

In four N-(4-halophenyl)-4-oxo-4H-chromene-3-carboxamides, halo = -F, -Cl, -Br and -I, the molecules are essentially planar and exhibit anti conformations with respect to the C—N rotamer of the amide and with cis geometries with respect to the relative positions of the C3arom—C2arom bond of the chromone ring and the carbonyl group of the amide.
Keywords: crystal structure, drug design, chromones, conformation, supramolecular structure
Abstract
Four N-(4-halophenyl)-4-oxo-4H-chromene-3-carboxamides (halo = F, Cl, Br and I), N-(4-fluorophenyl)-4-oxo-4H-chromene-3-carboxamide, C16H10FNO3, N-(4-chlorophenyl)-4-oxo-4H-chromene-3-carboxamide, C16H10ClNO3, N-(4-bromophenyl)-4-oxo-4H-chromene-3-carboxamide, C16H10BrNO3, N-(4-iodophenyl)-4-oxo-4H-chromene-3-carboxamide, C16H10INO3, have been structurally characterized. The molecules are essentially planar and each exhibits an anti conformation with respect to the C—N rotamer of the amide and a cis geometry with respect to the relative positions of the Carom—Carom bond of the chromone ring and the carbonyl group of the amide. The structures each exhibit an intramolecular hydrogen-bonding network comprising an N—H⋯O hydrogen bond between the amide N atom and the O atom of the carbonyl group of the pyrone ring, forming an S(6) ring, and a weak Carom—H⋯O interaction with the O atom of the carbonyl group of the amide as acceptor, which forms another S(6) ring. All four compounds have the same supramolecular structure, consisting of R 2 2(13) rings that are propagated along the a-axis direction by unit translation. There is π–π stacking involving inversion-related molecules in each structure.
Chemical context
Chromones are a group of natural and synthetic oxygen heterocyclic compounds having a high degree of chemical diversity that is frequently linked to a broad array of biological activities (Gaspar et al. 2014 ▸). Parkinson’s disease (PD) is a degenerative disorder of the central nervous system with an aetiology not yet completely clarified. There is no cure for PD, but medications, surgery and multidisciplinary management can provide relief from the symptoms. PD seems to be associated with a decrease in central levels of dopamine triggered by oxidative stress. These processes, among other factors, are mediated by the isoform B of the monoamino oxidase (MAO-B). Hence, the search for novel agents that can selectively inhibit MAO-B is of paramount relevance. In this context, the decoration of chromone, a privileged structure for the discovery and development of new chemical entities (NCEs), have led to the preparation of chromone carboxamides and to promising outcomes since preliminary data indicate that chromone-3-carboxamides are selective MAO-B inhibitors (Gaspar, Reis et al., 2011 ▸; Gaspar, Silva et al., 2011 ▸).
Previous results showed that the carbonyl group of the chromone moiety and the amide function play an important role in the establishment of hydrogen interactions with the MAO-B active pocket. In addition, the presence of a phenyl substituent attached to the amide seems to play a pivotal role in the potency conveyed by the ligand (Helguera et al., 2013 ▸). In this context, some N-(4-halophenyl)-4-oxo-4H-chromene-3-carboxamides (1)–(4), shown in the scheme, have been synthesized and structurally characterized in order to rationalize the structural factors that may affect the selectivity and the potency of their inhibitory activities towards MAO-B. These structures are compared with N-(4-phenyl)-4-oxo-4H-chromene-2-carboxamide and N-(4-bromophenyl)-4-oxo-4H-chromene-2-carboxamide, compounds (5) and (6) (Reis et al., 2013 ▸; Gomes et al., 2013 ▸), which do not show inhibitory activities against human MAO-B.
Structural commentary
The structural analysis of (1)–(4) confirmed them to be N-(4-halophenyl)-4-oxo-4H-chromene-3-carboxamides with halosubstituents F (Fig. 1 ▸), Cl (Fig. 2 ▸), Br (Fig. 3 ▸) and I (Fig. 4 ▸), respectively, as depicted in the scheme. Figs. 1 ▸–4 ▸
▸
▸ show the displacement ellipsoid diagrams with the adopted labelling schemes. All compounds crystallize in the space group P
 . Compounds (1) and (2) are isostructural, as are compounds (3) and (4). The cell lengths are very similar in each pair of compounds.
. Compounds (1) and (2) are isostructural, as are compounds (3) and (4). The cell lengths are very similar in each pair of compounds.
Figure 1.

A view of the asymmetric unit of (1), showing the atom-numbering scheme. Displacement ellipsoids are drawn at the 80% probability level. Dashed lines indicate the intramolecular contacts.
Figure 2.

A view of the asymmetric unit of (2), showing the atom-numbering scheme. Displacement ellipsoids are drawn at the 80% probability level. Dashed lines indicate the intramolecular contacts.
Figure 3.

A view of the asymmetric unit of (3), showing the atom-numbering scheme. Displacement ellipsoids are drawn at the 80% probability level. Dashed lines indicate the intramolecular contacts.
Figure 4.

A view of the asymmetric unit of (4), showing the atom-numbering scheme. Displacement ellipsoids are drawn at the 80% probability level. Dashed lines indicate the intramolecular contacts.
