

Abstract
Background
In patients with metastatic melanoma and KIT amplifications and/or mutations, therapy with imatinib mesylate may prolong survival. 18F-labeled 2-fluoro-2-deoxy-D-glucose (18F-FDG) PET/CT may be used to assess metabolic response. We investigated associations of metabolic response, mutational status, progression-free survival and overall survival in this population.
Methods
Baseline and 4-week follow-up 18F-FDG-PET/CT were evaluated in 17 patients with metastatic melanoma and KIT amplifications and/or mutations treated with imatinib in a multicenter phase II clinical trial. The maximum standardized uptake values (SUVmax) were measured in up to 10 lesions on each scan. Metabolic response was classified using modified EORTC criteria. Each patient had a diagnostic CT or MR at baseline, after 6 weeks of therapy and then at intervals of 2 months and anatomic response was classified using RECIST 1.0. Median follow-up was 9.8 months.
Results
Partial metabolic response (PMR), stable metabolic disease (SMD) and progressive metabolic disease (PMD) was seen in 5 (29%), 5 (29%), and 7 (41%) patients respectively. Five patients (29%) had a KIT mutation in exon 11, four of whom (80%) had PMR while 1 (20%) had SMD. Twelve patients (71%) did not have a KIT mutation in exon 11, and only 1 (8%) had PMR, 4 (33%) had SMD and 7 (58%) had PMD. There was agreement of metabolic and anatomic classification in 12 of 17 patients (71%). Four of 17 patients (24%) had PR on both metabolic and anatomic imaging and all had a KIT mutation in exon 11. Survival of patients with PMD was lower than with SMD or PMR.
Conclusions
Metabolic response by 18F-FDG-PET/CT is associated with mutational status in metastatic melanoma patients treated with imatinib. 18F-FDG-PET/CT may be a predictor of outcome, although a larger study is needed to verify this.
Clinical trial registration
Keywords: Melanoma, KIT mutation, 18F-FDG-PET/CT, Imatinib
Background
Malignant melanoma is a spectrum of diseases associated with several genetic aberrations. Mucosal and acral melanoma arising from sites such as the palms, soles and nail beds as well as cutaneous melanoma arising from chronically sun-damaged skin are forms of the disease with different but overlapping oncogenes and biologic behavior [1]-[5] that have recently been found to harbor KIT mutations [6]-[10]. The majority of these mutations occur in exon 11 at the juxtamembrane portion of the protein, which predicts responsiveness to imatinib mesylate [11].
Imatinib mesylate (Gleevec) is a protein-tyrosine kinase inhibitor that inhibits platelet-derived growth factor and stem cell factor mediated cellular processes. It can also inhibit proliferation and induce apoptosis in Bcr-Abl positive cells or cells with a KIT mutation including melanoma [12]. In the past, imatinib mesylate has been successfully used to treat patients with gastrointestinal stromal tumor (GIST), where malignancy is associated with a KIT mutation [13]-[15]. Imaging in patients with GIST often includes 18F-labeled 2-fluoro-2-deoxy-D-glucose (18F-FDG) positron emission tomography/computed tomography (PET/CT), which can show metabolically active disease and may be more effective for evaluating therapeutic response to tyrosine kinase inhibition than anatomic imaging [16]-[20]. Further, changes in tumor metabolism may predict outcome.
In GIST, the majority of KIT mutations occur in exon 11 near the N-terminus. In contrast, melanoma patients have a higher prevalence of KIT mutations in exon 11 near the C-terminus as well as KIT mutations in exon 13 and exon 17, which may confer resistance to imatinib therapy [7]. Although preliminary results in the literature suggest imatinib therapy may be helpful for the treatment of melanoma [21], this may not be the case for all patients with melanoma despite the presence of a KIT mutation. In particular, recent studies by Woodman et al. and Antonescu et al. suggest that the L576P mutation in exon 11, the most common KIT mutation in melanoma, may induce structural changes resulting in resistance to imatinib therapy compared with other tyrosine kinase inhibitors such as dasatinib [22]-[24].
We previously suggested that imatinib can be effective when melanoma tumors harbor KIT mutations [25]. The aim of this study was to provide a detailed sub-analysis of the 18F-FDG-PET/CT data acquired during the course of the previously reported clinical trial [25]. Specifically, our aim was to assess the metabolic response of patients with metastatic melanoma and a KIT mutation and/or amplification treated with imatinib using 18F-FDG-PET/CT and to investigate the association of 18F-FDG uptake with mutational status, time-to-progression (TTP) and overall survival (OS).
Methods
Study description
The institutional review boards of the sites participating in a phase II multicenter clinical trial of imatinib in metastatic melanoma with KIT amplifications and/or mutations approved the study before patient enrollment and continuing approval was maintained throughout the study (This clinical trial was approved by the Dana-Farber/Harvard Cancer Center IRB).
Prior to enrollment in the trial, KIT mutational status was determined by tumor biopsy, followed by polymerase chain reaction, high performance liquid chromatography and DNA sequencing, with amplicons arising from exons 9, 11, 13 and 17. KIT gene amplification was assessed by quantitative polymerase chain reaction, where the threshold for increased KIT copy number relative to normal tissue was established using the 95% confidence level according to Chebychev’s inequality and the threshold for positive was 5.29 copies of KIT relative to GAPDH. KIT gene copy number was measured in relationship to a chromosome 2 peri-centromeric probe, corresponding to a region rarely subject to gain or loss in melanoma. A sample (or histologically distinctive region) was scored as amplified if the ratio of probe/centromere was >1.5 (highly-amplified >5.0). Oncogene mutation screened was also done on pre-treatment tissue in all patients using a mass spectroscopy system (Sequenom, San Diego, CA) and a panel of 643 hotspot mutations across 53 cancer genes.
