

Abstract
Working with a combination of ProMOL (a plugin for PyMOL that searches a library of enzymatic motifs for local structural homologs), BLAST and Pfam (servers that identify global sequence homologs), and Dali (a server that identifies global structural homologs), we have begun the process of assigning functional annotations to the approximately 3,500 structures in the Protein Data Bank that are currently classified as having “unknown function”. Using a limited template library of 388 motifs, over 500 promising in silico matches have been identified by ProMOL, among which 65 exceptionally good matches have been identified. The characteristics of the exceptionally good matches are discussed.
Keywords: bioinformatics, catalytic motif, enzyme, ProMOL, protein function, PyMOL, structural biology
Introduction
Elucidating the functions of proteins is a core component of biochemistry, structural biology, and bioinformatics. Scientists in these disciplines seek to understand the relationship among protein sequence, structure, and function. A recent search identified approximately 3,500 structures of “unknown function” in the Protein Data Bank (PDB) [1, 2]. Software tools have been developed to relate sequence, structure and function. Several programs exist that can propose functional annotations for a specific target of interest. Sequence databases can be searched with tools such as BLAST [3] and HMMER [4] to identify sequence homologs. Databases, repositories, and servers such as UniProt [5], Pfam [6, 7], the Structural Biology Knowledgebase [8], Dali [9], and MarkUs [10] collect and display information from various sources that may be used to identify structural homologs and/or give functional insight.
We have developed the ProMOL [11] plugin for PyMOL [12], a tool used to explore the catalytic site structural homologies between proteins of known function and those for which functions are not yet known. ProMOL uses template-based alignment of these structures with a current library of 388 active site motifs as reported in the Catalytic Site Atlas [13]. We have applied this approach to examine structures in the Protein Data Bank which are listed as having “unknown function”. Although catalytic site structural homology alone is not sufficient to define the function of a protein, it provides one mechanism which, when combined with other structural and sequence motifs, can suggest candidates for experimental verification. We also applied three well-established methods for protein function assignment based on sequence (BLAST, Pfam) and global structure alignments (Dali) to gain more insight into the functions proposed by ProMOL. ProMOL is being developed collaboratively and distributed freely as open source software (http://sourceforge.net/projects/sbevsl/ or http://www.promol.org).
The aim of this study is to analyze all uncharacterized PDB entries using ProMOL to identify potential enzymatic functions for some of these structures. Targets showing high similarity to known catalytic sites using ProMOL were then analyzed using additional in silico methods (BLAST, Pfam, and Dali) in an attempt to identify potential enzymatic functions. Of the 3,437 PDB entries of “unknown function” that have been processed with the current ProMOL motifs, over 500 entries yielded high-probability matches (≥3-residue alignments with RMSD <10 Å for non-hydrogen atoms). There were 65 exceptional matches (3-residue alignments with RMSD ≤2.5 A or 4- and 5-residue alignments with RMSD ≤4.0 A) against the current motif library.
Methods
The search of the PDB for proteins of “unknown function” was performed with an advanced text search for entries containing “unknown function”. An additional search using the above criteria plus a deposition date between 1/1/2000 - 7/7/2014 was also performed, indicating that 96.4% of these structures have been generated since the advent of the Protein Structure Initiative (PSI) [14].
We have created a library of 388 motif templates containing well-defined enzyme active sites, as found in the Catalytic Site Atlas [13]. The motif templates span all six top-level EC number groups to at least the third level (e.g., 3.1.21.*) wherever structures in those groups are available. Structures were selected based on the availability of literature definitions of active sites in the Catalytic Site Atlas [15]. Two sets of motif templates make up the current library: roughly half were generated manually with the Motif Maker tool in ProMOL [11], which can be used to create motif templates based on residue name, residue number and chain identifier for the residues in the active site of an enzyme structure found in the PDB. The remaining motif templates were generated automatically with a script that tests the motif template against itself, homologs and random PDB structures to provide sensitivity and specificity data for each new motif template [manuscript in preparation].
These structures were screened according to the protocol illustrated in Figure 1. The first step was to run all of the uncharacterized proteins through ProMOL to compare the identity and geometry of the amino acid residues in these structures with the motif library. Results which met the following criteria were subjected to further analysis: (1) 3-residue alignments with RMSD ≤ 2.5 Å (for non-hydrogen atoms) or (2) RMSD ≤ 4.0 Å (for non-hydrogen atoms) for ≥ 4-residue alignments (RMSD values were calculated in three ways: Cα used only the alpha carbons; Cα and Cβ used the alpha and beta carbons; and All used all the non-hydrogen atoms in the residues). In an attempt to narrow our search results to structures with reliable functional annotations, the sequences of hits from our initial search were used to search the PDB with BLAST. The protein sequences were also evaluated with Pfam to gain additional functional insight. Finally a global structural alignment was conducted with Dali. The results from these four approaches (ProMOL, BLAST, Pfam, and Dali) were then compared and evaluated to predict the most likely function for the proteins.
Figure 1.

Flowchart of the process used for characterization of proteins of “unknown function”. Although in silico characterization methods and results in the top box are the focus of this report, additional in silico (the second box), in biblio (third box) and in vitro steps (fourth box) are also illustrated.
Ligand binding was conducted with AutoDock Vina [16] according to the instructions on their web site (http://autodock.scripps.edu/). Once a protein had an assigned EC number, ligands that were bound to PDB entries under that same EC number were selected for in silico binding studies. Ligands and protein structures were converted to *.pdbqt files using AutoDockTools. The grid box for the enzyme active site was defined based on the location of catalytic residues predicted by ProMOL. AutoDock Vina was executed from the command prompt and the results were then visualized in PyMOL. Each docking experiment was repeated four times and the average free energy values are reported in Table 2.
Table 2.
