

Abstract
Recent thymic emigrants (RTEs) must undergo phenotypic and functional maturation to become long-lived mature naïve T cells. In CD4-cre NKAP conditional knockout mice, NKAP-deficient RTEs fail to complete T cell maturation. Here, we demonstrate that NKAP-deficient immature RTEs do not undergo apoptosis, but are eliminated by complement. C3, C4 and C1q are bound to NKAP-deficient peripheral T cells, demonstrating activation of the classical arm of the complement pathway. As thymocytes mature and exit to the periphery, they increase sialic acid incorporation into cell surface glycans. This is essential to peripheral lymphocyte survival, as stripping sialic acid with neuraminidase leads to the binding of natural IgM and complement fixation. NKAP-deficient T cells have a defect in sialylation on cell surface glycans, leading to IgM recruitment. We demonstrate that the defect in sialylation is due to aberrant α2,8-linked sialylation, and the expression of three genes (ST8sia1, ST8sia4 and ST8sia6) that mediate α2,8 sialylation are down regulated in NKAP-defcient RTEs. The maturation of peripheral NKAP-deficient T cells is partially rescued in a C3-deficient environment. Thus, sialylation during T cell maturation is critical to protect immature RTEs from complement in the periphery.
Introduction
T cell maturation occurs continuously after thymocytes pass positive selection. The maturation process for single-positive (SP) thymocytes starts with down-regulation of CD24 and CD69, and up-regulation of Qa2 and CD62L. Mature SP thymocytes (defined as CD24−Qa2+) express sphingosine-1-phosphate receptor 1 (S1P1) and gain the ability to egress from thymic cortico-medullary junctions (1, 2). In the periphery, newly generated T cells, designated as recent thymic emigrants(RTEs), continue post-thymic maturation in secondary lymphoid organs to acquire functional competency and enter the long-lived naïve T cell pool(3, 4). The mechanism of T cell maturation is poorly understood, in part due to the current inability to distinguish between RTEs and long-lived mature naïve T cells with surface markers. In addition, RTEs and long-lived mature naïve T cells share overlapping survival niches in the periphery (5). T cell maturation is not dependent on TCR engagement (3) or IL-7Rα signaling (6, 7), but requires interactions of RTEs within secondary lymphoid organs (4).
NKAP was identified in a genetic complementation screen for novel regulators of T cell activation. NKAP is a transcriptional repressor that binds to histone deacetylase 3 (HDAC3) and also corepressor interacting with RBP-Jκ (CIR), a component of the Notch corepressor complex(8). NKAP is required at several points in hematopoiesis. NKAP is essential for the double-negative 3 (DN3) to double-positive (DP) transition in early T cell development(8), for hematopoietic stem cell (HSC) maintenance and survival(9), and for invariant NKT (iNKT) cell development(10). NKAP was also the first molecule shown to be required for T cell maturation(11). When NKAP is conditionally deleted at the DP stage using CD4-cre, positive selection and egress of SP thymocytes occur normally, but NKAP-deficient RTEs fail to mature (11). The failure of T cell maturation in the absence of NKAP is cell intrinsic. CD4-cre NKAP cKO mice have a 20-fold decrease in the absolute number of splenic naïve T cells, and the cells in this population are almost entirely comprised of functionally and phenotypically immature RTEs(11).
The complement system is a component of the innate immune system that is activated in a cascade-like manner in the presence of pathogen-associated molecular patterns (PAMPs) such as interaction of C1 with antibodies (the classical pathway), activation of mannose-binding lectin (MBL) with pathogen-specific carbohydrate patterns (the lectin pathway), and the spontaneous hydrolysis of complement C3 (the alternative pathway). Deposition of C3b and C4b leads to opsonization for phagocytosis, and formation of a C3 convertase at the cell surface leads to direct lysis by the pore-forming membrane-attack complex (MAC)(12). Here, we show that NKAP-deficient RTEs do not die by apoptosis, but are eliminated by complement. C3 is deposited on the cell surface of NKAP-deficient but not wild-type (WT) RTEs. As WT RTEs mature, they upregulate the complement inhibitor CD55/DAF as well as increase the incorporation of sialic acids into cell surface glycoproteins which leads to the recruitment of the complement inhibitor Factor H(13). NKAP-deficient RTEs express less CD55, and have decreased incorporation of sialic acid in their cell surface glycoproteins as demonstrated by increased binding of the lectin peanut agglutinin (PNA). RTEs that lack NKAP have C4 and C1q deposited on their cell surface, in addition to C3, indicating activation of the classical arm of the complement pathway. Activation of the classical complement pathway is strongest when cells are bound by IgM. Interestingly, it had been previously shown that treatment of cells with neuraminidase, which cleaves sialic acid from the cell surface, leads to the binding of natural IgM antibodies which recognize carbohydrate moieities and complement fixation(14, 15). Consistent with decreased sialic acid incorporation in the absence of NKAP, we find that NKAP-deficient RTEs and mature naïve T cells are bound by IgM. Therefore, decreased expression or recruitment of complement inhibitors, coupled with activation of the classical complement pathway, effectively eliminate NKAP-deficient RTEs. Reconstitution of CD4-cre NKAP cKO bone marrow into a complement C3-deficient host partially rescued the maturation defect in NKAP-deficient RTEs, leading to increased proportions of long-lived mature naïve T cells. Thus, our results reveal a previously unappreciated role for complement in shaping the peripheral T cell pool by eliminating RTEs that fail to properly mature.
Materials and Methods
Mice
The floxed NKAP mice were generated in our laboratory as described previously (8). Rag1-GFP knock-in mice were generated and provided by Dr. Nobuo Sakaguchi (16). C57BL/6 (B6), B6.129X1-Baxtm1Sjk/J (Bax knockout), B6.Cg-Tg (LCKprBCL2L1) 12 Sjk/J (Bcl-xL transgenic) mice and B6; 129S4-C3tm1Crr/J (complement C3 knockout) mice were purchased from The Jackson Laboratory. B6.SJL mice were purchased from National Cancer Institute Frederick. Littermates were used whenever possible as controls, and if not available, age matched controls were used. WT littermates included either NKAP floxed/cre negative, NKAP WT/cre positive or NKAP WT/cre negative mice. All of these littermate controls were indistinguishable from WT B6 mice. For convenience, since NKAP is expressed in all of these littermate controls, we called them WT mice. All animal experiments were approved by and performed under the guidelines from the IACUC at Mayo Clinic. All mice were housed in a barrier facility and were analyzed between 8 to 12 weeks of age, unless otherwise specified.