The title compounds display similar structures, which are reflected in the molecular geometries and conformations; the values of the dihedral angles between the mean planes of the chromone ring and the exocyclic phenyl ring of the N-phenyl-4-oxo-4H-chromene-3-carboxamides are close to 2° in the case of the F, Cl pair [2.51 (3) and 1.95 (7)°, respectively,] and close to 5° for the Br, I pair [4.90 (10) and 5.37 (10)°, respectively]. In N-phenyl-4-oxo-4H-chromene-2-carboxamide (5) (Reis et al., 2013 ▸), the dihedral angle between the mean planes of the chromone ring and the phenyl ring is 6.57° and in N-(4-bromophenyl)-4-oxo-4H-chromene-2-carboxamide (6), the structural isomer of (3) (Gomes et al., 2013 ▸), the dihedral angle between the mean planes of the chromone ring and the phenyl ring is 5.0 (2)°. Selected dihedral angles are given in Table 1 ▸.
Table 1. Selected dihedral angles ().
1 is the dihedral angle between the mean planes of the chromene and phenyl rings and the phenyl ring. 2 is the dihedral angle between the mean plane of the chromone ring and the plane defined by atoms O2, C31 and N3. 3 is the dihedral angle between the mean planes of the phenyl ring and the plane defined by atoms O3, C31 and N3.
| Compound | 1 | 2 | 3 |
|---|---|---|---|
| (1) | 2.51(3) | 5.51(12) | 5.05(13) |
| (2) | 1.95(7) | 5.7(3) | 4.4(3) |
| (3) | 4.90(10) | 2.0(4) | 2.9(4) |
| (4) | 5.37(10) | 1.8(4) | 3.6(4) |
In (1) and (2), the maximum deviations from the mean plane of the 10 atoms of the chromone ring plus the three carboxamide atoms O3, C31 and N3, are 0.1220 (8) and 0.1319 (17) Å, respectively, both for atom O3 (r.m.s. deviations of fitted atoms = 0.0519 and 0.0571 Å, respectively). In (3) and (4), the deviations of O3 from the mean plane defined above are 0.0384 (14) and 0.0342 (15) Å, respectively (r.m.s. deviations of fitted atoms = 0.0314 Å in both compounds). In the case of (3) and (4), atom C2 shows the greatest deviation from the mean plane having deviations of 0.0569 (18) and 0.0596 (18) Å, respectively. These values indicate that the carboxamide groups are practically planar with the chromone ring, particularly in the case of the Br and I chromone carboxamide derivatives. This planarity may be related to the internal hydrogen-bond pattern in those molecules, which thus defines the molecular conformations.
The conformational features herein established are probably most relevant for the extrapolation of the inhibitory MAO-B activities of chromone carboxamides as they are related to the intermolecular forces responsible for enzyme–ligand binding affinity. The data can explain the MAO-B selectivity found for chromone-3-carboxamides (1)–(4), as opposed to the lack of activity presented by chromone-2-carboxamides (5) and (6). As seen in the scheme, (1)–(4) are N-(phenyl)-4-oxo-4H-chromene-3-carboxamides while (5) and (6) are N-(phenyl)-4-oxo-4H-chromene-2-carboxamides. As can be seen in Fig. 5 ▸, an anti conformation is adopted with respect to the C—N rotamer of the amide in all of the compounds. Nevertheless, due to the asymmetry of the chromone residue, the anti conformation can assume a cis (a) or trans (b) geometry with respect to the relative position of the carbonyl O atom of the carboxamide and the C2arom—C3arom bond of the chromone. Compounds (1)–(4) exhibit a cis relation between these bonds, as can be seen in the ellipsoid diagrams, Figs. 1 ▸–4 ▸ ▸ ▸. This molecular conformation permits the formation of two intramolecular hydrogen bonds, which generate a network that probably enhances their planarity. Details of the intramolecular hydrogen-bonding interactions are given in Tables 2 ▸ ▸ ▸ to 5 ▸. Specifically for each molecule, there is an intramolecular N—H⋯O hydrogen bond between the amide nitrogen and the oxygen atom of the carbonyl group, O4, of the chromone ring, forming an S(6) ring identified as ring C. In addition, the carbonyl oxygen of the amide, O3, acts as the acceptor for a weak interaction with an ortho hydrogen of the exocyclic phenyl ring, forming another S(6) ring, B. The corresponding trans structures (top right in Fig. 5 ▸) would probably only allow the formation of a weak hydrogen-bonding interaction with an ortho hydrogen atom of the exocyclic phenyl ring. It is interesting to compare the internal hydrogen-bonding network presented by the title compounds with those of the analogous 4-oxo-N-(substituted phenyl)-4H-chromene-2-carboxamides (Reis et al., 2013 ▸) and (Gomes et al., 2013 ▸), compounds (5) and (6). Previous studies concerning the structures of the chromone-2-carboxamides show that the majority have geometries similar to compound (5), e.g. as in (1)–(4), they assume a cis conformation, but this is not the case for (6), the bromo isomer of (3), as shown in Fig. 5 ▸ (bottom right). In spite of this, none of this type of derivative displays inhibitory activity towards the MAO-B isoenzyme. When the geometries of the relative positions of rings D and E of the chromone residue with respect to rings A and B are compared, it can be seen that the effect of the 2/3 positional isomerism is to ‘reflect’ their relative positions while the effect of the cis/trans conformations is a ‘twofold rotation’ of the rings around the Camide— Cchromone bond. Those particular differences in conformation may condition the ability for docking when pharmacological activities are considered.
