

Abstract
Background and objective
Heterozygous mutations in KCNA1 cause episodic ataxia type 1 (EA1), an ion channel disorder characterised by brief paroxysms of cerebellar dysfunction and persistent neuromyotonia. This paper describes four previously unreported families with EA1, with the aim of understanding the phenotypic spectrum associated with different mutations.
Methods
15 affected individuals from four families underwent clinical, genetic and neurophysiological evaluation. The functional impact of new mutations identified in the KCNA1 gene was investigated with in vitro electrophysiology and immunocytochemistry.
Results
Detailed clinical documentation, dating back to 1928 in one family, indicates that all patients manifested episodic ataxia of varying severity. Four subjects from three families reported hearing impairment, which has not previously been reported in association with EA1. New mutations (R167M, C185W and I407M) were identified in three out of the four families. When expressed in human embryonic kidney cells, all three new mutations resulted in a loss of Kv1.1 channel function. The fourth family harboured a previously reported A242P mutation, which has not been previously described in association with ataxia.
Conclusions
The genetic basis of EA1 in four families is established and this report presents the earliest documented case from 1928. All three new mutations caused a loss of Kv1.1 channel function. The finding of deafness in four individuals raises the possibility of a link between Kv1.1 dysfunction and hearing impairment. Our findings broaden the phenotypic range associated with mutations in KCNA1.
Keywords: Cerebellar Ataxia; Epilepsy; Neurogenetics; Neuromuscular; Neurophysiol, Clinical
Introduction
Voltage-gated potassium channels (Kv channels) play fundamental roles in controlling neuronal excitability. The main pore-forming channel is composed of four α subunits, of which approximately 40 subtypes are known, organised in 12 subfamilies (Kv1–Kv12). They include two subgroups relevant to disorders of peripheral nerve excitability, ataxia and epilepsy: fast potassium channels (Kv1), and slow potassium channels (Kv7). The Kv channel also contains four intracellular auxiliary β subunits. The specificity of α subunits for β subunits is conferred by the cytosolic N-terminus of the α subunit, and the combination of subunits influences the expression and localisation of Kv channels, as well as their gating and kinetic properties.1 2
Fast K+ channels that contain Kv1.1 subunits usually occur as heterotetramers in combination with Kv1.2 or Kv1.4 subunits. These channels are mostly closed at resting membrane potential and have a relatively low threshold for activation and rapid activation kinetics, but are slower than fast Na+ channels (hence the term ‘delayed rectifier’). K+ ion efflux, driven by the electrochemical trans-membrane gradient, has a hyperpolarising effect on the membrane potential and therefore limits neuronal excitability.3 4 In myelinated nerves, Kv1.1-containing K+ channels are densely expressed in the juxtaparanodal region of the axon.2 Activation of fast K+ channels reduces the resistance of the internodal membrane and limits axonal hyperexcitability after an action potential.5 6 Dysfunction is thus expected to lead both to excessive excitability of neurons and to an enhanced duration of action potentials, which may in turn cause excessive neurotransmitter release. These effects contribute to the repetitive activity and paraesthesiae elicited by the Kv blocker 4-aminopyridine.7 Of the many other K+ channels, slow K+ channels encoded by KCNQ genes are especially important in neurological disease, such as benign familial neonatal epilepsy.8 They co-assemble to form outward rectifying currents that helps stabilise the membrane potential. In neurones, these commonly exist as Kv7.2–Kv7.3 heteromers, which activate slowly and are non-inactivating.3 9 Slow K+ channels are highly expressed in myelinated nerve at the nodes of Ranvier and the axonal initial segments,10 11 and in the central nervous system they are abundant at the axonal initial segments.10
Heterozygous mutations in the KCNA1 gene (chromosome 12) encoding the α subunit of Kv1.1 result in episodic ataxia type 1.12 EA1 is a dominantly inherited condition characterised by brief attacks of midline cerebellar disturbance including limb ataxia, dysarthria, titubation, nystagmus, tremor and gait incoordination. Episodes may be triggered by startle, vigorous activity, change in posture (eg, from sitting to standing), emotion, hunger, alcohol or intercurrent illness. At the onset of events, some of our patients describe the sensation of a ‘click’ or a ‘shock’ in their head. Episodes last seconds to minutes and may be associated with a feeling of apprehension or anxiety. Cerebellar function is typically normal between attacks. However, almost all patients exhibit persistent neuromyotonia which can be confirmed on electromyography (EMG).13 14 An increased incidence of epilepsy has also been reported in association with EA1. Brain imaging in patients with EA1 is typically unremarkable.15–18