ProMOL assigned functions to structures for which BLAST, Pfam, and Dali did not suggest an assignment. There were fifteen instances in which ProMOL suggested a function for a structure, while the other three programs did not. RMSD values were calculated for all non-hydrogen atoms. Fourteen of fifteen alignments in ProMOL consisted of identical active site residue matches between the query and the motif template.
| Query ID | Motif ID | EC Number of Motif | RMSD: All (Å) | Function Assigned by ProMol | Liganda | ΔG (kcal/mol)b | Kd (μM) |
|---|---|---|---|---|---|---|---|
| 1OQ1 | 1E2T | 2.3.1.118 | 1.94 | transferase | KH2 | −5.8 | 57 |
| 2DBI | 1BP2 | 3.1.1.4 | 2.29 | hydrolase | 8IN | −6.1 | 34 |
| 2G6T | 1NAA | 1.1.99.18 | 2.13 | oxidoreductase | ABL | −6.1 | 34 |
| 2GFQ | 1D6O | 5.2.1.8 | 2.44 | isomerase | 1KY | −10.2 | 0.034 |
| 2P0O | 1K9Z | 3.1.3.7 | 0.93 | hydrolase | AMP | −7.2 | 5.3 |
| 2P0V | 1AAM | 2.6.1.1 | 1.96 | transferase | CBA | −6.8 | 10 |
| 2PK7 | 1PJH | 5.3.3.8 | 1.64 | isomerase | ADP | −5.1 | 180 |
| 2Q4K | 1OD1 | 3.4.23.22 | 2.08 | hydrolase | 0EO | −6.1 | 34 |
| 2XU2 | 1BMT | 2.1.1.13 | 1.52 | transferase | C2F | −5.7 | 67 |
| 3D19 | 1HY3 | 2.8.2.4 | 2.02 | transferase | EST | −9.5 | 0.11 |
| 3L6I | 1C4X | 3.7.1.8 | 2.14 | hydrolase | 22J | −4.8 | 305 |
| 3M3I | 4HOH | 3.1.27.3 | 2.69 | hydrolase | 2GP | −6.7 | 12 |
| 3NLC | 1BMT | 2.1.1.13 | 1.54 | transferase | SAH | −7.2 | 5.3 |
| 3RBY | 1BK7 | 3.1.27.1 | 2.15 | hydrolase | 5GP | −6.5 | 17 |
| 4GHB | 1PNO | 1.6.1.2 | 1.75 | oxidoreductase | NAP | −8.1 | 1.2 |
- KH2: 3-(1-methylpiperidinium-1-yl)propane-1-sulfonate
- 8IN: [3-(1-benzyl-3-carbamoylmethyl-2-methyl-1h-indol-5-yloxy)-propyl-]-phosphonic acid
- ABL: 5-amino-5-deoxy-cellobiono-1,5-lactam
- 1KY: 6-({(1S,5R)-3-[2-(3,4-dimethoxyphenoxy)ethyl]-2-oxo-3,9-diazabicyclo[3.3.1]non-9-yl}sulfonyl)-1,3-benzothiazol-2(3H)-one
- AMP: adenosine 5′-monophosphate
- CBA: N-pyridoxyl-2,3-dhydroxyaspartic acid-5-monophosphate
- ADP: adenosine 5′-diphosphate
- 0EO: (2S)-2-[[(3S,4S)-5-cyclohexyl-4-[[(4S,5S)-5-[(2-methylpropan-2-yl)oxycarbonylamino]-4-oxidanyl-6-phenyl-hexanoyl]amino]-3-oxidanyl-pentanoyl]amino]-4-methyl-pentanoic acid
- C2F: 5-methyl-5,6,7,8-tetrahydrofolic acid
- EST: estradiol
- 22J: (3E,5R)-5-fluoro-6-(2-fluorophenyl)-2,6-dioxohex-3-enoic acid
- 2GP: guanosine-2′-monophosphate
- SAH: S-adenosyl-L-homocysteine
- 5GP: guanosine-5′-monophosphate
- NAP: nicotinamide adenine dinucleotide phosphate
The binding energy as computed by Autodock Vina [16] was converted to a dissociation constant with the formula: Kd=exp(−ΔG/(R*T)) [http://autodock.scripps.edu/faqs-help/faq/how-autodock-4-converts-binding-energy-kcal-mol-into-ki]
Once a putative function is established, literature searches are conducted for enzymes from these families so that suitable assay conditions and substrates may be found. Plasmids that can express these proteins must be obtained from sources such as DNASU [17–19]. Substrates are then ordered from commercial suppliers such as Sigma-Aldrich. The protein can be expressed in E. coli and isolated using established methods of protein production and purification. The predicted molecular weight is confirmed using SDS-PAGE. Enzyme activity assays are then conducted using appropriate substrates.
Results and Discussion
As of 31 October 2014 there were 3,646 structures of “unknown function” among the more than 100,000 structures in the PDB. The initial search in May 2013 yielded 3,437 structures, which are described below; any newly deposited structures which are classified as “unknown function” will be characterized with new versions of ProMOL containing more motif templates. The 3,437 proteins were analyzed with ProMOL, BLAST, Pfam, and Dali and over 500 promising hits (15%) were identified. It should be noted that about 58% of current entries in the PDB have Enzyme Commission (EC) numbers; if the same probability exists for structures of unknown function, then roughly 2,000 of them would be expected to have enzyme function. One reason for this discrepancy is the limited number of motif templates that are currently available. As mentioned above, our motif template library is being expanded and will be used to further characterize the remaining structures of unknown function. It is also possible that a significant number of these proteins have enzymatic activity that has not been previously characterized and for which no homologous structures exist.
The set of 500 promising hits (≥3-residue alignment with RMSD <10.0Å) was further distilled down to 65 exceptional hits (3-residue alignment with RMSD <2.5A or ≥4-residue alignment with RMSD <4.0Å). These results are presented below in a series of tables and in the supplemental materials. Table 1 highlights results with agreement of functional annotations among ProMOL, BLAST, Pfam, and Dali. Table 2 summarizes the cases in which ProMOL provided promising alignments compared to inconclusive results provided by BLAST, Pfam and Dali. The results in Table 3 are cases where BLAST, Pfam and Dali suggested similar potential functions, while ProMOL suggested a clearly different function. Table 4 contains inconsistent results, i.e., the predicted functions from the four programs were not in agreement. Table 1S provides the full list of results for each of these comparisons,
Table 1.
