ✓ | ✓ | ✓ | 77.85 | 78.69 | 78.27 | |
| 6 | CHEMDNER | Patents | LingPipe | GENIA | ✗ | ✗ | ✗ | 72.66 | 52.97 | 61.27 |
| training & dev. | Cafetiere | OSCAR4 | ✓ | ✓ | ✓ | 73.43 | 57.91 | 64.75 | ||
| 7 | CHEMDNER | DDI | LingPipe | GENIA | ✗ | ✗ | ✗ | 76.52 | 75.00 | 75.75 |
| training & dev. | test | Cafetiere | OSCAR4 | ✓ | • | ✓ | 75.88 | 92.05 | 83.18 | |
| 8 | CHEMDNER | PK | LingPipe | GENIA | ✗ | ✗ | ✗ | 79.29 | 84.66 | 81.89 |
| training & dev. | Cafetiere | GENIA | ✓ | ✓ | ✓ | 79.83 | 88.34 | 83.87 | ||
| 9 | CHEMDNER | NaCTeM | LingPipe | GENIA | ✗ | ✗ | ✗ | 63.57 | 71.63 | 67.36 |
| training & dev. | Metabolites | Cafetiere | OSCAR4 | ✓ | ✓ | ✓ | 65.08 | 83.29 | 73.07 | |
The first row in each set corresponds to the baseline. Key: Cust. Feats. = Custom Features, Abbr. = Abbreviation recognition, Comp. = Chemical composition-based token relabelling; ✓ = enabled, ✗ = disabled, • = enabling or disabling makes no difference in performance.
The results of all 20 combinations, in each of the nine experimental set-ups described in Table 16, are provided in Additional file 3. The impact on performance of individually selecting a particular pre-processing analytic or enabling a specific post-processing heuristic can be easily observed from this file. For example, on the CHEMDNER test data, the ChER variant that employs Cafetiere Sentence Splitter, OSCAR4 Tokeniser, knowledge-rich features and abbreviation recognition for the CEM task obtains an F1 score of 86.65%. Replacing OSCAR4 Tokeniser with GENIA Tokeniser, however, leads to a 6-percentage point drop in F1 score (80.1%).
Conclusions
The exhaustive evaluation of our proposed tool ChER shows that in majority of cases the most optimal variant incorporates specialised pre-processing analytics (specifically, the Cafetiere sentence splitter and OSCAR4 tokeniser), knowledge-rich machine-learning features and a post-processing step for abbreviation recognition. In each experiment that we performed, comparison of the optimal combination with the baseline (i.e., the variant of the NER without any of our proposed additions) indicates noticeably better performance of the former over the latter. When compared to state-of-the-art methods, our solutions obtain competitive, if not superior, performance.
ChER with a statistical model learned from the training and development sets of the CHEMDNER corpus proved to achieve a satisfactory performance on a variety of corpora, regardless of document type and chemical subdomain, consistently outperforming the state of the art.
As our solutions are all accessible and usable via the Argo text mining platform, interested parties can replicate our results, if not introduce further improvements to our solution by exploring other analytics. Moreover, owing to the interoperable nature of Argo, our chemical entity recogniser, ChER, does not impose any restrictions in terms of input and output formats. It can be easily integrated as a semantic analytic in other text mining tasks such as document indexing and entity relation extraction.
Methods
Sequence labelling
In addressing the problem of named entity recognition, we employed a sequence labelling approach which involves the automatic assignment of labels to a given sequence of items, i.e., the ordered tokens in a sentence. The set of possible labels was defined by our chosen encoding scheme, the begin-inside-outside (BIO) representation. This scheme uses the labels 'B' and 'I' to indicate the beginning and continuing tokens of a chemical name, respectively, and 'O' to mark tokens which are not part of any chemical name. To transform the documents into this representation, the following pre-processing pipeline was applied on raw input text:
Sentence splitting
Text contained in each document was segmented by means of a sentence splitter. As described previously, the LingPipe Indo-European sentence model and Cafetiere sentence splitter were individually applied in this work.
Tokenisation
In segmenting each sentence into tokens, we utilised each of the GENIA and OSCAR4 tokenisers in our experiments.