Over 25 mutations have been identified in KCNA1, with functional studies demonstrating a loss of channel function through a dominant-negative mechanism.13 16 However, the full clinical spectrum of EA1 is unknown. Here we describe new clinical and genetic findings in four previously unreported families with EA1. In addition to typical features of EA1, several individuals reported hearing impairment. New heterozygous mutations in KCNA1 were identified in three of the four families. All three new mutations caused a loss of KV1.1 function. The fourth family carried the A242P mutation which has previously been reported in association with epilepsy and neuromyotonia but not with ataxia.16
Methods
Ethics approval was obtained from the Joint National Hospital for Neurology and Neurosurgery/University College London Ethics Committee and the University of Sydney Research Ethics Committee for this project. All subjects provided written informed consent. Patients were assessed clinically and, where possible, nerve conduction studies and EMG were performed according to a previously described protocol.13 Non-invasive nerve excitability studies were also performed in this cohort, and have been reported elsewhere.8
Gene sequencing
DNA was extracted from venous blood or salivary samples using standard procedures. PCR amplification of DNA was performed using newly designed primers and conditions (see online supplementary data; tables S1 and S2) for KCNA1 reported in NCBI (accession number NM000217). DNA sequencing was performed using ABI BigDye terminator sequencing kit (V.1.1) and an ABI 3730xl automated DNA sequencer. Analysis of results was performed using ABI Seqscape (V.3.5) software. DNA from 198 control chromosomes was sequenced for comparison.
Functional expression
Cell culture
Human embryonic kidney cells (HEK-293) were grown in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum and 1% glutamax at 37°C with 5% CO2. Cells were plated onto poly-D-lysine coated 10 mm cover slips. KCNA1 and green fluorescent protein (GFP) cDNA contained within pMT2LF were co-transfected using lipofectamine2000 (Invitrogen). A total of 4 µg of DNA was used. All recordings were made 36–72 h after transfection. To control for variability in batches of transfections, recordings from wild-type (wt) KCNA1-expressing cells were routinely intercalated with recordings from cells expressing mutant KCNA1.
Molecular biology
The identified human mutations were introduced into the corresponding positions of the wt Kv1.1 cDNA construct contained within the pMT2LF mammalian expression vector using a PCR-based site-specific mutagenesis strategy (Stratagene Quickchange). Mutant and wt clones were sequenced to verify the presence of the mutations. Primer sequences are available upon request.
Electrophysiology
Whole-cell patch clamp recordings were performed to record potassium currents at room temperature. The external solution contained (in mM): NaCl2 135; KCl 4; MgCl2 1; CaCl2 2; HEPES 10. The pipette solution contained (in mM): NaCl 135; KCl 2.5; EGTA 2; HEPES 10. Pipette resistance was 2–4 MΩ when filled with intracellular solution. Cells were held at −80 mV, and series resistance was not compensated. Cells with series resistance above 10 MΩ were discarded. A –P/4 protocol was used to subtract leak currents. Currents were recorded using an Axopatch 200B amplifier (Molecular Devices). Data were acquired and analysed using LabView software (V.8.0, National Instruments) with in-house acquisition programs (DMK). Data were sampled at 20 kHz and filtered at 5 kHz. Graphs were plotted using Sigmaplot software (V.8.0, SPSS). Statistical significance was determined using one-way analysis of variance.
Immunocytochemistry
HEK cells were co-transfected using lipofectamine with pMT2LF KCNA1 and pcDNA3.1 GFP constructs 48 h prior to immunocytochemistry. Cells were initially fixed for 5 min with 2% and then 4% of paraformaldehyde, followed by washing with 0.01% phosphate buffered saline solution (PBS). Cells were permeabilised with 0.1% Triton X-100 in PBS for 5 min and then incubated with 4% normal goat serum for 20 min. Cells were incubated with the primary antibody, 1:500 rabbit anti-human Kv1.1 (Abcam) which was mixed in 0.1% Triton X-100 in PBS for 1 h. After washing, cells were incubated with the secondary antibody, 1:1000 goat anti-rabbit alexa fluor 546 (Invitrogen) for 1 h, washed and mounted in polyvinyl alcohol with DABCO anti fading (Sigma). Images were acquired with a Leica confocal microscope.