Antibodies, lectins and reagents
Fluorophore-conjugated antibodies for FACS analysis were purchased from BD Bioscience, eBioscience, BioLegend and Tonbo Biosciences. Biotinylated antibodies for complement C3 (RMC11H9), complement C1q (RMC7H8) and complement C4 (RMC16D2) were purchased from Cedarlane. Plant lectins PNA (peanut aggutinin), SNBL (Sambucus nigra bark lectin) and MAL II (Maackia amurensis lectin II) were purchased from Vector Laboratory. Recombinant mouse Siglec-E Fc chimera was purchased from R&D systems.
Cell preparation, flow cytometry and cell sorting
Single-cell suspensions were obtained from thymus and spleens of wild-type B6, CD4-cre NKAP cKO, Bax KO, Bax KO/CD4-cre NKAP cKO, Bcl-xL tg, Bcl-xL tg/CD4-cre NKAP cKO, Rag1-GFP WT, Rag1-GFP CD4-cre NKAP cKO, and bone marrow chimeras. For the complement experiments, freshly harvested splenocytes were incubated in GVB++ buffer (Complement Technology) for 1 hour at room temperature prior FACS staining. For Annexin-V/DAPI staining, splenocytes were freshly harvested, resuspended in 1X binding buffer (eBioscience) and stained with fluorescence-conjugated Annexin-V for 30 minutes at 4°C, and cells stained with DAPI (invitrogen) in the last step. Stained cells were analyzed with a LSR II (BD Bioscience) flow cytometer and the data were analyzed with FlowJo 9.2 software (Tree Star). All data were doublet excluded with FSC-W/FSC-H and SSC-W/SSC-H before analysis, and dead cell were excluded using DAPI in all experiments except Annexin-V experiments. To obtain splenic CD4+Rag1-GFP+ T cells, non-CD4-expressing cells were eliminated by negative selection from splenic single-cell suspensions using a MACS separation kit (Miltenyi Biotec). Enriched CD4+ cells were stained and analyzed on a FACSAria cell sorter (BD Bioscience), and CD4+Rag1-GFP+ T cells were collected.
Real-time PCR analysis
RNA from FACS-sorted splenic CD4+CD8−Rag1-GFP+ T cells was extracted using a RNeasy mini kit according to the manufacturer’s instructions (QIAGEN). cDNA was generated and amplified with an Ovation PicoSL WTA kit (NuGen). TaqMan probes for detecting Bcl-2, Bcl-xL, A1, Mcl-1, Bak1, Bax, Bok, Bad, Bim, Bid, NKAP, ST8sia1, ST8sia4, ST8sia6 and 18S rRNA were purchased from Applied Biosystems, and reactions were analyzed on an ABI RT-PCR StepOne Plus System (Applied Biosystems). The relative expression level of mRNA was calculated via the 2ΔΔCT method. All data was normalized to the expression of one of the WT samples (=1).
Determination of serum IgG, IgM, C3, C1q, C4, C1INH and Factor H
Sera was isolated from 8 WT and 7 CD4-cre NKAP cKO mice between 11 and 14 weeks of age. The serum concentration of IgG, IgM, C3, C1q, C4, C1INH and Factor H were determined using murine-specific ELISA kits using the manufacturer’s instructions. Dilution factors were empirically determined to be within the lineage range of each assay. IgG and IgM concentrations were determined using a mouse clonotyping kit with IgM and IgG2a standards from Southern Biotechnology Associates. Complement factors were analyzed using kits from Abcam (C3), R&D Systems (C1q), and United States Biologicals (C1INH, C4 and Factor H).
The generation of radiation chimeras
CD45.2+/+ Rag1-GFP CD4-cre NKAP cKO mice were bred to B6.SJL (CD45.1+/+) to produce CD45.1+CD45.2+ Rag1-GFP WT and CD45.1+CD45.2+ Rag1-GFP CD4-cre NKAP cKO donor for bone marrow transplantation into CD45.2+/+ C3 KO mice. Recipient complement C3 KO mice were lethally irradiated with 1000 rads prior to injecttion with 4 X 106 bone marrow cells from donors. Chimeric mice received antibiotic enrofloxacin in the drinking water for 3 weeks and were analyzed after 10 weeks.
Statistical analysis
All statistical analyses were generated using student’s t tests using GraphPad Prism. Statistical significance was defined as p value < 0.05. Data are shown as mean ± SEM.
Results
The disappearance of NKAP-deficient immature RTEs is not due to apoptosis
Apoptosis is tightly controlled and is crucial for the development and maintenance of the immune system. The balance between anti-apoptotic and pro-apoptotic molecules results in cell survival or cell death (17). Our previous data demonstrated that overexpression of Bcl-2 could not rescue the T cell maturation defect in NKAP-deficient RTEs (11). However, as many Bcl-2 family members have non-redundant roles in lymphocyte survival at particular stages of development, dysreulated expression of a different Bcl-2 family member expression may have been responsible for the loss of NKAP-deficient immature RTEs. Using a Rag1-GFP reporter to mark RTEs, splenic CD4+GFP+RTEs from Rag1-GFP WT and Rag1-GFP CD4-cre NKAP cKO mice were sorted and examined for the expression of anti-apoptotic (Bcl-2, Bcl-xL, A1 and Mcl-1), pro-apoptotic (Bak, Bax and Bok), and BH3-only (Bad, Bim and Bid) Bcl-2 family members by Q-PCR. The expression of most Bcl-2 family members, including Bcl-2 and Mcl-1, was similar between WT RTEs and NKAP-deficient RTEs. However, RTEs from CD4-cre NKAP cKO mice had increased expression of Bax and decreased Bcl-xL expression (Fig. 1A). To determine whether altered expression of Bax or Bcl-xL led to the disappearance of NKAP-deficient RTEs, CD4-cre NKAP cKO mice were crossed to Bcl-xl transgenic mice (Fig. 1B) and Bax-deficient mice (Fig. 1C). However, neither loss of Bax nor overexpression of Bcl-xL rescued the lack of mature Qa2+ T cells in the thymus or spleen, or restored peripheral T cell numbers to normal. In addition, no differences were observed between WT and NKAP-deficient RTEs when apoptosis was examined using Annexin-V and DAPI (Fig. 1D) to identify early apoptotic (Annexin-V+DAPI−) or dead (DAPI+) cells. Thus, the disappearance of NKAP-deficient immature RTEs from the periphery is not due to apoptosis, indicating that another mechanism must be responsible.
