

Abstract

A mild method for regioselective formation of 1,5-substituted 1,2,3-triazoles is described. The zinc-mediated reaction works at room temperature and is successful across a wide range of azido/alkynyl substrates. Additionally, the triazole 4-position can be further functionalized through the intermediate aryl-zinc to accommodate a diverse three-component coupling strategy.
The 1,2,3-triazole has risen to prominence in recent years as a superbly versatile heterocycle, with the 1,4-isomer being readily prepared from azide and alkyne components using copper-catalyzed azide alkyne cycloaddition (CuAAC).1 This reaction reliably functions under mild conditions, displays superb substrate scope, and has driven a vast range of triazole application across the chemical, biological, and materials sciences.2 Methods for accessing the alternate 1,5-isomer, by contrast, are far less developed. The synthesis of both triazole geometrical isomers has conventionally been achieved using the thermal Huisgen cycloaddition between azides and alkynes to afford a mixture of the 1,4- or 1,5-substituted 1,2,3-triazoles.3 However, the separation of these products is frequently a tedious and sometimes insurmountable challenge.4 Existing methods for the exclusive construction of 1,5-triazoles require strongly basic conditions, utilizing alkali5 or magnesium6 acetylides, and have proven too demanding for many useful substrate classes. Alternatively, bulky ruthenium catalysts7 are capable of forming the desired 1,5-triazole (also 1,4,5-substituted triazoles). However, the cost of using a noble metal catalyst in this RuAAC procedure is an impediment to the development of a general, cost-effective application.8
With these concerns in mind, a milder and more economical route toward 1,5-substituted triazoles is sorely needed, as is a suitable method for the further functionalization of the 4-position. Such reactions could see significant application, as the alternative 1,5-linkage would afford molecules and materials with new and contrasting properties to 1,4-triazoles synthesized via CuAAC. In response to these demands we have investigated a zinc-mediated method for triazole synthesis, inspired by significant advances in the formation of zinc acetylides and their further reaction with carbonyl functional groups.9 It was anticipated that the less nucleophilic zinc reagents (with respect to magnesium or lithium) would permit a much wider substrate scope and permit further functionalization.
We quickly discovered that simple addition of stoichiometric ZnEt2 [1 M in hexanes] to a THF/toluene solution of alkyne 1 and azide 2 would exclusively form the desired 1,5-substituted triazole isomer (Scheme 1). Proof of the geometry was initially determined by comparison with the 1,4-isomer formed under CuAAC, followed by single crystal X-ray crystallography of 3a (Figure 1) and an analog (3x) subsequently prepared (vide infra). However, repeating the reaction with resynthesized 2 gave no reaction at all, with only starting materials observed. We surmised that a catalytic base could be required to form the zinc acetylide and that residual aniline from the azide synthesis10 was promoting the reaction. Addition of 10% N-methylimidazole (NMI) promptly restored reactivity, and screening could continue.11 A range of solvents were found to be suitable including CH2Cl2, 1,4-dioxane, MeCN, PhCF3, i-PrOAc, and PhMe; although THF afforded a better purity profile and is readily available anhydrously. A concentration of 0.125 M was utilized to ensure all the zinc species remained in solution, and a slight excess of alkyne and ZnEt2 were used to drive the reaction to completion. The reaction was typically complete after 18 h at ambient temperature or in 2 h at 100 °C in a microwave reactor. Yields were lower in the latter case due to the formation of an azide derived aniline byproduct (typically ∼10%). The final optimized conditions are outlined in Scheme 1 and afforded the 1,5-product (3) in an isolated yield of 75% on a 1 mmol scale. The reaction was then directly scaled up to 10 mmol and afforded just over of 2 g of 3a in a very similar 76% isolated yield.
Scheme 1. Optimized Reaction Conditions and Substrate Scope.

Standard conditions but 2.4 equiv azide and 3.0 equiv ZnEt2. b As standard but 72 h. c As standard but 2.5 equiv ZnEt2 and 72 h.
Figure 1.