Part-of-speech and chunk tagging. Each resulting token is automatically lemmatised and assigned tags which correspond to its part-of-speech (POS) and enclosing chunk. This information was supplied by the GENIA Tagger [39] which employs maximum entropy models in analysing both general and biomedical-domain documents. Shown in Table 8 are the lemmata, POS and chunk tags assigned to the tokens of the given sentence.
Our sequence labelling approach was realised as an application of the machine learning-based conditional random fields algorithm (CRFs). Given an item sequence, a CRF model predicts the most probable label sequence based on functions capturing characteristics of the current token and its context. These functions, typically referred to as features (discussed in detail in the Experiments section), are employed in both training and prediction phases. We built our named entity recognisers on top of the NERsuite package [48], an implementation of CRFs with a built-in extractor of features typically used in biomedical NER.
Evaluation metrics
We reported the effectiveness of our methods with the commonly used information retrieval metrics, namely, precision (P), recall (R) and F1 score defined as follows:
where TP, FP and FN are the numbers of, respectively, true positive, false positive and false negative recognitions. Intuitively, precision is the fraction of recognised entities that are correct, recall is the fraction of manually annotated entities that were recognised, and F1 is a balanced harmonic mean between the two. F1 represents a more conservative metric than the arithmetic average.
We note that all of the results reported in this paper, including those of the other chemical NER tools, were obtained using the evaluation tool provided by the BioCreative organisers [49]. The tool calculates the macro- and micro-averaged values of the aforementioned metrics.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
RB carried out the development and evaluation of the proposed methods for chemical NER optimisation. RR developed the various components for making the results of this work available and usable in Argo. Both RR and RB formulated the study. SA provided guidance in the evaluation of the work and coordinated the effort. All authors read and approved the final manuscript.
Supplementary Material
List of chemical affixes. A listing of the most common chemical prefixes and suffixes.
List of chemical element symbols. A listing of the chemical element symbols.
Chemical Entity Recogniser (ChER) Experiments. Tables containing the results of nine experiments, each comparing 20 combinations of the proposed methods.
Acknowledgements
The first author was financially supported by the Engineering Research and Development for Technology (ERDT) faculty development program. This work was also partially supported by Europe PubMed Central funders (led by Wellcome Trust).
Declaration
Funding for this article's publication was granted by the University of Manchester's Wellcome Trust award.
This article has been published as part of Journal of Cheminformatics Volume 7 Supplement 1, 2015: Text mining for chemistry and the CHEMDNER track. The full contents of the supplement are available online at http://www.jcheminf.com/supplements/7/S1.
References
- Davis AP, Wiegers TC, Johnson RJ, Lay JM, Lennon-Hopkins K, Saraceni-Richards C, Sciaky D, Murphy CG, Mattingly CJ. Text mining effectively scores and ranks the literature for improving chemical-gene-disease curation at the comparative toxicogenomics database. PLoS ONE. 2013;8(4):58201. doi: 10.1371/journal.pone.0058201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolářik C, Hofmann-Apitius M. In: Chemical Information Mining. Banville DL, editor. Chap 7. 2009. Linking Chemical and Biological Information with Natural Language Processing; pp. 123–150. [Google Scholar]
- Segura-Bedmar I Martínez P de Pablo-Sánchez C Extracting drug-drug interactions from biomedical texts BMC Bioinformatics 201011S-5920053276 [Google Scholar]
- Deftereos SN, Andronis C, Friedla EJ, Persidis A, Persidis A. Drug repurposing and adverse event prediction using high-throughput literature analysis. Wiley interdisciplinary reviews. Systems biology and medicine. 2011;3(3):323–34. doi: 10.1002/wsbm.147. [DOI] [PubMed] [Google Scholar]
- Li C, Liakata M, Rebholz-Schuhmann D. Biological network extraction from scientific literature: state of the art and challenges. Briefings in Bioinformatics. 2013. [DOI] [PubMed]
- Banville DL. Mining chemical structural information from the drug literature. Drug Discovery Today. 2006;11(1):35–42. doi: 10.1016/S1359-6446(05)03682-2. [DOI] [PubMed] [Google Scholar]
- Vazquez M, Krallinger M, Leitner F, Valencia A. Text mining for drugs and chemical compounds: Methods, tools and applications. Molecular Informatics. 2011;30(6-7):506–519. doi: 10.1002/minf.201100005. [DOI] [PubMed] [Google Scholar]
- Gurulingappa H, Mudi A, Toldo L, Hofmann-Apitius M, Bhate J. Challenges in mining the literature for chemical information. RSC Adv. 2013. pp. 16194–16211.
