

Abstract
The beta2-adrenergic receptor (β2AR) family, which is the largest family of cell surface receptors in humans. Extra attention has been focused on the human GPCRs because they have been studied as important protein targets for pharmaceutical drug development. In fact, approximately 40% of marketed drugs directly work on GPCRs. GPCRs respond to various extracellular stimuli, such as sensory signals, neurotransmitters, chemokines, and hormones, to induce structural changes at the cytoplasmic surface, activating downstream signaling pathways, primarily through interactions with heterotrimeric G proteins or through G-protein independent pathways, such as arrestin. Most GPCRs, except for rhodhopsin, which contains covalently linked 11 cis-retinal, bind to diffusible ligands, having various conformational states between inactive and active structures. The first human GPCR structure was determined using an inverse agonist bound β2AR in 2007 and since then, more than 20 distinct GPCR structures have been solved. However, most GPCR structures were solved as inactive forms, and an agonist bound fully active structure is still hard to obtain. In a structural point of view, β2AR is relatively well studied since its fully active structure as a complex with G protein as well as several inactive structures are available. The structural comparison of inactive and active states gives an important clue in understanding the activation mechanism of β2AR. In this review, structural features of inactive and active states of β2AR, the interaction of β2AR with heterotrimeric G protein, and the comparison with β1AR will be discussed.
Keywords: beta2-adrenergic receptor (β2AR), conformational change, crystal structure, G-protein coupled receptor (GPCR), heterotrimeric G protein
OVERALL STRUCTURE OF BETA2-ADRENERGIC RECEPTOR
Based on sequence similarity, GPCRs can be divided into four classes: class A, B, C, and F. The majority of GPCRs belong to class A, also referred to as rhodopsin type GPCRs. In 2000, the first GPCR structure was visualized by using bovine rhodopsin complexed with 11-cis-retinal and this structure has been used as an important template for GPCR modeling (Palczewski et al., 2000). The overall rhodopsin structure consists of seven transmembrane (TM) helices and three loop regions at the extracellular and the cytoplasmic sides. The ligand binding pocket of rhodopsin is formed by hydrophobic residues from TM5 and TM6 to stabilize the hydrocarbone backbone of retinal, which is covalently bound to Lys296 of TM7.
One of the common sequence motifs in rhodopsin type GPCRs is the D[E]RY motif on TM3, which forms an ionic lock by making a salt bridge between Arg3.50 of the D[E]RY motif and Asp/Glu6.30 of TM6. The ionic lock was suggested as a characteristic of inactive conformation of GPCR, to block the G protein binding at the cytoplasmic region. The other common motif is the NPXXY motif on TM7. In contrast to the ionic lock, which stabilizes inactive conformation, it has been suggested to play an important role in GPCR activation. Although rhodopsin structure provided the first structural aspects of GPCR, it was suggested that most other GPCRs, which interact with diffusible ligands with different efficacy, would have different structural features from rhodopsin since rhodopsin has a covalently bound ligand.
