

Abstract
Background
The GENCODE project has collected over 10,000 human long non-coding RNA (lncRNA) genes. However, the vast majority of them remain to be functionally characterized. Computational investigation of potential functions of human lncRNA genes is helpful to guide further experimental studies on lncRNAs.
Results
In this study, based on expression correlation between lncRNAs and protein-coding genes across 19 human normal tissues, we used the hypergeometric test to functionally annotate a single lncRNA or a set of lncRNAs with significantly enriched functional terms among the protein-coding genes that are significantly co-expressed with the lncRNA(s). The functional terms include all nodes in the Gene Ontology (GO) and 4,380 human biological pathways collected from 12 pathway databases. We successfully mapped 9,625 human lncRNA genes to GO terms and biological pathways, and then developed the first ontology-driven user-friendly web interface named lncRNA2Function, which enables researchers to browse the lncRNAs associated with a specific functional term, the functional terms associated with a specific lncRNA, or to assign functional terms to a set of human lncRNA genes, such as a cluster of co-expressed lncRNAs. The lncRNA2Function is freely available at http://mlg.hit.edu.cn/lncrna2function.
Conclusions
The LncRNA2Function is an important resource for further investigating the functions of a single human lncRNA, or functionally annotating a set of human lncRNAs of interest.
Background
Thousands of human long non-coding RNAs (lncRNAs) have been identified and emerging studies have revealed that lncRNAs play important roles in a wide range of biological processes [1,2] and diseases [3,4]. However, functions of most human lncRNAs are still elusive. Functions of a lncRNA may be determined by loss- and gain-of-function biological experiments [5,6]. However, this is not straightforward since it is difficult to knock down a lncRNA expressed as multiple isoforms. Alternatively, computational exploration of human lncRNA functions is helpful to guide further studies on lncRNAs.
Currently, computational investigation of lncRNA functions is still at its early development stage, since it is a considerable challenge due to the characteristics of lncRNAs, e.g., many lncRNA gene sequences are not conserved and do not contain conserved sequence motifs [7], which makes it difficult to infer potential functions of lncRNAs based on their sequences alone. In addition, few available molecular interaction data of new identified lncRNAs also hamper the lncRNA functional annotations [8,9].
Since genes with similar expression patterns across multiple conditions may share similar functions [10] or be involved in related biological pathways [11], identifying protein-coding genes that are co-expressed with lncRNAs may help to assign functions to the lncRNAs. By analyzing lncRNA-mRNA co-expression pattern, Guttman et al. identified several sets of mouse lncRNAs associated with protein-coding gene sets of distinct GO functional categories [12]. In addition, two recent studies separately constructed a mouse co-expressed lncRNA-mRNA network using mouse microarray data and assigned functions to 340 and 1,625 mouse lncRNAs [13,14].
Despite accumulating insights into the mouse lncRNA functions, more than 10,000 human lncRNAs remain to be functionally characterized. Firstly, given a single human lncRNA gene, it needs to be established whether it executes crucial biological functions. Secondly, given a set of human lncRNA genes such as differential lncRNAs between cancer and normal samples, it is an important downstream task to identify significantly enriched function terms. Thirdly, given an important functional term such as a Wnt signalling pathway, how to know which lncRNAs may be involved in the pathway.
Here, based on the expression correlation between lncRNAs and protein-coding genes inferred from RNA-seq data of 19 human normal tissues, we functionally annotated 9,625 human lncRNAs with significantly enriched functional terms among the co-expressed protein-coding genes, and developed a user-friendly web interface for the lncRNA community to obtain the lncRNAs associated with a specific functional term, the functional terms associated with a specific lncRNA, or to assign functions to a set of human lncRNAs of interest.
Methods
Data sources
We downloaded: (1) genomic coordinates of all human lncRNA genes and protein-coding genes from the GENCODE V15 [15], (2) paired-end RNA-Seq data of 19 human normal tissues from the Human Body Map 2 project (ArrayExpress accession no. E-MTAB-513) and another study (GEO accession no. GSE30554), (3) GO assignments for the proteins of the human UniProtKB Complete Proteome from the website of the Gene Ontology Project [16], (4) 4,380 human biological pathways from the ConsensusPathDB database which integrated 12 pathway databases [17].
Workflow of LncRNA2Function
The schematic workflow of lncRNA2Function is shown in Figure 1. Firstly, RNA-Seq reads sequenced in 19 human normal tissues were firstly mapped to human genome (hg19) using tophat with the default parameters [18], and expression values of all human lncRNA and protein-coding genes in the 19 tissues were computed using cufflinks with the default parameter [19]. Secondly, the Pearson Correlation Coefficients (PCC) of all lncRNA-mRNA gene pairs were computed, and a set of significantly co-expressed protein-coding genes was thus obtained for each human lncRNA (significant: the absolute value of the Pearson correlation coefficient >0.9 and adjusted P-value < 0.05). Thirdly, each lncRNA was functionally annotated with significantly enriched GO terms and biological pathways among the set of co-expressed protein-coding genes. Finally, a web interface was developed to facilitate researchers to browse or search the functions associated with a given lncRNA or lncRNAs associated with a specific function, or to functionally annotate a set of lncRNA genes of interest.
Figure 1.
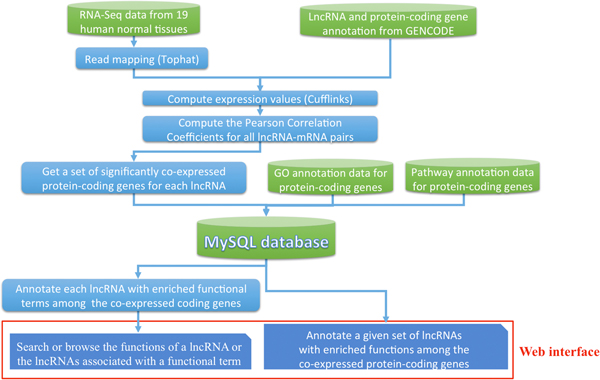
Schematic workflow of the LncRNA2Function.