Results
Clinical assessment
Fifteen patients from four families in three countries were studied (figure 1). All patients gave a history of brief episodes of cerebellar dysfunction beginning in childhood or adolescence (see online supplementary data, table S3). Clinical documentation of EA1 in Family C dates back to 1928 (figure 2). To our knowledge this is the earliest clinical record of EA1. Several interesting clinical features were noted. In most patients, the frequency of ataxic episodes reached a peak during adolescence. A common feature was exercise-induced ataxia, which would disrupt sporting activities and, in several patients, resulted in the inability to play sport or run at all. Vocation was significantly affected in four of 15 subjects. The proband in family C could not speak publically in his role as a journalist because of anxiety-induced attacks. The proband in family A, a naval officer, was required to significantly modify his role significantly because of stress-induced ataxia. This subject also described proximal muscle weakness during attacks, and ictal clinical assessment verified this. The proband in family D experienced seizures, which had not previously been described with this mutation. Four subjects from three families reported hearing impairment.
Figure 1.

Pedigree for families A, B, C and D. Black shapes denote affected family members. Asterisk indicates proband.
Figure 2.

Clinic letter dated 1928 describing assessment of subject II:6 in family C.
Clinical neurophysiology
Nerve conduction studies were performed in 10 of 15 affected individuals (see online supplementary data, table 4). Apart from one patient with a mild, unrelated neuropathy, routine sensory and motor nerve conduction studies in patients with EA1 showed normal motor conduction velocities and amplitudes. Spontaneous repetitive discharges after a single stimulus were seen in two of the 10 subjects. There was no evidence of denervation on EMG, and voluntary contraction produced a normal EMG interference pattern in patients. Neuromyotonia was evident in 14 of 15 patients on EMG or clinical examination. The degree of myokymia/neuromyotonia varied and was seen more often in facial muscles or small muscles of the hand. Not all muscles samples in affected patients displayed spontaneous discharges.
Molecular genetics
In family A, a new heterozygous T to G substitution was identified at nucleotide position 500. This results in the change of a positively charged arginine to a non-polar methionine at amino acid position 167 (R167M). This mutation is located in the N-terminus of the channel, at its junction with the transmembrane S1 domain. In family B, a new heterozygous C to G substitution was identified at nucleotide position 555, resulting in a substitution of cysteine to tryptophan at amino acid position 185 (C185W) in the S1 transmembrane domain. In family C, a new heterozygous T to G substitution was identified at nucleotide position 1221, resulting in the substitution of isoleucine to methionine at amino acid position 407 (I407M) in the S6 transmembrane domain, in close proximity to the start of the intracytoplasmic C-terminus. The amino acids affected by each newly described mutation were highly conserved across species (ENSEMBL genome browser; figure 3). These nucleotide changes were not observed in 198 control chromosomes. In family D, a heterozygous T to G substitution was identified at nucleotide position 500, resulting in the substitution of alanine to proline at amino acid 242 (A242P). This mutation has been previously reported in an unrelated kindred without ataxia.16
Figure 3.

(i) Electrophoretograms showing new mutations for families A, B and C. Red=thymine, green=adenosine, black=guanine, blue=cytosine. (ii) Conservation of KCNA1 amino acid sequence across species. KCNA1 amino acids 152–196 are shown with the S1 segment outlined in grey. Arrows A, R167; B, C185. (iii) Amino acids 381–413 with S6 segment outlined in grey. Arrow C: I407 in S6.
In vitro expression
Heterologous expression
No detectable potassium currents were observed when R167M or C185W were expressed in HEK cells. In contrast, expression of wt KV1.1 resulted in a slowly activating current evoked with depolarising steps (figure 4). When expressed in HEK cells, both the R167M and C185W mutants were detected with anti-KV1.1 antibodies using immunofluorescence techniques. The distribution of immunofluorescence was comparable to that of wt KV1.1, implying normal trafficking to the HEK cell membrane. No intracellular aggregates were detected, confirming that the R167M and C185W mutants were not being retained in the cytoplasm (figure 5).
Figure 4.