Figure 5.

Anti-rotamer conformations around the C—N rotamer for the 3-carboxamides (top) and for the 2-carboxamide isomers (bottom), showing the relative positions of the C3arom—C2arom bond of the chromone ring with respect to the carboxylic group of the amide: cis (right) or trans (left) geometries.
Table 2. Hydrogen-bond geometry (, ) for (1) .
| DHA | DH | HA | D A | DHA |
|---|---|---|---|---|
| N3H3O4 | 0.896(17) | 1.901(17) | 2.7024(13) | 147.9(15) |
| C312H312O3 | 0.95 | 2.26 | 2.8714(15) | 122 |
| C2H2O4i | 0.95 | 2.45 | 3.1645(14) | 132 |
| C316H316O3ii | 0.95 | 2.46 | 3.3160(14) | 149 |
Symmetry codes: (i)  ; (ii)
; (ii)  .
.
Table 3. Hydrogen-bond geometry (, ) for (2) .
| DHA | DH | HA | D A | DHA |
|---|---|---|---|---|
| N3H3O4 | 0.85(3) | 1.92(3) | 2.680(3) | 148(3) |
| C312H312O3 | 0.95 | 2.29 | 2.892(3) | 121 |
| C2H2O4i | 0.95 | 2.47 | 3.194(3) | 133 |
| C316H316O3ii | 0.95 | 2.45 | 3.286(3) | 146 |
Symmetry codes: (i) ; (ii) .
Table 4. Hydrogen-bond geometry (, ) for (3) .
| DHA | DH | HA | D A | DHA |
|---|---|---|---|---|
| N3H3O4 | 0.86(2) | 1.95(2) | 2.695(2) | 145(2) |
| C312H312O3 | 0.95 | 2.26 | 2.877(2) | 129 |
| C2H2O4i | 0.95 | 2.41 | 3.167(2) | 137 |
| C316H316O3ii | 0.95 | 2.47 | 3.314(2) | 148 |
Symmetry codes: (i) ; (ii) .
Table 5. Hydrogen-bond geometry (, ) for (4) .
| DHA | DH | HA | D A | DHA |
|---|---|---|---|---|
| N3H3O4 | 0.92(2) | 1.89(2) | 2.6977(19) | 145(2) |
| C2H2O3 | 0.95 | 2.33 | 2.718(2) | 104 |
| C312H312O3 | 0.95 | 2.27 | 2.881(2) | 122 |
| C2H2O4i | 0.95 | 2.44 | 3.185(2) | 136 |
| C316H316O3ii | 0.95 | 2.49 | 3.312(2) | 145 |
Symmetry codes: (i) ; (ii) .
Supramolecular features
Intermolecular hydrogen-bonding information is given in Table 2 ▸ to 5. All compounds have the same supramolecular structure in which the C2—H2⋯O4(x + 1, y, z) and C316—H316⋯O3(x − 1, y, z) form 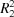 (13) ring structures, which are propagated along the a-axis direction by unit translation. Fig. 6 ▸ shows the Cl compound, (3), as an example.
(13) ring structures, which are propagated along the a-axis direction by unit translation. Fig. 6 ▸ shows the Cl compound, (3), as an example.
Figure 6.

The distorted ladder formed by linked (13) rings in compound (3). The chain runs parallel to the a axis. Hydrogen bonds are indicated by blue dashed lines. Hydrogen atoms not involved in the hydrogen bonding have been omitted for clarity. A similar structure is found for compound (1) and all the halo-substituted compounds. [Symmetry codes: (i) x + 1, y, z; (ii) x − 1, y, x.]
There is π–π stacking in each compound, involving inversion-related molecules in all compounds, Table 6 ▸.
Table 6. stacking (, ).
Cg1, Cg2, Cg3 and Cg7 [compound (6)] are the centroids of the rings containing atoms O1, C5, C311 and C211 [compound (6)], respectively. In contacts indicated *, the planes involved are inclined to each other, the perpendicular distance between the planes is an average value and the angle between the planes is given in place of a slippage. Only interplanar interactions with Cg Cg distances 4.0 and with angles between the planes of 10 are included.
| Compound | contact | distance | perp. dist. | angle between planes |
|---|---|---|---|---|
| (1) | Cg1Cg3iii | 3.5187(8) | 3.3226* | 1.77(6)* |
| Cg1Cg3iv | 3.543(8) | 3.3719* | 1.77(6)* | |
| (2) | Cg1Cg3v | 3.5341(17) | 3.3573* | 0.77(13)* |
| Cg2Cg3vi | 3.6691(17) | 3.3985* | 3.14(13)* | |
| (3) | Cg1Cg3v | 3.5464(11) | 3.3342* | 4.66(9)* |
| (4) | Cg1Cg3iii | 3.5721(11) | 3.3518* | 5.37(9) |
Symmetry codes: (iii) x+1, y+1, z+1; (iv) x, y+2, z; (v) x+1, y, z+1; (vi) x, y, z.