GO and pathway enrichment analysis of human lncRNAs
Given a single human lncRNA gene, we obtained a set of protein-coding genes that were significantly co-expressed with the lncRNA. The lncRNA was then functionally annotated with significantly enriched GO and pathway terms among the set of co-expressed protein-coding genes. The enrichment analysis was separately executed for each term (denoted as T), and a P-value of each term was calculated by the hypergeometric test:
| (1) |
Herein, N is the number of all protein-coding genes in human genome, M is the number of protein-coding genes that were annotated in the functional term T, n is the number of protein-coding genes that were significantly co-expressed with the lncRNA, and m is the number of protein-coding genes that were both significantly co-expressed with the lncRNA and annotated in the functional term T.
For each GO term, protein-coding genes directly belong to it as well as those belong to any of its offspring terms are all considered as its annotated genes. Since the statistical analysis is not appropriate to problems with small sample size, those GO and pathway terms with less than 5 annotated protein-coding genes and those lncRNAs with less than 5 co-expressed protein-coding genes were excluded form the enrichment analysis.
Given a set of human lncRNA genes of interest, LncRNA2Function first identify a set of protein-coding genes, each of which are significantly co-expressed with one or more of the given lncRNAs across 19 human normal tissues. Then, the set of lncRNAs are functionally annotated with the enriched GO and pathway terms among the set of co-expressed protein-coding genes. If researchers input a large number of lncRNAs, the LncRNA2Function may obtain thousands of co-expressed protein-coding genes, some of which are co-expressed with only one of the lncRNAs. To improve the accuracy of functional assignments to the set of lncRNAs, users can select the protein-coding genes that are co-expressed with at least K lncRNAs (the K can be assigned based on the size of the set of lncRNAs. The default K is 1).
There are two commonly used methods for controlling false discovery rate (FDR), the Benjamini-Yekutieli (BY) method [20] and the Benjamini-Hochberg (BH) method [21]. The former is suitable for positively related multiple hypothesis tests whereas the later is suitable for independent multiple hypothesis tests [22]. Since the hierarchical GO terms are often dependent, we chose the BY method to correct the P-values from the GO enrichment analysis, and the BH method to correct the P-values from the pathway enrichment analysis. The significant cut-off of corrected P-value was set as 0.05.
Results and discussion
Functional annotations of a single human lncRNA
We obtained 5,232,299 significantly co-expressed pairs between 9,625 human lncRNA genes and 10,919 protein-coding genes. Each of the 9,625 lncRNAs was functionally annotated with significantly enriched GO terms and biological pathways among its co-expressed protein-coding genes. Consequently, we obtained 614,174 associations between 5,735 lncRNA genes and 3,890 GO terms, and 240,050 associations between 6,062 lncRNAs and 3,034 biological pathways. To understand the major functions of lncRNAs, we ranked GO biological processes and biological pathways according to the number of lncRNAs associated with each of them. Among the top ranked 200 GO biological processes and pathways, we found that lncRNAs play roles in many important biological processes, including defense response to bacterium, DNA packaging, meiosis, developmental process, metabolic process, cell cycle process, cell adhesion, cell differentiation, Jak-STAT signaling pathway and PI3K-Akt signaling pathway. A part of the enriched functions of lncRNAs have been validated by published studies [23-26].
Case studies
Due to the lack of a large gold standard dataset of known human lncRNA functions, five well-studied lncRNAs were used as the examples to show the usefulness of LncRNA2Function.
Case study 1: HOTAIR
The HOTAIR is a well-studied lncRNA. Rinn et al. found that the HOTAIR interacts with the Polycomb repressive complex 2 (PRC2) to modify chromatin and repress transcription of the HOX genes, which regulate development [27]. Niinuma et al. revealed that overexpression of HOTAIR was strongly associated with high-risk grade and metastasis among gastrointestinal stromal tumors (GIST) specimens, and knockdown of HOTAIR suppressed GIST cell invasiveness [28]. In addition, Gupta et al. demonstrated that the lncRNA HOTAIR is increased in expression in primary breast tumors and metastases, and enforced expression of HOTAIR in epithelial cancer cells leaded to altered histone H3 lysine 27 methylation, gene expression, and increased cancer invasiveness and metastasis in a manner dependent on PRC2. Conversely, loss of HOTAIR can inhibit breast cancer invasiveness [26].
To examine whether our LncRNA2Function can functionally annotate the lncRNA HOTAIR with development and metastasis-related functional terms, we applied the LncRNA2Function to the HOTAIR, and found that it was annotated with 99 GO biological processes and 33 pathways (The significant Corrected P-value cutoff is 0.05). Of the 99 GO biological processes, 77.8% (77/99) are involved in the development and morphogenesis as expected (The top 20 GO development-related biological processes are shown in Table 1), and 9.1% (9/99) are involved in the cell invasion and metastasis, such as cell migration (GO:0016477), cell adhesion (GO:0007155), biological adhesion (GO:0022610) and cell motility (GO:0048870). In addition, Of the 33 biological pathways, 72.7% (24/33) are involved in the cell invasion and metastasis (Table 2), such as focal adhesion, beta1 integrin cell surface interactions, NCAM1 interactions, Syndecan-1-mediated signaling events, PI3K-Akt signaling pathway and cell surface interactions at the vascular wall. Taken together, these results demonstrated that our LncRNA2Function can successfully recall the known functions of a well-studied lncRNA HOTAIR and suggested that it is applicable to infer potential functions of new identified lncRNAs.
Table 1.
