

Abstract
The histone-like nucleoid structuring protein (H-NS) functions as a transcriptional silencer by binding to AT-rich sequences at bacterial promoters. However, H-NS repression can be counteracted by other transcription factors in response to environmental changes. The identification of potential toxic factors, the expression of which is prevented by H-NS could facilitate the discovery of new regulatory proteins that may contribute to the emergence of new pathogenic variants by anti-silencing. Vibrio cholerae hns mutants of the El Tor biotype exhibit altered virulence, motility and environmental stress response phenotypes compared to wild type. We used an RNA-seq analysis approach to determine the basis of the above hns phenotypes and identify new targets of H-NS transcriptional silencing. H-NS affected the expression of 18% of all predicted genes in a growth phase-dependent manner. Loss of H-NS resulted in diminished expression of numerous genes encoding methyl-accepting chemotaxis proteins as well as chemotaxis toward the attractants glycine and serine. Deletion of hns also induced an endogenous envelope stress response resulting in elevated expression of rpoE encoding the extracytoplamic sigma factor E (σE). The RNA-seq analysis identified new genes directly repressed by H-NS that can affect virulence and biofilm development in the El Tor biotype cholera bacterium. We show that H-NS and the quorum sensing regulator HapR silence the transcription of the vieSAB three-component regulatory system in El Tor biotype V. cholerae. We also demonstrate that H-NS directly represses the transcription of hlyA (hemolysin), rtxCA (the repeat in toxin or RTX), rtxBDE (RTX transport) and the biosynthesis of indole. Of these genes, H-NS occupancy at the hlyA promoter was diminished by overexpression of the transcription activator HlyU. We discuss the role of H-NS transcriptional silencing in phenotypic differences exhibited by V. cholerae biotypes.
Introduction
Cholera is an acute water-borne diarrheal disease caused by Vibrio cholerae of serogroups O1 and O139. V. cholerae O1 can be divided in two biotypes, classical and El Tor [1], which differ in the expression and regulation of major virulence factors [1–3]. One major difference is that El Tor biotype strains require special conditions for the in vitro expression of cholera toxin (CT) and the toxin co-regulated pilus (TCP), required for intestinal colonization [4]. Other regulators and virulence factors that differ in their expression between biotypes are the VieSAB three-component regulatory system, [3], hemolysin [1] and the repeat in toxin (RTX) [2].
The cholera bacterium has evolved to sense and effectively colonize disparate ecological niches, the aquatic environment and the human small intestine. In the aquatic environment, Vibrios are subject to numerous physical, chemical and biological stresses which include nutrient limitation, extreme temperatures, oxidative stress, bacteriophage infection and protozoan grazing [5–8]. During infection, Vibrios are subject to a myriad of additional host-induced stresses such as low pH, bile and the activity of antimicrobial peptides. Transitions between the aquatic environment and the human small intestine require the regulation, integration and fine tuning of numerous cellular processes, such as virulence gene expression, motility and biofilm development. The formation of biofilm communities is critical for V. cholerae survival and persistence in nature as well as in disease transmission [5, 9–11].
The histone-like nucleoid structuring protein (H-NS) is a nucleoid associated protein and transcriptional repressor [12]. H-NS belongs to a family of small nucleoid associated proteins that include the factor for inversion stimulation (FIS), and the integration host factor (IHF) [13]. H-NS consists of an N-terminal oligomerization domain connected by a flexible linker to a nucleic acid binding domain [12–15]. Both DNA binding and oligomerization are required for the biological activities of H-NS, which include DNA condensation and the regulation of transcription [16, 17]. In transcription regulation, H-NS negatively affects gene expression by binding to promoters exhibiting AT-rich, highly curved DNA regions [18–20]. Repression by H-NS can be relieved in response to environmental cues that activate the expression of other regulators whose binding site overlaps that of H-NS [13, 21]. For example, transcriptional silencing of V. cholerae tcpA and ctxA promoters by H-NS is antagonized by the AraC-like transcriptional regulator ToxT and the IHF [22–24]. Also, H-NS repression at the flrA and rpoN promoters in late stationary phase cells is counteracted by RpoS and IHF [25].