Human β2AR was first identified in the 1990s but its structural study hadn’t begun until 2007. Unlike rhodopsin, β2AR shows conformational instability, suggested by its agonist independent basal activity. Also, β2AR has a much longer flexible intracellular loop 3 (IL3), which could be an obstacle for crystallization. A high affinity inverse agonist, carazolol, was used to stabilize the inactive conformation of β2AR and the flexibility of IL3 was reduced by making a complex with IL3-specific Fab fragment or by replacing it with T4 lysozyme (T4L) (Fig. 1A) (Cherezov et al., 2007; Rosenbaum et al., 2007; Rasmussen et al., 2007). Although the β2AR-Fab complex structure didn’t resolve the carazolol binding site, the native conformation around IL3 showed that the ionic lock was broken in its inactive structure, explaining why β2AR shows basal activity even in the presence of an inverse agonist. High resolution structure of β2AR with T4L fusion was obtained from the crystals in lipid cubic phase (LCP). The T4L region greatly facilitates crystallization by making favorable crystal packing interaction of T4L with the extracellular loop regions of neighboring β2AR. LCP crystallization method and T4L fusion strategy are now commonly used for GPCR structure determination. High resolution structure of β2AR provides invaluable information on the ligand binding site of β2AR. Carazolol forms hydrogen bonding inter actions with Asp1133.32, Asn3127.39, and Tyr3167.43 and has hydrophobic interactions with Val1143.33, Phe2906.52, and Phe1935.32 of β2AR (Fig. 1B). Its binding site is partially overlapped with that of retinal in rhodopsin. The β-ionone ring of retinal probes deep inside rhodopsin to interact with Trp2866.48, which is known as the “toggle switch” for receptor activation. In contrast, carazolol cannot reach deep enough to interact with the toggle switch. The ligand binding site of β2AR is relatively open to the solvent which enables a ligand to diffuse in and out easily. In the case of retinal-bound rhodopsin, direct access to the ligand binding site is restricted by extracellular loop 2 (ECL2), which forms a β sheet above the retinal binding site by interacting with the N-terminus. ECL2 in β2AR, which doesn’t make any direct contact with the N-terminus, contains an alpha helix and a disulfide bond between Cys1844.76 and Cys1905.29. Another disulfide bond between Cys1915.30 and Cys1063.25 from TM3 contributes to the stabilization of ECL2. A 2.4Å resolution structure of T4L fusion of β2AR clearly showed water mediated hydrogen bonding network between TM residues. These water-filled, loosely packed regions may allow for conformational changes upon activation.
Fig. 1.

Carazolol bound inactive structures of β2AR. (A) Crystal structures of carazolol bound β2AR with T4L fusion (left) and that complexed with Fab (right) are shown in green and cyan, respectively (pdb ID: 2RH1, 2R4R). T4L and Fab are colored in grey and carazolol is shown as yellow spheres. (B) Close view of the carazolol binding site in β2AR-T4L. Carazolol is shown as yellow sticks and β2AR amino acids making polar and hydrophobic interactions are labeled in black and blue, respectively.
Other inactive β2AR structures in a complex with the partial inverse agonist, timolol or antagonist, alprenolol, have been reported and their overall structural folds are maintained with minor structural rearrangements of the ligand binding site to accommodate different chemical properties of ligands (Hanson et al., 2008; Wacker et al., 2010). While the hydrogen bond network with Asp1133.32, Asn3127.39, and Tyr3167.43 of β2AR is conserved in the interactions of carazolol, timolol and alprenolol, additional hydrogen bonding interactions and hydrophobic interactions are varied for each ligand, which could be related to the strength of inverse agonism or antagonism.
AGONIST INDUCED CONFORMATIONAL CHANGE OF BETA2-ADRENERGIC RECEPTOR
Since the inactive structure of β2AR was first reported in 2007, lots of effort had been made to determine the agonist bound active conformation of β2AR. One of the approaches was to design the covalently bound agonist to stabilize the agonist bound active form of β2AR. For this purpose, Cys was incorporated into residue 93 of β2AR instead of His, to make a disulfide bond with an agonist, FAUC50 (Rosenbaum et al., 2011). Structural study of β2AR with the covalently linked agonist discovered an interesting result that the agonist alone was not sufficient to stabilize the active conformation of β2AR, which was unexpected since the structure of metarhodopsin II showed the active state conformation, like outward movement of the cytopalsmic end of TM6, in the absence of a cytoplasmic binding partner. In 2011, two active structures of β2AR bound to a high affinity agonist (BI-167107) were determined using either Nb80 (nanobody 80) or Gs protein bound to the cytoplasmic side of β2AR (Figs. 2A and 2B) (Rasmussen et al., 2011a; 2011b). RMSD evaluation found that the structural difference between Nb80 bound and Gs bound β2AR was minimal (Fig. 2C). Detailed structural analysis of G protein bound β2AR will be discussed later.
Fig. 2.

Agonist bound active structures of β2AR. (A) The structure of active conformation of β2AR with strong agonist, BI-167107, stabilized by Nb80 is shown in orange, with BI-167107 in yellow and Nb80 in grey (pdb ID: 3P0G). (B) The overall structure of the β2AR-Gs complex, omitting T4L at the N-terminus of β2AR and Nb35 complexed with this complex to stabilize Gs (pdb ID: 3SN6). Gαs, Gβ, and Gγ are colored in blue, light purple and purple, respectively. (C) Two structures of active β2AR, complexed with Nb80 and Gs, are superimposed.
