

Abstract
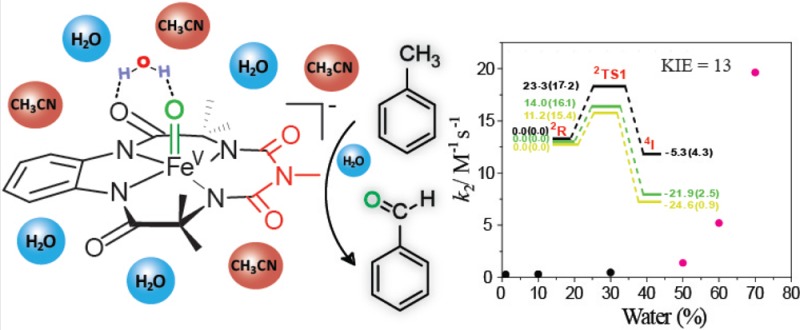
The presence of an FeV(O) species has been postulated as the active intermediate for the oxidation of both C–H and C=C bonds in the Rieske dioxygenase family of enzymes. Understanding the reactivity of these high valent iron–oxo intermediates, especially in an aqueous medium, would provide a better understanding of these enzymatic reaction mechanisms. The formation of an FeV(O) complex at room temperature in an aqueous CH3CN mixture that contains up to 90% water using NaOCl as the oxidant is reported here. The stability of FeV(O) decreases with increasing water concentration. We show that the reactivity of FeV(O) toward the oxidation of C–H bonds, such as those in toluene, can be tuned by varying the amount of water in the H2O/CH3CN mixture. Rate acceleration of up to 60 times is observed for the oxidation of toluene upon increasing the water concentration. The role of water in accelerating the rate of the reaction has been studied using kinetic measurements, isotope labeling experiments, and density functional theory (DFT) calculations. A kinetic isotope effect of ∼13 was observed for the oxidation of toluene and d8-toluene showing that C–H abstraction was involved in the rate-determining step. Activation parameters determined for toluene oxidation in H2O/CH3CN mixtures on the basis of Eyring plots for the rate constants show a gain in enthalpy with a concomitant loss in entropy. This points to the formation of a more-ordered transition state involving water molecules. To further understand the role of water, we performed a careful DFT study, concentrating mostly on the rate-determining hydrogen abstraction step. The DFT-optimized structure of the starting FeV(O) and the transition state indicates that the rate enhancement is due to the transition state’s favored stabilization over the reactant due to enhanced hydrogen bonding with water.
Short abstract
An FeV(O) complex was generated at room temperature in H2O/CH3CN mixtures (<1 to 90% H2O) from a biuret-modified FeIII−TAML complex upon the addition of 1.1 equiv of NaOCl. The rate of tolune oxidation was accelerated up to 60 times upon increasing the concentration of water.
Introduction
Metalloenzymes use oxidants like O2 and H2O2 to catalyze oxidation reactions that exhibit exquisite substrate specificity and selectivity and operate under mild conditions through inherently “green” processes.1 Examples of such catalysts include cytochrome P450 and peroxidases, enzymes that use an iron(IV) oxoporphyrin radical cation intermediate to catalyze the oxidation of various organic substrates selectively and efficiently.2 Other non-heme-based monooxygenases, such as α-KG dioxygenase, catalyze the oxidation of C–H bonds by an FeIV(O) intermediate.3 In the Rieske dioxygenase family of enzymes, FeV(O) has been postulated to be the active intermediate reactive species for carrying out oxidation of both C–H and C=C bonds.4 Although several models of FeIV(O) intermediates have been synthesized, and their reactivity for oxidizing C–H bonds has been studied in detail,5 very few studies have explored the reactivity of FeV(O) toward the oxidation of C–H bonds.6
Recently, we reported for the first time the quantitative formation of a well-defined FeV(O) complex at room temperature using biuret-modified Fe–TAML7 and mCPBA in CH3CN.8 FeV(O) was characterized by UV–vis, X-band electron paramagnetic resonance (EPR), Mössbauer, and high resolution mass spectrometry (HR-MS). It has also been shown to catalyze the oxidation of C–H bonds with abstraction of the hydrogen atom being the rate-determining step. Because the iron monooxygenase reactions occur in water,2d it is important to understand the effect of water on the rate of reaction for the oxidation of C–H bonds by FeV(O). Water has been shown to play a critical role during oxidation of organic substrates in heme-containing enzymes such as cytochrome P450.9 The formation of FeIV(O) by Fe–TAML in water has been previously reported.10 In this Article, we verify the stability of FeV(O) in H2O/CH3CN mixtures containing up to 90% water. The effect of water on the reactivity of FeV(O) during C–H activation has also been studied both experimentally and theoretically. We also report that the rate of oxidation of toluene with FeV(O) in an H2O/CH3CN mixture is enhanced up to 60-fold, likely attributable to preferential stabilization of the transition state during C–H abstraction due to hydrogen bonding with water molecules.
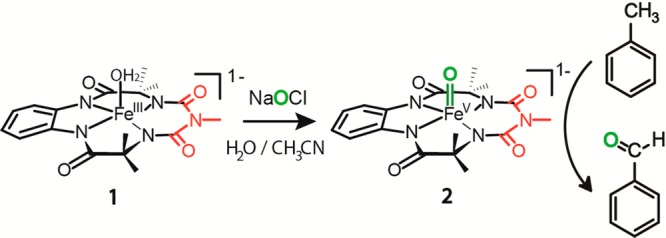
Scheme 1. Formation of FeV(O) Species in an H2O/CH3CN Mixture and Its Reaction toward Toluene.