The top 20 biological processes assigned to the development-regulating HOTAIR by LncRNA2Function.
| GO term | Background frequency | Sample frequency | P-value | Corrected P-value |
|---|---|---|---|---|
| System development | 3253/20447 | 38/74 | 1.63E-12 | 2.37E-08 |
| Anatomical structure morphogenesis | 1884/20447 | 28/74 | 2.45E-11 | 7.10E-08 |
| Tissue development | 1183/20447 | 23/74 | 1.33E-11 | 7.10E-08 |
| Embryonic skeletal system development | 120/20447 | 10/74 | 1.73E-11 | 7.10E-08 |
| Anatomical structure development | 3717/20447 | 39/74 | 2.07E-11 | 7.10E-08 |
| Skeletal system development | 388/20447 | 14/74 | 1.01E-10 | 2.44E-07 |
| Organ morphogenesis | 790/20447 | 18/74 | 2.89E-10 | 4.73E-07 |
| Multicellular organismal development | 3830/20447 | 38/74 | 2.56E-10 | 4.73E-07 |
| Developmental process | 4248/20447 | 40/74 | 2.93E-10 | 4.73E-07 |
| Organ development | 2271/20447 | 29/74 | 3.63E-10 | 5.27E-07 |
| Skeletal system morphogenesis | 189/20447 | 10/74 | 1.54E-09 | 1.72E-06 |
| Multicellular organismal process | 5336/20447 | 44/74 | 1.39E-09 | 1.72E-06 |
| Single-multicellular organism process | 5125/20447 | 43/74 | 1.51E-09 | 1.72E-06 |
| Extracellular matrix organization | 204/20447 | 10/74 | 3.23E-09 | 3.28E-06 |
| Extracellular structure organization | 205/20447 | 10/74 | 3.39E-09 | 3.28E-06 |
| Head development | 52/20447 | 6/74 | 3.26E-08 | 2.96E-05 |
| Embryonic skeletal system morphogenesis | 91/20447 | 7/74 | 3.86E-08 | 3.30E-05 |
| Single-organism developmental process | 3161/20447 | 31/74 | 4.39E-08 | 3.54E-05 |
| Chordate embryonic development | 557/20447 | 13/74 | 9.05E-08 | 6.57E-05 |
| Embryo development ending in birth or egg hatching | 564/20447 | 13/74 | 1.05E-07 | 7.23E-05 |
Table 2.
The metastasis-associated HOTAIR was annotated with metastasis-related GO and pathway terms by LncRNA2Function.
| Database | Functional term | Background frequency | Sample frequency | P-value | Corrected P-value |
|---|---|---|---|---|---|
| GO | Locomotion | 1022/20447 | 14/74 | 1.53E-05 | 0.003417 |
| GO | Cell migration | 603/20447 | 10/74 | 6.07E-05 | 0.010887 |
| GO | Cell adhesion | 790/20447 | 11/74 | 1.21E-04 | 0.020655 |
| GO | Biological adhesion | 792/20447 | 11/74 | 1.24E-04 | 0.020663 |
| GO | Cell motility | 664/20447 | 10/74 | 1.34E-04 | 0.021695 |
| GO | Positive regulation of cell-cell adhesion | 33/20447 | 3/74 | 2.30E-04 | 0.032376 |
| PID | Beta1 integrin cell surface interactions | 75/20447 | 7/74 | 9.92E-09 | 2.22E-06 |
| Reactome | Extracellular matrix organization | 102/20447 | 7/74 | 8.54E-08 | 9.57E-06 |
| KEGG | ECM-receptor interaction | 110/20447 | 7/74 | 1.44E-07 | 1.07E-05 |
| INOH | Integrin | 141/20447 | 7/74 | 7.82E-07 | 4.01E-05 |
| Wikipathways | Focal Adhesion | 203/20447 | 7/74 | 8.79E-06 | 2.46E-04 |
| KEGG | Focal adhesion | 219/20447 | 7/74 | 1.44E-05 | 3.58E-04 |
| PID | Beta3 integrin cell surface interactions | 47/20447 | 4/74 | 2.51E-05 | 5.61E-04 |
| PID | Syndecan-1-mediated signaling events | 50/20447 | 4/74 | 3.21E-05 | 6.53E-04 |
| PID | Integrin cell surface interactions | 58/20447 | 4/74 | 5.78E-05 | 0.001079 |
| PID | Integrins in angiogenesis | 73/20447 | 4/74 | 1.42E-04 | 0.002454 |
| Reactome | Integrin cell surface interactions | 88/20447 | 4/74 | 2.93E-04 | 0.004373 |
| KEGG | PI3K-Akt signaling pathway | 361/20447 | 7/74 | 3.29E-04 | 0.004606 |
| Reactome | Cell surface interactions at the vascular wall | 104/20447 | 4/74 | 5.53E-04 | 0.006566 |
| Reactome | Signaling by PDGF | 187/20447 | 5/74 | 5.86E-04 | 0.006566 |
| Reactome | NCAM1 interactions | 45/20447 | 3/74 | 5.79E-04 | 0.006566 |
| Reactome | NCAM signaling for neurite out-growth | 72/20447 | 3/74 | 0.002268 | 0.023101 |
| Reactome | Platelet Adhesion to exposed collagen | 22/20447 | 2/74 | 0.002848 | 0.025533 |
| PID | VEGFR3 signaling in lymphatic endothelium | 25/20447 | 2/74 | 0.003673 | 0.031645 |
| Reactome | Basigin interactions | 26/20447 | 2/74 | 0.003969 | 0.032935 |
| KEGG | TGF-beta signaling pathway | 92/20447 | 3/74 | 0.004537 | 0.036298 |
| PID | Wnt signaling network | 29/20447 | 2/74 | 0.004924 | 0.038039 |
| Reactome | Degradation of the extracellular matrix | 32/20447 | 2/74 | 0.005974 | 0.043169 |
| Reactome | Activation of Matrix Metalloproteinases | 32/20447 | 2/74 | 0.005974 | 0.043169 |
| PID | Alpha4 beta1 integrin signaling events | 34/20447 | 2/74 | 0.006725 | 0.046835 |
Case study 2: HCP5
The lncRNA HCP5 was found to be associated with AIDS [29-31]. Rodriguez-Novoa et al. analyzed a total of 245 HIV patients and found a good correlation between HLA-B*5701 and HCP5 (negative and positive predictive values of 100% and 93%, respectively). Colombo et al. analyzed that 1,103 singles infected with human immunodeficiency virus (HIV) and concluded that HCP5 genotyping could serve as a simple screening tool for ABC-HSR, particularly in settings where sequence-based HLA typing is not available.