Cases in which ProMOL, BLAST, Pfam, and Dali all yielded similar functional annotations for a given query. The items in the column entitled, “Function Assigned by ProMOL”, were determined based on the most general classification of the motif with which the query structure was aligned. The PDB IDs of the motif templates that yielded these alignments can be found in Table 1S. There are thirteen query structures to which all four programs assigned functions that were under the same top-level EC number. There are more alignments (eighteen) than structures (thirteen) because ProMOL returned multiple alignments for several query structures. Fourteen of eighteen alignments in ProMOL consisted of identical active site residue matches between the query and the motif template.
| Query ID | Function Assigned by ProMOL | EC Number Assigned by ProMOL | Common function from Dali, BLAST, Pfam | EC Number Assigned by Dali, BLAST, Pfam |
|---|---|---|---|---|
| 1MK4a | transferase | 2.4.2.31 | acetyltransferase | 2.3 |
| 2AQW | lyase | 4.1.1.23 | orotidine-5′-phosphate decarboxylase | 4.1.1.23 |
| 2I3D | hydrolase | 3.7.1.8 or 3.4.11.5 | alpha/beta hydrolase | 3 |
| 2O14 | hydrolase | 3.1.1.47 or 3.4.11.5 | esterase or lipase | 3.1 |
| 2PW6 | oxidoreductase | 1.11.1.6 | dioxygenase | 1.13.11 or 1.14 |
| 2R8B | hydrolase | 3.4.11.5 or 3.1.1.3 | hydrolase or esterase | 3 or 3.1.1 |
| 2YYS | hydrolase | 3.7.1.9 | hydrolase or proline iminopeptidase | 3 or 3.4.11.5 |
| 3CBW | hydrolase | 3.2.1.31 | beta-mannanase or glycosyl hydrolase family 26 | 3.2 |
| 3DS8 | hydrolase | 3.1.1.3 | alpha/beta hydrolase or esterase | 3 |
| 3H04 | hydrolase | 3.4.11.5 or 3.7.1.8 or 3.4.21.26 | alpha/beta hydrolase or esterase | 3 or 3.1.1 |
| 3L1W | hydrolase | 3.1.11.2 | endonuclease/exonuclease | 2 or 3.1 |
| 4DIU | hydrolase | 3.4.11.5 | hydrolase or carboxylesterase or lipase | 3 or 3.1.1 |
| 4Q7Q | hydrolase | 3.1.1.47 | GDSL-like lipase | 3.1.1 |
The citations for all of the PDB structures in this manuscript can be found in Table 1S.
Table 3.
Query structures for which BLAST, Pfam, and Dali provided a consensus EC number while ProMOL reported a conflicting result. There were thirty structures for which all four programs gave promising results. These are the instances in which BLAST, Pfam, and Dali all returned similar results, but ProMOL did not. Twenty-eight of thirty-seven alignments in ProMOL consisted of identical active site residue matches between the query and the motif template.
| Query ID | Function Assigned by ProMOL | EC Number Assigned by ProMOL | Common function from Dali, BLAST, Pfam | EC Number Assigned by Dali, BLAST, Pfam |
|---|---|---|---|---|
| 1IUY | hydrolase | 3.2.1.15 | cullin family (ligase) | 6 |
| 1K77 | hydrolase | 3.1.1.29 | xylose isomerase; epimerase | 5.3.1.22 |
| 1NOG | oxidoreductase | 1.1.1.158 | adenosyltransferase | 2.5 |
| 1VQW | lyase | 4.2.1.24 | flavin monooxygenase | 1.14 |
| 1YEM | oxidoreductase | 1.1.1.85 | adenylate cyclase | 4.6.1.1 |
| 1YEY | hydrolase | 3.1.3.7 or 3.1.21.1 | mandelate racemase | 5.1.2.2 |
| 1YRE | hydrolase | 3.5.1.5 | acetyltransferase | 2.3 |
| 1YX3 | hydrolase | 3.2.1.14 | sulfite reductase | 1.8 |
| 1Z40 | transferase | 2.7.11.13 | apical membrane antigen 1 | N/Aa |
| 2DAF | hydrolase | 3.7.1.8 | ubiquitin-like domain | 1, 2, or 3 |
| 2DC4 | isomerase | 5.4.99.5 | adenylate cyclase | 4.6.1.1 |
| 2DEC | hydrolase | 3.5.2.6 | phosphosugar isomerase | 5.3 |
| 2F06 | oxidoreductase | 1.5.1.34 | aspartokinase | 2.7.2.4 |
| 2GGE | ligase | 6.3.2.9 | isomerase; mandelate racemase | 5 or 5.1.2.2 |
| 2KFL | transferase | 2.6.1.16 | prion protein | N/Aa |
| 2Q4D | hydrolase | 3.7.1.9 | lysine decarboxylase | 4.1.1.18 |
| 2R85 | hydrolase | 3.5.1.11 | formate-phosphoribosylaminoimidazole carboxamide ligase | 6.3 |
| 2YWO | transferase | 2.3.1.118 | thiol-disulfide isomerase; alkyl hydroperoxide reductase | 5.3.1.11 |
| 2Z0J | oxidoreductase | 1.1.1.158 | 2-phosphosulpholactate phosphatase | 3 |
| 2Z6V | isomerase | 5.3.3.10 | sulfotransferase | 2.8 |
| 2ZBV | hydrolase | 3.2.1.45 | S-adenosyl-1-methionine hydroxide adenosyltransferase | 2.5.1 |
| 3EC6 | isomerase | 5.3.3.1 | general stress protein; pyridoxamine 5′-phosphate oxidase | N/A or 1 |
| 3E8X | isomerase | 5.1.3.20 | oxidoreductase or NADP binding | 1 |
| 3FLJ | hydrolase | 3.1.3.7 | SnoaL_2 domain or steroid delta-isomerase | 5 |
| 3NRN | transferase | 2.1.1.184 | oxidoreductase | 1 |
| 3RE2 | transferase | 2.3.3.1 | menin | N/Aa |
| 3S5E | lyase | 4.2.1.24 | mitochondrial frataxin | N/Aa |
| 3TB2 | hydrolase | 3.1.3.7 | 1-cys peroxidoxin | N/Aa |
| 4ILS | hydrolase | 3.8.1.5 | alanine racemase | 5.1.1.1 |
| 4J10 | hydrolase | 3.5.1.77 | secreted protein | N/Aa |
Dali, BLAST and Pfam all agreed on the putative nonenzymatic functions for these protein structures; N/A indicates that no EC number is provided in these cases.
Table 4.