Experimental Methods
Materials
Biuret-amide-modified Fe–TAML (1) was synthesized as we have previously reported.757FeCl2 was purchased from Trace Sciences International Corporation (Canada). Aqueous sodium hypochlorite (reagent grade, Aldrich, available chlorine 4.00–4.99%) was used as received and quantified by iodiometry. Acetonitrile (HPLC grade, Aldrich) was used after passing through an activated neutral alumina column. D2O (99.9 atom % D) was used as received (Aldrich). 18O-enriched water (98%) was procured from the Shanghai Research Institute of Chemical Industry (China). Deionized water was used to make all of the stock solutions for the reaction and kinetic runs. Toluene (Aldrich, 99.8%) was passed through activated neutral alumina and distilled prior to use. All reactions were carried out without any special precautions under atmospheric conditions unless otherwise specified.
General Instrumentation
X-band EPR spectra were recorded on a Bruker 300 spectrometer equipped with an Oxford ESR-910 liquid helium cryostat. For both instruments, the microwave frequency was calibrated with a frequency counter and the magnetic field with an NMR gaussmeter. Mössbauer spectra were recorded with two spectrometers using a Janis Research Super Vari-temp Dewar. UV–vis spectral studies were carried out using an Agilent diode array 8453 spectrophotometer with an attached electrically controlled thermostat. Gas chromatography (GC) was performed on a PerkinElmer Arnel Clarus 500 instrument equipped with a hydrogen flame ionization detector; BP20 columns (polar) (12 m × 0.53 mm × 1 μm) were used with helium as the carrier gas at a flow rate of 1 mL min–1. GC-MS was performed on an Agilent 5977A mass selective detector interfaced with an Agilent 7890B gas chromatograph using an HP-5ms capillary column (30 m × 0.32 mm × 0.25 μm, J&W Scientific). HR-MS was performed in a Thermo Scientific Q-Exactive Orbitrap analyzer using an electrospray ionization source connected with a C18 column (150 m × 4.6 mm × 8 μm).
EPR Spectroscopy
The sample for EPR analysis was prepared by adding 10 μL of 40 mM NaOCl to 190 μL of 2 mM biuret-modified Fe–TAML (1) in an ice bath; the spectrum was taken at 21 K. The signal was quantified relative to a Cu–EDTA spin standard. A modulation frequency of 100 kHz was used for the EPR spectra. The EPR simulation software (SpinCount) was written by one of the authors.11 The software diagonalizes the spin Hamiltonian (eq i), where S is the total spin of the complex (unless explicitly stated otherwise), and the parameters have the usual definitions. The quantitative simulation was a least-squares fit of the experimental spectra generated with consideration for the intensity factor, which allows for the computation of simulated spectra at a specified sample concentration.
| i |
Mössbauer Spectroscopy
57Fe-enriched 1 was prepared following the procedure used for the synthesis of 1; in this case, 57FeCl3 was used in place of 56FeCl2 (70% yield). The corresponding 57Fe-enriched 2 (2 mM) was quantitatively prepared by reacting 1 with 2 equiv of NaOCl in an ice bath; spectra were recorded at 4.2 K. The isomer shift was reported relative to the Fe metal. Simulation of the Mössbauer spectra was calculated with least-squares fitting using the program SpinCount and the standard spin Hamiltonian (eq ii).
| ii |
Preparation of FeV(O) (2) in Different H2O/CH3CN Mixtures
NaOCl (1.1 equiv) was added to a 1 × 10–4 M solution of biuret-modified Fe–TAML (1) in different H2O/CH3CN mixtures (10, 30, 50, 70, and 90% water); UV−vis spectra of FeV(O) (2) were recorded at 25 °C.
Stability of FeV(O) (2) in Different H2O/CH3CN Mixtures
The rate constant (k5/4,3) (Table 1) for the spontaneous reduction of FeV to FeIV/FeIII in H2O/CH3CN mixtures at 25 °C was measured by the initial rate approach. The quantitative formation of FeV was carried out by adding 1.1 equiv of NaOCl in solution to 1. The rate of spontaneous reduction of FeV to FeIV/FeIII was monitored by measuring the decrease in absorbance at 613 nm (λmax for FeV). Linear correlation of the initial rate against [FeV] was found, which indicated first-order dependency on FeV (i.e., rate = k5/4,3[FeV]).
Table 1. Stability and Reactivity of the FeV(O) Intermediate for Toluene Oxidation in Various H2O/CH3CN Mixtures.
| percentage of water in the H2O/CH3CN mixture (%) | stability (k5/4,3, s–1) | reactivity (k2, M–1 s–1) | yield (% benzaldehyde) |
|---|---|---|---|
| <1 | 2.4 × 10–5 | 0.27 | 23 |
| 10 | 2.8 × 10–5 | 0.31 | 21 |
| 30 | 3.1 × 10–5 | 0.47 | 19 |
| 50 | 6.5 × 10–5 | 1.4 | 20 |
| 60 | 7.6 × 10–5 | 5.2 | 20 |
| 70 | 1.1 × 10–4 | 19.6 | 20 |
| 90 | 3.2 × 10–4 |
Reactivity of FeV(O) (2) toward Toluene Oxidation
Kinetics were monitored in either kinetic mode or scanning spectral kinetics mode of the spectrophotometer using a 1.0 cm quartz cell at 375 nm (the isosbestic points of FeIV and FeIII species) at 25.0 °C.12 All of the kinetic experiments were carried out in H2O/CH3CN mixtures where the percentage of water present varied as required per experiment. During kinetic measurements, the concentration of the FeV(O) complex (10–4 M) was kept constant while the substrate concentration was varied. A pseudo-first-order rate constant (kobs) was calculated at the isosbestic wavelength by nonlinear curve fitting [At = Aα – (Aα – A0)e(−kobst)] and was in good agreement with the rate constant value (within 5% error). The resulting kobs values correlated linearly with the substrate concentration to give the second-order rate constant k2 (Table 1).
Product Quantification
Gas chromatography (GC) was used for product quantification. Excess substrate (100 μL, 10–1 M) was added to a freshly prepared 1 mL solution of 2 (generated by the reaction of 1 (10–4 M) and NaOCl (1.1 × 10–4 M)) at room temperature under an argon atmosphere. After completion of the reaction (determined by UV–vis spectroscopy), the internal standard bromobenzene was added, and the reaction mixture was immediately passed through an alumina column. Product (% yield) was quantified by GC using the calibration curve obtained with authentic benzaldehyde and bromobenzene.