Mutations that inactivate hns are highly pleiotropic [26, 27]; the mutants form small colonies in agar plates; exhibit diminished growth rate, and altered cell morphology. The El Tor biotype hns mutants express CT and TCP under ToxR non-permissive conditions (LB, 30°C) [27, 28]. Although El Tor biotype hns mutants are flagellated, they show reduced motility in swarm agar plates [25]. In addition, we have shown that hns mutants exhibit enhanced resistance to low pH and hydrogen peroxide [27]. To determine the basis of the hns mutant altered virulence gene expression, its reduced swarm halo in soft agar plates, and altered stress responses [25, 27], we conducted an RNA-seq analysis of an El Tor biotype hns mutant in exponential and stationary phases. Here we show that H-NS affects the expression of 18% of the V. cholerae genome in a growth phase-dependent manner. Loss of H-NS resulted in diminished chemotaxis and induced an endogenous envelope/oxidative stress. Further, we demonstrate that H-NS directly represses the expression of (i) the vieSAB operon, hemolysin, and the RTX toxin, and (ii) indole biosynthesis, an activator of biofilm development.
Materials and Methods
Strains, plasmids, primers and culture media
The strains, plasmids, and oligonucleotide primers used in this study are listed and briefly described in Tables 1 and 2. V. cholerae and Escherichia coli strains were grown in LB medium at 37°C with agitation (225 rpm). E. coli strain TOP10 (Invitrogen) and S17–1λpir [29] were used for plasmid propagation and cloning. To measure the expression of CT genes, V. cholerae strains were grown in LB medium or AKI as described in [30]. To measure chemotaxis, V. cholerae strains were grown in TG medium (1% tryptone, 0.5% NaCl and 0.5% glycerol) at 30°C. Culture media were supplemented with ampicillin (Amp, 100 μg/mL), kanamycin (Km, 25 μg/mL), chloramphenicol (Cm, 2.5 μg/mL), streptomycin (Str, 100 μg/mL), 2,2′-dipyridyl (0.2 mM), polymyxin B (PolB, 100 units/mL), isopropyl-β-D-thiogalactopyranoside (IPTG, 20 μg/mL), L-arabinose (0.02% w/v) or 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-Gal, 20 μg/mL) as required and described in the figure legends.
Table 1. Strains and Plasmid.
| Strain or plasmid | Description | Source or reference |
|---|---|---|
| Strains | ||
| C7258 | Wild type O1; El Tor biotype | Peru, 1991 |
| C7258ΔlacZ | C7258 lacZ deletion mutant | [27] |
| AJB51ΔlacZ | C7258ΔlacZ ΔhapR | [27] |
| O395ΔlacZ | Classical biotype O395 lacZ deletion mutant | [35] |
| AJB608 | O395ΔlacZ Δhns::km | This study |
| AJB80 | C7258 ΔlacZ Δhns::km | [27] |
| AJB700 | C7258 ΔlacZ ΔhapR Δhns::km | This study |
| C7258HNS-FLAG | C7258 hns::hns-FLAG | [25] |
| AJB606 | C7258 ΔlacZΔvieA | This study |
| AJB607 | C7258 ΔlacZ Δhns::km ΔvieA | This study |
| AJB600 | C7258 ΔlacZ ΔvieSAB | This study |
| AJB601 | C7258 ΔlacZ Δhns::km ΔvieSAB | This study |
| AJB602 | C7258 ΔlacZ ΔhlyU::cm | This study |
| AJB603 | C7258 ΔlacZ Δhns::km ΔhlyU::cm | This study |
| WL7258ΔlacZ | C7258 ΔlacZ Δcrp | [27] |
| AJB604 | C7258 ΔlacZ Δcrp Δhns::Km | This study |
| Plasmids | ||
| pCR2.1 | Bacterial expression vector for TA cloning (AmpR) | Invitrogen |
| pBAD33 | pACYC184 ori araC paraBAD, CmR | [45] |