To assess whether the HCP5 can be correctly predicted to have immune-related functions, we applied our LncRNA2Function to it and found that HCP5 was annotated with 549 GO biological processes terms and 270 biological pathways. As expected, most of them are indeed immune system and response functional terms, which are strongly associated with the development of AIDS. The top 20 GO biological terms assigned to the HCP5 are shown in Table 3 while the top 20 biological pathways assigned to the HCP5 are shown in Table 4.
Table 3.
The top 20 biological processes assigned to the AIDS-related lncRNA HCP5 by LncRNA2Function.
| GO term | Background frequency | Sample frequency | P-value | Corrected P-value |
|---|---|---|---|---|
| Immune system process | 1581/20447 | 208/458 | 1.0E-109 | 3.49E-105 |
| Immune response | 867/20447 | 148/458 | 1.57E-90 | 2.70E-86 |
| Defense response | 968/20447 | 144/458 | 2.37E-79 | 2.72E-75 |
| Regulation of immune system process | 879/20447 | 131/458 | 1.16E-71 | 9.95E-68 |
| Regulation of immune response | 527/20447 | 105/458 | 9.62E-70 | 6.62E-66 |
| Response to stimulus | 6195/20447 | 312/458 | 9.35E-64 | 5.36E-60 |
| Cell activation | 557/20447 | 89/458 | 3.11E-50 | 1.53E-46 |
| Leukocyte activation | 344/20447 | 73/458 | 5.00E-50 | 2.15E-46 |
| Regulation of response to stimulus | 2379/20447 | 173/458 | 1.23E-48 | 4.72E-45 |
| Positive regulation of immune system process | 522/20447 | 84/458 | 1.39E-47 | 4.77E-44 |
| Response to stress | 2747/20447 | 181/458 | 4.76E-45 | 1.26E-41 |
| Signal transduction | 3612/20447 | 205/458 | 4.25E-42 | 1.04E-38 |
| Positive regulation of immune response | 331/20447 | 63/458 | 4.17E-40 | 9.56E-37 |
| Cellular response to stimulus | 4596/20447 | 231/458 | 5.11E-40 | 1.10E-36 |
| Lymphocyte activation | 276/20447 | 58/458 | 1.81E-39 | 3.66E-36 |
| Innate immune response | 474/20447 | 72/458 | 6.13E-39 | 1.17E-35 |
| Positive regulation of response to stimulus | 1154/20447 | 106/458 | 6.43E-37 | 1.16E-33 |
| T cell activation | 176/20447 | 46/458 | 5.40E-36 | 9.28E-33 |
| Single organism signaling | 4081/20447 | 208/458 | 1.23E-35 | 1.92E-32 |
| Immune response-regulating signaling pathway | 218/20447 | 49/458 | 6.76E-35 | 1.01E-31 |
Table 4.
The top 20 pathways assigned to AIDS-related lncRNA HCP5 by our LncRNA2Function.
| Pahtway database |
Pahtway name | Background frequency | Sample frequency | P-value | Corrected P-value |
|---|---|---|---|---|---|
| Reactome | Immune System | 1177/20447 | 98/458 | 1.66E-30 | 2.14E-27 |
| KEGG | Natural killer cell mediated cytotoxicity | 219/20447 | 38/458 | 6.00E-23 | 3.87E-20 |
| PID | Immunoregulatory interactions between a Lymphoid and a non-Lymphoid cell | 104/20447 | 28/458 | 1.06E-22 | 4.54E-20 |
| Reactome | Immunoregulatory interactions between a Lymphoid and a non-Lymphoid cell | 202/20447 | 35/458 | 3.36E-21 | 1.08E-18 |
| Reactome | Adaptive Immune System | 772/20447 | 66/458 | 4.68E-21 | 1.21E-18 |
| NetPath | TCR | 252/20447 | 36/458 | 6.84E-19 | 1.47E-16 |
| KEGG | Chemokine signaling pathway | 195/20447 | 30/458 | 7.56E-17 | 1.39E-14 |
| PID | Generation of second messenger molecules | 15/20447 | 11/458 | 7.96E-16 | 1.28E-13 |
| KEGG | Osteoclast differentiation | 174/20447 | 27/458 | 2.20E-15 | 3.16E-13 |
| PID | TCR signaling in naive CD4+ T cells | 80/20447 | 19/458 | 1.03E-14 | 1.32E-12 |
| PID | TCR signaling in naive CD8+ T cells | 63/20447 | 17/458 | 2.63E-14 | 3.09E-12 |
| PID | IL12-mediated signaling events | 81/20447 | 18/458 | 1.80E-13 | 1.94E-11 |
| KEGG | Cytokine-cytokine receptor interaction | 291/20447 | 31/458 | 6.97E-13 | 6.42E-11 |
| Reactome | Innate Immune System | 542/20447 | 43/458 | 6.96E-13 | 6.42E-11 |
| KEGG | Hematopoietic cell lineage | 114/20447 | 20/458 | 9.26E-13 | 7.97E-11 |
| KEGG | T cell receptor signaling pathway | 116/20447 | 20/458 | 1.30E-12 | 1.05E-10 |
| Reactome | Cell surface interactions at the vascular wall | 104/20447 | 19/458 | 1.62E-12 | 1.23E-10 |
| PID | Cell surface interactions at the vascular wall | 42/20447 | 13/458 | 4.26E-12 | 3.06E-10 |
| Reactome | Class A/1 (Rhodopsin-like receptors) | 319/20447 | 31/458 | 7.85E-12 | 5.33E-10 |
| PID | Fc-epsilon receptor I signaling in mast cells | 64/20447 | 15/458 | 8.34E-12 | 5.38E-10 |
Case study 3: HULC
The lncRNA HULC is highly upregulated in liver cancer and plays an important role in tumorigenesis [32]. Depletion of HULC resulted in a significant deregulation of several genes involved in liver cancer [33], and colorectal carcinomas that metastasize to the livers but not to lymph nodes experience an up-regulation of HULC in all the samples tested (n = 8), with a strong-to-moderate expression in six out of eight [34].