Computational Methods
All of the minima and transition states reported in this study were fully optimized at the density functional theory (DFT)-UB3LYP/6-31G*, LANL2DZ (Fe) level of theory13 using the Gaussian09 suite of quantum chemical programs.14 The stationary points on the potential energy surface were characterized by evaluating the vibrational frequencies. The transition states were characterized by a single imaginary frequency. The zero point vibrational energy corrections and thermal corrections were applied to the “bottom-of-the-well” values to obtain values for the Gibbs free energy at 298.15 K.
The experimental results suggest that the solvent plays a vital role in the rate-determining step, as the hydroxylation reaction of toluene by [FeV(O)(biuret–TAML)]− is favored by increasing the polarity of the solvent. The electrostatic effect of the solvent dielectric on the reactive species was determined by full geometry optimizations at the UB3LYP/6-31G*, LANL2DZ (Fe) level of theory in the dielectric continuum of acetonitrile and water using the conductor-like polarizable continuum model.15 Charge stabilization through hydrogen-bonding interactions with explicitly added water molecules was studied by spotting the water-bound reactants, transition states, and intermediates in the gas phase for the rate-determining hydrogen abstraction step. This solvent model has recently been employed in several DFT studies.16
Recent literature suggests that improved treatment of hydrogen bond interactions could be achieved with the M06 functional.17 Therefore, single point energies were calculated at the ROM062X/6-31G*, LANL2DZ (Fe) level of theory for the species involved in the rate-determining step (RDS) for UB3LYP-optimized structures. These energy values have better concurrence with the experimental findings. All of the values reported at the ROM062X/6-31G*, LANL2DZ (Fe) level of theory are single point energies. To have a fair comparison between UB3LYP and ROM062X energies, only the electronic energy values describing the effect of the solvent have been reported in this Article. All assessments on the role of the solvent are thus made on the basis of their relative electronic energy values.
All of the energies were calculated with respect to optimized structures with all of the reactants at a finite distance from each other. This approach is purported to minimize errors in the computation of the translational entropy in determining the relative free energy values. Intermediates were optimized by keeping FeIV(OH) and the toluene radical together in the relative vicinity of each other in one structure. The same strategy was employed for obtaining the optimized geometries of the final product. Such strategies have been employed in the recent past in other computational reports.18
Results and Discussion
Water-soluble NaOCl (household bleach) was used as an oxidant for the formation of FeV(O). NaOCl has been used as a co-oxidant for a variety of metal-catalyzed oxidation reactions, such as olefin epoxidation.19 Addition of NaOCl to 1 showed the same UV–vis spectral features (Figure 1A) previously observed using mCPBA in organic media (with both 0.5 and 1.1 equiv of oxidant). The EPR and Mössbauer spectra (Figure 1C and D, respectively) of this species determined by the addition of 1.1 equiv of NaOCl to 1 are similar to the spectra of the FeV(O) species we reported previously.8 The g tensors of the species observed in the X-band EPR spectra from our previous study were 1.983, 1.935, and 1.726 with mCPBA as the oxidant and are 1.977, 1.888, and 1.720 in the present work using NaOCl as the oxidant. Spin quantification of the observed signal indicated quantitative conversion of 1 to the FeV(O) species. The Mössbauer spectra showed a single quadrupole doublet with an isomer shift of −0.45 mm/s and quadrupole splitting of 4.27 mm/s for the FeV(O) species, which are within the error compared to the parameters of the species from mCPBA.8
Figure 1.

(A) UV–vis spectral changes of 1 (10–4 M) (orange) upon the addition of 0.5 equiv of NaOCl (5 × 10–5 M) in CH3CN, forming the μ-Oxo-FeIV dimer species (violet). Addition of another 0.5 equiv of NaOCl (5 × 10–5 M) to the preformed μ-Oxo-FeIV dimer species produces the spectrum of FeV(O) (2, green). (B) UV–vis spectra of FeV(O) in different H2O/CH3CN mixtures. (C) EPR spectra of FeV(O) (2) in acetonitrile (2 mM) at 21 K. Black = experimental, red = simulated. (D) Mössbauer spectra of 57Fe-enriched [FeV(O)] ([2]) in acetonitrile (2 mM) at 4.2 K. The solid lines are spectral simulations.
We then proceeded to evaluate the formation of FeV(O) in H2O/CH3CN mixtures by monitoring its characteristic spectra through UV–vis spectroscopy. It was observed that in H2O/CH3CN mixtures with 10, 30, 50, 70, and 90% water, the spectral characteristics were identical to that of fully characterized FeV(O) in acetonitrile medium (Figure 1B). Formation of 2 in a 30:70 H2O/CH3CN mixture was also confirmed by HR-MS spectra (Figure SI 1 in the Supporting Information). At 100% water, the spectrum of FeV(O) was not observed; instead, a violet species was observed whose UV–vis spectrum was similar to that of the μ-Oxo-FeIV dimer species.8,12,20 The EPR spectrum of complex 2 in 50:50 H2O/CH3CN showed a species characteristic of the S = 1/2 species of FeV(O).21 The slight difference observed from the spectrum in pure CH3CN may be due to hydrogen bonding of water with the FeV(O) complex, as has been proposed below.