| pBAD-HlyU-HIS | pBAD33 containing the hlyU ORF 3’ expressed from the arabinose promoter | This study |
| pCAT | Chloramphenicol acetyl transferase (cat) gene in pUC219 | B. Kan |
| pUC-VieA-UP | 0.56 kb XbaI-SalI DNA fragment 5’ of vieA in pUC19 | This study |
| pUC-VieA-DN | 0.57 kb SalI-SphI DNA fragment 3’ of vieA in pUC19 | This study |
| pUCΔVieA | XbaI-SalI and SalI-SphI fragments flanking vieA in pUC19 | |
| pCVDΔVieA | XbaI-SphI fragment harboring vieA deletion in pCVD442 | |
| pUC-VieSAB-UP | 0.52 kb XbaI-SalI DNA fragment 5’ of vieSAB in pUC19 | This study |
| pUC-VieSAB-DN | 0.47 kb SalI-SphI DNA fragment 3’ of vieSAB in pUC19 | This study |
| pUCΔVieSAB | XbaI-SalI and SalI-SphI fragments flanking vieSAB in pUC19 | This study |
| pCVDΔVieSAB | XbaI-SphI fragment harboring vieSAB deletion in pCVD442 | This study |
| pUC-HlyU-UP | 0.52 kb SacI-BamHI DNA fragment 5’ of hlyU in pUC19 | This study |
| pUC-HlyU-DN | 0.48 kb BamHI-SphI DNA fragment 3’ of hlyU in pUC19 | This study |
| pUCΔHlyU | SacI-BamHI and BamHI-SphI fragments flanking hlyU in pUC19 | This study |
| pUCΔHlyU- Cm | 0.8 kb BamHI fragment encoding cat gene in pUCΔHlyU | This study |
| pCVDΔHlyU- Cm | SacI-SphI fragment harboring vieSAB deletion and cat insertion in pCVD442 | This study |
| pCVDΔHNSK | DNA fragment carrying hns deletion and km insertion in pCVD442 | [27] |
| pTT3 | XbaI-PstI fragment encoding rrnBT 1 T 2 transcription terminator in pUC19 | [38] |
| pTT3VieSABET | 633 bp SphI-HindIII El Tot biotype vieSAB promoter fragment in pTT3 | This study |
| pTT3VieSABCL | 633 bp SphI-HindIII classical biotype vieSAB promoter fragment in pTT3 | This study |
| pTT3HlyA | 540 bp SphI-HindIII hlyA promoter fragment in pTT3 | This study |
| pTT3RtxCA | 250 bp SphI-HindIII rtxCA promoter fragment in pTT3 | This study |
| pTT3RtxBDE | 230 bp SphI-HindIII rtxBDE promoter fragment in pTT3 | This study |
| pTT3RpoEP1 | 190 bp SphI-HindIII rpoE P1 promoter fragment in pTT3 | This study |
| pTT3RpoEP2 | 180bp SphI-HindIII rpoE P2 promoter fragment in pTT3 | This study |
| pKRZ1 | Plasmid source for promoterless lacZ gene | [39] |
| pVieSABET-LacZ | 1040 bp KpnI-HindIII rrnBT 1 T 2-vieSAB ET cassette ligated to promoterless lacZ gene in pKRZ1 | This study |
| pVieSABCL-LacZ | 1040 bp KpnI-HindIII rrnBT 1 T 2-vieSAB CL cassette ligated to promoterless lacZ gene in pKRZ1 | This study |
| pHlyA-LacZ | 1001 bp KpnI-HindIII rrnBT 1 T 2-hlyA cassette fused to lacZ gene | This study |
| pRtxCA-LacZ | 704 bp KpnI-HindIII fragment rrnBT 1 T 2-rtxCA cassette fused to lacZ gene | This study |
| pRtxBDE-LacZ | 684 bp KpnI-HindIII rrnBT 1 T 2-rtxBDE cassette fused to lacZ gene | This study |
| pRpoEP1-LacZ | 641 bp KpnI-HindIII rrnBT 1 T 2-rpoE P1 cassette ligated lacZ gene | This study |
| pRpoEP2-LacZ | 625 bp KpnI-HindIII rrnBT 1 T 2-rpoE P2 cassette ligated to lacZ gene | This study |
| pTT5 | KpnI-BamHI rrnBT 1 T 2 transcription terminator fragment in pUC19 | This study |
| pTT5TnaA | 1140 bp BamHI-XbaI tnaA promoter fragment in pTT5 | This study |
| pTnaA-LacZ | 1570 bp KpnI-XbaI rrnBT 1 T 2-tnaA cassette ligated to lacZ gene | This study |
| pCTX-LacZ | rrnBT 1 T 2-ctxA promoter fragment ligated to lacZ gene | [35] |
| pRpoE-LacZ | rrnBT 1 T 2 and rpoE P1P2 promoter in tandem fused to lacZ gene | [35] |
Table 2. Primers.