Seventeen patients had baseline and follow-up 18F-FDG-PET/CT after one month of therapy. Metabolic tumor response was classified using modified European Organisation for Research and Treatment of Cancer (EORTC) criteria [26]. Anatomic tumor response was classified according to the best response achieved using Response Evaluation Criteria in Solid Tumors (RECIST) 1.0 applied to diagnostic CT or MR images obtained after 6 weeks of therapy and then at intervals of 2 months. The clinical trial methods have been previously described [25]. Choi criteria was not performed as the CT scans were acquired according to each institution’s standard of care in this multicenter trial with no standardization of the dosage and timing of contrast media [17].
PET/CT acquisition and analysis
Patients fasted for 6 hours prior to the 18F-FDG-PET/CT. Each 18F-FDG-PET/CT was acquired approximately 60 minutes (mean: 62 min, range: 56 min – 70 min) after the intravenous administration of approximately 20 mCi 18F-FDG (mean: 20.1 mCi, range 17.6 mCi – 22.1 mCi). Whole-body imaging was performed from the skull vertex through the toes using a combined PET/CT scanner with images corrected for detector efficiency, attenuation, scatter, decay and random coincidences. Consistency was maintained across participating institutions by adherence to a standard quality assurance program at each site in accordance with NCI consensus guidelines [27]. All 18F-FDG-PET/CT images acquired were transferred to the Dana-Farber Cancer Institute after de-identification for central quality control and analysis. Two nuclear medicine physicians evaluated each study for extent and change of 18F-FDG-avid disease without knowledge of the mutational analysis, RECIST classification, or outcome. Final results were based on consensus. For each subject, up to 10 target lesions with the greatest FDG-avidity were identified on the baseline scans and analyzed on both the baseline and follow-up studies. Tumor FDG uptake was quantified using the maximum standardized uptake value (SUVmax). For each subject, the SUVmax of all target lesions was summed at each time point and the relative change in the summed SUVmax was used to assess metabolic response where: partial metabolic response (PMR) ≤ −25% < stable metabolic disease (SMD) < +25% ≤ progressive metabolic disease (PMD). The appearance of a new FDG-avid lesion in the post-treatment scans supersedes the relative change and results in PMD. Conversely, the complete resolution of all FDG-avid lesions in the post-treatment scans results in complete metabolic response (CMR).
Statistical methods
The best overall response rate was defined as the proportion of patients with at least partial response as best response to therapy. The disease-control rate was defined as the proportion of patients who achieved complete response, partial response, or stable disease lasting at least 12 weeks as their best response to therapy. The agreement of response- and disease-control rate classifications based on metabolic response and RECIST 1.0 best overall response was conducted using McNemar’s test. The relationship between overall metabolic response and mutational status was explored using Fisher’s exact test. As there was not sufficient documentation of the location of biopsied lesions in this multi-center study, the overall metabolic response of the patient based on all lesions was compared to mutational status rather than the individual response of the biopsied lesion.
Time-to-progression (TTP) was defined as the time interval from study enrollment to the date of first documented disease progression. The follow-up of patients who died without disease progression was censored at the date of death; follow-up of patients who had not progressed at the time of the analysis was censored at the date of the last study assessment. Overall survival (OS) was defined as the time from study enrollment to death from any cause. The follow-up of patients who were alive at the time of data analysis was censored at the last date of vital status.
The distributions of TTP and OS are presented using the method of Kaplan-Meier, with point-wise, 95% confidence intervals estimated using log(−log(survival)) methodology. Comparisons of TTP and OS according to type of melanoma or mutational status were made using the log-rank test. To avoid bias, comparisons of TTP and OS according to metabolic response classification were conducted using one-month conditional landmark analyses [28]. Patients who were alive and progression-free at the time of the one-month metabolic tumor assessment were followed forward in time; patients with rapid anatomic progression of disease within one month were removed. Thus, 3 patients with rapid, anatomic progression prior to the one-month 18F-FDG-PET/CT were removed from this portion of the analysis. TTP and OS were then compared according to metabolic response categories using the log-rank test. Statistical significance was defined as p < 0.05; there were no corrections for multiple comparisons.
Results
Nine medical centers participated in the phase II clinical trial between July 6, 2006 and March 1, 2011. Seventeen patients had baseline and follow-up 18F-FDG-PET/CT. Of the 17 patients with 18F-FDG-PET/CT included in the study, 7/17 (41%) had a KIT mutation, 5 of which were in exon 11 and 2 of which were in exon 13 or 17, while 10/17 (59%) had KIT amplification (Table 1). When there was both a KIT mutation and amplification, patients were classified as having a mutation for the purposes of analysis. At the time of enrollment, all patients had metastatic disease and a history of cancer-directed surgery. The median TTP was 4.7 months (95% CI: 1.3 to 7.4 months) and was independent of melanoma type (log-rank p = 0.98). TTP was similar for patients with a KIT mutation, or amplification (log-rank p = 0.33). Median OS was 12.9 months for patients with a KIT mutation in exon 11 (95% CI: 5.5 to ∞), 11.7 months for patients with a KIT gene amplification (95% CI: 2.8 to 16.2) and 10.4 months for patients with another type of mutation (95% CI: 1.5 to 19.3). Interestingly, the one patient with a KIT mutation in exon 17 did comparatively well with OS of 19.3 months (Table 1) while the 3 patients with the most rapid progression had amplification (TTP 1.0 and 0.9 months) and a KIT mutation in exon 13 (TTP 0.9 months).