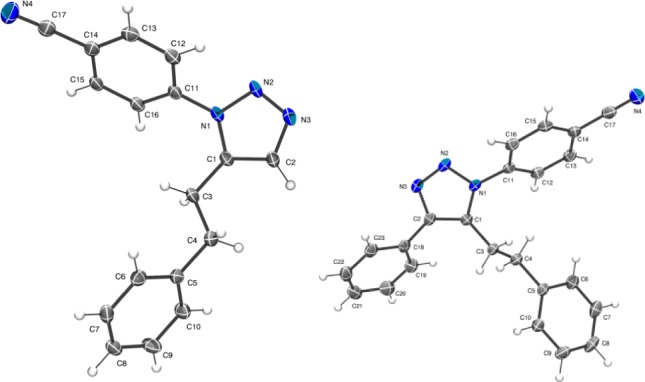
X-ray crystal structures of 3a and 3x proving the 1,5 and 1,4,5 configurations of the triazole products. Thermal ellipsoids at 50%.13
The alkyne substrate range encompasses both alkyl and aryl terminal alkynes (Scheme 1) including enyne (3d) and silylated (3e) functionalities. Further success was found with propargylic ethers (3g), esters (3i), and thioethers (3q) although 1,2-diphenyl acetylene, tosyl azide, and (iodoethynyl)benzene failed to provide the desired product. Important substrate classes unsuitable for the magnesium system were able to withstand our zinc mediated conditions including esters (entries 3f, 3i, and 3l), amides (3m), ketones (3n), nitriles, nitros (3k), aryl iodides (3o), heterocycles (3p and 3q), and ortho-substituents (3c and 3d). Diynes (3r) were also suitable starting materials, suggesting this method could find application in polymer or dendrimer synthesis.12 These substrates are not productive in the RuAAC method due to the formation of unreactive ruthenacycles.7
Alkyl azides were not generally suitable as substrates, although benzyl azide could be reacted in good yield using extended reaction times (72 h) at ambient temperature (3s). Efforts to accelerate the reaction through heating resulted in poor yields and significant decomposition of starting materials. Further difficult examples included substrates with free alcohols. Nevertheless, a successful reaction could be achieved by the addition of extra ZnEt2 and extended reaction times (72 h) at ambient temperature (3t and 3u). Formation of triazole 3tvia the zinc method is of particular interest, representing the successful functionalization of the hindered propargyl alcohol mestranol (a commercial estrogen) in excellent yield. The reaction evidently has the capacity for late stage elaboration of sterically hindered, chiral molecules and biologically important scaffolds.
Further insight into the mechanism of the reaction was discovered when the mixture was quenched with D2O/D3CCO2D rather than with NH4Cl (aq).141H NMR and LCMS determined an 89% deuterium incorporation at the triazole 4-position (3v, Table 1). Implying that a stoichiometric aryl-zinc intermediate (Scheme 2, 4) was formed in the reaction in a way analogous to the previously described magnesium methods.6 This aryl-zinc reagent (4) was then used for further elaboration to afford a number of 1,4,5-trisubstituted 1,2,3-triazoles and to demonstrate this technology’s potential for the synthesis of highly diverse libraries through three-component coupling. The initial exploration successfully incorporated bromine (3w) and thus offers a partner for palladium catalyzed cross-coupling at some later stage. Conversely, the aryl-zinc itself easily underwent palladium mediated cross-coupling with iodobenzene (3x) and thus offers a myriad of opportunities for biaryl synthesis. The ketone and alcohol products (3y and 3z) present significant opportunities for further molecular diversity through a ‘capping’ step to incorporate four distinct components in only a few steps.
Table 1. Further Functionalization of the Aryl Zinc Reagent (4).