| Primer | Sequence (5’→3’) |
|---|---|
| 70-F | GAAGCATGCCCGCATCGATTGAGCAGAC |
| 70-R | GCGAAGCTTTTTATATAGTGGTGAATGC |
| 70-P1-F1 | CCGCATCGATTGAGCAGACATG |
| 70-P1-R1 | TTTATATAGTGGTGAATGCAGCG |
| 357F | CCTACGGGAGGCAGCAG |
| 926R | CCGTCAATTCMTTTRAGT |
| 1448-F1 | GCAACCATCAGACAAATTGCAC |
| 1448-F2 | GTATTCCCTCAATTTCACACAGAT |
| 1448S-F | GAAGCATGCGCAACCATCAGACAAATTGC |
| 1448-R1 | AATGCATATCAGCGTGAGTCTTTC |
| 1448L-R | GCGAAGCTTAATGCATATCAGCGTGAGTC |
| 1449-F1 | ATCTGTGTGAAATTGAGGGAATAC |
| 1449S-F | GAAGCATGC ATCTGTGTGAAATTGAGGG |
| 1449-R1 | CTTCTCCATCATAAATTTCCCCAT |
| 1449-R2 | GTGCAATTTGTCTGATGGTTGC |
| 1449L-R | GCGAAGCTTCTTCTCCATCATAAATTTCC |
| CAT-3 | CGCGGATCCGATCGGCACGTAAGAGGTTC |
| CAT-4 | CGCGGATCCCGTAGCACCAGGCGTTTAAG |
| HlyA-F | GAAGCATGCGTCTTTAGAGGCTAAAATCTG |
| HlyA-F9 | GAGACACATGCAAAATGGGTATG |
| HlyA-R | GCGAAGCTTGCGCAACGATTGAGTTTTGG |
| HlyA-R7 | GCGCAACGATTGAGTTTTGGCAT |
| HlyA2-F2 | GGATATGCATTTCTGCTAAAAG |
| HlyA2-R2 | GCGCAACGATTGAGTTTTGG |
| HlyU-F1 | GCTCTAGATTTAGGATACATTTTTATGCCGTATTTAAAGGGGGC |
| HlyU-R1 | ACATGCATGCCTAATGATGATGATGATGATGCTGATTCGCCTGACAATAAAG |
| HlyU-U1 | GCCGAGCTCGTTCCAGGCAGTCGAACCG |
| HlyU-U2 | AAAGGATCCCGGCATTTTAATTCCAACCC |
| HlyU-D3 | GAAGGATCCGGCGAATCAGTAGGGTGGTC |
| HlyU-D4 | TAAGCATGCCCGCGAAGGCTCTTCGATAC |
| RnnB-F1 | CGGGGTACC GATTTTCAGCCTGATAC |
| RnnB-R1 | CGCGGATCC TGGCTTGTAGATATGAC |
| RpoE-F | GAAGCATGCCGGGTGAAAATGCTGCCTTG |
| RpoE-R | GCGAAGCTTTCCTATTGTTATTCCCCTAC |
| RpoE-P2-F2 | CGGGTGAAAATGCTGCCTTGAT |
| RpoE-P2-R2 | TCCTATTGTTATTCCCCTACCTTC |
| RpsM-F51 | GCAACTGCGTGATGGTGTAGCTAA |
| RpsM-R52 | GCTTGATCGGCTTACGCGGACC |
| TcpA-F1 | GTTCATAATTTCGATCTCCACTCCG |
| TcpA-R2 | GTTAACCACACAAAGTCACCTGCAA |
| TnaA-F3 | CCTGCGTAATCCCTTCTCGC |
| TnaA-R3 | GTGTGGTGCGTTTAACTGG |
| TnaA-F | CGCGGATCCATGGGTTGGTCTCGCGCTG |
| Try-R | CTAGTCTAGAGTGTGGTGCGTTTAACTG |
| VC1922-F61 | TAGAAGGTTGACGAAACAAGCAATCA |
| VC1922-R62 | GGTTCAACCACCATAGGTACGAGT |
| VieA-U1 | TGCTCTAGAGGGCTTTGAGCGCATGTTTG |
| VieA-U2 | ACGCGTCGACAGAAATCGCCTTACAACTCG |
| VieA-D1 | ACGCGTCGACCCATCCATCTGCGGCATTCG |
| VieA-D2 | ACATGCATGCCTTATACCTTAGCCAATTTG |
| VieS-F | GAAGCATGCGTCCTGATCTTGGGCATC |
| VieS-R | GCGAAGCTTGTCCCACCCCAGTAAGGC |
| VieS-F2 | GATATTGCTTGGGGTTGAAT |
| VieS-R2 | TGTCCCACCCCAGTAAGGC |
| VieSAB-U1 | TGCTCTAGATCAAAGCCTTACCGTGCATC |
| VieSAB-U2 | ACGCGTCGACTCTCGCTACTGGCCGACG |
| VieSAB-D1 | ACGCGTCGACCGCTTGCTCATTCGGATCCAAA |
| VieSAB-D2 | ACATGCATGCGTAATTACACAGTATATTTCCC |
Construction of mutants