To examine whether the HULC was predicted to have liver-related functions, we analyzed it using our lncRNA2Function. Expectedly, LncRNA2Function also works well to functionally annotate the HULC. The results showed that it was annotated with 373 GO biological processes and 383 biological pathways (the significant P-value cutoff is 0.05). Of the 373 GO biological processes and 383 pathways, over 80% are involved in the known liver-related biological functions, such as metabolic function, bile secretion, lipid transport and homeostasis, cholesterol homeostasis, regulation of blood coagulation, protein-lipid complex subunit organization, detoxification, Immune defense and complement activation. The Figure 2 shows the top 25 GO functional terms assigned to the HULC, and the Table 5 shows the top 20 pathways enriched in protein-coding genes that are co-expressed with the liver-related lncRNA HULC.
Figure 2.
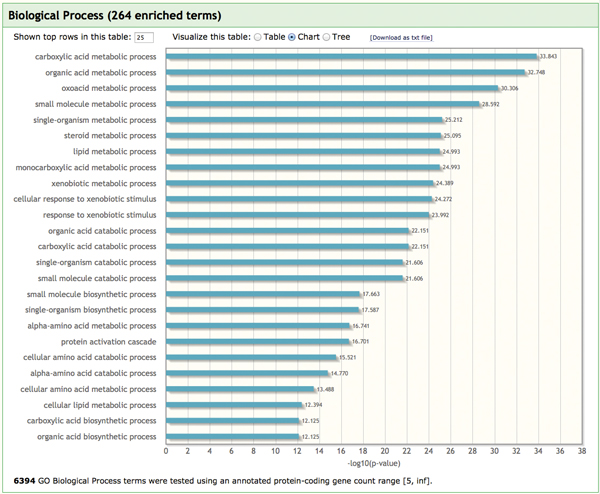
The top 25 statistically significant enriched GO biological processes assigned to liver-related HULC are associated with the metabolic function of liver.
Table 5.
Top 20 pathways enriched in protein-coding genes that are co-expressed with the liver-related lncRNA HULC.
| Pahtway database |
Pahtway name | Background frequency | Sample frequency | P-value | Corrected P-value |
|---|---|---|---|---|---|
| Reactome | Metabolism | 1394/20447 | 128/390 | 6.17E-54 | 6.23E-51 |
| KEGG | Metabolic pathways | 1256/20447 | 109/390 | 7.95E-43 | 4.01E-40 |
| KEGG | Retinol metabolism | 69/20447 | 29/390 | 5.66E-32 | 1.91E-29 |
| KEGG | Complement and coagulation cascades | 87/20447 | 30/390 | 6.18E-30 | 1.56E-27 |
| EHMN | Androgen and estrogen biosynthesis and metabolism | 90/20447 | 30/390 | 2.02E-29 | 4.07E-27 |
| Reactome | Biological oxidations | 151/20447 | 36/390 | 2.71E-29 | 4.55E-27 |
| KEGG | Drug metabolism - cytochrome P450 | 76/20447 | 28/390 | 5.84E-29 | 8.41E-27 |
| KEGG | Metabolism of xenobiotics by cytochrome P450 | 87/20447 | 29/390 | 1.78E-28 | 2.24E-26 |
| KEGG | Chemical carcinogenesis | 86/20447 | 28/390 | 3.33E-27 | 3.74E-25 |
| EHMN | Tyrosine metabolism | 113/20447 | 30/390 | 4.29E-26 | 4.33E-24 |
| EHMN | Xenobiotics metabolism | 54/20447 | 23/390 | 9.33E-26 | 8.56E-24 |
| Reactome | Metabolism of amino acids and derivatives | 159/20447 | 32/390 | 1.09E-23 | 9.20E-22 |
| Wikipathways | metapathway biotransformation | 190/20447 | 34/390 | 2.43E-23 | 1.88E-21 |
| EHMN | Linoleate metabolism | 75/20447 | 24/390 | 2.81E-23 | 2.03E-21 |
| Wikipathways | cytochrome P450 | 68/20447 | 23/390 | 5.40E-23 | 3.63E-21 |
| Wikipathways | Complement and Coagulation Cascades | 60/20447 | 22/390 | 5.97E-23 | 3.77E-21 |
| EHMN | Leukotriene metabolism | 102/20447 | 26/390 | 2.87E-22 | 1.70E-20 |
| Reactome | Phase 1 - Functionalization of compounds | 74/20447 | 23/390 | 5.01E-22 | 2.81E-20 |
| KEGG | Drug metabolism - other enzymes | 53/20447 | 20/390 | 2.85E-21 | 1.51E-19 |
| Reactome | Xenobiotics | 15/20447 | 13/390 | 3.68E-21 | 1.86E-19 |
Case study 4: H19
H19 is an important lncRNA that play roles in the infertility [35] and multiple cancers such as breast cancer [36,37], cervical cancer [38], liver cancer [39,40] and bladder cancer [41]. For example, Korucuoglu et al. revealed that H19 expression was lower in the infertility group as compared to the control group (4-fold change, P < 0.0001), and Lottin et al. showed that over-expression of H19 transcript is associated with cells exhibiting higher tumorigenic phenotypes and promotes tumor progression.
We applied the LncRNA2Function to the lncRNA H19 and found that it was annotated with 6 GO biological processes and 31 biological pathways. The GO terms includes female pregnancy (GO: 0007565), estrogen biosynthetic process (GO:0006703), growth hormone receptor signaling pathway (GO:0060396), cellular response to growth hormone stimulus (GO:0071378) and JAK-STAT cascade involved in growth hormone signaling pathway (GO:0060397), which suggest that H19 may play roles in infertility or breast cancer by participating in these biological processes. In addition, the cancer-related lncRNA H19 was correctly annotated with many important caner pathways, such as PI3K-Akt signaling pathway, GPCR signaling-G alpha s Epac and ERK pathway, Nuclear signaling by ERBB4 pathway, Akt signaling pathway and JAK-STAT-Core cancer pathway. These results suggest that our LncRNA2Function correctly recall the known functions of H19.