Synthesis and crystallization
The title compounds were obtained by synthetic strategies described elsewhere (Cagide et al., 2011 ▸). Chromone-3-carboxamides were synthesized using chromone-3-carboxylic acid as starting material which, after in situ activation with phosphorus(V) oxychloride (POCl3) in dimethylformamide, react with the different haloanilines. Recrystallization from dichloromethane afforded colourless plates whose dimensions are given in Table 7 ▸.
Table 7. Experimental details.
| (1) | (2) | (3) | (4) | |
|---|---|---|---|---|
| Crystal data | ||||
| Chemical formula | C16H10FNO3 | C16H10ClNO3 | C16H10BrNO3 | C16H10INO3 |
| M r | 283.25 | 299.70 | 344.16 | 391.15 |
| Crystal system, space group | Triclinic, P
|
Triclinic, P
|
Triclinic, P
|
Triclinic, P
|
| Temperature (K) | 100 | 100 | 120 | 120 |
| a, b, c () | 6.6213(5), 7.0517(5), 14.0864(10) | 6.6325(12), 7.0577(12), 14.671(3) | 6.6505(5), 9.3580(7), 11.0060(8) | 6.6750(5), 9.4166(7), 11.2673(8) |
| , , () | 101.957(7), 90.047(6), 106.657(7) | 103.536(7), 89.714(6), 105.589(7) | 100.280(6), 90.461(6), 100.884(6) | 100.974(6), 90.769(6), 100.062(6) |
| V (3) | 615.17(8) | 641.9(2) | 661.24(9) | 683.77(9) |
| Z | 2 | 2 | 2 | 2 |
| Radiation type | Mo K | Mo K | Mo K | Mo K |
| (mm1) | 0.12 | 0.31 | 3.12 | 2.35 |
| Crystal size (mm) | 0.46 0.32 0.02 | 0.17 0.17 0.04 | 0.58 0.18 0.06 | 0.46 0.22 0.05 |
| Data collection | ||||
| Diffractometer | Rigaku Saturn724+ | Rigaku AFC12 | Rigaku R-AXIS conversion | Rigaku R-AXIS conversion |
| Absorption correction | Multi-scan (CrystalClear-SM Expert; Rigaku, 2012 ▸) | Multi-scan (CrystalClear-SM Expert; Rigaku, 2012 ▸) | Multi-scan (CrystalClear-SM Expert; Rigaku, 2012 ▸) | Multi-scan (CrystalClear-SM Expert; Rigaku, 2012 ▸) |
| T min, T max | 0.949, 0.998 | 0.950, 0.988 | 0.265, 0.835 | 0.411, 0.892 |
| No. of measured, independent and observed [I > 2(I)] reflections | 8176, 2789, 2393 | 7435, 2265, 1668 | 9930, 3017, 2525 | 10032, 3095, 2819 |
| R int | 0.056 | 0.078 | 0.045 | 0.026 |
| (sin /)max (1) | 0.649 | 0.598 | 0.649 | 0.649 |
| Refinement | ||||
| R[F 2 > 2(F 2)], wR(F 2), S | 0.044, 0.135, 1.06 | 0.056, 0.145, 0.99 | 0.027, 0.058, 0.94 | 0.018, 0.044, 1.03 |
| No. of reflections | 2789 | 2265 | 3017 | 3095 |
| No. of parameters | 194 | 194 | 194 | 194 |
| H-atom treatment | H atoms treated by a mixture of independent and constrained refinement | H atoms treated by a mixture of independent and constrained refinement | H atoms treated by a mixture of independent and constrained refinement | H atoms treated by a mixture of independent and constrained refinement |
| max, min (e 3) | 0.41, 0.27 | 0.30, 0.65 | 0.53, 0.69 | 0.67, 0.32 |
Refinement
Crystal data, data collection and structure refinement details are summarized in Table 7 ▸. Amino H atoms were located in difference Fourier maps and were refined isotropically. All other H atoms were treated as riding atoms with C—H(aromatic) = 0.95 Å, U iso= 1.2Ueq(C).
Compounds (1) and (2), reduced cell: [a = 6.6325 (12), b = 0.0577 (12), c = 14.671 (3) Å, α = 76.464 (7), β = 89.714 (6), γ = 74.411 (7)°, V = 641.9 (2) Å3], have different reduced cells in which the x and z coordinates are comparable and the y coordinate of (2) is close to 1 − y of (1). For ease of comparison of the structures of (1) and (2), the refinement reported here was carried out for the non-reduced cell of (2) in which the α and γ angles were given the supplementary values of those of the reduced unit cell. The coordinates of (1) were used as starting values and the transformation matrix for the reduced to non-reduced cell was 0 0 0 1 0 0 0 . This gave the same final refinement values as those for the refinement with the reduced cell. Compounds (1) and (2) are therefore isostructural.