We then proceeded to evaluate the stability of the FeV(O) complex as a function of increasing water concentration. The stability was measured by monitoring the spontaneous self-reduction of FeV to FeIV/FeIII at room temperature using UV–vis spectroscopy at 613 nm (characteristic band of the FeV(O) species). The initial rate of decay at 613 nm displayed first-order dependency with respect to FeV(O), and the first-order rate constant (k5/4,3, s–1) was determined from the slope of the fitted straight line (Figure 2, Table 1). It was observed that the stability of FeV(O) decreased with increasing water concentration. At 90:10 H2O/CH3CN, the rate constant k5/4,3 was 10-fold higher than that observed in 100% CH3CN. This is likely due to the attack of water molecules on FeV(O), as we have recently proposed while studying photochemical water oxidation using 1.21 However, the stability at 70:30 H2O/CH3CN was only 4 times lower than that in 100% CH3CN, and the self reduction was much slower than what we observed for toluene oxidation by FeV(O) in 100% CH3CN.8 We therefore proceeded to evaluate the rate of toluene oxidation in H2O/CH3CN mixtures with the water concentration varied from <1 to 70% (Table 1).
Figure 2.
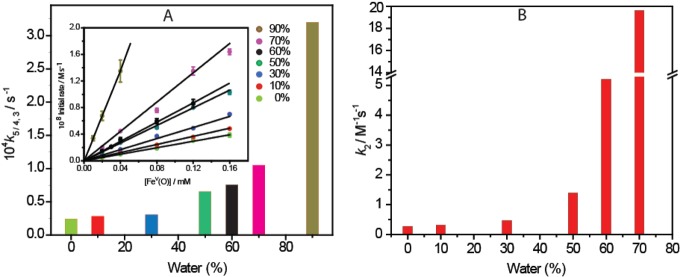
(A) First-order rate constant k5/4,3 varies with water concentration. Inset shows initial rate vs [FeV(O)]. (B) Second-order rate constant k2 for toluene oxidation vs the percentage of water content in the H2O/CH3CN mixture.
The ability of FeV(O) to facilitate toluene oxidation was studied under pseudo-first-order conditions at the isosbestic points of FeIII and FeIV interconversions (345 and 375 nm) at 25 °C, as has been shown before.8 The reaction was studied for different H2O/CH3CN mixtures (10, 30, 50, 60, and 70% H2O). The pseudo-first-order rate constant was determined from nonlinear curve fitting of the absorbance relative to time data using the equation At = Aα – (Aα – A0)e(−kobst) and exhibited good agreement with rate constant values within 5% error. The kobs correlated linearly with the toluene concentration (Figure SI 2, Supporting Information), and the second-order rate constant k2 (M–1 s–1) was calculated from the slope of the straight line that passed through the origin (Table 1, Figure 2B). At the end of the reaction, the parent FeIII complex was quantitatively regenerated as was observed from the UV–vis spectra (peak at 356 nm; Figure SI 3, Supporting Information) and HR-MS (m/z = 413.0716; Figure SI 4, Supporting Information). After completion of the reaction, product identification was carried out by GC-MS (Figure SI 5, Supporting Information) and HR-MS (Figure SI 6, Supporting Information). The results showed that benzaldehyde was formed, and the yield was quantified by GC to be ∼20–23% (theoretically, only 25% is expected because toluene oxidation to benzaldehyde is a four electron process and 50% of FeV(O) comproportionates with 1). The UV–vis scanning kinetics study showed that the starting FeV(O) species changed to the diamagnetic μ-Oxo-FeIV dimer species, which gradually regenerated parent FeIII complex 1 (Figure SI 3, Supporting Information). In all of the different H2O/CH3CN composite mixtures (10 to 70%), the rate of toluene oxidation (kobs) was much faster than the spontaneous reduction of FeV(O). The variation in k2 values indicated that the reactivity of FeV(O) increased with water concentration. Initially, the rate enhancements were modest, but it increased significantly when the water concentration exceeded 50%. At 70% water, k2 was around 60-fold higher than that in 100% CH3CN. Such an increase in the rate of oxidation of aromatic hydrocarbons by RuV(O) has been previously reported by Meyer et al.22
The role of H2O during C–H abstraction was then investigated. First, the direct role of water, such as its attack on the benzylic carbon of toluene to facilitate C–H bond cleavage, was investigated. Toluene oxidation was performed using a 50:50 H218O/CH3CN mixture so that the direct attack of H218O on the benzylic carbon would lead to the exclusive formation of 18O-benzaldehyde. However, the percentage of 18O incorporation (calculated on the basis of the relative abundance of 16O and 18O in the product by GC-MS) was found to be only 20%. This 18O-incorporated product was likely the result of (a) exchange of H218O with product benzaldehyde via the formation of a gem-diol and (b) oxygen atom exchange between FeV(O) (2) and H218O leading to the formation of small amounts of FeV(18O), which would lead to the incorporation of 18O-benzaldehyde.23 The absence of exclusive 18O-benzaldehyde formation indicates that direct attack of water is unlikely. Next, the kinetics of C–H abstraction was studied in mixed H2O/CH3CN solvents to evaluate if they differed from the kinetics in pure CH3CN that we have reported previously. Kinetic isotope effects (KIE) of 13 and 9 for the oxidation of toluene/toluene-d8 in 50:50 and 70:30 H2O/CH3CN (Figure SI 7, Supporting Information), respectively, were observed. This value is similar to the value determined for pure CH3CN (KIE of 9).8 This observation suggests that the rate-determining step in 50:50 H2O/CH3CN involves C–H abstraction, as has been reported for pure CH3CN.8 Further, a similar rate enhancement was also observed with 2,3-dimethylbutane, suggesting that this phenomenon was not limited to only toluene (Figure SI 8, Supporting Information). To further understand the role of water, the solvent kinetic isotope effect (SKIE) was evaluated with varying D2O/CH3CN mixtures as the solvent for toluene oxidation. A constant SKIE (kH2O/CH3CN/kD2O/CH3CN) value of 1.4–1.5 was observed in (H2O/D2O)/CH3CN mixtures with 10, 30, 50, and 70% H2O or D2O (Table 2 and Figure SI 9 in the Supporting Information), indicating the likely involvement of hydrogen bonding in the transition state.