FIGURE 1.

The disappearance of RTEs in CD4-cre NKAP cKO mice is not due to apoptosis.(A)Differential expression of Bcl-2 family members in RTEs from WT and CD4-cre NKAP cKO mice. Quantitative PCR analysis comparing mRNA levels of anti-apoptotic (Bcl-2, Bcl-xL, A1 and Mcl-1), pro-apoptotic (Bak1, Bax and Bok), and BH3-only (Bad, Bim and Bid) Bcl-2 family members in sorted splenic CD4+Rag1-GFP+ RTEs from Rag1-GFP WT or Rag1-GFP CD4-cre NKAP cKO mice is shown. The relative expression of these genes was normalized to 18S rRNA. Data are the average from 6 Rag1-GFP WT and 6 Rag1-GFP CD4-cre NKAP cKO mice from 2 independent experiments, with all data normalized to one of the WT samples. Error bars indicate SEM. Expression of Bok was not determined (ND) as it was below the level of detection. (B)The block in T cell maturation in the absence of NKAP cannot be rescued by a Bcl-xL transgene. CD4 SP thymocytes and naïve CD4+CD62L+CD44− splenic T cells were examined for T cell maturation using the markers CD24 and Qa2. A representative FACS analysis of thymic and splenic T cell populations in WT, CD4-cre NKAP cKO, Bcl-xL tg, and Bcl-xL tg/CD4-cre NKAP cKO mice is shown and is representative of 4WT, 4 CD4-cre NKAP cKO, 2 Bcl-xL tg, and 4 Bcl-xL tg/CD4-cre NKAP cKO mice analyzed in 3 independent experiments. (C) Loss of Bax in T cells from CD4-cre NKAP cKO mice does not rescue the maturation defect. Analysis of thymic T cell development and T cell maturation as in (B). A representative analysis in a CD4-cre NKAP cKO mouse, a Bax KO mouse, a Bax KO/CD4-cre NKAP cKO mouse and a WT littermate is shown. The experiments were performed on four-week-old mice due to the spontenous development of autoimmunity in the Bax KO background. Data are representative of 4 CD4-cre NKAP cKO, 3 Bax KO, 5Bax KO/CD4-cre NKAP KO mice and 5 WT littermates from 4 independent experiments. (D)Splenocytes were freshly harvested from Rag1-GFP WT and Rag1-GFP CD4-cre NKAP cKO mice and were stained with Annexin-V and DAPI. RTEs were defined as CD4+CD62L+CD44−Rag1-GFP+ T cells. Data shown are representative of 3 mice per group from 3 independent experiments.
Complement deposition is found in peripheral T cell populations of CD4-cre NKAP cKO mice
While apoptosis is one mechanism to eliminate developmentally arrested lymphocytes, complement-mediated opsonization and lysis also efficiently eliminates targeted cells. To determine if the lack of peripheral naïve T cells in CD4-cre NKAP cKO mice was due to complement activation, we examined complement C3 deposition on splenic T cells from Rag1-GFP WT and Rag1-GFP CD4-cre NKAP cKO mice. Interestingly, both CD4+ RTEs and mature naïve CD4+ T cells from Rag1-GFP CD4-cre NKAP cKO mice had substantial C3 deposition on the cell surface, as compared to either RTEs or mature naïve T cells from WT Rag1-GFP reporter mice (Fig. 2A). In particular, the majority of mature naïve CD4+ T cells from Rag1-GFP CD4-cre NKAP cKO mice had C3 on the cell surface while very few T cells from WT mice were bound by C3. This suggests that the increased complement C3 deposition on NKAP-deficient naïve T cells may be responsible for the loss of these cells. To determine which complement pathway is activated, we examined the deposition of other complement regulatory proteins, including deposition of C1q, which is the initiating event in the activation of the classical complement pathway. As shown in Figure 2B, C4 and C1q were also found on the surface of NKAP-deficient peripheral T cells, including both RTEs and mature naïve T cells. The serum concentration of several complement components were examined by ELISA. Very similar levels of C3, C1q, C4, C1INH or Factor H were observed in WT and CD4-cre NKAP cKO mice, indicating that increased complement deposition was not due to increased serum concentration of these proteins. Thus, the classical arm of the complement pathway is mobilized against NKAP-deficient T cells.
FIGURE 2.

Increased complement C3 and C4 deposition, and binding of complement C1q, is found in peripheral naïve T cell population of CD4-cre NKAP cKO mice. Splenocytes from Rag1-GFP WT and Rag1-GFP CD4-cre NKAP cKO mice were incubated in the GVB++ buffer prior to staining with anti-C3 or anti-C4 (A), or anti-C1q antibodies (B). Splenic CD4+CD44−CD62L+Rag1-GFP+ cells were defined as RTEs while CD4+CD44−CD62L+Rag1-GFP− cells were defined as mature naïve T cells (MNTs). Data shown are representative of at least 4 mice per group from 7 independent experiments. Quantitation from at least 7 Rag1-GFP WT and 7 Rag1-GFP CD4-cre NKAP cKO mice from at least 5 independent experiments is also shown. Error bars indicate SEM. Data were analyzed for significance by an unpaired Student’s t test. NS, not significant. (C) Serum concentrations of C3, C4, C1q, factor H and C1INH in 8 WT and 7 CD4-cre NKAP cKO mice were determined by ELISAs. The average concentration in μg/ml and the SEM for each is shown. The concentrations of these proteins in WT and CD4-cre NKAP cKO mice were very similar.