The comparison between a carazolol bound inactive structure and an agonist bound active structure shows that only little changes occur on the extracellular side of the receptor. In fact, the interaction pattern in the agonist-binding pocket differs only slightly between carazolol and BI-167107. The key change appears to be in the interaction with Ser2045.43 and Ser2075.46 on TM5. The hydrogen bonding between BI-167107 and the polar pocket residues, including the two serines, causes a 2Å inward movement of TM5 at position Ser2075.46, resulting in the rearrangement of the hydrophobic interaction network that Pro2115.50 forms with Ile1213.40 and Phe2826.44, causing the cytoplasmic end of TM6 to swing outward. The outward movement of TM6 is the largest change on the cytoplasmic side of β2AR that was brought on by the agonist binding and its outward displacement measures to be about 11Å in a nanobody bound structure and 14Å in a Gs protein-bound structure (Fig. 3). Its movement is accompanied by the outward movement of TM5 and a slight inward adjustment in the position of TM3 and TM7 to accommodate space for the interaction with nanobody or Gs protein. The outward movement of TM6 was observed in two opsin structures, in the absence and presence of the carboxy terminus of the Gα-subunit of transducin (Gαt).
Fig. 3.

Comparison of inactive and active structures of β2AR. The active conformation of β2AR from β2AR-Gs complex is colored in gold and the carazolol bound inactive structure is shown in green. Side and cytoplasmic views show outward movement of the cytoplasmic end of TM6, creating an opening for the interaction with G protein.
However, it should be taken into consideration that the crystal structures only show the most thermodynamically stable endpoint structures of agonist induced β2AR transformation. The crystal structure may be biased to one possible conformation out of many and the actual structural change that takes place as agonist binds is expected to be more dynamic than a rigid two-state model of activation and inactivation. Although the FAUC50 bound structure did not represent the fully active conformation, it showed hydrogen bonding interaction between the agonist and Ser 2035.42 and Ser 2075.46 on TM5, as seen in the BI-167107 ligand bound active structure. However, this interaction is not propagated to induce large structural changes at the cytoplasmic region, proposing the existence of multiple intermediate states between inactive and active structures.
Crystallographic study is not good enough to understand the dynamic structural features of β2AR and other biophysical analyses using NMR, HDX-MS and DEER spectroscopy have been implemented to elucidate the further details of the activation mechanism of β2AR. In 2013, Nygaard et al. (2013) used 13CH3ε-Met NMR spectroscopy to study the conformational change that occurs as the agonist binds. The NMR results generally agree with the crystal structures of β2AR but they give us more insight into the dynamics. Analysis of HSQC spectra of β2AR either with BI-167107 bound alone or with BI-167107 and Nb80 showed that even a strong agonist like BI-167107 was not enough to stabilize the active state, producing heterogeneous states of β2AR. Molecular dynamics (MD) simulation of an Nb80 bound active structure of β2AR showed that removal of Nb80 caused the structural transition into an inactive like conformation after 11 μs and inactive state was stably kept for a 30 μs simulation. These results explain why the crystal structure of the active state could not be obtained with the agonist alone. The suggested conformational link of Ile1213.40/Phe2826.44 between the agonist binding pocket and the cytoplasmic side was observed to be not very strong. Based on these findings, it appears that while BI-167107 binding destabilizes the inactive conformation and makes β2AR switch back and forth from inactive to active, thereby making the receptor more thermodynamically available for the activation, the interaction with Nb80 or possibly Gs protein at the cytoplasmic region finally leads to the active conformation.
After the success of capturing the active state of β2AR using a nanobody, another crystal structure of the active state β2AR bound to the relatively low-affinity endogenous agonist adrenaline was determined using an engineered high affinity nanobody (Nb6B9) (Ring et al., 2013). Nb6B9 was developed by a directed evolution method to improve the affinity of the original nanobody, Nb80. The comparison of the active structures of β2AR bound to two different agonists, adrenaline and BI-167107, which does not have catechol moiety, showed that the differences in the ligand size and chemical properties did not change the overall structure of β2AR. The smaller catechol ring of adrenaline induced a shift in the position of Asn2936.55 to maintain the hydrogen bonding interaction. However, the overall conformation change, that is, the outward movement of the cytoplasmic part of TM6, followed by repacking of the side chains of the transmembrane is preserved in adrenaline bound β2AR.