Divergent Results. It is impossible to assign these structures to consensus EC numbers based on ProMOL, BLAST, Pfam, and Dali results. All programs generated promising, yet varying, results. Six of seven alignments in ProMOL consisted of identical active site residue matches between the query and the motif template.
| Query | ProMOL: Assigned Function and EC | BLAST: Assigned Function and EC | Pfam: Assigned Function and EC | Dali: Assigned Function and EC |
|---|---|---|---|---|
| 1R3D | Hydrolase (3.1.1.3) | SHCHC synthase (4.2.99.20) | alpha/beta hydrolase (3) | SHCHC synthasea (4.2.99.20) |
| 2FBMb | lyase (4.2.1.24) | Y-like chromo domain (2.3.1.48) | enoyl-CoA hydratase (4.2.1.17) or isomerase (5.3.3.8) | Y-like chromadomainb (2.3.1.48) |
| 2I1S | isomerase (5.3.1.5) | plasmid pRiA4b ORF-3 family protein | plasmid pRiA4b ORF-3 family protein | No significant results |
| 3KK4 | hydrolase (3.1.3.7) | RHH-4 superfamily and COG4321 | RHH_4 super- family | No significant results |
| 3HFQ | Isomerase (5.5.1.5) | No significant results | 7-bladed beta- propeller lactonase family (EC 3.1.1.31) | No significant results |
| 3Q9D | hydrolase (3.2.2.1) | various hypothetical/ uncharacter-ized proteins and CT584 protein | no significant results | CT584 protein |
| 4EZI | hydrolase (3.4) | various hypothetical proteins | no significant results | esterase or lipase (3) |
For PDB entry 1R3D, the catalytic triad SER-HIS-ASP is typical of both alpha/beta hydrolases and SHCHC synthases [44].
For PDB entry 2FBM, ProMOL does not yet manage motifs of the size of the chromodomain. Regarding PDB entry 2FBM, many members of the crotonase (or enoyl-CoA hydratase) family have similar structures, but differing functions as they are from separate enzyme classes [45]. In vitro testing of these cases might prove the most interesting in establishing the value of each ProMOL functional assignment relative to BLAST, Pfam and Dali.
Uniform Function Assignment
Of the 65 structures, there were 13 cases in which ProMOL, BLAST, Pfam, and Dali all assigned similar probable functions, agreeing at least on the first digit in the EC number; in many cases agreement extended to additional digits. For example, in the case of 2AQW [20], all four programs assigned the structure to EC number 4.1.1.23. This level of agreement among the four bioinformatics programs indicates that 2AQW is a strong candidate for in vitro characterization.
High quality five-residue matches between a query structure and a motif template are rare, primarily because most enzyme motifs in the ProMOL library do not contain five residues within the active site. The alignments of 3L1W (query) [21] with 1AKO (motif template) [22] and 2O14 (query) [23] with 1BWR (motif template) [24] are uniquely interesting because both structures exhibit five-residue alignments with their respective motif templates and had RMSDs < 1.0 Å.
Figure 2A depicts the five-residue alignment of 3L1W (query) with the 1AKO motif template (hydrolase, EC 3.1.11.2). The RMSD for all non-hydrogen atoms / Cα / Cα + Cβ was 0.25 / 0.16 / 0.17 Å, respectively. Pfam, BLAST, and Dali all indicated that 3L1W was most likely an endonuclease, exonuclease, or phosphatase with a likely EC number either 2 or 3.1 (Tables 1 and 1S). The matching motif template, 1AKO, has been identified as an exonuclease III [22], which is involved in the removal of abasic sites in E. coli DNA. Finally, a global structural alignment using Dali (Figure 2B) reveals a fairly high level of full backbone structural conservation between 3L1W and 1AKO: Z-score 20.8, RMSD 2.9 Å (covering 213 residues of the 268 total residues in 1AKO with only 18% sequence identity).
Figure 2.
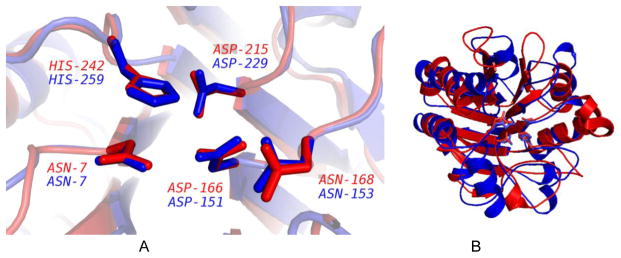
A. The five residues aligned by ProMOL are drawn for 3L1W (query/red) and 1AKO (motif template/blue). B. The superposition of 3L1W (red) and 1AKO (blue) was generated by Dali and visualized in PyMOL.
A second five-residue alignment was also found between 2O14 (query molecule) and 1BWR (motif template), an acetylhydrolase. The ProMOL alignment consisted of residues SER171, GLY209, ASN241, ASP339, and HIS342 (for the query, 2O14) and residues SER47, GLY74, ASN104, ASP192, and HIS195 (for the template, 1BWR). The RMSD values for all non-hydrogen atoms / Cα / Cα + Cβ were 0.58 / 0.51 / 0.71 Å, respectively. BLAST characterizes 2O14 as an esterase (EC 3.1.1), while Pfam characterizes it as a GDSL-like lipase/acylhydrolase (EC 3.1). Dali characterizes 2O14 as a rhamnogalacturonan acetylesterase, or putative lipase (EC 3.1). These results represent general agreement to the first two EC numbers, as hydrolases that act on ester linkages; in vitro characterization is needed to resolve the subtle differences.
Consistency between ProMOL, BLAST, Pfam, and Dali for these 13 targets makes the structures in Table 1 strong candidates for in vitro characterization. Some of these structures are currently being analyzed in our labs.
Unique ProMOL Assignments
Of the 65 structures of unknown function that generated significant “hits” when screened through ProMOL, there were fifteen cases in which BLAST, Pfam, and Dali all returned inconclusive results. These programs either assigned the structures to DUF (domain of unknown function), compared them to other uncharacterized or “hypothetical” proteins, had inconclusive matches, or simply returned no results. The results obtained with ProMOL are listed in Table 2. The first column is the structure of unknown function, while the second column provides the PDB ID for the motif template that provided the alignment results and the suggested function, followed by the EC number of the motif template, RMSD for all non-hydrogen atoms, and the proposed function, as determine by ProMOL. The last three columns reflect ligand binding to the query proteins.
To explore these unique ProMOL alignments more deeply, we selected ligands that were bound to PDB entries under the same EC number. Autodock Vina [16] was used to estimate the free energy and dissociation constants for binding these ligands to the query proteins. To see if these values were consistent with those found for the natural ligand-protein interactions, three ligands were chosen from Table 2 based on high (1KY), medium (AMP) and low (ADP) affinity to the query structures. Autodock Vina was then used to estimate the binding affinity for the ligands to the structures in their original PDB entries with these results.
Ligand 1KY binds to PDB entry 1D6O [25] with a free energy of −6.8 kcal/mol and 10 μM dissociation constant.