Table 2. SKIE Values [k2(H2O)/k2(D2O)] of FeV(O) for Toluene Oxidation in Various Mixtures of H2O and D2O in CH3CN.
| H2O or D2O (%) | k2 in H2O (M–1 s–1) | k2 in D2O (M–1 s–1) | k2(H2O)/k2(D2O) |
|---|---|---|---|
| 10 | 0.31 | 0.21 | 1.5 |
| 30 | 0.47 | 0.33 | 1.4 |
| 50 | 1.4 | 0.91 | 1.5 |
| 70 | 19.6 | 14.0 | 1.4 |
To have a better understanding of the transition state, variable-temperature (283–300 K) kinetic measurements (Table SI 1, Supporting Information) were performed for toluene oxidation to determine the activation parameters (Table 3, Figure 3) on the basis of Eyring plots for the rate constants. According to the activation parameters for toluene oxidation in CH3CN (ΔH° = 69.3 kJ mol–1, TΔS° = −7.6 kJ mol–1, 300 K) and 70:30 H2O/CH3CN (ΔH° = 51.1 kJ mol–1, TΔS° = −14.7 kJ mol–1, 300 K), the 60-fold increase in the reaction rate is mainly controlled by the favorable activation enthalpy terms (ΔH° gain of 18 kJ mol–1). In contrast, the contribution of the activation entropy terms (ΔS° loss of 7 kJ mol–1) to the transition states is negative upon the addition of water. Because the enthalpic gain is 3 times that of the entropic loss, a favorable ΔG° leads to rate acceleration upon the addition of water. The gain in enthalpy with a concomitant loss in entropy points to the formation of an ordered transition state involving water molecules. To more comprehensively understand the role of water in the reaction kinetics, we performed a careful density functional theory (DFT) study, concentrating mostly on the rate-determining hydrogen abstraction step.24
Table 3. Thermodynamic Parameter (ΔH°, ΔS°, and ΔG°) Values for Toluene Oxidation in CH3CN and in a 70:30 H2O/CH3CN Mixture in the Temperature Range of 283–300 K.
| ΔH° (kJ mol–1) | ΔS° (J K–1) | ΔG° (kJ mol–1) | |
|---|---|---|---|
| CH3CN (100%) | 69.3 | –25.3 | 61.7 |
| 70:30 H2O/CH3CN | 51.1 | –49.2 | 36.4 |
Figure 3.
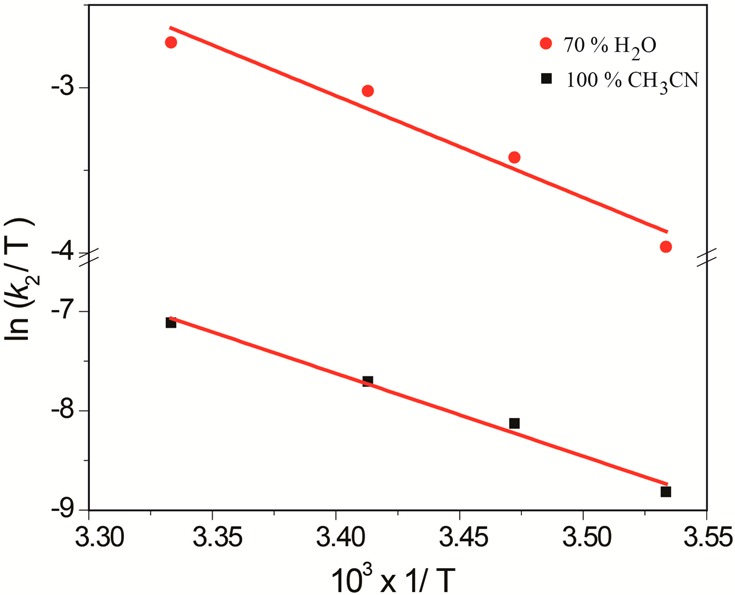
Plot of ln(k2/T) vs 1/T for toluene oxidation in 100% CH3CN and in 70:30 H2O/CH3CN in the temperature range of 283–300 K.
All of the stationary points were fully optimized at the DFT-UB3LYP/6-31G*, LANL2DZ (Fe) level of theory.13−15 The complete gas-phase energy profile clearly indicates that the intermediate with the quartet spin state is more stable than the doublet (Figure 4A and Table SI 3 in the Supporting Information). This result is further confirmed by the separate optimization of intermediate complex [FeIV(OH)]− in singlet and triplet spin states in both the gas phase and in an acetonitrile solvent. The triplet is stabilized by more than 15.0 kcal mol–1 compared to the singlet. The energy values are summarized in Table SI 4 of the Supporting Information. Because the reaction is favored by increasing the polarity (i.e., by increasing the relative concentration of water), we envisaged that water could affect the barrier through two separate mechanisms: by direct active participation in hydrogen abstraction (ruled out by the experimental kinetic isotope study) or by favored stabilization of the rate-determining transition state over that of the reactant.
Figure 4.

(A) Gas-phase free energy profile for the hydroxylation of toluene by 2 at the UB3LYP/6-31G*, LANL2DZ (Fe) level of theory. Violet and black colors represent energy profiles at S = 3/2 and S = 1/2 spin states, respectively. All of the values are in kcal mol–1. (B) Electronic energy profile for the rate-determining step of toluene hydroxylation catalyzed by 2. All of the values correspond to gas-phase data. Values outside and inside the parentheses correspond to the ROM062X/6-31G*, LANL2DZ (Fe) and UB3LYP/6-31G*, LANL2DZ (Fe) levels of theory, respectively. Black, green, and yellow colors represent the energy profile with zero, one, and two explicitly added water molecules, respectively. (i, ii, and iii) UB3LYP optimized transition state structures for the hydrogen atom abstraction rate-determining step (RDS) with zero, one, and two explicitly added water molecules, respectively. Hydrogen atoms not involved in the reaction coordinate are removed for clarity. All of the values are in kcal mol–1. All atom–atom distances are in Å.