NKAP-deficient T cells fail to upregulate incorporation of sialic acid at the cell surface, leading to IgM deposition
As thymocytes complete development before export to the periphery, they increase incorporation of sialic acid into glycoproteins and glycolipids at the cell surface(18). This addition of sialic acid is critical to lymphocyte survival in the periphery, as stripping of cell surface sialic acids by neuraminidase leads to the binding of natural IgM and complement fixation (14, 15). The lectin peanut agglutinin (PNA) recognizes unsialylated core 1-O-glycans, and the incorporation of sialic acids into cell surface glycans leads to decreased PNA binding. As WT thymocytes transit from DP to the mature SP stage, PNA binding decreases significantly, however, PNA binding remained high in NKAP-deficient semi-mature thymocytes and peripheral T cells (Fig. 3A), demonstrating that there is a defect in sialic acid incorporation. As neuraminidase treatment of lymphocytes leads to binding of natural IgM(14, 15), we examined whether NKAP-deficient peripheral T cells were bound by IgM. As shown and quantified in Figure 3B, substantial binding of IgM to NKAP-deficient RTEs and mature naïve T cells was found. The serum concentration of IgM and total IgG was examined by ELISA. No substantial differences in the concentration of IgM or total IgG were observed between WT and CD4-cre NKAP cKO mice, indicating that increased IgM deposition was not due to increased serum IgM concentration. Thus, the loss of NKAP during T cell maturation leads to defects in sialic acid incorporation, IgM binding and activation of the classical complement pathway. Interestingly, we did not find any deposition of IgM, C3, C1q or C4 on CD4-cre NKAP cKO thymocytes (Supplemental Figure 1). Thus, thymocytes that fail to initiate post-positive selection maturation become targets for elimination only after they are exported from the thymus and enter the circulation.
FIGURE 3.

Increased binding of PNA and IgM is found on T cells of CD4-cre NKAP cKO mice.(A)Representative flow cytometry analysis of PNA binding on thymocytes and splenocytes of Rag1-GFP WT (grey filled histogram) and Rag1-GFP CD4-cre NKAP cKO mice (black open histogram) is shown. Each cell population was gated on the following markers: DP thymoctyes (CD4+CD8+), semi-mature CD4 SP thymocytes (CD4+CD8−CD24hiRag1-GFP+), mature CD4 SP thymocytes (CD4+CD8−CD24loRag1-GFP+), RTEs (splenic CD4+CD44−CD62L+Rag1-GFP+), and MNTs (splenic CD4+CD44−CD62L+Rag1-GFP−). Below the FACS plots, the average MFI with SEM for WT and CD4-cre NKAP cKO mice is shown for each population. For the thymic analysis, 8 WT and 6 CD4-cre NKAP cKO mice were analyzed. For the splenic analysis, 7 WT and 7 CD4-cre NKAP cKO mice were analyzed. Statistical analysis of the differences in MFI was performed using students t test as denoted in the figure. (B) RTEs and MNTs from Rag1-GFP WT and Rag1-GFP CD4-cre NKAP cKO mice were stained with anti-IgM antibody. Cell populations were defined as above. A representative FACS analysis is shown, and IgM binding was quantified from least 8 mice per group from 6 independent experiments. Error bars indicate SEM. Data were analyzed for significance by Student’s t test. Serum concentrations of IgM and total IgG in 8 WT and 7 CD4-cre NKAP cKO mice were determined by ELISAs. The average and SEM for each is shown. The concentrations of IgG and IgM in serum were very similar in WT and CD4-cre NKAP cKO mice.
Defects in α2,8 sialic acid incorporation in the absence of NKAP
Sialic acids are added to glycans through either an α2,3-, α2,6- or α2,8-linkage by specific sialyltransferases (ST3Gals, ST6Gals/ST6GalNacs and ST8Sias, respectively). The lectin PNA generally recognizes core-1-O-glycans that lack terminal sialic acid, but does not discriminate between the types of sialic acid linkages. However, Sambucus nigra back lectin (SNBL) is highly specific for α2,6-linked sialic acids and Maackia amurensis lectin II (MAL II) is highly specific for α2,3-linked sialic acids. To determine which linkages were defective, NKAP-deficient RTEs were examined for association with SNBL and MAL II. As shown in Figure 4A, although PNA binding was increased in NKAP-deficient RTEs as compared to WT, there was similar binding with SNBL and MAL II, indicating that there is not a general defect in either α2,3- or α2,6-sialic acid linkages. Thus, the defects appears to be in the generation of α2,8-linked sialic acids, which are added by members of the ST8Sia family of sialyltransferases. Examination of the ImmGen database indicated that three members of this family, ST8Sia1, ST8Sia4 and ST8Sia6, are expressed in T cells. ST8Sia1 (also known as GD3 synthase) sialylates gangliosides, in particular GM3, to produce GD3. ST8Sia4 leads to the production of polysialic acid. ST8Sia6 mediates the transfer of two sialic acids preferentially onto O-linked glycosylations. Q-PCR demonstrated that mRNA expression of all three of these genes was decreased in NKAP-deficient RTEs compared to WT (Fig. 4B). Thus, in the absence of NKAP, expression of multiple ST8Sia sialyltransferases is decreased leading to a defect in α2,8-linked sialic acids on cell surface glycans, and this failure in sialic acid incorporation at the cell surface leads to IgM binding and activation of the classical complement pathway.
FIGURE 4.
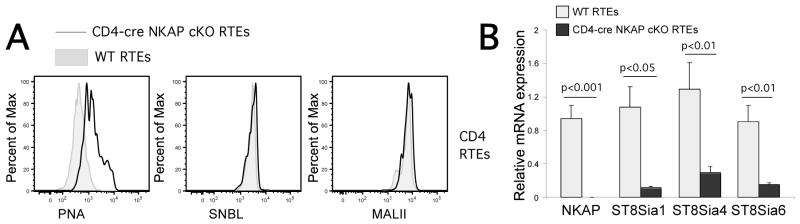
T cells from CD4-cre NKAP cKO mice show reduced sialylation via α2,8-linkage on cell surface that leads to decreased binding of inhibitory receptor Siglec-E. A, Splenocytes were freshly harvested from Rag1-GFP WT (grey filled histogram) or Rag1-GFP CD4-cre NKAP cKO mice (black open histogram) and were stained with plant lectins PNA, SNBL, or MAL II. PNA recognizes unsialylated O-glycans. SNBL specifically recognizes sialic acids via α2,6-linkage while MAL II specifically recognizes sialic acids via α2,3-linkage. Data shown are representitive of at least 3 mice per group from 4 independent experiments. B, Quantitative PCR analysis comparing mRNA levels of NKAP, α2,8-sialyltransferases (ST8sia1, ST8sia4 and ST8sia6) in sorted splenic CD4+Rag1-GFP+ RTEs from Rag1-GFP WT or Rag1-GFP CD4-cre NKAP cKO mice is shown. The relative expression of these genes was normalized to 18S rRNA. Data shown is the mean ± SEM of 5 mice in each group, with all data normalized to one of the WT mice in each group. Data were analyzed for significance by an unpaired Student’s t test.