Table 1.
Patient characteristics and response assessment
| Sex | Age (yrs) | Genetic status | TTP (mo) | Survival (mo) | Metabolic response (modified EORTC) | Anatomic response (RECIST 1.0) |
|---|---|---|---|---|---|---|
| Female |
79 |
Exon 11 insertion PYD577-582 |
4.7 |
12.9 |
PMR |
PR |
| Female |
61 |
Exon 17 D820Y |
10.2 |
19. 3 |
SMD |
PR |
| Female |
69 |
Amplified |
7.4 |
8.8 |
SMD |
PD |
| Male |
57 |
Amplified |
5.0 |
11.5 |
PMD |
PD |
| Female |
62 |
Amplified |
2.6 |
11.9 |
SMD |
PD |
| Male |
69 |
Amplified |
5.8 |
16.2 |
PMR |
PD |
| Female |
81 |
Amplified |
1.0 |
3.8 |
PMD |
PD |
| Female |
72 |
Amplified |
1.3 |
9.8 |
PMD |
PD |
| Female |
55 |
Amplified |
10.6 |
18.3 |
SMD |
SD |
| Female |
59 |
Amplified |
0.9 |
2.8 |
PMD |
PD |
| Female |
59 |
Exon 11 L576P |
27.1 |
27.1 |
PMR |
PR |
| Female |
75 |
Amplified |
5.6 |
12.5 |
PMD |
SD |
| Female |
47 |
Exon 11 L576P |
2.6 |
7.3 |
PMR |
PR |
| Male |
84 |
Amplified |
1.6 |
21.8 |
PMD |
PD |
| Female |
53 |
Exon 11 deletion WKVVE557-560 |
10.6 |
24.3 |
SMD |
SD |
| Male |
66 |
Exon 13 K642E |
0.9 |
1.5 |
PMD |
PD |
| Female | 66 | Exon 11 L576P | 3.4 | 5.5 | PMR | PR |
PMR = Partial Metabolic Response, SMD = Stable Metabolic Disease, PMD = Progressive Metabolic Disease, PR = Partial Response, SD = Stable Disease, PD = Progressive Disease, TTP = Time to Progression.
Figure 1 shows waterfall plots of metabolic response by lesion and patient using 18F-FDG-PET/CT. PMR was seen in 5 of 17 patients (29%), SMD was seen in 5 of 17 patients (29%), and PMD was seen in 7 of 17 patients (41%). Figure 2 shows illustrative images of a patient with both PMR and anatomic PR compared with a patient with both PMD and anatomic PD after 1 month of therapy. There was agreement of metabolic and anatomic response classification in 12 of 17 patients (71%, 95% CI: 44% to 90%). For the 5 patients where metabolic and anatomic classification did not agree, the response to therapy using RECIST 1.0 and anatomic imaging after 6 weeks of therapy was slightly more likely to show PD than metabolic imaging obtained after 4 weeks of therapy.
Figure 1.
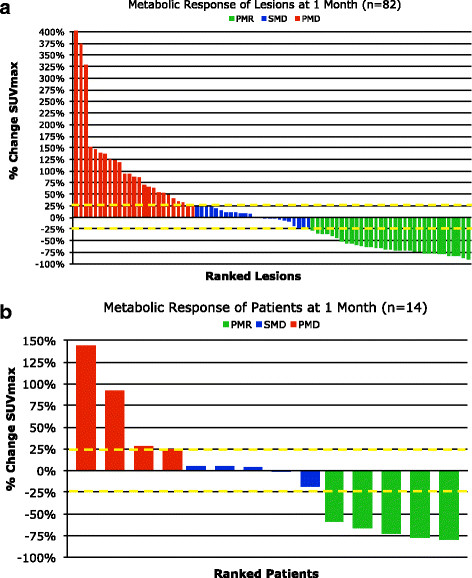
Waterfall plots of metabolic response by lesion (a) and patient (b). Three subjects were classified as PMD based on the presence of new lesions rather than % change SUVmax & have not been included in the waterfall plot above.
Figure 2.
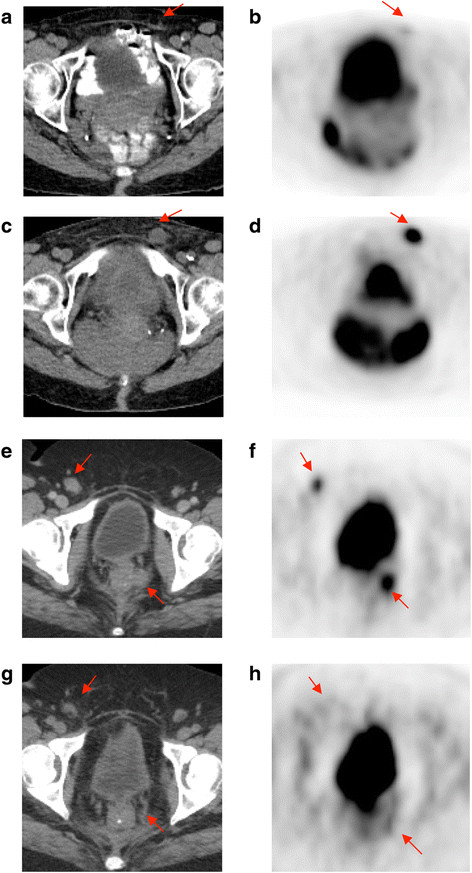
Representative18F-FDG-PET/CT images showing progression in a patient without an exon 11 KIT mutation (a-d) and response in a patient with an exon 11 KIT mutation (e-h) at baseline (a-b, e-f) and after 1 month of therapy (c-d, g-h). Red arrows point to sites of disease. Reprinted with permission. © (2013) American Society of Clinical Oncology. All rights reserved. From: Hodi S, et al. J Clin Oncol. 31(26), 2013:3182-3190.