To construct vieA deletion mutants, we amplified DNA fragments flanking the vieA locus (VC1652) from C7258 genomic DNA using primer pairs VieA-U1/ VieA-U2 and VieA-D1/VieA-D2 (Table 2). The PCR products were sequentially cloned in pUC19 to yield pUCΔVieA and confirmed by DNA sequencing. The chromosomal DNA fragment harboring the vieA deletion was subcloned in the suicide vector pCVD442 [31] to yield pCVDΔVieA. This vector was transferred by conjugation to strains C7258ΔlacZ and AJB80 (Δhns) (Table 1) and exconjugants were selected in LB agar containing Amp and PolB. Then, the vieA deletion mutants AJB606 and AJB607 were isolated by sucrose selection as previously described [25, 27, 32–34]. Similarly, for the construction of a vieSAB deletion mutant, we amplified DNA fragments flanking the entire vieSAB cluster (VC1651-VC1652-VC1653) using primer pairs VieSAB-U1/VieSAB-U2 and VieSAB-D1/VieSAB-D2 (Table 2). The PCR products were sequentially cloned in pUC19 to yield pUCΔvieSAB and the chromosomal DNA fragment harboring the vieSAB deletion was subsequently moved to pCVD442 to yield pCVDΔvieSAB. This plasmid was transferred by conjugation to strains C7258ΔlacZ and AJB80 and strains AJB600 and AJB601 harboring vieSAB deletions were isolated by sucrose selection. An identical strategy was used to construct V. cholerae ΔhlyU mutants, except that primer pairs HlyU-U1/HlyU-U2 and HlyU-D3/HlyU-D4 were used and the chloramphenicol acetyl transferase (cat) gene conferring chloramphenicol resistance was inserted in place of the deleted hlyU DNA. The cat gene was amplified with primers CAT-3 and CAT-4 from plasmid pCAT kindly provided by B. Kan (China CDC Beijing). The hlyU deletion/insertion was introduced into strains C7258ΔlacZ and AJB80 by conjugation as described above. Finally, the segregants AJB602 and AJB603, harboring the hlyU deletions and cat insertions, were isolated by sucrose selection. A mutant with a deletion of crp encoding the cAMP receptor protein (CRP) and hns (AJB604) was constructed by conjugal transfer of the suicide vector pCVDΔHNSK [27] to the crp mutant WL7258ΔlacZ [27] followed by sucrose selection. Similarly, a mutant lacking the quorum sensing regulator HapR and hns was constructed by conjugal transfer of pCVDΔHNSK to strain AJB51ΔlacZ resulting in strain AJB700. Finally, we constructed an hns mutant in a classical biotype background. To this end, we transferred the suicide vector pCVDΔHNSK [27] to strain O395ΔlacZ [35]. Exconjugants were selected in LB medium containing Amp and Str and strain AJB608 isolated as described above.