Complement activation is exacerbated by a defect in CD55 upregulation during T cell maturation in CD4-cre NKAP cKO mice
Cells de-activate complement through the recruitment of soluble inhibitors. Factor H is recruited to cells by sialic acid(19), which should be decreased on NKAP-deficient peripheral T cells and thus should fail to inhibit complement mediated deposition due to activation of the classical pathway. In addition, lymphocytes express additional complement regulatory proteins including CD55 (also known as DAF, decay accelerating factor) and Crry (homology to human CD46). CD55 expression is upregulated concurrently with egress of mature thymocytes into the periphery (20), although CD55-deficiency does not lead to peripheral T cell lymphopenia(21). Conditional deletion of Crry in the T cell lineage does not alter T cell development, although peripheral T cell numbers are severely reduced (22). Consistent with the ImmGen consortium expression data, we found that expression of CD55 on the cell surface increased concurrently with T cell maturation (Figure5) in Rag1-GFP WT mice. NKAP-deficient T cells, however, had decreased expression of CD55 in RTEs and mature naïve T cells (Figure5). Crry expression is not modulated during T cell development, and was unaffected by NKAP-deficiency. Although Crry is an inhibitor of the classical complement pathway and is expressed at normal levels in NKAP-deficient RTE and long-lived naïve T cells, it must be not able to effectively block complement deposition on these cells. The failure of NKAP-deficient RTEs to appropriately upregulate expression of CD55 during maturation or recruit Factor H due to a defect in sialic acid incorporation in cell surface glycoproteins likely synergizes with antibody-mediated complement deposition to accelerate complement fixation and lysis.
FIGURE 5.

The expression of CD55 is induced during T cell maturation, and peripheral T cells from CD4-cre NKAP cKO mice have a defect in upregulation of CD55. Representative flow cytometry analysis of CD55 (A) and Crry (B) binding on thymocytes and splenocytes of Rag1-GFP WT (grey filled histogram) and Rag1-GFP CD4-cre NKAP cKO mice (black open histogram) is shown. Populations were defined as in Fig. 1. Below the FACS plots, the average MFI with SEM for WT and CD4-cre NKAP cKO mice is shown for each population. For CD55, 7 WT and 7 CD4-cre NKAP cKO mice were analyzed for each population. For Crry, at least 3 WT and 3 CD4-cre NKAP cKO mice were analyzed for each population. Statistical analysis of the differences in MFI was performed using student’s t test as denoted in the figure.
Use of CD55 and CD45RB to define RTE in the periphery
Although CD31 expression can be used as one way to distinguish RTEs in humans(23), there has not been a way to identify murine RTEs through cell surface markers, necessitating the use of Rag1-GFP reporters. As CD55 and CD45RB expression increases concurrently with maturation, we examined whether expression of these molecules could be used to identify RTEs. As shown in Figure 6, peripheral CD4 T cells from Rag1-GFP mice were examined for expression of CD55 and CD45RB. Rag1-GFP+ RTEs were predominantly CD45RBloCD55lo and Rag1-GFP− MNTs were predominantly CD45RBhiCD55hi. If we first gated naïve peripheral CD4 T cells based on expression of CD45RB and CD55, the majority of CD45RBloCD55lo cells were RTEs and the CD45RBhiCD55hi population was almost exclusively comprised of MNTs. Naïve CD4 T cells from Rag1-GFP CD4-cre NKAP cKO mice were primarily CD45RBloCD55lo as compared to WT, consistent with being comprised almost primarily of RTEs. Thus, decreased expression of CD45RB and CD55 in peripheral T cells can be used to identify a population that is highly enriched for RTEs, and alterations in the relative proportion of T cells expressing CD45RB and CD55 would indicate an imbalance between RTEs and MNTs within the naïve T cell pool.
FIGURE 6.

Identification of RTEs using CD55 and CD45RB. Splenic naïve CD4+ T cells from Rag1-GFP WT and Rag1-GFP CD4-cre NKAP cKO mice were stained with a combination of CD55 and CD45RB antibodies. In the first and third row, naïve CD4+ T cells from Rag1-GFP WT or Rag1-GFP CD4-cre NKAP cKO mice respectively, were first gated on GFP+ or GFP−, and CD55 and CD45RB expression in RTEs or mature naïve CD4+ T cells were analyzed. In the second and fourth row, naïve CD4+ T cells from Rag1-GFP WT or Rag1-GFP CD4-cre NKAP cKO mice respectively, were first gated based on CD55 and CD45RB expression, and then analyzed for the expression of Rag1-GFP. The CD55loCD45RBlo cells represent a majority of RTEs while the CD55hiCD45RBhi cells consist primarily of mature naïve CD4+ T cells. Data shown are representative of eight mice per group from eight independent experiments.
Increased survival of NKAP-deficient mature naïve T cells in C3-deficient mice
If the loss of NKAP-deficient peripheral T cells is due to complement activation, then the development of these cells in a C3-deficient host may rescue their survival. To test this, lethally irradiated complement C3 KO mice were reconstituted with bone marrow from Rag1-GFP WT or Rag1-GFP CD4-cre NKAP cKO mice. After 10 weeks, CD4+ RTEs from Rag1-GFP WT/C3 KO bone marrow chimeras or Rag1-GFP CD4-cre NKAP cKO/C3 KO bone marrow chimeras were compared to unmanipulated Rag1-GFP WT or Rag1-GFP CD4-cre NKAP cKO mice (representative FACS analysis is shown in Figure 7A with quantitation in Figure 7B). No differences were observed in T cell maturation between T cells that developed in either a WT or C3-deficient environment. However, in contrast to Rag1-GFP CD4-cre NKAP cKO mice in which the naïve T cell compartment is comprised almost entirely of RTEs, approximately half of the naïve T cells from the Rag1-GFP CD4-cre NKAP cKO/C3 KO chimeras were GFP− mature naïve T cells. In addition, CD4+ RTEs and mature naïve CD4+ T cells from Rag1-GFP CD4-cre NKAP cKO/C3 KO chimeras had increased expression of CD55 and Qa2. In particular, the mature naïve T cells from Rag1-GFP CD4-cre NKAP cKO/C3 KO chimeras had approximately 3-fold higher expression of CD55 as compared to WT RTE or long lived naïve T cells, indicating that increased expression of CD55 may confer a selective advantage. However, C3-deficiency did not completely eliminate the defect in maturation in NKAP-deficient RTEs. This may be because C3-deficiency would curtail, but not block, formation of the MAC, as C5 convertase activity is displaced by thrombin(24)and the C4b2a (another C3 convertase, activated by the classical complement pathway), albeit inefficiently(25). Thus, complement-mediated lysis could still occur. However, the partial effect, and increased expression of CD55 by the NKAP-deficient mature naïve T cells in the C3-deficient chimera, demonstrates the importance of this pathway to the loss of NKAP-deficient peripheral T cells.