AMP binds to PDB entry 1K9Z [26] with a free energy of −8.5 and 0.60 μM dissociation constant.
ADP binds to PDB entry 1PJH [27] with a free energy of −5.2 and 160 μM dissociation constant.
Based on these values, the range of dissociation constants for the ligand in Table 2 support biologically significant interactions that warrant further study. The binding of one ligand, nicotinamide adenine dinucleotide phosphate (NAP), to the query structure 4GHB [28] is further explored in Figure 3A and Figure 3B. This is a medium affinity case when binding NAP to 1PNO [29], and AutoDock predicts bindings of NAP to 4GHB in the vicinity of residues TYR-57, ARG-91 and TYR-188 as predicted, but also at two other nearby binding sites as shown in Figure 3B. The best affinity for the site involving TYR-57, ARG-91 and TYR-188 was −6,9 kcal/mol (Kd = 8,8 μM) achieved with the seventh of twenty models, while a better affinity of −7.6 kcal/mol (Kd = 2.7 μM) was found at a site more than 11 Å distant from the first model, and a credible affinity of −6.6 kcal/mol (Kd = 15 μM) at a third remote site. Thus, while the predicted function might be confirmed in future experiments, the multiple sites, the lack of specificity in binding orientation, and the pore-like conformation of the quaternary structure of the 4GHB dodecamer suggest that another possibility to explore is transmembrane transport of NAP [L.C. Andrews, private communication].
Figure 3.
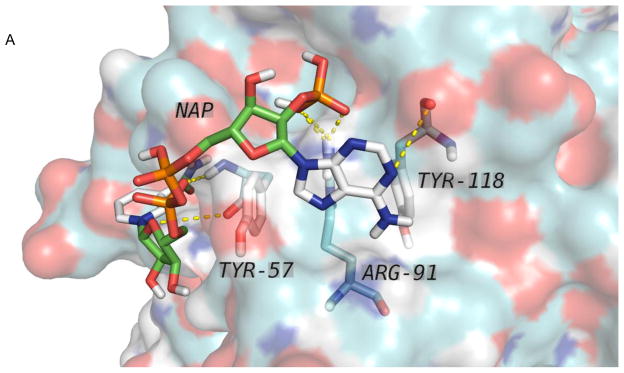
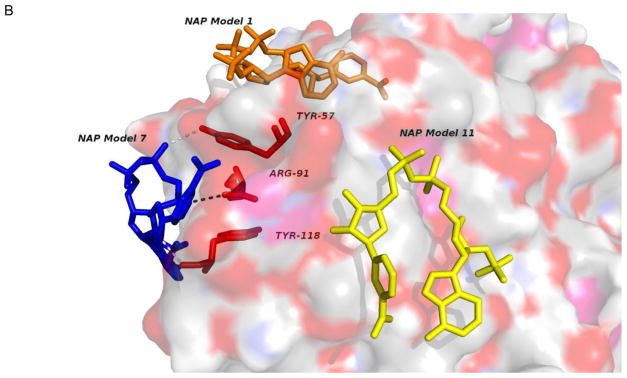
Predicted ligand binding. A. The binding of nicotinamide adenine dinucleotide phosphate (NAP) to three residues in the proposed active site of PDB entry 4GHB was predicted by AutoDock Vina at a binding site involving TYR-57, ARG-91 and TYR-188 as predicted, but also (B) at two or more additional adjacent binding sites not involving those residues.
With these fifteen structures, ProMOL provided encouraging leads for exploring the function of these structures, while the other programs did not provide testable hypotheses.
Conflicting Function Assignment
Table 3 contains thirty structures, which gave promising alignments from all four programs. The noteworthy feature is that results from BLAST, Pfam and Dali were consistent, but ProMOL gave distinctly different results. In all but one case, there is poor agreement even to the first digit of the EC numbers.
Because Dali, BLAST and Pfam all suggested that PDB entry 1K77 [30] is a xylose isomerase or an epimerase with a 4-digit EC number 5.3.1.22, while ProMOL suggested EC number 3.1.1.29, we were particularly interested in the alignments of 1K77 in Table 4. The alignment is also shown in Figure 4. Based on the results in the other programs, ProMOL was used to compare 1K77 against all of our EC 5 motif templates. ProMOL revealed an alignment between 1K77 and one EC5 motif template: 1D6O, a peptidylprolyl isomerase EC 5.2.1.8. However, the RMSD values were greater than the cutoff values that were deemed reliable for alignments in ProMOL (non-hydrogen atoms/ Cα / Cα + Cβ of 3.04/3.32/2.97 Å).
Figure 4.
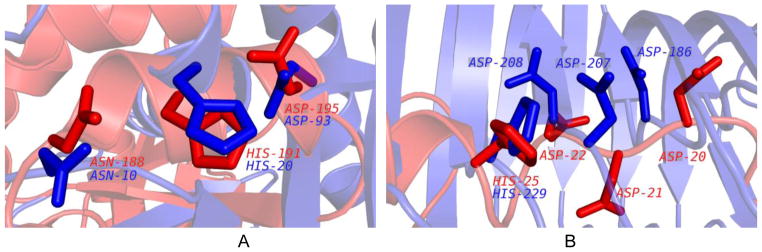
The significance of visual alignment. A. The aligned residues are shown for 1K77 (query/red; ASN188, HIS191, and ASP195) and for 2PTH (motif template/blue; ASN10, HIS20, and ASP93). The RMSD values for all non-hydrogen atoms / Cα / Cα + Cβ were 1.91 / 1.18 / 1.30 Å, respectively. B. The aligned residues shown for 1IUY (query/red; ASP20, ASP21, ASP22, and HIS25 and 1NHC (motif template/blue; ASP186, ASP207, ASP208, and HIS229). The RMSD values for all non-hydrogen atoms / Cα / Cα + Cβ were 1.93 / 1.64 / 1.56 Å, respectively.
The importance of visual comparison
The ProMOL three-residue alignment between 1K77 (query structure) and 2PTH (motif template) [31] is shown in Figure 4A. 2PTH is a peptidyl-tRNA hydrolase with an EC number of 3.1.1.29. The RMSD value for Cα of the aligned residues is 1.18 Å and the visual alignment is reasonably good, with the three aligned residues having the side chains in similar orientations. The alignment of 1IUY [32] and 1NHC [33] shown in Figure 4B had similar RMSD values to the alignment of 1K77 and 2PTH, but the alignment of 1K77 and 2PTH is significantly better, based on visual inspection.