Two possible means for the differential stabilization of the transition state and reactant with increasing concentrations of water were investigated: (a) a bulk solvation effect (macrosolvation) through the solvent dielectric and (b) a solute–solvent interaction through hydrogen bonding interactions (microsolvation). Only a marginal decrease in the rate-determining barrier (RDB) was obtained by changing the dielectric of the medium (Table SI 5, Supporting Information) from acetonitrile to pure water, which suggests that bulk solvation has a nominal effect on the rate enhancement with increasing water concentrations. On the other hand, microsolvation through the explicit consideration of one and two water molecules was found to be the primary reason for the decreased RDB (Figure 4B and Table SI 6 in the Supporting Information). A clear decrease in the rate-determining barrier (by 9.3 kcal mol−1) at the ROM062X level, which is known to provide better treatment of hydrogen bonds,17 upon the inclusion of one explicit water molecule, and a further decrease of 2.8 kcal mol−1 upon the inclusion of a second explicit water molecule corroborate the experimental findings.
Our calculations show that water is capable of making a hydrogen bond with the oxo group of the FeV(O) complex. At the same time, the FeV(O) complex exhibits other hydrophilic hydrogen bonding interactive sites, as well those that can compete with the oxo group. However, the increasing concentration of water enhances the possibility of Fe(oxo)–water interactions. It also increases the chance of having more water in the active site of the FeV(O) complex. Careful examination of the water-bound reactant structures implies that the Fe–O bond length increases with an increase in the number of water molecules hydrogen bonded with the oxo group in the active site of FeV(O) (2) (Table SI 7 and Figure SI 10, Supporting Information). An increase of 0.053 Å in the Fe–O bond length upon the inclusion of two explicit water molecules in the reactant structure suggests that the increasing concentration of water leads to a decrease in the Fe–O bond order of FeV(O) (Table SI 7, Supporting Information). This means that the bond dissociation energy of the Fe–O bond decreases. We also know that elongation of a chemical bond is associated with a decrease in the overlap of the involved orbitals, which usually causes weakening (and thus destabilization) of the chemical bond, and thus makes the bond (and eventually the molecule via that bond) more prone to dissociation upon chemical attack. On the other hand, we can see that the Fe–O bond is elongated upon consideration of explicit water molecules in the transition state structures as well. However, this elongation decreases upon the addition of additional explicit water molecules (0.12, 0.092, and 0.087 Å for zero, one, and two explicit water molecules, respectively, with respect to the corresponding reactants). The extent of elongation of the Fe–O bond in the transition state compared to that of the corresponding reactants can be directly correlated with the barrier according to the “extended activation strain model” proposed by Jong et al.25 This model proposes that smaller deformation in the structure of the involved species results in less strain energy, which leads to a lower activation barrier. We should also note that the Fe–O bond in the equilibrium geometry of the reactant with two explicitly added water molecules is significantly longer than in the bare reactant, which causes the strain energy to increase to a lesser extent as this bond expands in the activated complex compared to that of the bare reactant without any explicit water included. Hence, the smaller difference between the transition state and reactant geometries, as well as the change that occurs when the Fe–O bond has been weakened significantly by solute–solvent interactions through hydrogen bonding upon the inclusion of explicit water molecules, together leads to a smaller barrier and an enhanced rate of reaction. This indicates that the bond dissociation energy of the Fe–O bond should decrease. A theoretical study on a related FeIII(O) system by Borovik et al. confirms that the bond dissociation energy of the Fe–O bond decreases upon the inclusion of hydrogen bonds from solvent water.26 Their study further supports the notion that the Fe–O bond length increases upon the inclusion of explicit water molecules. They have also shown that the increase in bond length is associated with a decrease in the percentage of covalent character of the Fe–O bond. This suggests that the ionic character of the Fe–O bond in the transition state should be greater as the Fe–O bond becomes distended. This elongation is 0.12 Å in the structure without consideration of explicit water. The greater ionic character of the Fe–O bond explains the stronger hydrogen bonding with water in the transition state relative to that of the reactant.
To understand the differential stabilization of water-bound reactants and transition states in more detail, we looked at the Mulliken population analysis27 of all species involved in the RDS (Table SI 8, Supporting Information). The oxo atom in Fe–O in the transition state having a greater negative charge than in the reactant indicates the possibility of a better electrostatic interaction (i.e., stronger hydrogen bond) with water in the transition state compared to that with the reactant. Consequently, the barrier decreases with an increasing number of hydrogen bonds, thereby providing an explanation for the effect of microsolvation on the activity of the FeV(O) system.
Conclusions
In summary, we have demonstrated for the first time the formation of an FeV(O) complex in H2O/CH3CN mixtures. This complex was synthesized by the reaction of biuret-modified Fe–TAML (1) and NaOCl in H2O/CH3CN mixtures with up to 90% water. The stability of the FeV(O) complex decreases with increasing water content. This complex is reasonably stable, allowing for studies on the rate of oxidation of C–H bonds. We show that for toluene oxidation in 70:30 H2O/CH3CN mixtures, the rate of toluene oxidation increases 60-fold. The increase in activity for FeV(O) upon the addition of water is due to the preferential stabilization of the transition state by water during the C–H abstraction process. We believe that this study will help to elucidate the role of water molecules in the active site of Fe-based enzymes.
Acknowledgments
S.S.G. acknowledges DST, New Delhi (Grant EMR/2014/000106) for funding. M.P.H. acknowledges NIH Grant GM077387 and NSF Grant CHE1126268 for support. K.K.S., M.K.T., M.G., and C.P. acknowledge CSIR (Delhi) for their fellowships. B.B.D. also acknowledges CSIR for his SRA position. S.S.G. and M.G. thank Professor T. J. Collins for hosting M.G. at Carnegie Mellon University.