The diversity of the agonists makes the study of β2AR activation even more challenging. Recent work with MD simulations and mass spectroscopy on the ligand specificity of the receptor suggests that the different ligands form different hydrogen bonding network with β2AR and the key residues involved in the interaction may also vary. More detailed account of conformational change during activation will have to be worked on further.
INTERACTION WITH G PROTEIN
G protein is a heterotrimeric protein with Gα, Gβ and Gγ subunits. Among these, only Gα has been shown to make direct contact with β2AR. Gα consists of two domains: GαsRas, related to Ras family, and GαsAH, a small globular domain with α helices. The interface between the two forms the guanidine binding site, surrounded by p-loop, switch I, switch II and switch III motifs. It is not clear whether the inactive G protein which is bound to GDP, is precoupled to β2AR before agonist binds but it is known that the agonist binding to β2AR makes the receptor more thermodynamically available for G protein binding. The activated β2AR of the complex with G protein induces a conformational change in the G protein to release GDP and bind GTP. The GTP bound Gα subunit dissociates from Gβγ subunits and the separate Gα and Gβγ subunits interact with effectors, such as adenylate cyclase and calcium channel, to propagate the GPCR signaling.
A long waited complex structure of agonist bound active β2AR and Gs was published in 2011 (Rasmussen et al., 2011b). It shows the interaction between activated β2AR and nucleotide-free Gs protein. Interestingly, it was observed that GαsAH is largely displaced with respect to GαsRas in nucleotide-free state compared to nucleotide-bound state (Fig. 4A). Although the crystal structure shows GαsAH in only one orientation, the displacement is likely to be more flexible as no nucleotide is present to hold the two domains together. The most noticeable characteristic at the interface between G protein and β2AR is the α5 helix of GαsRas domain. The carboxy end of α5 helix is clearly pushed more into the transmembrane core of the receptor when Gs is activated (Fig. 4B). Fusing this C-terminal end of Gs to the receptor was enough to mimic the increased agonist affinity of the β2AR-Gs complex, further reinforcing the fact that the helix is the key motif to initiate the interaction with β2AR. The importance of this helix was well established previously by mutational studies.
Fig. 4.

Interaction of heterotrimeric Gs with the cytoplasmic region of agonist bound β2AR. (A) The crystal structure of Gαs in a complex with guanosine 5′-O-(3-thio-triphosphate) (GTPγS) (pdb ID = 1AZT) is colored in pink and GTPγS is shown as blue and red spheres. The β2AR-Gs complex is colored as in Fig. 3B and only the cytoplasmic region of β2AR and Gαs are shown for simplicity. Structural alignment of Gαs shows that GαsAH is largely displaced with respect to GαsRas in the nucleotide-free state compared to the nucleotide-bound state. (B) The interface between the G protein and β2AR is shown. The carboxy end of the α5 helix, circled in red, is clearly pushed into the trans-membrane pocket of β2AR. In contrast, Gβγ subunits do not make direct contact with β2AR.
Dynamic view of the interaction was also obtained from hydrogen-deuterium exchange mass spectrometry (HDX-MS) and single-particle electron microscopy (EM) whose data agree well with the crystal structure (Chung et al., 2011). EM, again shows the separation of GαsAH from GαsRas when the nucleotide or its substituent is absent. The high flexibility of the GαsAH is also suggested by HDX-MS data. HDX-MS measures the deuterium-hydrogen exchange rate to see how exposed the surface is. The exchange rate at the interface between the GαsRas and the GαsAH domain, including the nucleotide-binding site, increases in the loss of GDP. HDX-MS also showed a large increase in the exchange rate in the β1-strand, a feature that was not evident in the crystal structure. The conformation of β1-strand did not appear to alter much in the crystal. The β1-strand interacts with the ICL2 of the activated receptor and is expected to have a role in linking the interaction with the receptor and the release of the nucleotide through P-loop. The well conserved R31KKK45 motif that forms the hydrogen-bonding with β1-strand is disturbed when the β2AR-Gs complex forms. To generalize the idea that the large structural change of GαsAH happens upon binding to activated GPCR, more structural data of G protein bound active GPCR should be required.