In another case, the alignment of 2KFL [34] and 1MOQ [35] (glucosamine 6-phosphate synthase; EC 2.6.1.16) yielded good RMSD values for an alignment of three of four residues (all non-hydrogen atoms/ Cα / Cα + Cβ were 0.34 / 0.28 / 0.29 Å, respectively), but the visual alignment was much less convincing. BLAST, Dali and Pfam all predict that 2KFL is a prion protein. This is strongly supported by the Dali alignment (Figure 5) of 2KFL with 1QM2 [36] (a prion protein fragment), supporting the function proposed in Dali over the function proposed in ProMOL.
Figure 5.

Superposition of 2KFL (red) and 1QM2 (human prion protein fragment 121 – 230, blue). The image was generated from the Dali website.
The proposed functions from Table 3 are clearly not as promising as those in Table 1 and Figure 2. Visual inspection is essential in these cases before deciding on next steps and the results found in ProMOL need to be carefully scrutinized and compared to the results found with the other programs before deciding the best approach for in vitro characterization.
Divergent Results with the Four Programs
Table 4 lists seven proteins that gave grouped results. For 1R3D [37], ProMOL and Pfam suggest a hydrolase, while BLAST and Dali suggest a 2-succinyl-6-hydroxy-2,4-cyclohexadiene-1-carboxylate (SHCHC) synthase EC 4.2.99.2. Likewise for 2FBM [38], ProMOL and Pfam agree on lyase activity (EC 4), while BLAST and Dali place the structure under the transferase EC number 2. For two structures (2I1S [39] and 3KK4 [40]), the only agreement was between BLAST and Pfam. Only ProMOL and Pfam proposed functions for 3HFQ [41]; this is discussed in more detail below and in Figure 6. In the case of 3Q9D [42], there was no agreement among the programs and in the final case (4EZI [43]), ProMOL and Dali gave similar results.
Figure 6.

Four-residue alignment of 3HFQ (query/red) with 1JOF (motif template/blue). A. The order of residues and the spacing between residues in the sequences of both structures is nearly identical. B. The sequences of 3HFQ and 1JOF were aligned with BLASTP and the largest alignment is displayed. The catalytic residues shown in 6A are highlighted in bold.
In one intriguing case from Table 4, PDB entry 3HFQ aligns very well with chain A of motif template 1JOF [46], an isomerase with EC number of 5.5.1.5, with a Cα RMSD value of 0.56 Å. The initial ProMOL alignment between 3HFQ and 1JOF lacked the second arginine residue in 3HFQ. Alignment of the two sequences with Clustal Omega [47, 48] revealed that the 3HFQ contained ARG259, which aligned with ARG274 in the sequence of 1JOF. When the precision factor in ProMOL was relaxed from 1.0 to 1.1, ARG259 in 3HFQ aligned very closely with ARG274 in 1JOF (Figure 6).
While the Protein Data Bank characterizes 3HFQ as a protein of unknown function, it labels the molecule as “uncharacterized protein Ip_2219,” with an EC number 3.1.1.31, based on sequence alignment [49]. BLAST and Dali did not suggest a function for 3HFQ, but Pfam agreed with the PDB assignment in its suggestion that 3HFQ is a member of the 7-bladed beta-propeller lactonase family (EC 3.1.1.31). The Gene Ontology project has also placed 3HFQ under the EC number 3.1.1.31 [50]. It should be noted that InterPro [51] includes the lactonases (EC 3.1.1.31) and muconate lactonizing enzymes (EC 5.5.1.5) in the same entry as 7-bladed beta-propeller fold, IPR019405. Furthermore, of the ten structures in the PDB with EC 3.1.1.31, only 3HFQ and 1RI6 [52] demonstrated alignment with 1JOF (EC 5.5.1.5). The in vitro study of 3HFQ should help to resolve the correct EC number.
A manual BLASTP alignment of 3HFQ versus 1JOF provides insight into the lack of BLAST search results for 3HFQ, yielding poor alignment (21% identity) between the overall sequences. The ProMOL 1JOF motif active site residues align in BLASTP with the proposed active site residues in 3HFQ (Figure 6C, bold text), in runs of one to two sequence-aligned residues. This is a compelling example of the value of structural alignment.
Conclusions
With its current library of motifs, ProMOL has shown value in a significant, but limited, range of applications. The limitations can be addressed, in part, by extending the size of the motif library to cover larger motifs, such as the chromodomain, and more types of motifs, such as metal-ion-containing motifs. However, it will always have difficulty in identifying function based on global structure. When a ProMOL functional characterization is consistent with the BLAST, Pfam and Dali characterizations, as seen for the structures in Table 1, it is reasonable to assume that the expense of verification or rebuttal in the wet lab is justified. The structures in Table 2 for which ProMOL found active site structural homology, but whose peptide sequence alignments did not yield significant results, require investigations into the possibility of convergent evolution. When a ProMOL functional characterization differs from the unanimous conclusions of the more global methods, this suggests that wet lab tests must do more than simply verify or refute a hypothetical characterization, because the possibility of multiple functions within a single protein or a sub-function intrinsic to the overall function of a protein must be considered. Table 3 outlines thirty such results. The structures in Table 4, for which there are conflicting results among ProMOL, BLAST, Pfam and Dali also require further, careful investigation.
ProMOL is a promising tool to determine the function of uncharacterized proteins, particularly in concert with other sequence and global alignment tools. ProMOL provides useful information because of its unique characterization approach in which it evaluates the three-dimensional alignment of active site residues. However, without additional in silico and in vitro structural and functional protein analysis data, ProMOL is not a sufficient tool to explicitly determine protein function. There are constant updates to the program, which serve to improve its accuracy and reliability. Nevertheless, due to the ambiguous nature of enzymatic active sites, it is crucial to further investigate any “good hit” generated by ProMOL.
For the cases in which ProMOL results are in strong agreement with BLAST, Pfam, and Dali results, there is a high probability of being able to determine protein function experimentally. The fact that four different in silico approaches for protein analysis yielded similar conclusions means that those query macromolecules possess a significant number of the major characteristics of motifs from those corresponding classifications.