Figure 3 shows the conditional landmark analyses of TTP and OS by metabolic response. Patients classified with PMR or SMD had significantly longer TTP than those with PMD (6.6 months vs. 3.3 months; log-rank p = 0.04). However, there was no evidence of a difference in overall survival (log-rank p = 0.53).
Figure 3.
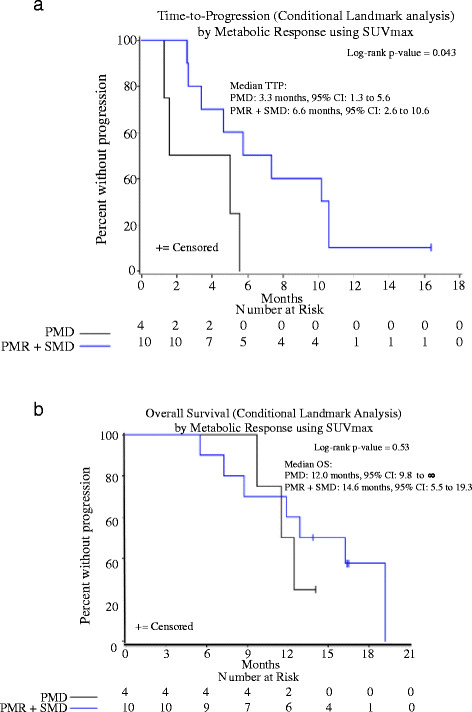
Time-to-progression (a) and overall survival (b) by metabolic response (Met).
There was an association between metabolic response and KIT mutational status (Fisher’s exact p-value = 0.03). Five of 17 patients (29%, 95% CI: 10% to 56%) had a KIT mutation in exon 11. Of these 5 patients, 4 (80%) had PMR and 1 (20%) had SMD. Twelve of 17 patients (71%) did not have a KIT mutation in exon 11 (2 with other mutation and 10 KIT amplified), and only 1 (8%) had PMR, 4 (33%) had SMD and 7 (58%) had PMD. Four of 17 patients (24%) had PMR and PR with anatomic imaging and all of these patients had a KIT mutation in exon 11. At the time of the data analysis, 4 of 17 patients remained alive and 2 of them (50%) had a KIT mutation in exon 11.
Discussion
In 2013, it is estimated that 76,690 new cases of malignant melanoma were diagnosed in the United States and there were 9,480 deaths from the disease [29]. Since prognosis depends on disease histology and the extent of metastases, among other factors, anatomic and metabolic imaging play a key role in patient evaluation. Specifically, ultrasound and lymphoscintigraphy are helpful for the evaluation of lymph node involvement [30],[31]. Diagnostic CT is used for the detection of metastases particularly to the lungs, lymph nodes and liver while MRI is used to evaluate the brain, meninges and spinal cord as well as to clarify indeterminate CT lesions [32]. The role of 18F-FDG-PET/CT has evolved in recent years. 18F-FDG-PET/CT is more accurate than anatomic imaging alone for the detection of metastases [32]-[35], and can lead to change in patient management [36],[37]. Surgical excision may be curative for disease confined to a thin lesion (<1 mm); however the prognosis for patients with metastases remains dismal [38].
There are few treatment options available for patients with metastatic melanoma [39]-[42]. Recently, immunotherapy has shown promising results. In particular, ipilimumab, a human monoclonal antibody that blocks cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), has been associated with improved survival in patients with metastatic melanoma [43]. Targeted therapy for patients with specific oncogenic mutations including BRAF and NRAS has also been used. For example, vemurafenib, a selective inhibitor of the BRAF serine-threonine kinase enzyme has been linked to improved rates of overall survival in patients with the BRAF V600E mutation [44]. KIT mutations have been found in the rare subgroup of patients with mucosal melanoma, acral melanoma and melanoma arising on chronically sun damaged skin, which has raised the possibility of utilizing imatinib mesylate for targeted therapy in these types of melanoma.
Imatinib mesylate is a tyrosine kinase inhibitor that has previously been used to treat patients with GIST [14],[15]. These patients have a spectrum of neoplastic disease ranging from benign to malignant, depending on the tumor size, mitotic activity, anatomic site of origin and the presence of a KIT mutation [45]. It is thought that the KIT mutation promotes activation of tyrosine kinase and results in tumorigenesis [13]. Unfortunately, patients often develop secondary resistance to imatinib mesylate within 2 years of treatment, likely due to the presence of new KIT mutations in existing tumor cells. Although 18F-FDG-PET/CT is not used to make the initial diagnosis, it is helpful for monitoring therapy response and can detect therapy resistant tumors by demonstrating re-emergence of glycolytic activity and resulting increased FDG uptake. 18F-FDG-PET/CT has been used to evaluate response to molecular targeted therapy in several cancers including melanoma [46].