STRUCTURAL COMPARISON WITH BETA1-ADRENERGIC RECEPTOR
The beta adrenergic receptor family includes 3 different subtypes, β1AR, β2AR, and β3AR. Turkey β1AR structure was first determined in 2008 using a thermostable mutant (Warne et al., 2008). Since then, several β1AR structures have been determined with various antagonists, partial-antagonists or the agonists bound and most recently oligomeric ligand-free structure was published (Huang et al., 2013; Moukhametzianov et al., 2011; Warne et al., 2011; 2012). The protein sequence identity between human β2AR and β1AR is about 67% in the TM regions. As expected from high sequence similarity, the overall structures of the two receptors are very similar. The sequence identity of amino acids constituting the ligand-binding pocket is also very high, although the two receptors still exhibit different ligand specificity and function. Based on current structural data, the structural basis for this difference is subtle at the extracellular region including the ligand binding site. More differences can be observed on the intracellular side, especially, ICL2 (Fig. 5).
Fig. 5.
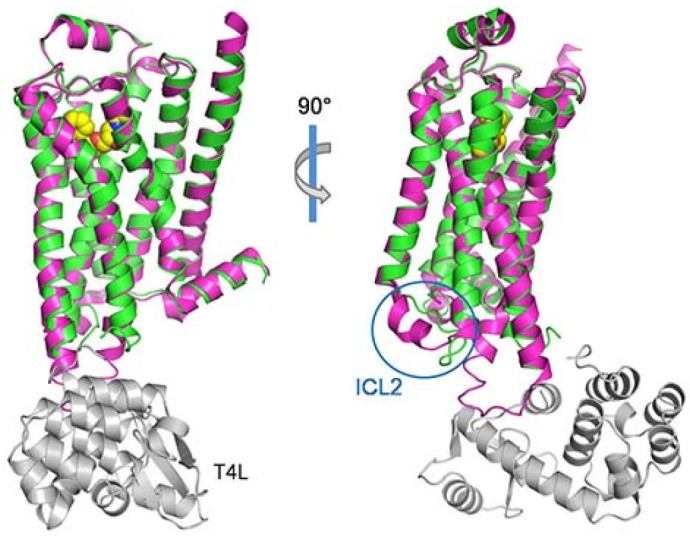
The superposition of β1AR from the antagonist bound inactive structure into the carazolol bound β2AR with T4L fusion. The structure of β1AR (pdb ID: 2YCW) is shown in magenta and the β2AR structure is colored as in Fig. 1A. The two structures are very similar except for the intracellular loop 2 (ICL2), circled in blue. Unlike in β2AR, ICL2 forms an α helix in β1AR.
Unlike in β2AR, ICL2 forms an α helix in β1AR, which interacts with the D(E)RY motif in TM3. As mentioned earlier, this highly conserved motif, called the ionic lock salt bridge, has been hypothesized to stabilize the inactive structure of GPCR, based on the structure of rhodopsin, but the structural data of β1AR and β2AR dispute this proposition. The salt bridge is absent in the inverse agonist-bound inactive structure of β2AR and in some of the antagonist-bound β1AR but it is present in the ligand-free basal state conformation. It seems that the inactive conformation of the two receptors can cope with both the situations which reinforces how dynamic and flexible GPCR structures really are. TM6 of β1AR has been shown to take only two conformations either bent or straight. It was proposed that the bent TM6 is associated with an ionic lock while the straight TM6 implies a broken ionic lock. However the ligand-free structure shows that the ionic lock can also be present with the straight TM6 (Huang et al., 2013). The agonist-bound β1AR does not exhibit the striking outward movement of TM6 and TM5 as seen in active β2AR structure, and it could be that the crystal structure was resolved in the absence of a G protein or its substituent, such as nanobody in the case of β2AR, to stabilize the activated conformation at the cytoplasmic side. It was shown that agonist binding was not enough to fully stabilize the active conformation of β2AR, and it is probably the same for β1AR. However, agonist binding induces a 1Å contraction of the ligand binding pocket, associated with the rotamer conformation changes of side chains Ser2125.43 and Ser2155.46. The changed rotamer conformation strengthens the TM5-TM6 interaction but weakens the TM4-TM5 interaction that may lead up to the outward movement of TM5 and TM6 as observed in β2AR.