(A) Voltage step protocol and representative current traces elicited by applying a voltage step from a holding potential of −80 mV. (B) Peak current density measured following depolarisation to ±10 mV (SEM). (C) Voltage dependence of activation estimated from tail currents measured at −80 mV following depolarising steps to different potentials obtained in cells transfected with wt (n=7), I407 M (n=8), wt:I407M (n=5), wt:R167M (n=7) and wt:C185W (n=5). Symbols as in panel A. Tail currents were sampled 1 ms after return to −80 mV. Error bars are SEM. Recordings were made using low potassium internal solution.
Figure 5.
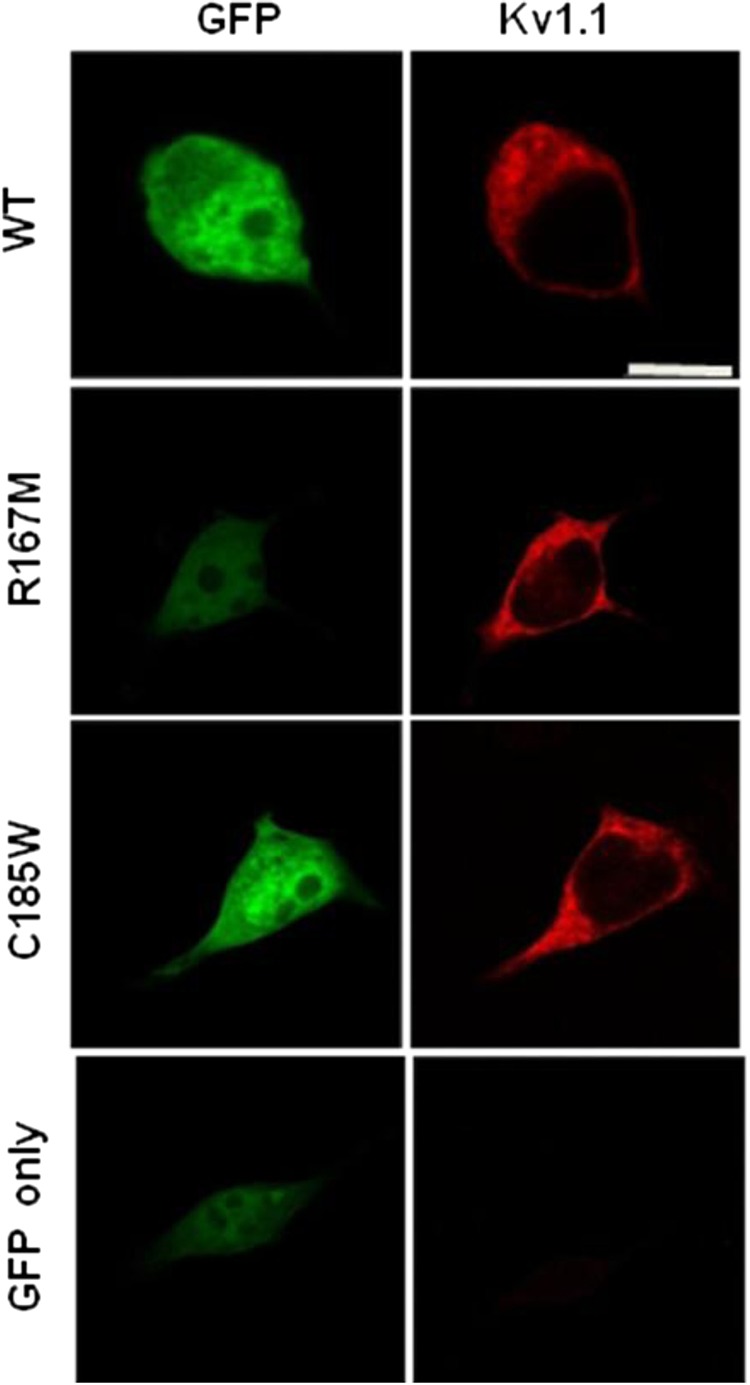
The Kv1.1 antibody detects the channel and the mutants, R167M and C185W in transfected human embryonic kidney (HEK) cells. The antibody does not detect any Kv1.1 channel in the HEK cells transfected with green fluorescent protein only (bottom panel).