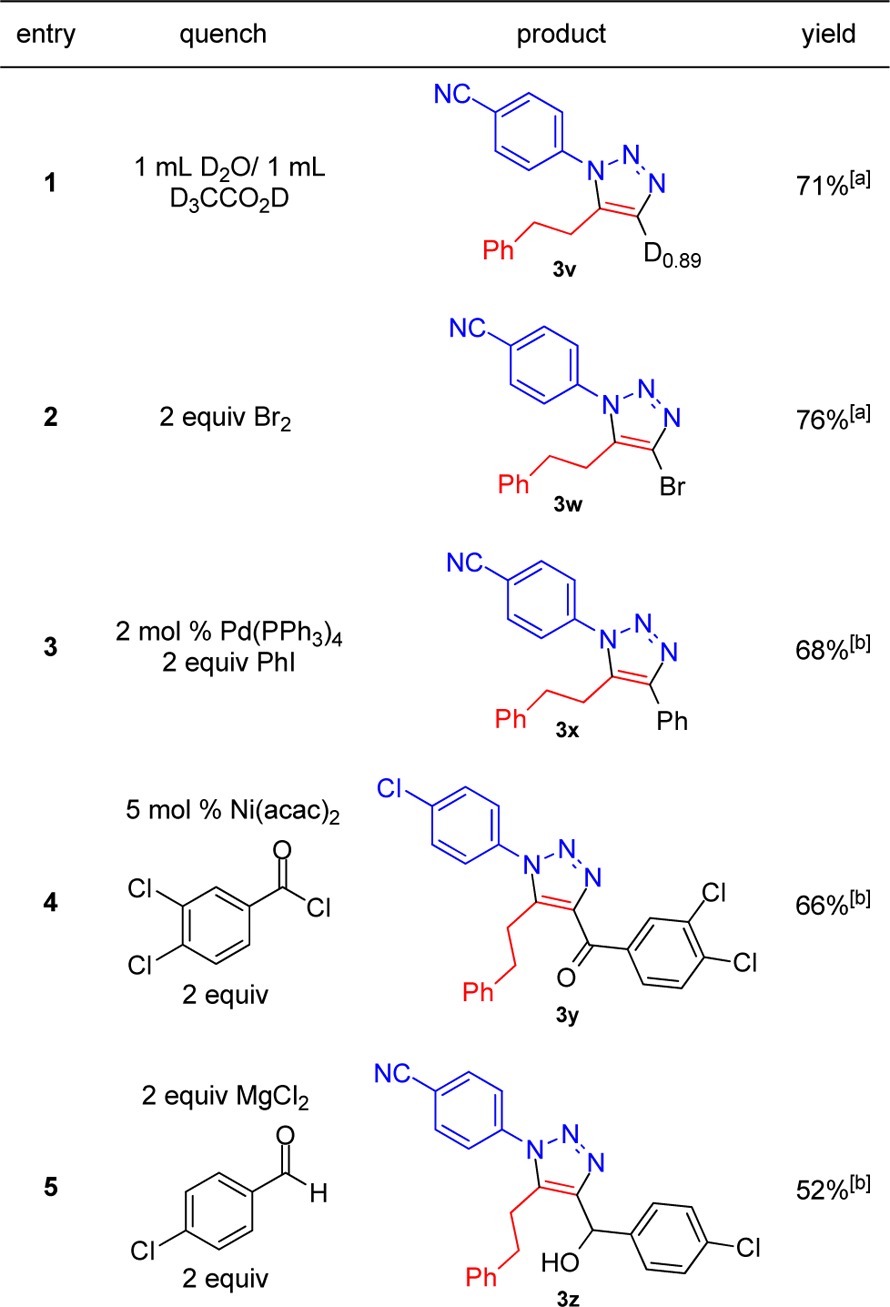
Standard conditions with quench added directly.
Standard conditions with third component addition to reaction as a THF solution and stirred for 18 h at rt.
Scheme 2. Proposed Mechanistic Pathway.

With our accrued observations we have proposed a reaction mechanism in Scheme 2 to help explain the observed reactivity of this system. It is reasonable to assume from previous reports that the transformation passes through the initial metalation of the alkyne-H, mediated by the amine base, to form the zinc acetylide 5.9f,9g,11 Reversible precoordination between the azide and zinc acetylide could be expected to occur before the [3 + 2]-cycloaddition can take place, explaining the necessity for stoichiometric quantities of ZnEt2 in the reaction and the formation of the aryl-zinc intermediate 4. Harnessing the further reactivity of this aryl-zinc species (4) has been demonstrated by the trapping experiments set out in Table 1.