In our study, the median TTP for patients with melanoma and a KIT genetic aberration (mutation or amplification) was 4.7 months (95% CI: 1.3 to 7.4 months) and was independent of melanoma type (log-rank p = 0.98) or KIT aberration sub-type (log-rank p = 0.20). On 18F-FDG-PET/CT there was a spectrum of disease response to therapy at 1 month, as shown in Figure 1. Five of 17 patients (29%) responded to therapy, 5 of 17 patients (29%) had stable disease and 7 of 17 patients (41%) showed disease progression. As shown in Figure 3, the metabolic response to therapy on 18F-FDG-PET/CT could be used to detect therapy-resistant tumors. Specifically, patients with progressive metabolic disease on 18F-FDG-PET/CT had a median TTP on anatomic imaging that was approximately 3.3 months shorter than patients with SMD or PMR (log-rank p-value = 0.04). Further, the overall survival of patients with progressive disease on 18F-FDG-PET/CT was approximately 3 months shorter than patients with SMD or PMR. This modest improvement in overall survival following metabolic response to therapy may be due, in part, to secondary drug resistance.
Tumor metabolic change on 18F-FDG-PET/CT was also associated with KIT mutational status. The majority of patients with a KIT mutation in exon 11 had PMR while the majority of patients without a KIT mutation in exon 11 had PMD. All of the patients with both PMR and anatomic PR had a KIT mutation in exon 11.
The principal limitation of this study is the sample size. Only 17 patients had evaluable 18F-FDG-PET/CT at baseline and after 1 month of therapy and only 5 patients had a KIT mutation in exon 11. Three of 5 patients with a KIT mutation (60%) had the L576P mutation, which the literature suggests may have only limited response to imatinib therapy. Although 12 patients did not have a KIT mutation in exon 11, several of these patients had KIT amplification. The overall survival was only slightly lower for patients with KIT amplification compared with patients who had a KIT mutation. Nevertheless, our results suggest a significant correlation between metabolic response and exon 11 KIT mutation and suggest that metabolic change on 18F-FDG-PET/CT may detect therapy-resistant disease. Evaluation of a larger patient population will be needed to confirm the statistical significance of the parameters we have studied.
Conclusions
Metabolic response by 18F-FDG-PET/CT is associated with exon 11 KIT mutational status in patients with mucosal melanoma, acral melanoma or melanoma arising on chronically sun damaged skin treated with imatinib. 18F-FDG-PET/CT may be a predictor of clinical outcome, although a larger study is needed to verify this.
Competing interests
The following authors have held consultant or advisory roles with the companies listed: F. Stephen Hodi, Novartis; Jeffrey S. Weber, Novartis; Thomas F. Gajewski, Bristol-Myers Squibb, Roche/Genentech, GlaxoSmithKline, Abbvie, Jounce Therapeutics.
The following authors have received research funding from the companies listed: F. Stephen Hodi, Novartis, Pfizer; Rene Gonzalez, Novartis; Thomas F. Gajewski, Bristol-Myers Squibb, Roche/Genentech, Eisai, Merck, Incyte; Steven J. O’Day, Novartis; Kevin B. Kim, Novartis; Jeffrey T. Yap, Novartis; Annick D. Van den Abbeele, Novartis.
Authors’ contributions
KZ contributed to the concept and design of the study, analysis and interpretation of data, and drafted and revised the manuscript, has given final approval of the version to be published, and agrees to be accountable for all aspects of the work. JY contributed to the concept and design of the study, analysis and interpretation of data, and revised the manuscript, has given final approval of the version to be published, and agrees to be accountable for all aspects of the work. AGH contributed to the concept and design of the study, analysis and interpretation of data, and revised the manuscript, has given final approval of the version to be published, and agrees to be accountable for all aspects of the work. JW contributed to the concept and design of the study, acquisition and interpretation of data, and revised the manuscript, has given final approval of the version to be published, and agrees to be accountable for all aspects of the work. RG contributed to the concept and design of the study, acquisition and interpretation of data, and revised the manuscript, has given final approval of the version to be published, and agrees to be accountable for all aspects of the work. TG contributed to the concept and design of the study, acquisition and interpretation of data, and revised the manuscript, has given final approval of the version to be published, and agrees to be accountable for all aspects of the work. SO contributed to the concept and design of the study, acquisition and interpretation of data, and revised the manuscript, has given final approval of the version to be published, and agrees to be accountable for all aspects of the work. KK contributed to the concept and design of the study, acquisition and interpretation of data, and revised the manuscript, has given final approval of the version to be published, and agrees to be accountable for all aspects of the work. SH contributed to the concept and design of the study, acquisition and interpretation of data, and revised the manuscript, has given final approval of the version to be published, and agrees to be accountable for all aspects of the work. AVDA contributed to the concept and design of the study, analysis and interpretation of data, and revised the manuscript, has given final approval of the version to be published, and agrees to be accountable for all aspects of the work. All authors read and approved the final manuscript.
Acknowledgements
We are extremely grateful to patients who participated in the trial and their families for contributing their time and effort. We would like to thank Jill Berkowitz, Leonid Syrkin, Tricia Locascio, Amanda Abbott and all those in the research teams who worked to make this paper possible.
Contributor Information
Katherine Zukotynski, Email: katherine.zukotynski@utoronto.ca.