Mutant and wt KV1.1 were co-expressed in HEK cells. Tail currents recorded at −80 mV following repolarisation from different voltages were used to characterise the voltage dependence of activation. Both wt:R167M and wt:C185W resulted in K+ currents with a significantly smaller peak current density compared with wt alone (wt alone, 57.9±9.0 pA/pF, n=7; wt:R167M, 16.4±2.4 pA/pF, n=8, p<0.001; wt:C185W, 17.3±4.1 pA/pF, n=5, p<0.001). When co-expressed with wt, mutants also caused a significant positive shift in the V1/2 Max of the voltage dependence of channel activation (wt:R167M= +10.0 mV; wt:C185W= +4.4 mV, p<0.05; wt alone V1/2MAX=−32.8±0.9 mV).
Expression of I407M in HEK cells resulted in potassium currents with a significantly smaller current density compared to wt KV1.1 alone (I407M: 17.8±3.9 pA/pF, n=7, p<0.001). The I407M mutant also caused a large positive shift in the voltage dependence of activation (+21.4 mV, p<0.05) compared with wt Kv1.1. When I407M was coexpressed with wt KV,1.1, the current density was similar to I407M alone and significantly lower than wt alone (25.2±6.5 pA/pF, n=8, p<0.0001). Cells expressing wt and I407M also exhibited a shift in the voltage dependence of activation (as measured from the V1/2Max; +10.9 mV, p<0.05). In summary, expression of all three mutants, R167M, C185W and I407M is consistent with a pathogenic loss of function, a change in activation threshold and a dominant-negative effect of the mutant on wt KV1.1 subunits (figure 4).
Discussion
Episodic ataxia type 1 typically presents with brief episodes of cerebellar dysfunction with full resolution of cerebellar features interictally. Continuous neuromyotonia is seen in most patients, although the degree of neuromyotonia can vary widely. This study describes four unreported families with a phenotype of EA1. Several interesting new clinical observations are documented. Although seemingly a benign disorder, attacks of cerebellar dysfunction can have a significant impact on vocational duties, participation in sport or daily activities. It is often the limitation of function resulting from attacks that prompted patients to seek medical assessment.
Clinical features
The proband in family A described proximal muscle weakness during an attack, which is rarely reported in EA1. The proband in family D experienced seizures, and this adds to the weight of evidence implicating this mutation specifically, and KV1.1 dysfunction in general, in epilepsy. This mutation has previously been described in association with epilepsy and neuromyotonia alone,16 implying that other factors, genetic or environmental, can affect the phenotype. Assessment for epilepsy should be considered for all patients with EA1, in light of the increased incidence of epilepsy in this disorder.
The presence of deafness in four patients is of particular interest as KV1.1 is expressed in the auditory brainstem. Studies in KV1.1 null mice have demonstrated altered auditory processing, including a failure to follow high-frequency sound stimulation.19 20 Furthermore, deafness is associated with mutations in other potassium channel genes.21 Although the four individuals in this study have not had audiological assessments, the finding suggests a link between KV1.1 dysfunction and hearing impairment.
Neurophysiological studies
Published data on routine sensory and motor nerve conduction studies in patients with EA1 show normal motor conduction velocities and amplitudes,17 18 22–24 as was the case with our cohort. Normal EMG interference pattern during voluntary contraction was seen in patients, and interictal neuromyotonia was readily detectable in all but one patient, as is documented in the literature.16 22 23 Nerve excitability studies reported in this cohort were able to detect changes specific to loss of fast K+ channel function in vivo, and changes in axonal excitability are sufficient to differentiate them from normal controls with 100% sensitivity.14
Genetics
New KCNA1 mutations were identified in three of the families. Evidence supporting pathogenicity of these mutations includes the observations that all three mutations alter amino acids in conserved regions of the gene; they segregate with the EA1 phenotype and were absent in control chromosomes.