FIGURE 7.

The defect in T cell maturation of CD4-cre NKAP cKO RTEs is partially rescued in complement C3 knockout bone marrow chimeric mice.(A)Radiation chimeras were generated by injecting lethally irradiated complement C3 KO mice (CD45.2+/+) with either Rag1-GFP WT (CD45.1+CD45.2+) or Rag1-GFP CD4-cre NKAP cKO (CD45.1+CD45.2+) bone marrow. After 10 weeks, T cell development and maturation was analyzed. A representative FACS analysis of splenic T cell populations in a Rag1-GFP WT, a Rag1-GFP WT/C3 KO chimera, a Rag1-GFP CD4-cre NKAP cKO, and a Rag1-GFP CD4-cre NKAP cKO/C3 KO chimera is shown. Data shown are representative of 6 Rag1-GFP WT, 4 Rag1-GFP WT/C3 KO chimeras, 3 Rag1-GFP CD4-cre NKAP cKO and 4 Rag1-GFP CD4-cre NKAP cKO/C3 KO chimeras from 4 independent experiments. The percentage of mature naïve CD4+ T cells (B) and the MFIs of CD55 and Qa2in mature naïve CD4+ T cells (C) or RTEs (D)were averaged from 6 Rag1-GFP WT, 4 Rag1-GFP WT/C3 KO chimeras, 3 Rag1-GFP CD4-cre NKAP cKO and 4 Rag1-GFP CD4-cre NKAP cKO/C3 KO chimeras. The MFI data were normalized to the expression of CD55 or Qa2 in one of the unmanipulated Rag1-GFP WT mice. Error bars indicate SEM.
Decreased binding of Siglec-E in NKAP-deficient T cells may also excerabate clearance due to opsonization
The incomplete rescue of NKAP-deficient T cells in the absence of C3 may also be due to opsonization of NKAP-deficient T cells. In the absence of C3, deposition of IgM, C1q and C4 still occurs (data not shown). The decision of innate immune cells such as macrophages and NK cells to kill targets involves the combination of both activating and inhibitory signals, including inhibitory signals through Siglecs. Siglecs are a family of inhibitory receptors that recognize specific sialic acid modifications. Within the Siglec family, Siglec-E (in mouse, homologous to Siglec-7 in humans) prefers α2,8-linked sialic acids, but also can bind to α2,3-, α2,6-linked sialic acids (26). Using recombinant Siglec-E-Fc fusion proteins, we demonstrate that the presence of Siglec-E ligands increases concurrently with T cell maturation and is lower in NKAP-deficient thymocytes and peripheral T cells (Figure 8). Siglec-E is expressed on neutrophils, macrophages and a subset of mature NK cells (26). Therefore, opsonization and elimination by complement may also synergize with a defect in Siglec-E binding driving the clearance of NKAP-deficient RTEs and mature naïve T cells.
FIGURE 8.
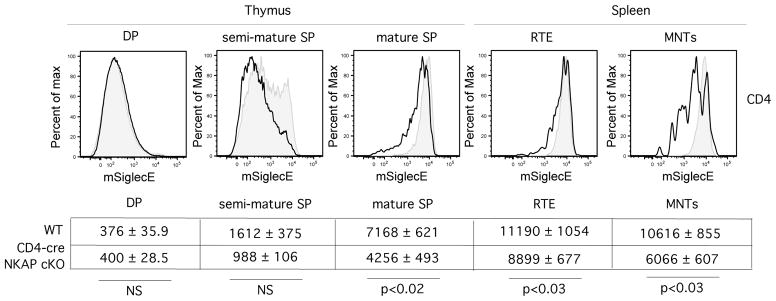
Ligands for Siglec-E are induced during T cell maturation. Thymocytes and splenocytes from Rag1-GFP WT (grey filled histogram) or Rag1-GFP CD4-cre NKAP cKO (black open histogram) mice were stained with Siglec-E Fc fusion protein. Mouse Siglec-E preferrentially binds to α2,8-linked sialic acids over α2,3- or α2,6-linkages. Cell populations were defined as in Fig. 2. Below the FACS plots, the average MFI with SEM for WT and CD4-cre NKAP cKO mice is shown for each population. For thymic analysis, 3 WT and 4 CD4-cre NKAP cKO mice were analyzed for each population. For the splenic analysis, 5 WT and 4 CD4-cre NKAP cKO mice were analyzed for each population. Statistical analysis of the differences in MFI was performed using student’s t test.
Discussion
Previously, we demonstrated that the transcriptional repressor NKAP is absolutely required for T cell maturation. In CD4-cre NKAP cKO mice, SP thymocytes successfully transit through positive and negative selection, egress from the thymus, enter peripheral lymphoid organs, but then fail to mature. Thus, the peripheral naïve T cell pool is comprised predominantly of RTEs. Here, we show that the disappearance of immature NKAP-deficient RTEs is not due to apoptosis, but is due to a defect in sialylation of cell surface glycans leading to recruitment of natural IgM and activation of the classical arm of the complement pathway. This is exacerbated by decreased expression of CD55 which is normally upregulated during maturation. Using CD55 and CD45RB as markers for maturation, we showed that CD45RBloCD55locells are Rag1-GFP+ RTEs within the naïve T cell pool, thus generating a method for their identification independent of a Rag1-GFP reporter. While there was significantly more complement bound by NKAP-deficient RTEs as compared to WT RTEs, the WT RTEs did bind complement, and to an extent that decreased significantly as they progressed to the mature naïve T cell pool. The defect in sialylation in NKAP-deficient T cells also leads to decreased expression of ligands for the inhibitor receptor Siglec-E, thus exacerbating removal of these cells by antibody- and/or complement-mediated opsonization. Thus, there are multiple mechanisms which simultaneously and independently lead to the rapid destruction of NKAP-deficient T cells. It is not surprising, then, that there was not a complete rescue when bone marrow chimeras were generated by transferring Rag1-GFP CD4-cre NKAP cKO hematopoietic progenitors into C3-deficient hosts.