- Grego T, Pesquita C, Bastos HP, Couto FM. Chemical Entity Recognition and Resolution to ChEBI. ISRN Bioinformatics. 2012;2012:9. doi: 10.5402/2012/619427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett P, Batchelor C, Teufel S. Proceedings of the Workshop on BioNLP 2007: Biological, Translational, and Clinical Language Processing. BioNLP '07. Association for Computational Linguistics, Stroudsburg, PA, USA; 2007. Annotation of chemical named entities; pp. 57–64. [Google Scholar]
- Chemistry Using Text Annotations. http://nactem.ac.uk/cheta Accessed: October 2013.
- Rebholz-Schuhmann D, Yepes J, Jose A, Van Mulligen EM, Kang N, Kors J, Milward D, Corbett P, Buyko E, Beisswanger E, Hahn U. CALBC silver standard corpus. Journal of Bioinformatics and Computational Biology. 2010;8(1):163–79. doi: 10.1142/S0219720010004562. [DOI] [PubMed] [Google Scholar]
- Kolářik C, Klinger R, Friedrich CM, Hofmann-Apitius M, Fluck J. Chemical names: Terminological resources and corpora annotation. Proceedings of the Workshop on Building and Evaluating Resources for Biomedical Text Mining. LREC. 2008. pp. 51–58.
- Fraunhofer SCAI Corpora for Chemical Entity Recognition. http://www.scai.fraunhofer.de/chem-corpora.html Accessed: October 2013.
- Corbett P, Copestake A. Cascaded classifiers for confidence-based chemical named entity recognition. BMC Bioinformatics. 2008;9(Suppl 11):4. doi: 10.1186/1471-2105-9-S11-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocktäschel T, Weidlich M, Leser U. ChemSpot: a hybrid system for chemical named entity recognition. Bioinformatics. 2012;28(12):1633–1640. doi: 10.1093/bioinformatics/bts183. [DOI] [PubMed] [Google Scholar]
- Hettne KM, Stierum RH, Schuemie MJ, Hendriksen PJM, Schijvenaars BJA, van Mulligen EM, Kleinjans J, Kors JA. A dictionary to identify small molecules and drugs in free text. Bioinformatics. 2009;25(22):2983–2991. doi: 10.1093/bioinformatics/btp535. [DOI] [PubMed] [Google Scholar]
- Lafferty JD, McCallum A, Pereira FCN. Proceedings of the Eighteenth International Conference on Machine Learning. ICML '01. Morgan Kaufmann Publishers Inc., San Francisco, CA, USA; 2001. Conditional Random Fields: Probabilistic Models for Segmenting and Labeling Sequence Data; pp. 282–289. [Google Scholar]
- Krallinger M, Leitner F, Rabal O, Vazquez M, Oyarzabal J, Valencia A. CHEMDNER: The drugs and chemical names extraction challenge. J Cheminform. 2015;7(Suppl 1):S1. doi: 10.1186/1758-2946-7-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krallinger M, Rabal O, Leitner F, Vazquez M, Salgado D, Lu Z, Leaman R, Lu Y, Ji D, Lowe DM, Sayle RA, Batista-Navarro RT, Rak R, Huber T, Rocktaschel T, Matos S, Campos D, Tang B, Xu H, Munkhdalai T, Ryu KH, Ramanan SV, Nathan S, Zitnik S, Bajec M, Weber L, Irmer M, Akhondi SA, Kors JA, Xu S, An X, Sikdar UK, Ekbal A, Yoshioka M, Dieb TM, Choi M, Verspoor K, Khabsa M, Giles CL, Liu H, Ravikumar KE, Lamurias A, Couto FM, Dai H, Tsai RT, Ata C, Can T, Usie A, Alves R, Segura-Bedmar I, Martinez P, Oryzabal J, Valencia A. The CHEMDNER corpus of chemicals and drugs and its annotation principles. J Cheminform. 