Construction of vieSAB-, tnaA-, hlyA-, rtxCA-, rtxBDE-, and rpoE-lacZ promoter fusions
To construct a vieSAB-lacZ promoter fusion, we amplified a 633 bp fragment containing the vieSAB promoter of strain C7258ΔlacZ and O395ΔlacZ with primers VieS-F and VieS-R. For the hlyA-lacZ promoter fusion, a 540 bp fragment containing the hlyA promoter was amplified with primers HlyA-F and HlyA-R. A 250 bp rtxCA promoter fragment [36] was amplified with primers 1449S-F and 1449L-R. To construct the rtxBDE-lacZ promoter fusion, a 230 bp fragment containing the rtxBDE promoter [36] was amplified with primers 1448S-F and 1448L-R. For the rpoE P1-lacZ and rpoE P2-lacZ promoter fusions, a 190-bp fragment containing the rpoE P1 promoter and a 180-bp fragment containing the rpoE P2 promoter as described elsewhere [37] were amplified with primer combinations 70-F/70-R and RpoE-F/RpoE-R, respectively. The promoter fragments were inserted downstream of the rrnBT1T2 transcription terminator in plasmid pTT3 [38] to generate pTT3VieSABET, pTT3VieSABCL, pTT3HlyA, pTT3RtxCA, pTT3RtxBDE, pTT3RpoEP1, and pTT3RpoEP2, respectively. Finally, the terminator-promoter fragments were inserted upstream of a promoterless lacZ gene in plasmid pKRZ1 [39] to generate pVieSAB-LacZET, pVieSAB-LacZCL, pHlyA-LacZ, pRtxCA-LacZ, pRtxBDE-LacZ, pRpoEP1-LacZ and pRpoEP2-LacZ (Table 1). To construct a tnaA-lacZ promoter fusion, we first amplified the rrnBT1T2 transcription terminator from plasmid pTT3 with primers RnnB-F1and RnnB-R1 and re-inserted it in pUC19 to generate pTT5. Then, a 1140 bp fragment containing the tnaA promoter and transcribed leader region [40] was amplified with primers TnaA-F and Try-R. The promoter fragment was inserted downstream of the rrnBT1T2 transcription terminator in plasmid pTT5 to generate pTT5TnaA. Finally, the terminator-promoter cassette was inserted upstream of a promoterless lacZ gene in plasmid pKRZ1 to generate pTnaA-LacZ. The construction of plasmids pCTX-lacZ containing a ctxA-lacZ promoter fusion and pRpoE-LacZ containing a both rpoE P1 and P2 promoters ligated to a promoterless lacZ gene has been described previously [35]. The lacZ promoter fusions were introduced into strains C7258ΔlacZ (WT), AJB80 (Δhns), AJB602 (ΔhlyU), AJB603 (ΔhnsΔhlyU), WL7258ΔlacZ (Δcrp), AJB604 (ΔcrpΔhns) AJB606 (ΔvieA), AJB607 (ΔhnsΔvieA), AJB600 (ΔvieSAB), AJB601 (ΔhnsΔvieSAB), O395ΔlacZ, and AJB608 (Δhns) by electroporation.
Total RNA extraction and removal of ribosomal RNA
Strains C7258ΔlacZ and AJB80 were grown in 50 mL of LB medium at 37°C with agitation to optical densities at 600 nm (OD600) of 0.5 and 2.0. Each culture was divided into 5 mL aliquots and the cells were harvested by centrifugation at 4,000 × g for 10 min at room temperature. The pellets were resuspended in 5 mL of RNAlater (Invitrogen) and agitated on a rotator for 10 min at room temperature. The cells were collected by centrifugation at 4,000 × g for 10 min and resuspended in 5 mL of RNAlater. Then, cell pellets corresponding to 1 mL aliquots were collected by centrifugation for 10 min at 4,000 × g. Total RNA was extracted using RNeasy Plus Mini Kit (Qiagen). To this end, the cell pellets were resuspended in 200 μL of bacterial lysis buffer [30 mM Tris-HCl pH 8.0, 1 mM EDTA, 15 mg/mL lysozyme (Sigma-Aldrich)], supplemented with 15 μL of proteinase K (20 mg/mL, Qiagen), and the samples were incubated at room temperature for 10 min with vortexing every 2 min. Qiagen RLT Plus buffer (750 μL) containing 1% v/v 2-mercaptoethanol was added to each sample and the tubes were briefly vortexed. The bacterial lysates were homogenized using QIAshredder spin columns (Qiagen) and genomic DNA removed using gDNA Eliminator spin columns (Qiagen). Finally, total RNA was recovered using the RNeasy spin column method (Qiagen) following the manufacturer’s protocol. RNA integrity was determined by formaldehyde agarose gel electrophoresis and the RNA was stored at -80°C. Contamination with