In conclusion we present a significant addition to the regioselective formation of 1,5-substiuted 1,2,3-triazoles, a method that has proved successful across a wide range of azido/alkynyl substrates. Additionally, the 4-position can be further functionalized through the intermediate aryl-zinc to accommodate a diverse three-component coupling strategy. The inherently benign nature and efficient construction of these triazoles make this protocol ideal for both library synthesis and the late stage functionalization of complex molecules. Equally, the procedure is operationally straightforward, eminently scalable, and expected to be of interest across the chemical community.
Acknowledgments
We thank the Wellcome Trust (Grant WT094899MA) and the EPSRC (Leadership Fellowship to MFG) for funding. Dr. Achim Schnaufer (University of Edinburgh) and Prof. Rommie Amaro (University of California, San Diego) are thanked for helpful discussions.
Supporting Information Available
Experimental procedures and full spectroscopic data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- a Rostovtsev V. V.; Green L. G.; Fokin V. V.; Sharpless K. B. Angew. Chem., Int. Ed. 2002, 41, 2596–2599. [DOI] [PubMed] [Google Scholar]; b Tornøe C. W.; Christensen C.; Meldal M. J. Org. Chem. 2002, 67, 3057–3064. [DOI] [PubMed] [Google Scholar]
- a Moses J. E.; Moorhouse A. D. Chem. Soc. Rev. 2007, 36, 1249–1262. [DOI] [PubMed] [Google Scholar]; b Meldal M. Macromol. Rapid Commun. 2008, 29, 1016–1051. [Google Scholar]; c Meldal M.; Tornøe C. W. Chem. Rev. 2008, 108, 2952–3015. [DOI] [PubMed] [Google Scholar]; d Baxendale I. R.; Ley S. V.; Mansfield A. C.; Smith C. D. Angew. Chem., Int. Ed. 2009, 48, 4017–4021. [DOI] [PubMed] [Google Scholar]; e Agalave S. G.; Maujan S. R.; Pore V. S. Chem.—Asian J. 2011, 6, 2696–2718. [DOI] [PubMed] [Google Scholar]; f Beale T. M.; Bond P. J.; Brenton J. D.; Charnock-Jones D. S.; Ley S. V.; Myers R. M. Bioorg. Med. Chem. 2012, 20, 1749–1759. [DOI] [PubMed] [Google Scholar]; g Lewandowski B.; De Bo G.; Ward J. W.; Papmeyer M.; Kuschel S.; Aldegunde M. J.; Gramlich P. M. E.; Heckmann D.; Goldup S. M.; D’Souza D. M.; Fernandes A. E.; Leigh D. A. Science 2013, 339, 189–193. [DOI] [PubMed] [Google Scholar]; h Thirumurugan P.; Matosiuk D.; Jozwiak K. Chem. Rev. 2013, 113, 4905–4979. [DOI] [PubMed] [Google Scholar]
- Huisgen R. Angew. Chem., Int. Ed. 1963, 2, 565–598. [Google Scholar]
- Landge K. P.; Seo Y. W.; Kwak J.; Park W. K.; Gong J. Y.; Lee H. Y.; Koh H. Y. Bull. Korean Chem. Soc. 2011, 32, 3101–3104. [Google Scholar]
- a Kwok S. W.; Fotsing J. R.; Fraser R. J.; Rodionov V. O.; Fokin V. V. Org. Lett. 2010, 12, 4217–4219. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Meza-Aviña M. E.; Patel M. K.; Lee C. B.; Dietz T. J.; Croatt M. P. Org. Lett. 2011, 13, 2984–2987. [DOI] [PubMed] [Google Scholar]; c Wu L.; Chen Y.; Tang M.; Song X.; Chen G.; Song X.; Lin Q. Synlett 2012, 1529–1533. [Google Scholar]; d Hong L.; Lin W.; Zhang F.; Liu R.; Zhou X. Chem. Commun. 2013, 49, 5589–5591. [DOI] [PubMed] [Google Scholar]