Case study 5: PCA3
The lncRNA prostate cancer antigen 3 (PCA3) is a highly specific biomarker upregulated and plays crucial roles in prostate cancer (PCa) [42-45]. Clarke et al. found that up-regulation of two new PCA3 isoforms in PCa tissues improves discrimination between PCa and benign prostatic hyperplasia (BPH). In 2012, the US Food and Drug Administration approved the use of the lncRNA PCA3 for the detection of prostate cancer.
To test whether our LncRNA2Function can annotate the PCA3 with prostate-related functions, we applied the LncRNA2Function to the PCA3. LncRNA2Function first identified 77 protein-coding genes that are co-expressed with the PCA3 and then annotated it with only one pathway named 'Regulation of Androgen receptor activity' (corrected P-value: 0.020385). This pathway has 62 genes, which includes 4 protein-coding genes that are co-expressed with the PCA3. These four genes are HOXB13, KLK3, KLK2 and SPDEF that have been validated to be useful in the diagnosis and monitoring of prostatic carcinoma and be suitable target for developing specific cancer therapies. Consequently, lncRNA2Function can correctly predict the functions of PCA3 by its co-expressed protein-coding genes.
Functional annotation for a set of human lncRNAs
High-throughput genomic technologies like lncRNA microarray and RNA-Seq usually generate hundreds of candidate lncRNA genes of interest, such as a cluster of co-expressed lncRNA genes across multiple conditions or a set of differentially expressed lncRNAs between cancer and normal samples. To manually map each lncRNA to functional terms is by far a simple task. Therefore, how to identify significantly enriched functions among the set of lncRNAs is an important downstream task for interpreting high-throughput experimental data.
As a proof-of-concept, a set of liver-specific lncRNAs and a set of heart-specific lncRNAs inferred from RNA-Seq data of 19 human normal tissues were used as examples to show the functionality of our lncRNA2Function system in annotating a set of lncRNAs of interest, respectively. As expected, lncRNA2Function correctly assigned the functional terms to the two distinct sets of lncRNAs. Users can test these two sets or their own lncRNA sets at our 'LncRNA set analyzer' web interface http://mlg.hit.edu.cn/lncrna2function/lncrna_enrich.jsp.
Web interface of LncRNA2Function
To facilitate researchers to access the functional annotations of lncRNA genes, we developed a web interface named 'LncRNA annotation browser', which is a user-friendly interface to browse or search lncRNAs associated with a specific functional term, or functional terms associated with a given lncRNA. To enable researchers to analyze a set of lncRNA genes of their interest, we implemented a web interface titled 'LncRNA set analyzer', which can help investigators to annotate a set of lncRNAs with Gene Ontology and 4,380 biological pathways curated from 12 pathway databases. In addition, we developed a web interface titled 'LncRNA expression viewer' to facilitate investigators to graphically view the expression dynamics of genes across multiple human normal tissues. Users can not only view expression value of a single lncRNA or protein-coding gene across 19 human normal tissues, but also simultaneously view the expression index of both lncRNA and protein-coding genes to learn about whether they are co-expressed across the 19 tissues. Furthermore, we provide a submission page that allows other researchers to submit known functional annotations of lncRNAs that are not documented in our LncRNA2Function system (Figure 3). They do not have to be an author on the original study to submit a record. Once approved by the submission review committee, the submitted records will be made available to the public in the coming release. LncRNA2Function is freely accessible at http://mlg.hit.edu.cn/lncrna2function.
Figure 3.
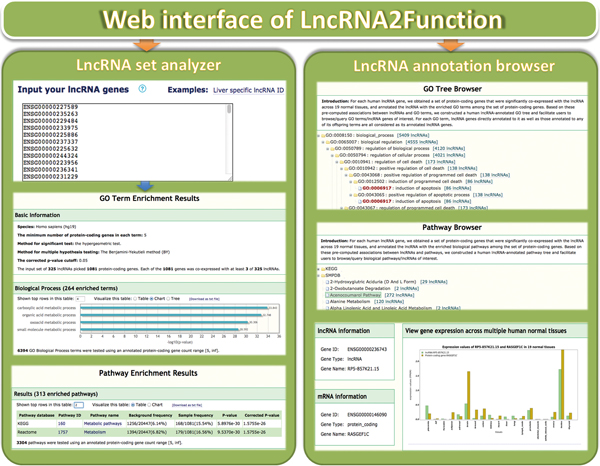
Screenshot of web interface of LncRNA2Function.
Conclusions
Thousands of human lncRNAs have been identified in recent several years, while the vast majority of the lncRNAs remain to be functionally characterized. In this study, we functionally annotate 9,625 human lncRNAs with the enriched functions among the protein-coding genes that are co-expressed with each lncRNA. Furthermore, we developed a web interface, which facilitates researchers to search the functions of a specific lncRNA or the lncRNAs associated with a given functional term, or annotate functionally a set of human lncRNAs of interest. The lncRNA2Function will become an important tool for investigating functions of human lncRNAs.
Competing interests
The authors declare that they have no competing interests
Authors' contributions
YW and YL conceived and designed the experiments. QJ, RM, XW, SJ, TZ, RT and JP performed the experiments and analyzed the data. QJ and JW designed and developed the web interface. QJ, YW and YL wrote the paper.
Contributor Information
Qinghua Jiang, Email: qhjiang@hit.edu.cn.
Rui Ma, Email: ruima@hit.edu.cn.
Jixuan Wang, Email: coderChase@gmail.com.
Xiaoliang Wu, Email: wuxiaoliang@hit.edu.cn.
Shuilin Jin, Email: jinsl@hit.edu.cn.