2015;7(Suppl 1):S2. doi: 10.1186/1758-2946-7-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rak R, Batista-Navarro RT, Carter J, Rowley A, Ananiadou S. Processing biological literature with customizable web services supporting interoperable formats. Database. 2014;2014:064. doi: 10.1093/database/bau064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista-Navarro RTB, Rak R, Ananiadou S. Chemistry-specific features and heuristics for developing a CRF-based chemical named entity recogniser. Proceedings of the Fourth BioCreative Challenge Evaluation Workshop. 2013;2:55–59. [Google Scholar]
- Nobata C, Dobson PD, Iqbal SA, Mendes P, Tsujii J, Kell DB, Ananiadou S. Mining metabolites: extracting the yeast metabolome from the literature. Metabolomics. 2011;7(1):94–101. doi: 10.1007/s11306-010-0251-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OSCAR4. https://bitbucket.org/wwmm/oscar4/wiki/Home Accessed: October 2013.
- ChemSpot. https://github.com/rockt/ChemSpot Accessed: October 2013.
- Hastings J, de Matos P, Dekker A, Ennis M, Harsha B, Kale N, Muthukrishnan V, Owen G, Turner S, Williams M, Steinbeck C. The ChEBI reference database and ontology for biologically relevant chemistry: enhancements for 2013. Nucleic Acids Research. 2012. [DOI] [PMC free article] [PubMed]
- Wishart DS, Knox C, Guo AC, Eisner R, Young N, Gautam B, Hau DD, Psychogios N, Dong E, Bouatra S, Mandal R, Sinelnikov I, Xia J, Jia L, Cruz JA, Lim E, Sobsey CA, Shrivastava S, Huang P, Liu P, Fang L, Peng J, Fradette R, Cheng D, Tzur D, Clements M, Lewis A, De Souza A, Zuniga A, Dawe M, Xiong Y, Clive D, Nazyrova A, Shaykhutdinov R, Li L, Vogel HJ, Forsythe I. HMDB: a knowledgebase for the human metabolome. Nucleic Acids Research. 2009;37(suppl 1):603–610. doi: 10.1093/nar/gkn810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NaCTeM Metabolite and Enzyme Corpus. http://www.nactem.ac.uk/metabolite-corpus Accessed: October 2013.
- Grego T, Pęzik P, Couto FM, Rebholz-Schuhmann D. Proceedings of the 10th International Work-Conference on Artificial Neural Networks: Part II: Distributed Computing, Artificial Intelligence, Bioinformatics, Soft Computing, and Ambient Assisted Living. IWANN '09. Springer, Berlin, Heidelberg; 2009. Identification of chemical entities in patent documents; pp. 942–949. [Google Scholar]
- Patents Gold Standard Annotations. http://chebi.cvs.sourceforge.net/viewvc/chebi/chapati/patentsGoldStandard Accessed: October 2013.
- Herrero-Zazo M, Segura-Bedmar I, Martínez P, Declerck T. The ddi corpus: An annotated corpus with pharmacological substances and drug-drug interactions. Journal of Biomedical Informatics. 2013;46(5):914–920. doi: 10.1016/j.jbi.2013.07.011. [DOI] [PubMed] [Google Scholar]
- Corpora for Drug-Drug Interaction Extraction. http://labda.inf.uc3m.es/doku.php?id=en:labda_ddicorpus Accessed: October 2013.
- Wu H-Y, Karnik S, Subhadarshini A, Wang Z, Philips S, Han X, Chiang C, Liu L, Boustani M, Rocha L, Quinney S, Flockhart D, Li L. An integrated pharmacokinetics ontology and corpus for text mining. BMC Bioinformatics. 2013;14(1):35. doi: 10.1186/1471-2105-14-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pharmacokinetics Corpus. http://rweb.compbio.iupui.edu/corpus Accessed: October 2013.