Supplementary Material
Crystal structure: contains datablock(s) 1, 2, 3, 4, global. DOI: 10.1107/S2056989014027054/lh5743sup1.cif
Structure factors: contains datablock(s) 1. DOI: 10.1107/S2056989014027054/lh57431sup2.hkl
Structure factors: contains datablock(s) 2. DOI: 10.1107/S2056989014027054/lh57432sup3.hkl
Structure factors: contains datablock(s) 3. DOI: 10.1107/S2056989014027054/lh57433sup4.hkl
Structure factors: contains datablock(s) 4. DOI: 10.1107/S2056989014027054/lh57434sup5.hkl
Supporting information file. DOI: 10.1107/S2056989014027054/lh57431sup6.cml
Supporting information file. DOI: 10.1107/S2056989014027054/lh57432sup7.cml
Supporting information file. DOI: 10.1107/S2056989014027054/lh57433sup8.cml
Supporting information file. DOI: 10.1107/S2056989014027054/lh57434sup9.cml
Additional supporting information: crystallographic information; 3D view; checkCIF report
Acknowledgments
The authors thank the National Crystallographic Service, University of Southampton for the data collection, (3a) and (3c), and for their help and advice (Coles & Gale, 2012 ▸). Thanks are also due the Foundation for Science and Technology (FCT) of Portugal (PEst-C/QUI/UI0081/2013). FC’s (SFRH/BPD/74491/2010) grant is also supported by the FCT.
supplementary crystallographic information
Crystal data
| C16H10INO3 | Z = 2 |
| Mr = 391.15 | F(000) = 380 |
| Triclinic, P1 | Dx = 1.900 Mg m−3 |
| a = 6.6750 (5) Å | Mo Kα radiation, λ = 0.71075 Å |
| b = 9.4166 (7) Å | Cell parameters from 9236 reflections |
| c = 11.2673 (8) Å | θ = 1.8–27.5° |
| α = 100.974 (6)° | µ = 2.35 mm−1 |
| β = 90.769 (6)° | T = 120 K |
| γ = 100.062 (6)° | Plate, colourless |
| V = 683.77 (9) Å3 | 0.46 × 0.22 × 0.05 mm |
Data collection
| Rigaku RAXIS conversion diffractometer | 3095 independent reflections |
| Radiation source: Sealed Tube | 2819 reflections with I > 2σ(I) |
| Graphite Monochromator monochromator | Rint = 0.026 |
| Detector resolution: 10.0000 pixels mm-1 | θmax = 27.5°, θmin = 2.2° |
| profile data from ω–scans | h = −7→8 |
| Absorption correction: multi-scan (CrystalClear-SM Expert; Rigaku, 20112) | k = −12→11 |
| Tmin = 0.411, Tmax = 0.892 | l = −14→14 |
| 10032 measured reflections |
Refinement
| Refinement on F2 | 0 restraints |
| Least-squares matrix: full | Hydrogen site location: mixed |
| R[F2 > 2σ(F2)] = 0.018 | H atoms treated by a mixture of independent and constrained refinement |
| wR(F2) = 0.044 | w = 1/[σ2(Fo2) + (0.026P)2] where P = (Fo2 + 2Fc2)/3 |
| S = 1.03 | (Δ/σ)max = 0.002 |
| 3095 reflections | Δρmax = 0.67 e Å−3 |
| 194 parameters | Δρmin = −0.32 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| I314 | 0.09431 (2) | 0.28556 (2) | 0.97093 (2) | 0.02278 (5) | |
| O1 | 0.88205 (19) | 0.94124 (14) | 0.35573 (11) | 0.0188 (3) | |
| O3 | 0.7838 (2) | 0.68549 (15) | 0.60794 (12) | 0.0219 (3) | |
| O4 | 0.28854 (19) | 0.79085 (14) | 0.42255 (11) | 0.0199 (3) | |
| N3 | 0.4362 (2) | 0.65097 (16) | 0.58280 (13) | 0.0152 (3) | |
| H3 | 0.337 (3) | 0.678 (2) | 0.5384 (19) | 0.017 (5)* | |
| C2 | 0.8340 (3) | 0.85191 (19) | 0.43445 (15) | 0.0166 (3) | |
| H2 | 0.9434 | 0.8230 | 0.4731 | 0.020* | |