T cell express three members of the α2,8 sialyltransferase family: ST8Sia1, ST8Sia4, and St8Sia6. Mice deficient in ST8Sia1 and ST8Sia4 have been generated. ST8Sia1 adds sialic acid to cell surface glycolipids, and is also known as GD3 synthase, as it is required for the generation of GD3 ganglioside. ST8Sia1 knockout mice do not have a defect in the thymic or splenic T cell populations, albeit there is a slight decrease in the ratio (but not absolute numbers) of CD8+ T cells to CD4+ T cells(27). ST8Sia4 adds polysialic acid (PSA) to glycoproteins. Mice deficient in ST8Sia4 have a 30% reduction in thymic cellularity due to decreased entrance of progenitors to the thymus, but once they have successfully entered, T cell development and export to the periphery is normal (28). ST8Sia6 is the only family whose expression increases concurrently with maturation (ImmGen), and whose expression correlates with the generation of ligands for Siglec-E (Fig. 8), which specifically binds to α2,8 sialic acids as well as correlates with the loss of ligands for PNA (Fig. 3). The expression of ST8Sia1 and ST8Sia4 change by less than two-fold throughout T cell development and maturation (ImmGen). Thus, ST8Sia6 may be the critical sialyltransferase that is required during T cell maturation. Retroviral transduction of ST8Sia6 into hematopoietic progenitors leads to a complete block in T cell development although myeloid cells and B cells are produced (FCH and VSS, unpublished results), and therefore we could not access whether ectopic retroviral expression of ST8Sia6 could rescue the block in T cell maturation in the absence of NKAP. The examination of the role of ST8Sia6 in T cell maturation awaits development of new genetic models, which is currently being pursued.
NKAP-deficient RTEs fail to upregulate CD55 and the α2,8-sialyltransferase ST8Sia6, which we believe contributes to their rapid elimination in the periphery via C3-dependent and C3-independent mechanisms. It is not known whether NKAP directly regulates expression of ST8Sia6 or CD55, or whether the loss of NKAP leads to a developmental block in T cell maturation leading to a failure to upregulate these genes. However, our results indicate that if WT T cells fail to receive the appropriate maturation signals, one consequence would be a failure to induce expression of ST8Sia6 and CD55. The loss of ST8Sia would result in decreased cell surface sialylation, leading to recruitment of natural IgM and initiation of the classical complement pathway. The complement inhibitory protein Factor H binds to sialic acid on the cell surface, which thus would also be decreased. Failure to turn on CD55 would also promote complement activation. Thus, newly produced T cells that fail to undergo maturation would also fail to upregulate ST8Sia6 and CD55, leading to their rapid elimination by complement. This suggests a mechanism to prevent failed RTEs from entering the long-lived naïve T cell pool.
Supplementary Material
Acknowledgments
We would like to thank Dr. Nobuo Sakaguchi for the Rag1-GFP knock-in mice.
Abbreviations
- BMC
bone marrow chimeras
- C1INH
C1 inhibitor
- cKO
conditional knockout
- DAF
decay accelerating factor
- DN
double-negative
- DP
double-positive
- MAL II
Maackia amurensis lectin II
- MNT
mature naïve T cells
- PNA
peanut agglutinin
- RTEs
recent thymic emigrants
- SNBL
Sambucus nigra bark lectin
- SP
single-positive
- tg
transgenic
Footnotes
This work was supported by the US National Institutes of Health (R01 AI083279) to V.S. Shapiro.
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, Allende ML, Proia RL, Cyster JG. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355–360. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 2.Zachariah MA, Cyster JG. Neural crest-derived pericytes promote egress of mature thymocytes at the corticomedullary junction. Science. 2010;328:1129–1135. doi: 10.1126/science.1188222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boursalian TE, Golob J, Soper DM, Cooper CJ, Fink PJ. Continued maturation of thymic emigrants in the periphery. Nat Immunol. 2004;5:418–425. doi: 10.1038/ni1049. [DOI] [PubMed] [Google Scholar]
- 4.Houston EG, Nechanitzky R, Fink PJ. Contact with Secondary Lymphoid Organs Drives Post-thymic T cell Maturation. J Immunol. 2008;181:5213–5217. doi: 10.4049/jimmunol.181.8.5213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Houston EG, Jr, Higdon LE, Fink PJ. Recent thymic emigrants are preferentially incorporated only into the depleted T-cell pool. Proc Natl Acad Sci U S A. 2011;108:5366–5371. doi: 10.1073/pnas.1015286108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Houston EG, Jr, Boursalian TE, Fink PJ. Homeostatic signals do not drive post-thymic T cell maturation. Cell Immunol. 2012;274:39–45. doi: 10.1016/j.cellimm.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weinreich MA, Jameson SC, Hogquist KA. Postselection thymocyte maturation and emigration are independent of IL-7 and ERK5. J Immunol. 2011;186:1343–1347. doi: 10.4049/jimmunol.1002238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pajerowski AG, Nguyen C, Aghajanian H, Shapiro MJ, Shapiro VS. NKAP is a transcriptional represor of Notch signaling and is required for T cell development. Immunity. 2009;30:696–707. doi: 10.1016/j.immuni.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pajerowski AG, Shapiro MJ, Gwin K, Sundsbak R, Nelson-Holte M, Medina KL, Shapiro VS. Adult hematopoietic stem cells require NKAP for maintenance and survival. Blood. 2010;116:2684–2693. doi: 10.1182/blood-2010-02-268391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thapa P, Das J, McWilliams D, Shapiro M, Sundsbak R, Nelson-Holte M, Tangen S, Anderson J, Desiderio S, Hiebert S, Sant’Angelo DB, Shapiro VS. The transcriptional repressor NKAP is required for the development of iNKT cells. Nat Commun. 2013;4:1582. doi: 10.1038/ncomms2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hsu F-C, Pajerowski AG, Nelson-Holte M, Sundsbak R, Shapiro VS. NKAP is required for T cell maturation and the acquisition of functional competency. J Exp Med. 2011;208:1291–1304. doi: 10.1084/jem.20101874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 13.Kazatchkine MD, Fearon DT, Austen KF. Human alternative complement pathway: membrane-associated sialic acid regulates the competition between B and beta1 H for cell-bound C3b. J Immunol. 1979;122:75–81. [PubMed] [Google Scholar]