Functional expression
All three mutations demonstrated a loss of KV1.1 channel function via a dominant-negative mechanism which is well established in EA1. The finding of three new mutations in this single-exon gene suggests that private family mutations may be common in this disorder. Although heterologous expression studies do not fully reveal the roles of KV1.1 in neuronal excitability, we have recently shown that nerve excitability measurement by threshold tracking provides direct evidence that impaired KV1.1 function underlies the hyperexcitability of motor axons in individuals with EA1.14
Paroxysmal nature of EA1
The reason for the intermittent manifestation of EA1 is not known. Peripheral nerve excitability in this cohort is increased14 and a mouse model of EA1 has suggested excessive GABA release at basket cell terminals in the cerebellar cortex.25 However, this does not explain the relationship to precipitants, nor how the ictus spreads to involve the circuitry underlying cerebellar coordination throughout the body. Studies of EA1 in twins have shown that variability in phenotype is a result of non-genetic factors.26 Progressive fixed or cerebellar syndromes are rarely seen in patients with EA1. This is in contrast to patients with episodic ataxia type 2 (EA2), which is a dominantly inherited disorder due to mutations in CACNA1A encoding the presynaptic Ca2+ channel Cav2.1 Patients with EA2 have long episodes of ataxia, lasting hours or days, and may develop fixed or progressive cerebellar features that can be detected interictally. They do not have neuromyotonia. EA2 is allelic with familial hemiplegic migraine and spinocerebellar ataxia type 6 (SCA6), and there is phenotypic overlap between these disorders.13
Treatment of EA1
Treatment for patients with EA1 is directed at symptom management. Owing to the brevity of events, many patients do not require medication for the ataxic events. Patients often learn to modify their behaviour to avoid triggering episodes. Furthermore, many report diminished frequency of episodes after adolescence. Given the reduced fast K+ currents in these patients, an agent that enhanced the current might be clinically useful. However, while there are blockers of fast potassium channel function (aminopyridines), there are no clinically useful agents that can increase this current, unlike the situation with slow potassium channels, where retigibine can enhance the current. Accordingly, therapies are designed to modulate nerve excitability and possibly have indirect effects on fast K+ channel function. If episodes are uncontrolled, acetazolamide may diminish frequency and severity of episodes. The severity of neuromyotonia is variable and may not require treatment but carbamazepine, phenytoin and sulthiame have been reported to be efficacious.17 18 Finally, overexpression of wt Kv1.1 with a lentiviral vector has recently been used successfully in a rodent model of neocortical epilepsy,27 potentially offering the prospect of a targeted correction of the genetic defect in EA1. However, gene delivery to a large area of the cerebellum for a relatively benign condition presents substantial challenges.
Supplementary Material
Footnotes
Contributors: Study concept and design: SET, SR, DB, MGH, DMK. Acquisition of data: SET, SR, TG, DB, SS, CMS, RWL. Analysis and interpretation of data: SET, SR, SVT, DMK, SS, RWL, DB. PG, MGH. Drafting of the manuscript: SET, SR, DB, DMK, MGH. Critical revision of the manuscript for important intellectual content: SET, SR, DMK, SS, MGH.
Funding: This work was supported by the Guarantors of Brain the British Medical Association Vera Down Fellowship (ST), the Charities Aid Foundation Patrick Berthoud Fellowship (ST), the University of Sydney (ST) and the UK Brain Research Trust (ST), Wellcome Trust Research Training Fellowship (SR), UK Medical Research Council grant number G0601943 (ST, SR, SS, MGH, DMK), The National Health and Medical Research Council of Australia (ST, DB and CMS) and the Consortium for Clinical Investigation of Neurological Channelopathies (CINCH) supported by NIH RU54 RR019482 (ST, SR, SVT, DMK, MGH).
Competing interests: None.
Ethics approval: University of Sydney Human Research Ethics Committee, Joint National Hospital for Neurology and Neurosurgery and Institute of Neurology Ethics Committee.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Vacher H, Mohapatra DP, Trimmer JS. Localization and targeting of voltage-dependent ion channels in mammalian central neurons. Physiol Rev 2008;88:1407–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scherer SS, Arroyo EJ. Recent progress on the molecular organization of myelinated axons. J Peripher Nerv Syst. 2002;7:1–12. [DOI] [PubMed] [Google Scholar]