Supporting Information Available
HR-MS spectra, GC-MS traces, kinetic traces, UV–vis spectral kinetics, KIEs, optimized reactant structures, DFT data, and xyz coordinates for all stationary points of the optimized structures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Costas M.; Mehn M. P.; Jensen M. P.; Que L. Chem. Rev. 2004, 104, 939. [DOI] [PubMed] [Google Scholar]; b Que L.; Tolman W. B. Nature 2008, 455, 333. [DOI] [PubMed] [Google Scholar]
- a Meunier B.; de Visser S. P.; Shaik S. Chem. Rev. 2004, 104, 3947. [DOI] [PubMed] [Google Scholar]; b Ortiz de Montellano P. R. Chem. Rev. 2010, 110, 932. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Rittle J.; Green M. T. Science 2010, 330, 933. [DOI] [PubMed] [Google Scholar]; d Denisov I. G.; Makris T. M.; Sligar S. G.; Schlichting I. Chem. Rev. 2005, 105, 2253. [DOI] [PubMed] [Google Scholar]; e Groves J. T. J. Inorg. Biochem. 2006, 100, 434. [DOI] [PubMed] [Google Scholar]
- a Krebs C.; Fujimori D. G.; Walsh C. T.; Bollinger J. M. Acc. Chem. Res. 2007, 40, 484. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Xue G.; Wang D.; De Hont R.; Fiedler A. T.; Shan X.; Münck E.; Que L. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 20713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chakrabarty S.; Austin R. N.; Deng D.; Groves J. T.; Lipscomb J. D. J. Am. Chem. Soc. 2007, 129, 3514. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ferraro D. J.; Gakhar L.; Ramaswamy S. Biochem. Biophys. Res. Commun. 2005, 338, 175. [DOI] [PubMed] [Google Scholar]
- a McDonald A. R.; Que L. Coord. Chem. Rev. 2013, 257, 414. [Google Scholar]; b Nam W.; Lee Y.-M.; Fukuzumi S. Acc. Chem. Res. 2014, 47, 1146. [DOI] [PubMed] [Google Scholar]; c Fukuzumi S.; Morimoto Y.; Kotani H.; Naumov P.; Lee Y.-M.; Nam W. Nat. Chem. 2010, 2, 756. [DOI] [PubMed] [Google Scholar]; d Que L. Acc. Chem. Res. 2007, 40, 493. [DOI] [PubMed] [Google Scholar]; e Rohde J.-U.; In J.-H.; Lim M. H.; Brennessel W. W.; Bukowski M. R.; Stubna A.; Münck E.; Nam W.; Que L. Science 2003, 299, 1037. [DOI] [PubMed] [Google Scholar]; f Kaizer J.; Klinker E. J.; Oh N. Y.; Rohde J.-U.; Song W. J.; Stubna A.; Kim J.; Münck E.; Nam W.; Que L. J. Am. Chem. Soc. 2004, 126, 472. [DOI] [PubMed] [Google Scholar]
- a de Oliveira F. T.; Chanda A.; Banerjee D.; Shan X.; Mondal S.; Que L.; Bominaar E. L.; Münck E.; Collins T. J. Science 2007, 315, 835. [DOI] [PubMed] [Google Scholar]; b Lyakin O. Y.; Bryliakov K. P.; Britovsek G. J. P.; Talsi E. P. J. Am. Chem. Soc. 2009, 131, 10798. [DOI] [PubMed] [Google Scholar]; c McDonald A. R.; Que L. Nat. Chem. 2011, 3, 761. [DOI] [PubMed] [Google Scholar]; d Prat I.; Mathieson J. S.; Güell M.; Ribas X.; Luis J. M.; Cronin L.; Costas M. Nat. Chem. 2011, 3, 788. [DOI] [PubMed] [Google Scholar]
- Panda C.; Ghosh M.; Panda T.; Banerjee R.; Sen Gupta S. Chem. Commun. (Cambridge, U.K.) 2011, 47, 8016. [DOI] [PubMed] [Google Scholar]
- Ghosh M.; Singh K. K.; Panda C.; Weitz A.; Hendrich M. P.; Collins T. J.; Dhar B. B.; Sen Gupta S. J. Am. Chem. Soc. 2014, 136, 9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Chen H.; Makino M.; Shiro Y.; Nagano S.; Asamizu S.; Onaka H.; Shaik S. J. Am. Chem. Soc. 2009, 131, 6748. [DOI] [PubMed] [Google Scholar]
- Chanda A.; Shan X.; Chakrabarti M.; Ellis W. C.; Popescu D. L.; de Oliveira F. T.; Wang D.; Que L.; Collins T. J.; Münck E.; Bominaar E. L. Inorg. Chem. 2008, 47, 3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golombek A. P.; Hendrich M. P. J. Magn. Reson. 2003, 165, 33. [DOI] [PubMed] [Google Scholar]
- Kundu S.; Thompson J. V. K.; Ryabov A. D.; Collins T. J. J. Am. Chem. Soc. 2011, 133, 18546. [DOI] [PubMed] [Google Scholar]