| C3 | 0.6432 (3) | 0.79929 (17) | 0.46363 (14) | 0.0144 (3) | |
| C4 | 0.4692 (3) | 0.83443 (18) | 0.40253 (14) | 0.0144 (3) | |
| C4A | 0.5235 (3) | 0.92875 (18) | 0.31366 (14) | 0.0149 (3) | |
| C5 | 0.3733 (3) | 0.9701 (2) | 0.24576 (16) | 0.0193 (4) | |
| H5 | 0.2334 | 0.9352 | 0.2557 | 0.023* | |
| C6 | 0.4280 (3) | 1.0611 (2) | 0.16480 (16) | 0.0221 (4) | |
| H6 | 0.3257 | 1.0865 | 0.1176 | 0.026* | |
| C7 | 0.6338 (3) | 1.1164 (2) | 0.15168 (16) | 0.0217 (4) | |
| H7 | 0.6696 | 1.1811 | 0.0971 | 0.026* | |
| C8 | 0.7853 (3) | 1.0777 (2) | 0.21751 (16) | 0.0211 (4) | |
| H8 | 0.9249 | 1.1154 | 0.2094 | 0.025* | |
| C8A | 0.7265 (3) | 0.98182 (19) | 0.29605 (15) | 0.0167 (3) | |
| C311 | 0.3707 (3) | 0.56515 (18) | 0.66915 (14) | 0.0147 (3) | |
| C312 | 0.5034 (3) | 0.52301 (19) | 0.74728 (15) | 0.0170 (3) | |
| H312 | 0.6466 | 0.5501 | 0.7426 | 0.020* | |
| C313 | 0.4242 (3) | 0.4409 (2) | 0.83219 (15) | 0.0183 (3) | |
| H313 | 0.5134 | 0.4119 | 0.8857 | 0.022* | |
| C314 | 0.2148 (3) | 0.40150 (18) | 0.83828 (15) | 0.0167 (3) | |
| C315 | 0.0823 (3) | 0.44141 (19) | 0.75977 (15) | 0.0178 (3) | |
| H315 | −0.0608 | 0.4127 | 0.7637 | 0.021* | |
| C316 | 0.1603 (3) | 0.52325 (19) | 0.67578 (15) | 0.0171 (3) | |
| H316 | 0.0702 | 0.5512 | 0.6221 | 0.020* | |
| C31 | 0.6294 (3) | 0.70661 (18) | 0.55846 (15) | 0.0154 (3) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| I314 | 0.02679 (8) | 0.02415 (7) | 0.01880 (6) | 0.00303 (5) | 0.00371 (4) | 0.00890 (4) |
| O1 | 0.0120 (6) | 0.0209 (6) | 0.0242 (6) | 0.0014 (5) | 0.0022 (5) | 0.0074 (5) |
| O3 | 0.0143 (6) | 0.0284 (7) | 0.0258 (7) | 0.0053 (5) | −0.0013 (5) | 0.0108 (5) |
| O4 | 0.0116 (6) | 0.0256 (7) | 0.0248 (6) | 0.0024 (5) | 0.0016 (5) | 0.0116 (5) |
| N3 | 0.0136 (7) | 0.0169 (7) | 0.0164 (7) | 0.0040 (6) | −0.0002 (5) | 0.0057 (5) |
| C2 | 0.0139 (8) | 0.0165 (8) | 0.0192 (8) | 0.0035 (6) | −0.0004 (6) | 0.0027 (6) |
| C3 | 0.0138 (8) | 0.0129 (8) | 0.0158 (7) | 0.0027 (6) | 0.0005 (6) | 0.0005 (6) |
| C4 | 0.0132 (8) | 0.0146 (8) | 0.0151 (7) | 0.0027 (6) | 0.0006 (6) | 0.0018 (6) |
| C4A | 0.0157 (9) | 0.0141 (8) | 0.0146 (7) | 0.0031 (6) | 0.0021 (6) | 0.0017 (6) |
| C5 | 0.0169 (9) | 0.0217 (9) | 0.0195 (8) | 0.0036 (7) | 0.0019 (6) | 0.0045 (6) |
| C6 | 0.0260 (10) | 0.0246 (9) | 0.0186 (8) | 0.0088 (8) | 0.0007 (7) | 0.0078 (7) |
| C7 | 0.0283 (10) | 0.0197 (9) | 0.0198 (8) | 0.0066 (7) | 0.0080 (7) | 0.0078 (6) |
| C8 | 0.0202 (10) | 0.0193 (9) | 0.0236 (9) | 0.0024 (7) | 0.0066 (7) | 0.0047 (7) |
| C8A | 0.0161 (9) | 0.0155 (8) | 0.0181 (8) | 0.0035 (6) | 0.0026 (6) | 0.0016 (6) |
| C311 | 0.0166 (9) | 0.0127 (8) | 0.0145 (7) | 0.0034 (6) | 0.0011 (6) | 0.0014 (6) |
| C312 | 0.0147 (9) | 0.0183 (8) | 0.0186 (8) | 0.0041 (7) | −0.0002 (6) | 0.0038 (6) |
| C313 | 0.0199 (9) | 0.0198 (8) | 0.0164 (8) | 0.0065 (7) | −0.0023 (6) | 0.0038 (6) |
| C314 | 0.0203 (9) | 0.0148 (8) | 0.0149 (7) | 0.0018 (7) | 0.0026 (6) | 0.0039 (6) |
| C315 | 0.0148 (9) | 0.0199 (9) | 0.0183 (8) | 0.0030 (7) | 0.0018 (6) | 0.0029 (6) |
| C316 | 0.0164 (9) | 0.0171 (8) | 0.0184 (8) | 0.0055 (7) | −0.0014 (6) | 0.0027 (6) |