- 3.Baker M, Bostock H, Grafe P, et al. Function and distribution of three types of rectifying channel in rat spinal root myelinated axons. J Physiol 1987;383:45–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwarz JR, Reid G, Bostock H. Action potentials and membrane currents in the human node of Ranvier. Pflügers Arch 1995;430:283–92. [DOI] [PubMed] [Google Scholar]
- 5.Bostock H, Cikurel K, Burke D. Threshold tracking techniques in the study of human peripheral nerve. Muscle Nerve 1998;21:137–58. [DOI] [PubMed] [Google Scholar]
- 6.Burke D, Kiernan MC, Bostock H. Excitability of human axons. Clin Neurophysiol 2001;112:1575–85. [DOI] [PubMed] [Google Scholar]
- 7.Kocsis JD, Waxman SG, Hildebrand C, et al. Regenerating mammalian nerve fibres: changes in action potential waveform and firing characteristics following blockage of potassium conductance. Proc R Soc Lond B Biol Sci 1982;217:77–87. [DOI] [PubMed] [Google Scholar]
- 8.Tomlinson SE, Bostock H, Grinton B, et al. In vivo loss of slow potassium channel activity in individuals with benign familial neonatal epilepsy in remission. Brain 2012;135:3144–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bostock H, Baker M. Evidence for two types of potassium channel in human motor axons in vivo. Brain Res 1988;462:354–8. [DOI] [PubMed] [Google Scholar]
- 10.Devaux JJ, Kleopa KA, Cooper EC, et al. KCNQ2 is a nodal K+ channel. J Neurosci 2004;24:1236–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwarz JR, Glassmeier G, Cooper EC, et al. KCNQ channels mediate IKs, a slow K+ currentregulating excitability in the rat node of Ranvier. J Physiol 2006; 573:17–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Browne DL, Gancher ST, Nutt JG, et al. Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1. Nat Genet 1994;8:136–40. [DOI] [PubMed] [Google Scholar]
- 13.Tomlinson SE, Hanna MG, Kullmann DM, et al. Clinical neurophysiology of the episodic ataxias: insights into ion channel dysfunction in vivo. Clin Neurophysiol 2009;120:1768–76. [DOI] [PubMed] [Google Scholar]
- 14.Tomlinson SE, Tan SV, Kullmann DM, et al. Nerve excitability studies characterize Kv1.1 fast potassium channel dysfunction in patients with episodic ataxia type 1. Brain 2010;133:3530–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zuberi SM, Eunson LH, Spauschus A, et al. A novel mutation in the human voltage-gated potassium channel gene (Kv1.1) associates with episodic ataxia type 1 and sometimes with partial epilepsy. Brain 1999;122:817–25. [DOI] [PubMed] [Google Scholar]
- 16.Eunson LH, Rea R, Zuberi SM, et al. Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability. Ann Neurol 2000;48:647–56. [PubMed] [Google Scholar]
- 17.Brunt ER, Van Weerden TW. Familial paroxysmal kinesigenic ataxia and continuous myokymia. Brain 1990;113:1361–82. [DOI] [PubMed] [Google Scholar]
- 18.Vaamonde J, Artieda J, Obeso JA. Hereditary paroxysmal ataxia with neuromyotonia. Mov Disord 1991;6:180–2. [DOI] [PubMed] [Google Scholar]
- 19.Brew HM, Hallows JL, Tempel BL. Hyperexcitability and reduced low threshold potassium currents in auditory neurons of mice lacking the channel subunit Kv1.1. J Physiol 2003;548:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kopp-Scheinpflug C, Fuchs K, Lippe WR, et al. Decreased temporal precision of auditory signaling in Kcna1-null mice: an electrophysiological study in vivo. J Neurosci 2003;23:9199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maljevic S, Wuttke TV, Seebohm G, et al. Kv7 channelopathies. Pflugers Arch 2010;460:277–88. [DOI] [PubMed] [Google Scholar]
- 22.Van Dyke DH, Griggs RC, Murphy MJ, et al. Hereditary myokymia and periodic ataxia. J Neurol Sci 1975;25:109–18. [DOI] [PubMed] [Google Scholar]
- 23.Hanson PA, Martinez LB, Cassidy R. Contractures, continuous muscle discharges, and titubation. Ann Neurol 1977;1:120–4. [DOI] [PubMed] [Google Scholar]
- 24.Gancher ST, Nutt JG. Autosomal dominant episodic ataxia: a heterogeneous syndrome. Mov Disord 1986;1:239–53. [DOI] [PubMed] [Google Scholar]
- 25.Herson PS, Virk M, Rustay NR, et al. A mouse model of episodic ataxia type-1. Nat Neurosci 2003;6:378–83. [DOI] [PubMed] [Google Scholar]
- 26.Graves TD, Rajakulendran S, Zuberi SM, et al. Nongenetic factors influence severity of episodic ataxia type 1 in monozygotic twins. Neurology 2010;27:367–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wykes RC, Heeroma JH, Mantoan L, et al. Optogenetic and potassium channel gene therapy in a rodent model of focal neocortical epilepsy. Sci Transl Med 2012;4:161ra152. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.