The extracellular side of β2AR and β1AR is almost identical, including the three extracellular loops. ECL2 has an α helix which β2AR and β1AR share but rhodopsin does not, suggesting that this structure might be involved in interacting with diffusible, reversible binding of the ligand. However, there are differences in amino acid sequences of ECL2 between β2AR and β1AR, suggesting that ECL2 may be involved in ligand specificity. How the subtype selectivity works would depend on the ligands. That is, cyanopindolo and carazolol, both bind to all βARs with high affinity but some other ligands preferentially bind to either β1AR or β2AR. Structural analysis of cyanopindolo bound β1AR and carazolol bound β2AR showed that two residues, Val1724.56 and Phe3257.35 in β1AR, and Thr1644.56 and Tyr3087.35 in β2AR, are different among the amino acids, positioned within the 8Å distance from the ligand binding pocket, and they may provide a different polar environment for the ligand. Development of subtype specific ligand is pharmacologically important and more structural and biochemical data of βARs with highly selective ligands will shed light on the structure-based design of novel subtype specific ligand.
DISEASE-RELATED β2AR MUTATIONS
The β2AR is involved in various diseases as it is widely distributed in our body. Asthma, heart failure and Alzheimer’s are some of the well-studied diseases in which β2AR is known to be an important drug target. For example, a class of β2AR agonists, such as Albuterol and Salmeterol, is in current clinical use to treat asthma.
There are not many mutational studies done that give much insight on a molecular level how a disease is related to the structural features of β2AR. Most of the studies were on β2AR polymorphism. The β2AR is coded by the ADRB2 gene, which has three polymorphism sites, Arg16Gly, Glu27Gln, and Thr164Ile. Among these, Thr164Ile is very rare, so it is of little importance clinically, even though it may be harmful. The mutated β2AR shows reduced adenylate cyclase activity suggesting that the mutation has somehow decreased the efficacy of signal transduction. Residue 164 is in the middle of TM4 and changing threonine to isoleucine would have increased the hydrophobicity of the helix. Although it is not one of the helices involved in the ionic lock or undergoing large motion during activation, it is probably important in holding the structure in that particular form. The significance of Arg16Gly, Glu27Gln polymorphism is controversial although some results show different response to drugs and different susceptibility to some diseases. Unfortunately, the structural information of the N-terminal end of β2AR is not available due to its flexibility. Only one crystal structure resolved the N-terminal region starting from residue 23 but it is not possible to tell what the significance of Glu27 is from this structure. Although it appears that the ligand binding site is mostly composed of the extracellular loops and extracellular ends of TMs, it is possible that the N-terminus may have a role in regulating the activity of β2AR. More structural, biophysical and mutational work would have to be done to validate the idea.
CONCLUSION
The β2AR structure was the first human GPCR structure to be discovered. As a widely expressed receptor involved in the well-known flight-or-fight system, its significance in our physiology and health cannot be understated. Its structures are actively investigated to screen for better drugs with better subtype specificity and to explain the varying response of the receptor to different ligands. All crystal structures of β2AR determined so far show the orthosteric ligand binding site, but molecular modeling and docking simulation propose that there could be secondary allosteric ligand binding site. The development of a selective allosteric modulator is becoming a novel approach for drug discovery. One important question is whether ligand binding at an allosteric region induces different conformational states. Another important aspect of ligand binding concerns ligand specific biased signaling pathway. That is, between the G-protein pathway and the arrestin pathway, some ligands prefer one over the other. The crystal structures of β1AR complexed with the biased agonists, bucindolol and carvedilol, were determined using a thermostable mutant of β1AR (Warne et al., 2012). Both ligands are known to activate the arrestin pathway but function as either inverse or partial agonists of the G protein pathway. However, the crystal structures didn’t show any significant differences from those of β1AR bound to nonbiased antagonists, except for the extended ligand binding site of both ligands containing bulky aromatic moieties. It is possible that the additional interactions at the ligand binding region may induce subtle conformational changes, which were not detected in the crystal structure of thermostable β1AR mutant.