| C31 | 0.0161 (9) | 0.0140 (8) | 0.0157 (7) | 0.0036 (6) | 0.0007 (6) | 0.0010 (6) |
Geometric parameters (Å, º)
| I314—C314 | 2.1023 (17) | C6—C7 | 1.401 (3) |
| O1—C2 | 1.340 (2) | C6—H6 | 0.9500 |
| O1—C8A | 1.376 (2) | C7—C8 | 1.385 (3) |
| O3—C31 | 1.229 (2) | C7—H7 | 0.9500 |
| O4—C4 | 1.243 (2) | C8—C8A | 1.394 (2) |
| N3—C31 | 1.357 (2) | C8—H8 | 0.9500 |
| N3—C311 | 1.406 (2) | C311—C312 | 1.397 (2) |
| N3—H3 | 0.92 (2) | C311—C316 | 1.398 (3) |
| C2—C3 | 1.352 (2) | C312—C313 | 1.395 (3) |
| C2—H2 | 0.9500 | C312—H312 | 0.9500 |
| C3—C4 | 1.460 (2) | C313—C314 | 1.388 (3) |
| C3—C31 | 1.497 (2) | C313—H313 | 0.9500 |
| C4—C4A | 1.469 (2) | C314—C315 | 1.389 (2) |
| C4A—C8A | 1.390 (2) | C315—C316 | 1.383 (2) |
| C4A—C5 | 1.404 (2) | C315—H315 | 0.9500 |
| C5—C6 | 1.377 (3) | C316—H316 | 0.9500 |
| C5—H5 | 0.9500 | ||
| C2—O1—C8A | 118.48 (14) | C7—C8—H8 | 121.0 |
| C31—N3—C311 | 128.55 (15) | C8A—C8—H8 | 121.0 |
| C31—N3—H3 | 114.0 (15) | O1—C8A—C4A | 121.30 (15) |
| C311—N3—H3 | 117.4 (15) | O1—C8A—C8 | 116.04 (16) |
| O1—C2—C3 | 125.61 (16) | C4A—C8A—C8 | 122.66 (17) |
| O1—C2—H2 | 117.2 | C312—C311—C316 | 119.80 (16) |
| C3—C2—H2 | 117.2 | C312—C311—N3 | 123.60 (16) |
| C2—C3—C4 | 119.41 (15) | C316—C311—N3 | 116.59 (15) |
| C2—C3—C31 | 115.53 (15) | C313—C312—C311 | 119.53 (17) |
| C4—C3—C31 | 125.07 (15) | C313—C312—H312 | 120.2 |
| O4—C4—C3 | 124.22 (16) | C311—C312—H312 | 120.2 |
| O4—C4—C4A | 121.28 (16) | C314—C313—C312 | 119.87 (16) |
| C3—C4—C4A | 114.49 (15) | C314—C313—H313 | 120.1 |
| C8A—C4A—C5 | 118.05 (16) | C312—C313—H313 | 120.1 |
| C8A—C4A—C4 | 120.59 (16) | C313—C314—C315 | 120.84 (16) |
| C5—C4A—C4 | 121.35 (16) | C313—C314—I314 | 120.05 (13) |
| C6—C5—C4A | 120.26 (18) | C315—C314—I314 | 119.08 (13) |
| C6—C5—H5 | 119.9 | C316—C315—C314 | 119.42 (17) |
| C4A—C5—H5 | 119.9 | C316—C315—H315 | 120.3 |
| C5—C6—C7 | 120.37 (18) | C314—C315—H315 | 120.3 |
| C5—C6—H6 | 119.8 | C315—C316—C311 | 120.53 (16) |
| C7—C6—H6 | 119.8 | C315—C316—H316 | 119.7 |
| C8—C7—C6 | 120.65 (17) | C311—C316—H316 | 119.7 |
| C8—C7—H7 | 119.7 | O3—C31—N3 | 124.75 (16) |
| C6—C7—H7 | 119.7 | O3—C31—C3 | 121.00 (16) |
| C7—C8—C8A | 117.95 (17) | N3—C31—C3 | 114.25 (15) |
| C8A—O1—C2—C3 | −1.7 (2) | C4—C4A—C8A—C8 | −176.55 (16) |
| O1—C2—C3—C4 | 3.0 (3) | C7—C8—C8A—O1 | 177.28 (15) |
| O1—C2—C3—C31 | −177.33 (15) | C7—C8—C8A—C4A | −2.4 (3) |
| C2—C3—C4—O4 | 179.64 (16) | C31—N3—C311—C312 | 0.3 (3) |
| C31—C3—C4—O4 | 0.0 (3) | C31—N3—C311—C316 | 179.48 (16) |
| C2—C3—C4—C4A | −0.9 (2) | C316—C311—C312—C313 | −0.7 (2) |
| C31—C3—C4—C4A | 179.46 (15) | N3—C311—C312—C313 | 178.37 (16) |
| O4—C4—C4A—C8A | 177.21 (15) | C311—C312—C313—C314 | 0.1 (3) |
| C3—C4—C4A—C8A | −2.3 (2) | C312—C313—C314—C315 | 0.8 (3) |
| O4—C4—C4A—C5 | −1.7 (3) | C312—C313—C314—I314 | −177.36 (13) |
| C3—C4—C4A—C5 | 178.81 (15) | C313—C314—C315—C316 | −1.0 (3) |
| C8A—C4A—C5—C6 | −0.2 (3) | I314—C314—C315—C316 | 177.17 (12) |
| C4—C4A—C5—C6 | 178.65 (16) | C314—C315—C316—C311 | 0.3 (3) |
| C4A—C5—C6—C7 | −1.7 (3) | C312—C311—C316—C315 | 0.5 (3) |