- 14.Ray PK, Sundaram K. Cytolysis of neuraminidase-treated autochthonous lymphoid cells by autologous serum. Clin Exp Immunol. 1975;19:529–532. [PMC free article] [PubMed] [Google Scholar]
- 15.Schlesinger M, Bekesi JG. Natural autoantibodies cytotoxic for thymus cells and for neuraminidase-treated leukemia cells in the sera of normal AKR mice. J Natl Cancer Inst. 1977;59:945–950. doi: 10.1093/jnci/59.3.945. [DOI] [PubMed] [Google Scholar]
- 16.Igarashi H, Kuwata N, Kiyota K, Sumita K, Suda T, Ono S, Bauer SR, Sakaguchi N. Localization of recombination activating gene 1/green fluorescent protein (RAG1/GFP) expression in secondary lymphoid organs after immunization with T-dependent antigens in rag1/gfp knockin mice. Blood. 2001;97:2680–2687. doi: 10.1182/blood.v97.9.2680. [DOI] [PubMed] [Google Scholar]
- 17.Opferman JT, Korsmeyer SJ. Apoptosis in the development and maintenance of the immune system. Nat Immunol. 2003;4:410–415. doi: 10.1038/ni0503-410. [DOI] [PubMed] [Google Scholar]
- 18.Moody AM, North SJ, Reinhold B, Van Dyken SJ, Rogers ME, Panico M, Dell A, Morris HR, Marth JD, Reinherz EL. Sialic acid capping of CD8beta core 1-O-glycans controls thymocyte-major histocompatibility complex class I interaction. J Biol Chem. 2003;278:7240–7246. doi: 10.1074/jbc.M210468200. [DOI] [PubMed] [Google Scholar]
- 19.Meri S, Pangburn MK. Discrimination between activators and nonactivators of the alternative pathway of complement: regulation via a sialic acid/polyanion binding site on factor H. Proc Natl Acad Sci U S A. 1990;87:3982–2986. doi: 10.1073/pnas.87.10.3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mingueneau M, Kreslavsky T, Gray D, Heng T, Cruse R, Ericson J, Bendall S, Spitzer MH, Nolan GP, Kobayashi K, von Boehmer H, Mathis D, Benoist C, Best AJ, Knell J, Goldrath A, Jojic V, Koller D, Shay T, Regev A, Cohen N, Brennan P, Brenner M, Kim F, Rao TN, Wagers A, Rothamel K, Ortiz-Lopez A, Bezman NA, Sun JC, Min-Oo G, Kim CC, Lanier LL, Miller J, Brown B, Merad M, Gautier EL, Jakubzick C, Randolph GJ, Monach P, Blair DA, Dustin ML, Shinton SA, Hardy RR, Laidlaw D, Collins J, Gazit R, Rossi DJ, Malhotra N, Sylvia K, Kang J, Fletcher A, Elpek K, Bellemare-Pelletier A, Malhotra D, Turley S. The transcriptional landscape of alphabeta T cell differentiation. Nat Immunol. 2013;14:619–632. doi: 10.1038/ni.2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miwa T, Sun X, Ohta R, Okada N, Harris CL, Morgan BP, Song WC. Characterization of glycosylphosphatidylinositol-anchored decay accelerating factor (GPI-DAF) and transmembrane DAF gene expression in wild-type and GPI-DAF gene knockout mice using polyclonal and monoclonal antibodies with dual or single specificity. Immunology. 2001;104:207–214. doi: 10.1046/j.0019-2805.2001.01280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miwa T, Zhou L, Kimura Y, Kim D, Bhandoola A, Song WC. Complement-dependent T-cell lymphopenia caused by thymocyte deletion of the membrane complement regulator Crry. Blood. 2009;113:2684–2694. doi: 10.1182/blood-2008-05-157966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kimmig S, Przybylski GK, Schmidt CA, Laurisch K, Mowes B, Radbruch A, Thiel A. Two subsets of naive T helper cells with distinct T cell receptor excision circle content in human adult peripheral blood. J Exp Med. 2002;195:789–794. doi: 10.1084/jem.20011756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, Gebhard F, Younger JG, Drouin SM, Wetsel RA, Ward PA. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. 2006;12:682–687. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- 25.Rawal N, Pangburn MK. Formation of high affinity C5 convertase of the classical pathway of complement. J Biol Chem. 2003;278:38476–38483. doi: 10.1074/jbc.M307017200. [DOI] [PubMed] [Google Scholar]
- 26.Zhang JQ, Biedermann B, Nitschke L, Crocker PR. The murine inhibitory receptor mSiglec-E is expressed broadly on cells of the innate immune system whereas mSiglec-F is restricted to eosinophils. Eur J Immunol. 2004;34:1175–1184. doi: 10.1002/eji.200324723. [DOI] [PubMed] [Google Scholar]
- 27.Okada M, Itoh Mi M, Haraguchi M, Okajima T, Inoue M, Oishi H, Matsuda Y, Iwamoto T, Kawano T, Fukumoto S, Miyazaki H, Furukawa K, Aizawa S. b-series Ganglioside deficiency exhibits no definite changes in the neurogenesis and the sensitivity to Fas-mediated apoptosis but impairs regeneration of the lesioned hypoglossal nerve. J Biol Chem. 2002;277:1633–1636. doi: 10.1074/jbc.C100395200. [DOI] [PubMed] [Google Scholar]
- 28.Drake PM, Stock CM, Nathan JK, Gip P, Golden KP, Weinhold B, Gerardy-Schahn R, Bertozzi CR. Polysialic acid governs T-cell development by regulating progenitor access to the thymus. Proc Natl Acad Sci U S A. 2009;106:11995–12000. doi: 10.1073/pnas.0905188106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.