Jeffrey T Yap, Email: jeffrey.yap@hci.utah.edu.
Anita Giobbie-Hurder, Email: agiohur@jimmy.harvard.edu.
Jeffrey Weber, Email: Jeffrey.Weber@moffitt.org.
Rene Gonzalez, Email: Rene.Gonzalez@ucdenver.edu.
Thomas F Gajewski, Email: tgajewsk@medicine.bsd.uchicago.edu.
Steven O’Day, Email: stevenjoday@gmail.com.
Kevin Kim, Email: kkim@mdanderson.org.
F Stephen Hodi, Email: Stephen_Hodi@dfci.harvard.edu.
Annick D Van den Abbeele, Email: abbeele@dfci.harvard.edu.
References
- Batsakis JG, Suarez P. Mucosal melanomas: a review. Adv Anat Pathol. 2000;7(3):167–180. doi: 10.1097/00125480-200007030-00006. [DOI] [PubMed] [Google Scholar]
- Tomicic J, Wanebo HJ. Mucosal melanomas. Surg Clin North Am. 2003;83(2):237–252. doi: 10.1016/S0039-6109(02)00100-7. [DOI] [PubMed] [Google Scholar]
- Mendenhall WM, Amdur RJ, Hinerman RW, Werning JW, Villaret DB, Mendenhall NP. Head and neck mucosal melanoma. Am J Clin Oncol. 2005;28(6):626–630. doi: 10.1097/01.coc.0000170805.14058.d3. [DOI] [PubMed] [Google Scholar]
- Stalkup JR, Orengo IF, Katta R. Controversies in acral lentiginous melanoma. Dermatol Surg. 2002;28(11):1051–1059. doi: 10.1046/j.1524-4725.2002.02082.x. [DOI] [PubMed] [Google Scholar]
- Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, Cho KH, Aiba S, Bröcker EB, LeBoit PE, Pinkel D, Bastian BC. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–2147. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006;24(26):4340–4346. doi: 10.1200/JCO.2006.06.2984. [DOI] [PubMed] [Google Scholar]
- Beadling C, Jacobsen-Dunlop E, Hodi FS, Le C, Warrick A, Patterson J, Town A, Harlow A, 3rd Cruz F, Azar S, Rubin BP, Muller S, West R, Heinrich MC, Corless CL. KIT gene mutations and copy number in melanoma subtypes. Clin Cancer Res. 2008;14(21):6821–6828. doi: 10.1158/1078-0432.CCR-08-0575. [DOI] [PubMed] [Google Scholar]
- Kong Y, Si L, Zhu Y, Xu X, Corless CL, Flaherty KT, Li L, Li H, Sheng X, Cui C, Chi Z, Li S, Han M, Mao L, Lu A, Guo J. Large-scale analysis of KIT aberrations in Chinese patients with melanoma. Clin Cancer Res. 2011;17:1684–1691. doi: 10.1158/1078-0432.CCR-10-2346. [DOI] [PubMed] [Google Scholar]
- Carvajal RD, Antonescu CR, Wolchok JD, Chapman PB, Roman RA, Teitcher J, Panageas KS, Busam KJ, Chmielowski B, Lutzky J, Pavlick AC, Fusco A, Cane L, Takebe N, Vemula S, Bouvier N, Bastian BC, Schwartz GK. KIT as a therapeutic target in metastatic melanoma. JAMA. 2011;305:2327–2334. doi: 10.1001/jama.2011.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handolias D, Salemi R, Murray W, Tan A, Liu W, Viros A, Dobrovic A, Kelly J, McArthur GA. Mutations in KIT occur at low frequency in melanomas arising from anatomical sites associated with chronic and intermittent sun exposure. Pigment Cell Melanoma Res. 2010;23:210–215. doi: 10.1111/j.1755-148X.2010.00671.x. [DOI] [PubMed] [Google Scholar]
- Frost MJ, Ferrao PT, Hughes TP, Ashman LK. Juxtamembrane mutant V560GKit is more sensitive to Imatinib (STI571) compared with wild-type c-kit whereas the kinase domain mutant D816VKit is resistant. Mol Cancer Ther. 2002;1:1115–1124. [PubMed] [Google Scholar]
- Jiang X, Zhou J, Yuen NK, Yuen NK, Corless CL, Heinrich MC, Fletcher JA, Demetri GD, Widlund HR, Fisher DE, Hodi FS. Imatinib targeting of KIT-mutant oncoprotein in melanoma. Clin Cancer Res. 2008;14:7726–7732. doi: 10.1158/1078-0432.CCR-08-1144. [DOI] [PubMed] [Google Scholar]
- Hirota S, Isozaki K, Moriyama Y, Hashimoto K, Nishida T, Ishiguro S, Kawano K, Hanada M, Kurata A, Takeda M, Muhammad Tunio G, Matsuzawa Y, Kanakura Y, Shinomura Y, Kitamura Y. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):577–580. doi: 10.1126/science.279.5350.577. [DOI] [PubMed] [Google Scholar]
- Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, Heinrich MC, Tuveson DA, Singer S, Janicek M, Fletcher JA, Silverman SG, Silberman SL, Capdeville R, Kiese B, Peng B, Dimitrijevic S, Druker BJ, Corless C, Fletcher CD, Joensuu H. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- Blanke CD, Demetri GD, von Mehren M, Heinrich MC, Eisenberg B, Fletcher JA, Corless CL, Fletcher CD, Roberts PJ, Heinz D, Wehre E, Nikolova Z, Joensuu H. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26:620–625. doi: 10.1200/JCO.2007.13.4403. [DOI] [PubMed] [Google Scholar]
- Antoch G, Kanja J, Bauer S, Kuehl H, Renzing-Koehler K, Schuette J, Bockisch A, Debatin JF, Freudenberg LS. Comparison of PET, CT, and dual-modality PET/CT imaging for monitoring of imatinib (STI571) therapy in patients with gastrointestinal stromal tumors. J Nucl Med. 2004;45(3):357–365. [PubMed] [Google Scholar]