- a Krasiński A.; Fokin V. V.; Sharpless K. B. Org. Lett. 2004, 6, 1237–1240. [DOI] [PubMed] [Google Scholar]; b Akao A.; Tsuritani T.; Kii S.; Sato K.; Nonoyama N.; Mase T.; Yasuda N. Synlett 2007, 31–36. [Google Scholar]
- a Zhang L.; Chen X.; Xue P.; Sun H. H. Y.; Williams I. D.; Sharpless K. B.; Fokin V. V.; Jia G. J. Am. Chem. Soc. 2005, 127, 15998–15999. [DOI] [PubMed] [Google Scholar]; b Boren B. C.; Narayan S.; Rasmussen L. K.; Zhang L.; Zhao H.; Lin Z.; Jia G.; Fokin V. V. J. Am. Chem. Soc. 2008, 130, 8923–8930. [DOI] [PubMed] [Google Scholar]; Correction:; J. Am. Chem. Soc. 2008, 130, 14900. [Google Scholar]; c Lamberti M.; Fortman G. C.; Poater A.; Broggi J.; Slawin A. M. Z.; Cavallo L.; Nolan S. P. Organometallics 2012, 31, 756–767. [Google Scholar]
- a Majireck M. M.; Weinreb S. M. J. Org. Chem. 2006, 71, 8680–8683. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Oppilliart S.; Mousseau G.; Zhang L.; Jia G.; Thuéry P.; Rousseau B.; Cintrat J.-C. Tetrahedron 2007, 63, 8094–8098. [Google Scholar]; c Nulwala H.; Takizawa K.; Odukale A.; Khan A.; Thibault R. J.; Taft B. R.; Lipshutz B. H.; Hawker C. J. Macromolecules 2009, 42, 6068–6074. [Google Scholar]; d Zhang J.; Kemmink J.; Rijkers D. T. S.; Liskamp R. M. J. Chem. Commun. 2013, 49, 4498–4500. [DOI] [PubMed] [Google Scholar]
- a Li Z.; Upadhyay V.; DeCamp A. E.; DiMichele L.; Reider P. J. Synthesis 1999, 1453–1458. [Google Scholar]; b Pu L.; Yu H.-B. Chem. Rev. 2001, 101, 757–824. [DOI] [PubMed] [Google Scholar]; c Boyall D.; Frantz D. E.; Carreira E. M. Org. Lett. 2002, 4, 2605–2606. [DOI] [PubMed] [Google Scholar]; d Zani L.; Alesi S.; Cozzi P. G.; Bolm C. J. Org. Chem. 2006, 71, 1558–1562. [DOI] [PubMed] [Google Scholar]; e Yang F.; Xi P.; Yang L.; Lan; Xie R.; You J. J. Org. Chem. 2007, 72, 5457–5460. [DOI] [PubMed] [Google Scholar]; f Turlington M.; Pu L. Synlett 2012, 649–684. [Google Scholar]; g Trost B. M.; Bartlett M. J.; Weiss A. H.; J. von Wangelin A.; Chan V. S. Chem.—Eur. J. 2012, 18, 16498–16509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barral K.; Moorhouse A. D.; Moses J. E. Org. Lett. 2007, 9, 1809–1811. [DOI] [PubMed] [Google Scholar]
- a Clegg W.; García-Álvarez J.; García-Álvarez P.; Graham D. V.; Harrington R. W.; Hevia E.; Kennedy A. R.; Mulvey R. E.; Russo L. Organometallics 2008, 27, 2654–2663. [Google Scholar]; b Du Y.; Turlington M.; Zhou X.; Pu L. Tetrahedron Lett. 2010, 51, 5024–5027. [Google Scholar]
- Astruc D.; Liang L.; Rapakousiou A.; Ruiz J. Acc. Chem. Res. 2011, 45, 630–640. [DOI] [PubMed] [Google Scholar]
- CCDC 931414 and CCDC 931415 contain the crystallographic data for 3a and 3x respectively. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre viawww.ccdc.cam.ac.uk/data_request/cif. ORTEP-3 was used to produce the thermal ellipsoid plots: Farrugia L. J.J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar]
- Akula H. K.; Lakshman M. K. J. Org. Chem. 2012, 77, 8896–8904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.