- Rak R, Rowley A, Black W, Ananiadou S. Argo: an integrative, interactive, text mining-based workbench supporting curation. Database : The Journal of Biological Databases and Curation. 2012. p. 010. [DOI] [PMC free article] [PubMed]
- Kolluru B, Hawizy L, Murray-Rust P, Tsujii J, Ananiadou S. Using workflows to explore and optimise named entity recognition for chemistry. PLoS ONE. 2011;6(5):20181. doi: 10.1371/journal.pone.0020181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alias-I: LingPipe 4.1.0. http://alias-i.com/lingpipe Accessed: July 2013.
- Cafetiere English Sentence Detector. http://metashare.metanet4u.eu/repository/browse/u-compare-cafetiere-english-sentence-detector/aff1ddc0bc8911e1a404080027e73ea259aeca28412944ea97f7b2580a41caec/# Accessed: October 2013.
- Tsuruoka Y, Tateisi Y, Kim J-D, Ohta T, McNaught J, Ananiadou S, Tsujii J. Developing a Robust Part-of-Speech Tagger for Biomedical Text. In: Advances in Informatics - 10th Panhellenic Conference on Informatics. LNCS, Springer, Volos, Greece. 2005;3746:382–392. [Google Scholar]
- Jessop DM, Adams SE, Willighagen EL, Hawizy L, Murray-Rust P. OSCAR4: a flexible architecture for chemical text-mining. Journal of Cheminformatics. 2011;3(1):41. doi: 10.1186/1758-2946-3-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki N. CRFsuite: a fast implementation of Conditional Random Fields (CRFs) http://www.chokkan.org/software/crfsuite Accessed: July 2013.
- Knox C, Law V, Jewison T, Liu P, Ly S, Frolkis A, Pon A, Banco K, Mak C, Neveu V, Djoumbou Y, Eisner R, Guo AC, Wishart DS. DrugBank 3.0: a comprehensive resource for 'omics' research on drugs. Nucleic acids research. 2011. pp. 1035–41. [DOI] [PMC free article] [PubMed]
- Davis AP, Murphy CG, Johnson R, Lay JM, Lennon-Hopkins K, Saraceni-Richards C, Sciaky D, King BL, Rosenstein MC, Wiegers TC, Mattingly CJ. The Comparative Toxicogenomics Database: update 2013. Nucleic Acids Research. 2012. [DOI] [PMC free article] [PubMed]
- Bolton EE, Wang Y, Thiessen PA, Bryant SH. PubChem: Integrated Platform of Small Molecules and Biological Activities. Annual Reports in Computational Chemistry. 2008;4 [Google Scholar]
- American Chemical Society. Registry file basic name segment dictionary. Technical report. 1993.
- Schwartz AS, Hearst MA. A simple algorithm for identifying abbreviation definitions in biomedical text. Pacific Symposium on Biocomputing. 2003. pp. 451–462. [PubMed]
- Segura-Bedmar I, Martínez P, Herrero Zazo M. Second Joint Conference on Lexical and Computational Semantics (*SEM), Volume 2: Proceedings of the Seventh International Workshop on Semantic Evaluation (SemEval 2013) Association for Computational Linguistics, Atlanta, Georgia, USA; 2013. SemEval-2013 Task 9: Extraction of Drug-Drug Interactions from Biomedical Texts (DDIExtraction 2013) pp. 341–350. [Google Scholar]
- Cho H-C, Okazaki N, Miwa M, Tsujii J. NERsuite: a named entity recognition toolkit. https://github.com/nlplab/nersuite Accessed: July 2013.
- Leitner F. BioCreative II.5 Evaluation Library. http://www.biocreative.org/resources/biocreative-ii5/evaluation-library Accessed: August 2013.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of chemical affixes. A listing of the most common chemical prefixes and suffixes.
List of chemical element symbols. A listing of the chemical element symbols.
Chemical Entity Recogniser (ChER) Experiments. Tables containing the results of nine experiments, each comparing 20 combinations of the proposed methods.