Jiajie Peng, Email: jiajiepeng@hit.edu.cn.
Renjie Tan, Email: 11b903027@hit.edu.cn.
Tianjiao Zhang, Email: ztj.hit@gmail.com.
Yu Li, Email: liyugene@hit.edu.cn.
Yadong Wang, Email: ydwang@hit.edu.cn.
Acknowledgements
The Natural Science Foundation of China (NSFC) [61102149, 61173085], Fundamental Research Funds for the Central Universities [HIT NSRIF. 2010057, HIT BRETIII.201219] and the China National 863 High-Tech Program (2012AA02A602, 2012AA020404 and 2012AA02A601). Funding for open access publication: NSFC [61102149]
This article has been published as part of BMC Genomics Volume 16 Supplement 3, 2015: Selected articles from the 10th International Symposium on Bioinformatics Research and Applications (ISBRA-14): Genomics. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcgenomics/supplements/16/S3.
References
- Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK, Munson G, Young G, Lucas AB, Ach R, Bruhn L. et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature. 2011;477(7364):295–300. doi: 10.1038/nature10398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova N, Zamudio JR, Jong RM, Soukup D, Resnick R, Sarma K, Ward AJ, Raj A, Lee JT, Sharp PA. et al. LincRNA-p21 activates p21 in cis to promote Polycomb target gene expression and to enforce the G1/S checkpoint. Molecular cell. 2014;54(5):777–790. doi: 10.1016/j.molcel.2014.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Wang Z, Wang D, Qiu C, Liu M, Chen X, Zhang Q, Yan G, Cui Q. LncRNADisease: a database for long-non-coding RNA-associated diseases. Nucleic acids research. 2013;41(D1):D983–986. doi: 10.1093/nar/gks1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnhill LM, Williams RT, Cohen O, Kim Y, Batova A, Mielke JA, Messer K, Pu M, Bao L, Yu AL. et al. High Expression of CAI2, a 9p21-Embedded Long Noncoding RNA, Contributes to Advanced-Stage Neuroblastoma. Cancer research. 2014;74(14):3753–3763. doi: 10.1158/0008-5472.CAN-13-3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter S, Aiello D, Atianand MK, Ricci EP, Gandhi P, Hall LL, Byron M, Monks B, Henry-Bezy M, Lawrence JB. et al. A long noncoding RNA mediates both activation and repression of immune response genes. Science. 2013;341(6147):789–792. doi: 10.1126/science.1240925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Goff LA, Trapnell C, Alexander R, Lo KA, Hacisuleyman E, Sauvageau M, Tazon-Vega B, Kelley DR, Hendrickson DG. et al. Long noncoding RNAs regulate adipogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(9):3387–3392. doi: 10.1073/pnas.1222643110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nature reviews Genetics. 2009;10(3):155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Molecular cell. 2011;44(4):667–678. doi: 10.1016/j.molcel.2011.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon MD, Wang CI, Kharchenko PV, West JA, Chapman BA, Alekseyenko AA, Borowsky ML, Kuroda MI, Kingston RE. The genomic binding sites of a noncoding RNA. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(51):20497–20502. doi: 10.1073/pnas.1113536108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Hsu AK, Sajdak J, Qin J, Pavlidis P. Coexpression analysis of human genes across many microarray data sets. Genome research. 2004;14(6):1085–1094. doi: 10.1101/gr.1910904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(25):14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP. et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458(7235):223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Gao L, Liao Q, Xiao H, Ma X, Yang X, Luo H, Zhao G, Bu D, Jiao F. et al. Long non-coding RNAs function annotation: a global prediction method based on bi-colored networks. Nucleic acids research. 2013;41(2):e35. doi: 10.1093/nar/gks967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Q, Liu C, Yuan X, Kang S, Miao R, Xiao H, Zhao G, Luo H, Bu D, Zhao H. et al. Large-scale prediction of long non-coding RNA functions in a coding-non-coding gene co-expression network. Nucleic acids research. 2011;39(9):3864–3878. doi: 10.1093/nar/gkq1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome research. 2012;22(9):1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamburov A, Stelzl U, Lehrach H, Herwig R. The ConsensusPathDB interaction database: 2013 update. Nucleic acids research. 2013. pp. D793–800. [DOI] [PMC free article] [PubMed]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome biology. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature biotechnology. 2010;28(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini YaY D. the control of false discovery rate in multiple testing under dependency. Ann Stat. 2001;29:1165–1168. doi: 10.1214/aos/1013699998. [DOI] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B. 1995. pp. 289–300.