| C5—C6—C7—C8 | 1.6 (3) | N3—C311—C316—C315 | −178.64 (15) |
| C6—C7—C8—C8A | 0.4 (3) | C311—N3—C31—O3 | 2.5 (3) |
| C2—O1—C8A—C4A | −1.8 (2) | C311—N3—C31—C3 | −177.41 (15) |
| C2—O1—C8A—C8 | 178.47 (15) | C2—C3—C31—O3 | 2.3 (2) |
| C5—C4A—C8A—O1 | −177.34 (15) | C4—C3—C31—O3 | −178.01 (15) |
| C4—C4A—C8A—O1 | 3.7 (2) | C2—C3—C31—N3 | −177.74 (14) |
| C5—C4A—C8A—C8 | 2.4 (3) | C4—C3—C31—N3 | 1.9 (2) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N3—H3···O4 | 0.92 (2) | 1.89 (2) | 2.6977 (19) | 145 (2) |
| C2—H2···O3 | 0.95 | 2.33 | 2.718 (2) | 104 |
| C312—H312···O3 | 0.95 | 2.27 | 2.881 (2) | 122 |
| C2—H2···O4i | 0.95 | 2.44 | 3.185 (2) | 136 |
| C316—H316···O3ii | 0.95 | 2.49 | 3.312 (2) | 145 |
Symmetry codes: (i) x+1, y, z; (ii) x−1, y, z.
References
- Cagide, F., Reis, J., Gaspar, A. & Borges, F. (2011). Tetrahedron Lett. 52, 6446–6449.
- Coles, S. J. & Gale, P. A. (2012). Chem. Sci. 3, 683–689.
- Gaspar, A., Matos, M. J., Garrido, J., Uriarte, E. & Borges, F. (2014). Chem. Rev. 114, 4960–4992. [DOI] [PubMed]
- Gaspar, A., Reis, J., Fonseca, A., Milhazes, N., Viña, D., Uriarte, E. & Borges, F. (2011). Bioorg. Med. Chem. Lett. 21, 707–709. [DOI] [PubMed]
- Gaspar, A., Silva, T., Yáñez, M., Vina, D., Orallo, F., Ortuso, F., Uriarte, E., Alcaro, S. & Borges, F. (2011). J. Med. Chem. 54, 5165–5173. [DOI] [PubMed]
- Gomes, L. R., Low, J. N., Cagide, F., Gaspar, A., Reis, J. & Borges, F. (2013). Acta Cryst. B69, 294–309. [DOI] [PubMed]
- Helguera, A. M., Pérez-Garrido, A., Gaspar, A., Reis, J., Cagide, F., Vina, D., Cordeiro, M. N. D. S. & Borges, F. (2013). Eur. J. Med. Chem. 59, 75–90. [DOI] [PubMed]
- Hübschle, C. B., Sheldrick, G. M. & Dittrich, B. (2011). J. Appl. Cryst. 44, 1281–1284. [DOI] [PMC free article] [PubMed]
- Macrae, C. F., Edgington, P. R., McCabe, P., Pidcock, E., Shields, G. P., Taylor, R., Towler, M. & van de Streek, J. (2006). J. Appl. Cryst. 39, 453–457.
- McArdle, P., Gilligan, K., Cunningham, D., Dark, R. & Mahon, M. (2004). CrystEngComm, 6, 30–309.
- Oszlányi, G. & Sütő, A. (2004). Acta Cryst. A60, 134–141. [DOI] [PubMed]
- Reis, J., Gaspar, A., Borges, F., Gomes, L. R. & Low, J. N. (2013). Acta Cryst. C69, 1527–1533. [DOI] [PubMed]
- Rigaku (2012). CrystalClear-SM Expert. Rigaku Corporation, Tokyo, Japan.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) 1, 2, 3, 4, global. DOI: 10.1107/S2056989014027054/lh5743sup1.cif
Structure factors: contains datablock(s) 1. DOI: 10.1107/S2056989014027054/lh57431sup2.hkl
Structure factors: contains datablock(s) 2. DOI: 10.1107/S2056989014027054/lh57432sup3.hkl
Structure factors: contains datablock(s) 3. DOI: 10.1107/S2056989014027054/lh57433sup4.hkl
Structure factors: contains datablock(s) 4. DOI: 10.1107/S2056989014027054/lh57434sup5.hkl
Supporting information file. DOI: 10.1107/S2056989014027054/lh57431sup6.cml
Supporting information file. DOI: 10.1107/S2056989014027054/lh57432sup7.cml
Supporting information file. DOI: 10.1107/S2056989014027054/lh57433sup8.cml
Supporting information file. DOI: 10.1107/S2056989014027054/lh57434sup9.cml
Additional supporting information: crystallographic information; 3D view; checkCIF report