- Choi H, Charnsangavej C, de Castro FS, Tamm EP, Benjamin RS, Johnson MM, Macapinlac HA, Podoloff DA. CT evaluation of the response of gastrointestinal stromal tumors after imatinib mesylate treatment: a quantitative analysis correlated with FDG PET findings. AJR Am J Roentgenol. 2004;183(6):1619–1628. doi: 10.2214/ajr.183.6.01831619. [DOI] [PubMed] [Google Scholar]
- Van den Abbeele AD. F18-FDG-PET provides early evidence of biological response to STI571 in patients with malignant gastrointestinal stromal tumors (GIST) for GIST Collaborative PET Study Group (Dana-Farber Cancer Institute, Boston, Massachusetts; OHSU, Portland, Oregon, Helsinki University Central Hospital, Turku University Central Hospital, Finland, Novartis Oncology) Proc Am Soc Clin Oncol. 2001;20:362a. [Google Scholar]
- Van den Abbeele AD, Badawi RD. Use of positron emission tomography in oncology and its potential role to assess response to Imatinib mesylate therapy in gastrointestinal stromal tumors (GIST) Eur J Cancer. 2002;38(Suppl 5):S60–S65. doi: 10.1016/S0959-8049(02)80604-9. [DOI] [PubMed] [Google Scholar]
- Holdsworth CH, Badawi RD, Manola JB, Kijewski MF, Israel DA, Demetri GD, Van den Abbeele AD. CT and PET: early prognostic indicators of response to imatinib mesylate in patients with gastrointestinal stromal tumor. AJR Am J Roentgenol. 2007;189(6):W324–W330. doi: 10.2214/AJR.07.2496. [DOI] [PubMed] [Google Scholar]
- Satzger I, Kuttler U, Volker B, Schenck F, Kapp A, Gutzmer R. Anal mucosal melanoma with KIT-activating mutation and response to imatinib therapy- case report and review of the literature. Dermatology. 2010;220:77–81. doi: 10.1159/000265558. [DOI] [PubMed] [Google Scholar]
- Woodman S, Trent J, Stemke-Hale K, Lazar AJ, Pricl S, Pavan GM, Fermeglia M, Gopal YN, Yang D, Podoloff DA, Ivan D, Kim KB, Papadopoulos N, Hwu P, Mills GB, Davies MA. Activity of desatinib against L576P KIT mutant melanoma: molecular, cellular, and clinical correlates. Mol Cancer Ther. 2009;8(8):2079–2085. doi: 10.1158/1535-7163.MCT-09-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluger HM, Dudek AZ, McCann C, Ritacco J, Southard N, Jilaveanu LB, Molinaro A, Sznol M. A phase 2 trial of dasatinib in advanced melanoma. Cancer. 2011;117(10):2202–2208. doi: 10.1002/cncr.25766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonescu CR, Busam KJ, Francone TD, Wong GC, Guo T, Agaram NP, Besmer P, Jungbluth A, Gimbel M, Chen CT, Veach D, Clarkson BD, Paty PB, Weiser MR. L576P KIT mutation in anal melanoma correlates with KIY protein expression and is sensitive to specific kinase inhibition. Int J Cancer. 2007;121:257–264. doi: 10.1002/ijc.22681. [DOI] [PubMed] [Google Scholar]
- Hodi S, Corless C, Giobbie-Hurder A, Fletcher JA, Zhu M, Marino-Enriquez A, Friedlander P, Gonzalez R, Weber JS, Gajewski TF, O'Day SJ, Kim KB, Lawrence D, Flaherty KT, Luke JJ, Collichio FA, Ernstoff MS, Heinrich MC, Beadling C, Zukotynski KA, Yap JT, Van den Abbeele AD, Demetri GD, Fisher DE. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun damaged skin. J Clin Oncol. 2013;31(26):3182–3190. doi: 10.1200/JCO.2012.47.7836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young H, Baum R, Cremerius U, Herholz K, Hoekstra O, Lammertsma AA, Pruim J, Price P. Measurement of clinical and subclinical tumour response using [18F]-fluorodeoxyglucose and positron emission tomography: review and 1999 EORTC recommendations. European Organization for Research and Treatment of Cancer (EORTC) PET Study Group. Eur J Cancer. 1999;35(13):1773–1782. doi: 10.1016/S0959-8049(99)00229-4. [DOI] [PubMed] [Google Scholar]
- Shankar LK, Hoffman JM, Bacharach S, Graham MM, Karp J, Lammertsma AA, Larson S, Mankoff DA, Siegel BA, Van den Abbeele A, Yap J, Sullivan D. Consensus recommendations for the use of 18F-FDG PET as an indicator of therapeutic response in patients in National Cancer Institute Trials. J Nucl Med. 2006;47(6):1059–1066. [PubMed] [Google Scholar]
- Giobbie-Hurder A, Gelber RD, Regan MM. Challenges of guarantee-time bias. J Clin Oncol. 2013;31(23):2963–2969. doi: 10.1200/JCO.2013.49.5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- http://seer.cancer.gov/statfacts/html/melan.html SEER Stat Fact Sheets: Melanoma of the Skin. [] Accessed 31 Mar 2014.