For most of the query structures, ProMOL generated a result that differed from results produced by BLAST, Pfam, and Dali. In some of these cases (Table 2), BLAST, Pfam and Dali collectively returned insignificant or no results. In others (Table 3), the latter three programs all agreed. In a third set of predictions (Table 4), individual results were incompatible. In all three of these instances it is difficult to draw conclusions, and further investigation into the bases for these discrepancies is warranted. The results for ligand binding with Autodock Vina shown in Table 2 offer significant guidance for substrate selection for future in vitro characterization studies.
By building on and extending current best practices, the methodology we are following shows promise of providing both more specificity and more reliability than existing approaches for ascribing function to proteins for which functions are not yet known experimentally. In addition to helping identify the functions of the large number of proteins of unknown function, this approach seems likely to help achieve a clearer understanding of structure-function relationships and to improve, codify and simplify existing laboratory practice in this important aspect of health-related research.
Supplementary Material
Acknowledgments
NIGMS 2R15GM078077-02, NIGMS 3R15GM078077-02S1, IGMS 3R15GM078077-02S2, Dowling College, Rochester Institute of Technology. We would like to thank the following team members who supported our efforts on this project: (from RIT) Weinishet Tedla-Boyd, Tananda Richards; (from Dowling College) Mogjan Asadi, Limone Rosa.
Our thanks to Frances C. Bernstein for editorial assistance.
Footnotes
Compliance with Ethical Standards
None of the authors have reported any conflicts of interest in the completion of the research described in this manuscript.
References
- 1.Bernstein FC, Koetzle TF, Williams GJB, et al. The Protein Data Bank: a computer-based archival file for macromolecular structures. J Mol Biol. 1977;112:535–542. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 2.Berman HM, Westbrook J, Feng Z, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Consortium TU. The Universal Protein Resource (UniProt) Nucleic Acids Res. 2008;36:D190–D195. doi: 10.1093/nar/gkm895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sonnhammer EL, Eddy SR, Durbin R. Pfam: a comprehensive database of protein domain families based on seed alignments. Proteins. 1997;28:405–420. doi: 10.1002/(sici)1097-0134(199707)28:3<405::aid-prot10>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 7.Finn RD, Miller BL, Clements J, Bateman A. iPfam: a database of protein family and domain interactions found in the Protein Data Bank. Nucleic Acids Res. 2014;42:D364–373. doi: 10.1093/nar/gkt1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gifford LK, Carter LG, Gabanyi MJ, et al. The Protein Structure Initiative Structural Biology Knowledgebase Technology Portal: a structural biology web resource. J Struct Funct Genomics. 2012;13:57–62. doi: 10.1007/s10969-012-9133-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holm L, Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischer M, Zhang QC, Dey F, et al. MarkUs: a server to navigate sequence-structure-function space. Nucleic Acids Res. 2011;39:W357–W361. doi: 10.1093/nar/gkr468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanson B, Westin C, Rosa M, et al. Estimation of protein function using template-based alignment of enzyme active sites. BMC Bioinformatics. 2014;15:87. doi: 10.1186/1471-2105-15-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delano WL. The PyMOL Molecular Graphics System. Schrodinger, LLC; San Carlos, CA, USA: [Google Scholar]
- 13.Porter CT. The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res. 2004;32:129D–133. doi: 10.1093/nar/gkh028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berman HM, Westbrook JD, Gabanyi MJ, et al. The protein structure initiative structural genomics knowledgebase. Nucleic Acids Res. 2009;37:D365–D368. doi: 10.1093/nar/gkn790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Torrance JW, Bartlett GJ, Porter CT, Thornton JM. Using a Library of Structural Templates to Recognise Catalytic Sites and Explore their Evolution in Homologous Families. J Mol Biol. 2005;347:565–581. doi: 10.1016/j.jmb.2005.01.044. [DOI] [PubMed] [Google Scholar]
- 16.Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seiler CY, Park JG, Sharma A, et al. DNASU plasmid and PSI:Biology-Materials repositories: resources to accelerate biological research. Nucleic Acids Res. 2014;42:D1253–D1260. doi: 10.1093/nar/gkt1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cormier C, Park J, Fiacco M, et al. PSI:Biology-materials repository: a biologist’s resource for protein expression plasmids. J Struct Funct Genomics. 2011;12:55–62. doi: 10.1007/s10969-011-9100-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cormier C, Mohr S, Zuo D, et al. Protein Structure Initiative Material Repository: an open shared public resource of structural genomics plasmids for the biological community. Nucleic Acids Res. 2010;38:D743–749. doi: 10.1093/nar/gkp999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vedadi M, Lew J, Artz J, et al. Genome-scale protein expression and structural biology of Plasmodium falciparum and related Apicomplexan organisms. Mol Biochem Parasitol. 2005;151:100–110. doi: 10.1016/j.molbiopara.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 21.Tan K, Rakowski E, Jedrzejczak R, Joachimiak A. The crystal structure of a functionally unknown conserved protein from. Enterococcus faecalis. 2009:V583. doi: 10.2210/pdb3l1w/pdb. [DOI] [Google Scholar]
- 22.Mol CD, Kuo C-F, Thayer MM, et al. Structure and function of the multifunctional DNA-repair enzyme exonuclease III. Nature. 1995;374:381–386. doi: 10.1038/374381a0. [DOI] [PubMed] [Google Scholar]
- 23.Kuzin AP, Chen Y, Seetharaman J, et al. X-Ray structure of the hypothetical protein YXIM_BACsu from. Bacillus subtilis. 2006 doi: 10.2210/pdb2o14/pdb. [DOI] [Google Scholar]