One of the important discoveries from structural studies for the last 7 years was the dynamic conformation of β2AR. In addition to crystallographic studies of β2AR, biophysical approaches like NMR, HDX-MS and MD simulation have allowed us to move away from the simple on-and-off model of activation and inactivation. Varying degree of functional activation can be achieved through its dynamic structure, in contrast to the relatively rigid rhodopsin structure. To understand the mechanism of how diverse ligands act on the same receptor but transmit different downstream signals, more structural, biophysical and biochemical studies need to be done. Various GPCR-G protein complex structures are required to explain G protein specificity and flexible C-terminal region, which involves multiple phosphorylation sites and arrestin binding sites needs more focus as well. Much work remains to be done but the β2AR structural studies have formed a stepping stone for a better understanding and advancing the structural studies of GPCR family members.
Acknowledgments
This work was supported by the Seoul National University Research Grant and Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2013R1A1A2061541).
REFERENCES
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KY, Rasmussen SG, Liu T, Li S, DeVree BT, Chae PS, Calinski D, Kobilka BK, Woods VL, Jr, Sunahara RK. Conformational changes in the G protein Gs induced by the beta2 adrenergic receptor. Nature. 2011;477:611–615. doi: 10.1038/nature10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EY, Velasquez J, Kuhn P, Stevens RC. A specific cholesterol binding site is established by the 2.8 A structure of the human beta2-adrenergic receptor. Structure. 2008;16:897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Chen S, Zhang JJ, Huang XY. Crystal structure of oligomeric beta1-adrenergic G protein-coupled receptors in ligand-free basal state. Nat Struct Mol Biol. 2013;20:419–425. doi: 10.1038/nsmb.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moukhametzianov R, Warne T, Edwards PC, Serrano-Vega MJ, Leslie AG, Tate CG, Schertler GF. Two distinct conformations of helix 6 observed in antagonist-bound structures of a beta1-adrenergic receptor. Proc Natl Acad Sci USA. 2011;108:8228–8232. doi: 10.1073/pnas.1100185108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygaard R, Zou Y, Dror RO, Mildorf TJ, Arlow DH, Manglik A, Pan AC, Liu CW, Fung JJ, Bokoch MP, et al. The dynamic process of beta(2)-adrenergic receptor activation. Cell. 2013;152:532–542. doi: 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, et al. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011a;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011b;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring AM, Manglik A, Kruse AC, Enos MD, Weis WI, Garcia KC, Kobilka BK. Adrenaline-activated structure of beta2-adrenoceptor stabilized by an engineered nanobody. Nature. 2013;502:575–579. doi: 10.1038/nature12572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- Rosenbaum DM, Zhang C, Lyons JA, Holl R, Aragao D, Arlow DH, Rasmussen SG, Choi HJ, Devree BT, Sunahara RK, et al. Structure and function of an irreversible agonist-beta(2) adrenoceptor complex. Nature. 2011;469:236–240. doi: 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D, Fenalti G, Brown MA, Katritch V, Abagyan R, Cherezov V, Stevens RC. Conserved binding mode of human beta2 adrenergic receptor inverse agonists and antagonist revealed by X-ray crystallography. J Am Chem Soc. 2010;132:11443–11445. doi: 10.1021/ja105108q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T, Serrano-Vega MJ, Baker JG, Moukhametzianov R, Edwards PC, Henderson R, Leslie AG, Tate CG, Schertler GF. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T, Moukhametzianov R, Baker JG, Nehme R, Edwards PC, Leslie AG, Schertler GF, Tate CG. The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature. 2011;469:241–244. doi: 10.1038/nature09746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T, Edwards PC, Leslie AG, Tate CG. Crystal structures of a stabilized beta1-adrenoceptor bound to the biased agonists bucindolol and carvedilol. Structure. 2012;20:841–849. doi: 10.1016/j.str.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]