- Shaffer JP. Controlling the false discovery rate with constraints: the Newman-Keuls test revisited. Biom J. 2007;49:136–143. doi: 10.1002/bimj.200610297. [DOI] [PubMed] [Google Scholar]
- Qureshi IA, Mehler MF. Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nature reviews Neuroscience. 2012;13(8):528–541. doi: 10.1038/nrn3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Yuan B, Flygare J, Lodish HF. Long noncoding RNA-mediated anti-apoptotic activity in murine erythroid terminal differentiation. Genes & development. 2011;25(24):2573–2578. doi: 10.1101/gad.178780.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Abdelmohsen K, Srikantan S, Yang X, Martindale JL, De S, Huarte M, Zhan M, Becker KG, Gorospe M. LincRNA-p21 suppresses target mRNA translation. Molecular cell. 2012;47(4):648–655. doi: 10.1016/j.molcel.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL. et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464(7291):1071–1076. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E. et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 2007;129(7):1311–1323. doi: 10.1016/j.cell.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niinuma T, Suzuki H, Nojima M, Nosho K, Yamamoto H, Takamaru H, Yamamoto E, Maruyama R, Nobuoka T, Miyazaki Y. et al. Upregulation of miR-196a and HOTAIR drive malignant character in gastrointestinal stromal tumors. Cancer research. 2012;72(5):1126–1136. doi: 10.1158/0008-5472.CAN-11-1803. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Novoa S, Cuenca L, Morello J, Cordoba M, Blanco F, Jimenez-Nacher I, Soriano V. Use of the HCP5 single nucleotide polymorphism to predict hypersensitivity reactions to abacavir: correlation with HLA-B*5701. The Journal of antimicrobial chemotherapy. 2010;65(8):1567–1569. doi: 10.1093/jac/dkq204. [DOI] [PubMed] [Google Scholar]
- Colombo S, Rauch A, Rotger M, Fellay J, Martinez R, Fux C, Thurnheer C, Gunthard HF, Goldstein DB, Furrer H. et al. The HCP5 single-nucleotide polymorphism: a simple screening tool for prediction of hypersensitivity reaction to abacavir. The Journal of infectious diseases. 2008;198(6):864–867. doi: 10.1086/591184. [DOI] [PubMed] [Google Scholar]
- Yoon W, Ma BJ, Fellay J, Huang W, Xia SM, Zhang R, Shianna KV, Liao HX, Haynes BF, Goldstein DB. A polymorphism in the HCP5 gene associated with HLA-B*5701 does not restrict HIV-1 in vitro. AIDS. 2010;24(1):155–157. doi: 10.1097/QAD.0b013e32833202f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Liu X, Wu H, Ni P, Gu Z, Qiao Y, Chen N, Sun F, Fan Q. CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic acids research. 2010;38(16):5366–5383. doi: 10.1093/nar/gkq285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panzitt K, Tschernatsch MM, Guelly C, Moustafa T, Stradner M, Strohmaier HM, Buck CR, Denk H, Schroeder R, Trauner M. et al. Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology. 2007;132(1):330–342. doi: 10.1053/j.gastro.2006.08.026. [DOI] [PubMed] [Google Scholar]
- Matouk IJ, Abbasi I, Hochberg A, Galun E, Dweik H, Akkawi M. Highly upregulated in liver cancer noncoding RNA is overexpressed in hepatic colorectal metastasis. European journal of gastroenterology & hepatology. 2009;21(6):688–692. doi: 10.1097/MEG.0b013e328306a3a2. [DOI] [PubMed] [Google Scholar]
- Korucuoglu U, Biri AA, Konac E, Alp E, Onen IH, Ilhan MN, Turkyilmaz E, Erdem A, Erdem M, Menevse S. Expression of the imprinted IGF2 and H19 genes in the endometrium of cases with unexplained infertility. European journal of obstetrics, gynecology, and reproductive biology. 2010;149(1):77–81. doi: 10.1016/j.ejogrb.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Lottin S, Adriaenssens E, Dupressoir T, Berteaux N, Montpellier C, Coll J, Dugimont T, Curgy JJ. Overexpression of an ectopic H19 gene enhances the tumorigenic properties of breast cancer cells. Carcinogenesis. 2002;23(11):1885–1895. doi: 10.1093/carcin/23.11.1885. [DOI] [PubMed] [Google Scholar]
- Berteaux N, Aptel N, Cathala G, Genton C, Coll J, Daccache A, Spruyt N, Hondermarck H, Dugimont T, Curgy JJ. et al. A novel H19 antisense RNA overexpressed in breast cancer contributes to paternal IGF2 expression. Molecular and cellular biology. 2008;28(22):6731–6745. doi: 10.1128/MCB.02103-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douc-Rasy S, Barrois M, Fogel S, Ahomadegbe JC, Stehelin D, Coll J, Riou G. High incidence of loss of heterozygosity and abnormal imprinting of H19 and IGF2 genes in invasive cervical carcinomas. Uncoupling of H19 and IGF2 expression and biallelic hypomethylation of H19. Oncogene. 1996;12(2):423–430. [PubMed] [Google Scholar]
- Song JY, Lee JH, Joe CO, Lim DS, Chung JH. Retrotransposon-specific DNA hypomethylation and two-step loss-of-imprinting during WW45 haploinsufficiency-induced hepatocarcinogenesis. Biochemical and biophysical research communications. 2011;404(2):728–734. doi: 10.1016/j.bbrc.2010.12.052. [DOI] [PubMed] [Google Scholar]
- Matouk IJ, DeGroot N, Mezan S, Ayesh S, Abu-lail R, Hochberg A, Galun E. The H19 non-coding RNA is essential for human tumor growth. PloS one. 2007;2(9):e845. doi: 10.1371/journal.pone.0000845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaegh GW, Verkleij L, Vermeulen SH, den Heijer M, Witjes JA, Kiemeney LA. Polymorphisms in the H19 gene and the risk of bladder cancer. European urology. 2008;54(5):1118–1126. doi: 10.1016/j.eururo.2008.01.060. [DOI] [PubMed] [Google Scholar]
- Clarke RA, Zhao Z, Guo AY, Roper K, Teng L, Fang ZM, Samaratunga H, Lavin MF, Gardiner RA. New genomic structure for prostate cancer specific gene PCA3 within BMCC1: implications for prostate cancer detection and progression. PloS one. 2009;4(3):e4995. doi: 10.1371/journal.pone.0004995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klecka J, Holubec L, Pesta M, Topolcan O, Hora M, Eret V, Finek J, Chottova-Dvorakova M, Babjuk M, Novak K. et al. Differential display code 3 (DD3/PCA3) in prostate cancer diagnosis. Anticancer research. 2010;30(2):665–670. [PubMed] [Google Scholar]
- Tinzl M, Marberger M, Horvath S, Chypre C. DD3PCA3 RNA analysis in urine--a new perspective for detecting prostate cancer. European urology. 2004;46(2):182–186. doi: 10.1016/j.eururo.2004.06.004. discussion 187. [DOI] [PubMed] [Google Scholar]
- Haese A, de la Taille A, van Poppel H, Marberger M, Stenzl A, Mulders PF, Huland H, Abbou CC, Remzi M, Tinzl M. et al. Clinical utility of the PCA3 urine assay in European men scheduled for repeat biopsy. European urology. 2008;54(5):1081–1088. doi: 10.1016/j.eururo.2008.06.071. [DOI] [PubMed] [Google Scholar]