- a Perdew J. P.; Chevary S. H.; Vosko K. A.; Jackson K. A.; Pederson M. R.; Singh D. J.; Fiolhais C. Phys. Rev. B 1992, 46, 6671. [DOI] [PubMed] [Google Scholar]; b Perdew J. P.; Chevary S. H.; Vosko K. A.; Jackson K. A.; Pederson M. R.; Singh D. J.; Fiolhais C. Phys. Rev. B 1993, 48, 4978. [DOI] [PubMed] [Google Scholar]; c Perdew J. P.; Burke K.; Wang Y. Phys. Rev. B 1996, 54, 16533. [DOI] [PubMed] [Google Scholar]; d Adamo C.; Barone V. J. Chem. Phys. 1998, 108, 664. [Google Scholar]; e Becke A. D. J. Chem. Phys. 1993, 98, 5648. [Google Scholar]; f Lee C.; Yang W.; Parr R. G. Phys. Rev. B 1988, 37, 785. [DOI] [PubMed] [Google Scholar]; g Roothan C. C. J. Rev. Mod. Phys. 1960, 362179. [Google Scholar]; h McWeeny R.; Dierksen G. J. Chem. Phys. 1968, 49, 4852. [Google Scholar]; i Pople J. A.; Nesbet R. K. J. Chem. Phys. 1954, 22, 571. [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, revision B.01; Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]
- a Barone V.; Cossi M. J. Phys. Chem. A 1998, 102, 1995. [Google Scholar]; b Cossi M.; Rega N.; Scalmani G.; Barone V. J. Comput. Chem. 2003, 24, 669. [DOI] [PubMed] [Google Scholar]
- a Kong S.; Evanseck J. D. J. Am. Chem. Soc. 2000, 122, 10418. [Google Scholar]; b Di Valentin C.; Freccero M.; Zanaletti R.; Sarzi-Amad M. J. Am. Chem. Soc. 2001, 123, 8366. [DOI] [PubMed] [Google Scholar]; c Chandrasekhar J.; Shariffskul S.; Jorgensen W. L. J. Phys. Chem. B 2002, 106, 8087. [Google Scholar]; d Domingo L. R.; Andrés J. J. Org. Chem. 2003, 68, 8662. [DOI] [PubMed] [Google Scholar]; e Freccero M.; Di Valentin C.; Sarzi-Amad M. J. Am. Chem. Soc. 2003, 125, 3544. [DOI] [PubMed] [Google Scholar]; f Mujika J. I.; Mercero J. M.; Lopez X. J. Am. Chem. Soc. 2005, 127, 4445. [DOI] [PubMed] [Google Scholar]; g Saettel N. J.; Wiest O. Tetrahedron 2006, 62, 6490. [Google Scholar]; h Gordillo R.; Dudding T.; Anderson C. D.; Houk K. N. Org. Lett. 2007, 9, 501. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Shi F.-Q.; Li X.; Xia Y.; Zhang L.; Yu Z.-X. J. Am. Chem. Soc. 2007, 129, 15503. [DOI] [PubMed] [Google Scholar]; j Patil M. P.; Sunoj R. B. Chem.—Eur. J. 2008, 14, 10472. [DOI] [PubMed] [Google Scholar]
- a Zhao Y.; Truhlar D. G. Theor. Chem. Acc. 2008, 120, 215. [Google Scholar]; b Zhao Y.; Truhlar D. G. J. Phys. Chem. A 2006, 110, 13126. [DOI] [PubMed] [Google Scholar]; c Zhao Y.; Truhlar D. G. J. Chem. Phys. 2006, 125, 194101. [DOI] [PubMed] [Google Scholar]
- a Kelly E.; Seth M.; Ziegler T. J. Phys. Chem. A 2004, 108, 2167. [Google Scholar]; b Yin H.; Wang D.; Valiev M. J. Phys. Chem. A 2011, 115, 12047. [DOI] [PubMed] [Google Scholar]; c Williams V. M.; Kong J. R.; Ko B. J.; Mantri Y.; Brodbelt J. S.; Baik M.-H.; Krische M. J. J. Am. Chem. Soc. 2009, 131, 16054. [DOI] [PMC free article] [PubMed] [Google Scholar]; d van Rensburg W. J.; Grové C.; Steynberg J. P.; Stark K. B.; Huyser J. J.; Steynberg P. J. Organometallics 2004, 23, 1207. [Google Scholar]; e Qi Y.; Dong Q.; Zhong L.; Liu Z.; Qiu P.; Cheng R.; He X.; Vanderbilt J.; Liu B. Organometallics 2010, 29, 1588. [Google Scholar]; f Bagno A.; Kantlehner W.; Kress R.; Saielli G.; Stoyanov E. J. Org. Chem. 2006, 71, 9331. [DOI] [PubMed] [Google Scholar]; g Li J.-N.; Pu M.; Ma C.-C.; Tian Y.; He J.; Evans D. G. J. Mol. Catal. A: Chem. 2012, 359, 14. [Google Scholar]; h Kumawat J.; Gupta V. K.; Vanka K. Eur. J. Inorg. Chem. 2014, 10.1002/ejic.201402180. [DOI] [Google Scholar]
- a Meunier B.; Guilmet E.; De Carvalho M. E.; Poilblanc R. J. Am. Chem. Soc. 1984, 106, 6668. [Google Scholar]; b Yoon H.; Burrows C. J. J. Am. Chem. Soc. 1988, 110, 4087. [DOI] [PubMed] [Google Scholar]
- Ghosh A.; de Oliveira F. T.; Yano T.; Nishioka T.; Beach E. S.; Kinoshita I.; Münck E.; Ryabov A. D.; Horwitz C. P.; Collins T. J. J. Am. Chem. Soc. 2005, 127, 2505. [DOI] [PubMed] [Google Scholar]
- Panda C.; Debgupta J.; Diaz Diaz D.; Singh K. K.; Sen Gupta S.; Dhar B. B. J. Am. Chem. Soc. 2014, 136, 12273. [DOI] [PubMed] [Google Scholar]
- Thompson M. S.; Meyer T. J. J. Am. Chem. Soc. 1982, 104, 5070. [Google Scholar]
- Seo M. S.; In J.-H.; Kim S. O.; Oh N. Y.; Kim J. H. J.; Que L.; Nam W. Angew. Chem., Int. Ed. 2004, 43, 2417. [DOI] [PubMed] [Google Scholar]
- Shaik S.; Hirao H.; Kumar D. Acc. Chem. Res. 2007, 40, 532. [DOI] [PubMed] [Google Scholar]
- de Jong G. T.; Bickelhaupt F. M. ChemPhysChem 2007, 8, 1170. [DOI] [PubMed] [Google Scholar]
- Dey A.; Hocking R. K.; Larsen P.; Borovik A. S.; Hodgson K. O.; Hedman B.; Solomon E. I. J. Am. Chem. Soc. 2006, 128, 9825. [DOI] [PubMed] [Google Scholar]
- Mulliken R. S. J. Chem. Phys. 1955, 23, 1833. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.