- 24.Ho YS, Sheffield PJ, Masuyama J, et al. Probing the substrate specificity of the intracellular brain platelet-activating factor acetylhydrolase. Protein Eng. 1999;12:693–700. doi: 10.1093/protein/12.8.693. [DOI] [PubMed] [Google Scholar]
- 25.Burkhard P, Taylor P, Walkinshaw MD. X-ray structures of small ligand-FKBP complexes provide an estimate for hydrophobic interaction energies. J Mol Biol. 2000;295:953–962. doi: 10.1006/jmbi.1999.3411. [DOI] [PubMed] [Google Scholar]
- 26.Patel S, Albert A, Blundell TL. Hal2p: Ion selectivity and implications on inhibition mechanism. 2001 doi: 10.2210/pdb1k9z/pdb. [DOI] [Google Scholar]
- 27.Mursula AM, Hiltunen JK, Wierenga RK. Structural studies on delta(3)-delta(2)-enoyl-CoA isomerase: the variable mode of assembly of the trimeric disks of the crotonase superfamily. FEBS Lett. 2003;557:81–87. doi: 10.1016/S0014-5793(03)01450-9. [DOI] [PubMed] [Google Scholar]
- 28.Joint Center for Structural Genomics (JCSG) Crystal structure of a hypothetical protein (BACUNI_01323) from. Bacteroides uniformis. 2012 doi: 10.2210/pdb4ghb/pdb. ATCC 8492 at 2.32 A resolution. [DOI] [Google Scholar]
- 29.Sundaresan V, Yamaguchi M, Chartron J, Stout CD. Conformational Change in the NADP(H) Binding Domain of Transhydrogenase Defines Four States. Biochemistry (Mosc) 2003;42:12143–12153. doi: 10.1021/bi035006q. [DOI] [PubMed] [Google Scholar]
- 30.Kim Y, Skarina T, Beasley S, et al. Crystal structure of Escherichia coli EC1530, a glyoxylate induced protein YgbM. Proteins. 2001;48:427–430. doi: 10.1002/prot.10160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmitt E, Mechulam Y, Fromant M, et al. Crystal structure at 1.2 A resolution and active site mapping of Escherichia coli peptidyl-tRNA hydrolase. EMBO J. 1997;16:4760–4769. doi: 10.1093/emboj/16.15.4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inoue M, Kigawa T, Yokoyama S. Solution structure of the cullin-3 homologue. 2002 doi: 10.2210/pdb1iuy/pdb. [DOI] [Google Scholar]
- 33.Van Pouderoyen G, Snijder HJ, Benen JA, Dijkstra BW. Structural insights into the processivity of endopolygalacturonase I from Aspergillus niger. FEBS Lett. 2002;554:462–466. doi: 10.1016/S0014-5793(03)01221-3. [DOI] [PubMed] [Google Scholar]
- 34.Christen B, Hornemann S, Damberger FF, Wuthrich K. Prion Protein NMR Structure from Tammar Wallaby (Macropus eugenii) Shows that the beta2-alpha2 Loop Is Modulated by Long-Range Sequence Effects. J Mol Biol. 2009;389:833–845. doi: 10.1016/j.jmb.2009.04.040. [DOI] [PubMed] [Google Scholar]
- 35.Teplyakov A, Obmolova G, Badet-Denisot MA, et al. Involvement of the C terminus in intramolecular nitrogen channeling in glucosamine 6-phosphate synthase: evidence from a 1.6 A crystal structure of the isomerase domain. Structure. 1997;6:1047–1055. doi: 10.1016/S0969-2126(98)00105-1. [DOI] [PubMed] [Google Scholar]
- 36.Zahn R, Liu A, Luhrs T, et al. NMR Solution Structure of the Human Prion Protein. Proc Natl Acad Sci. 1999;97:145. doi: 10.1073/PNAS.97.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gorman J, Shapiro L. Structural Genomics target NYSGRC-T920 related to A/B hydrolase fold. 2003 doi: 10.2210/pdb1r3d/pdb. [DOI] [Google Scholar]
- 38.Min JR, Antoshenko T, Hong W, et al. Crystal Structure of Acetyltransferases domain of Human Testis-specific chromodomain protein Y 1. 2005 doi: 10.2210/pdb2fbm/pdb. [DOI] [Google Scholar]
- 39.Nocek B, Borovilos M, Clancy S, Joachimiak A. Crystal structure of hypothetical protein MM_3350 from Methanosarcina mazei. Go1. 2006 doi: 10.2210/pdb2i1s/pdb. [DOI] [Google Scholar]
- 40.Chang C, Chhor G, Cobb G, Joachimiak A. Crystal structure of uncharacterized protein BP1543 from Bordetella pertussis Tohama I. 2009 doi: 10.2210/pdb3kk4/pdb. [DOI] [Google Scholar]
- 41.Vorobiev S, Scott L, Schauder C, et al. PDB ID: 3HFQ Crystal structure of the lp_2219 protein from Lactobacillus plantarum 2011 [Google Scholar]
- 42.Stone CB, Sugiman-Marangos SN, Junop MS, Mahony JB. Crystal Structure of Cpn0803 from C. pneumoniae. 2011 doi: 10.2210/pdb3q9d/pdb. [DOI] [Google Scholar]
- 43.Joint Center for Structural Genomics (JCSG) Crystal structure of a hypothetical protein (lpg1103) from Legionella pneumophila subsp. pneumophila str. Philadelphia 1 at 1.15 A resolution. 2012 doi: 10.2210/pdb4ezi/pdb. [DOI] [Google Scholar]
- 44.Jiang M, Chen X, Wu X-H, et al. Catalytic Mechanism of SHCHC Synthase in the Menaquinone Biosynthesis of Escherichia coli: Identification and Mutational Analysis of the Active Site Residues. Biochemistry (Mosc) 2009;48:6921–6931. doi: 10.1021/bi900897h. [DOI] [PubMed] [Google Scholar]
- 45.Holden HM, Benning MM, Haller T, Gerlt JA. The Crotonase Superfamily: Divergently Related Enzymes That Catalyze Different Reactions Involving Acyl Coenzyme A Thioesters. Acc Chem Res. 2001;34:145–157. doi: 10.1021/ar000053l. [DOI] [PubMed] [Google Scholar]
- 46.Kajander T, Merckel MC, Thompson A, et al. The structure of Neurospora crassa 3-carboxy-cis, cis-muconate lactonizing enzyme, a beta propeller cycloisomerase. Structure. 2001;10:483–492. doi: 10.1016/S0969-2126(02)00744-X. [DOI] [PubMed] [Google Scholar]
- 47.Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goujon M, McWilliam H, Li W, et al. A new bioinformatics analysis tools framework at EMBL–EBI. Nucleic Acids Res. 2010;38:W695–W699. doi: 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kleerebezem M, Boekhorst J, van Kranenburg R, et al. Complete genome sequence of Lactobacillus plantarum WCFS1. Proc Natl Acad Sci. 2003;100:1990–1995. doi: 10.1073/pnas.0337704100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.The Gene Ontology Consortium . Gene Ontology Annotations and Resources. Nucleic Acids Res. 2013;41:D530–D535. doi: 10.1093/nar/gks1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hunter S, Jones P, Mitchell A, et al. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res. 2012;40:D306–D312. doi: 10.1093/nar/gkr948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lima CD, Kniewel R, Solorzano V, Wu J. Structure of a putative 7-bladed propeller isomerase. 2003 doi: 10.2210/pdb1ri6/pdb. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.