- Mohr P, Eggermont AMM, Hauschild A. Buzaid: staging of cutaneous melanoma. Ann Oncol. 2009;20(Suppl 6):vi14–vi21. doi: 10.1093/annonc/mdp256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belhocine TZ, Scott AM, Evan-Sapir E, Urbain JL, Essner R. Role of nuclear medicine in the management of cutaneous malignant melanoma. J Nucl Med. 2006;47:957–967. [PubMed] [Google Scholar]
- Gritters LS, Francis IR, Zasadny KR, Wahl RL. Initial assessment of positron emission tomography using 2-fluorine-18-fluoro-2-deoxy-D-glucose in the imaging of malignant melanoma. J Nucl Med. 1993;34:1420–1427. [PubMed] [Google Scholar]
- Swetter SM, Carroll LA, Johnson DL, Segall GM. Positron emission tomography is superior to computed tomography for metastatic detection in melanoma patients. Ann Surg Oncol. 2002;9:646–653. doi: 10.1007/BF02574480. [DOI] [PubMed] [Google Scholar]
- Reinhardt M, Joe AY, Jaeger U, Huber A, Matthies A, Bucerius J, Roedel R, Strunk H, Bieber T, Biersack HJ, Tüting T. Diagnostic performance of whole-body dual modality 18F-FDG-PET/CT imaging for N- and M- staging of malignant melanoma: experience with 250 consecutive patients. J Clin Oncol. 2006;24:1178–1187. doi: 10.1200/JCO.2005.03.5634. [DOI] [PubMed] [Google Scholar]
- Bastiaannet E, Wobbes T, Hoekstra O, Hoekstra OS, van der Jagt EJ, Brouwers AH, Koelemij R, de Klerk JM, Oyen WJ, Meijer S, Hoekstra HJ. Prospective comparison of [18F] Fluorodeoxyglucose positron emission tomography and computed tomography in patients with melanoma with palpable lymph node metastases: diagnostic accuracy and impact on treatment. J Clin Oncol. 2009;27:4774–4780. doi: 10.1200/JCO.2008.20.1822. [DOI] [PubMed] [Google Scholar]
- Xing Y, Bronstein Y, Ross MI, Askew RL, Lee JE, Gershenwald JE, Royal R, Cormier JN. Contemporary diagnostic imaging modalities for the staging and surveillance of melanoma patients: a meta-analysis. J Natl Cancer Inst. 2011;103:129–142. doi: 10.1093/jnci/djq455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iagaru A, Quon A, Johnson D, Gambhir SS, McDougall IR. 2-Deoxy-2-[F-18]fluoro-D-glucose positron emission tomography/computed tomography in the management of melanoma. Mol Imaging Biol. 2007;9:50–57. doi: 10.1007/s11307-006-0065-0. [DOI] [PubMed] [Google Scholar]
- Gulec SA, Faries MB, Lee CC, Kirgan D, Glass C, Morton DL, Essner R. The role of fluorine-18 deoxyglucose positron emission tomography in the management of patients with metastatic melanoma: impact on surgical decision making. Clin Nucl Med. 2003;28:961–965. doi: 10.1097/01.rlu.0000099805.36471.aa. [DOI] [PubMed] [Google Scholar]
- Koh HK. Cutaneous melanoma. N Engl J Med. 1991;325(3):171–182. doi: 10.1056/NEJM199107183250306. [DOI] [PubMed] [Google Scholar]
- Cocconi G, Bella M, Calabresi F, Tonato M, Canaletti R, Boni C, Buzzi F, Ceci G, Corgna E, Costa P, Loticci R, Papadia F, Sofra MC, Bacchi M. Treatment of metastatic malignant melanoma with dacarbazine plus tamoxifen. N Engl J Med. 1992;327(8):516–523. doi: 10.1056/NEJM199208203270803. [DOI] [PubMed] [Google Scholar]
- Serrone L, Zeuli M, Sega FM, Cognetti F. Dacarbazine-based chemotherapy for metastatic melanoma: thirty-year experience overview. J Exp Clin Cancer Res. 2000;19(1):21–34. [PubMed] [Google Scholar]
- Tsao H, Atkins MB, Sober AJ. Management of cutaneous melanoma. N Engl J Med. 2004;351(10):998–1012. doi: 10.1056/NEJMra041245. [DOI] [PubMed] [Google Scholar]
- Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbé C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumabin patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O'Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364(26):2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMatteo RP. The GIST of targeted cancer therapy: a tumor (gastrointestinal stromal tumor), a mutated gene (c-kit), and a molecular inhibitor (STI571) Ann Surg Oncol. 2002;9(9):831–839. doi: 10.1007/BF02557518. [DOI] [PubMed] [Google Scholar]
- McArthur GA, Puzanov I, Amaravadi R, Ribas A, Chapman P, Kim KB, Sosman JA, Lee RJ, Nolop K, Flaherty KT, Callahan J, Hicks RJ. Marked, homogeneous, and early [18F]fluorodeoxyglucose-positron emission tomography responses to vemurafenib in BRAF-mutant advanced melanoma. J Clin Oncol. 2012;30(14):1628–1634. doi: 10.1200/JCO.2011.39.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]