DNA was further eliminated using the TURBO DNA-free kit (Invitrogen), which involves a second treatment with DNase for 30 min at 37°C. Reactions were terminated by addition of 0.2 volumes of the DNase inactivation reagent and RNA was purified using the Agencourt RNAClean XP kit (Beckman) following the manufacturer’s instructions. Total RNA was eluted in 60 μL of RNase-free water. The absence of DNA contamination was confirmed by PCR with 16S-specific primers 357F and 926R. The DNase treated RNA (2 μL) was added to each reaction in a final reaction volume of 20 μL. Each reaction was run in parallel to E. coli DNA (positive control) and nuclease-free water (negative control) with the following cycling conditions: 95°C for 1 min, 30 cycles of 95°C for 30 s, 50°C for 30 s, 68°C for 1 min. Next, rRNA was removed using Ribo-Zero Magnetic Kit (Epicentre) following the manufacturer’s instructions. Briefly, 6 μg of total RNA sample and 20 μL of Ribo-Zero rRNA Removal Solution were combined in a final reaction volume of 80 μL. Samples in Ribo-Zero rRNA removal solution were incubated at 68°C for 10 min followed by a 5 min incubation at room temperature. To remove the rRNA molecules from the mRNA, reactions mixtures were incubated with the magnetic beads provided in the kit, mixed and placed at room temperature for 5 min and then at 50°C for 5 min. The rRNA bound to the beads was then removed by magnetic separation. Finally, mRNA was purified using Agencourt RNAClean XP kit and eluted in 15 μL of RNase-free water.
RNA-seq and data analysis
RNA-seq was performed as described elsewhere [41] on the Illumina HiSeq2000 using the latest versions of sequencing reagents and flow cells providing up to 300 Gb of sequence information per cell. Briefly, the quality of the total RNA was assessed using the Agilent 2100 Bioanalyzer and samples were subsequently converted to cDNA. Libraries were constructed using the TruSeq library generation kits as per the manufacturer’s instructions (Illumina, San Diego, CA). The cDNA libraries were quantitated using qPCR in a Roche LightCycler 480 with the Kapa Biosystems kit for library quantitation (Kapa Biosystems, Woburn, MA) prior to cluster generation. Clusters were generated to yield approximately 725K—825K clusters/mm2. Paired end 2X50 bp sequencing runs were conducted to align the cDNA sequences to the reference genome. The TopHat software and the short read aligner Bowtie was used to align the raw RNA-seq fastq reads to the reference genome [42–44]. Transcripts assembly, abundance and evaluation of differential expression and regulation were accomplished using the Cufflinks software [42, 43]. Genes exhibiting a fold change ≥ ±2.0 and q-value < 0.05) were considered differentially expressed in the Δhns mutant.
Protein-DNA interaction assays
Electrophoresis mobility shift assay. Electrophoresis mobility shift assays (EMSA) were conducted using the second-generation digoxigenin (DIG) gel shift kit (Roche) as previously described [25, 32, 34]. Protein-DNA complexes were separated by electrophoresis in 5% Tris-borate-EDTA (TBE) polyacrylamide gels and transferred to nylon membranes, and DNA was visualized using an anti-DIG Fab fragment-AP conjugate, followed by chemiluminescence detection. The following primer combinations were used to amplify the promoter regions under study: VieS-F2 and VieS-R2 for vieSAB, TnaA-F3 and TnaA-R3 for tnaA, 1448-F1 and 1448-R1 for rtxBDE, 1449-F1 and 1449-R1 for rtxCA, HlyA2-F2 and HlyA2-R2 for hlyA, 70-P1-F1 and 70-P1-R1 for rpoE P1, RpoE-P2-F2 and RpoE-P2-R2 for rpoE P2, VC1922-F61 and VC1922-F62 for the negative control VC1922, and TcpA-F1 and TcpA-R2 for the positive control tcpA.
Chromatin immunoprecipitation. Occupation of the vieSAB, tnaA, rtx and hlyA promoters by H-NS in the cell was determined by chromatin immunoprecipitation (ChIP). To this end, strain C7258HNS-FLAG expressing an hns-FLAG allele from native transcription and translation signals [25, 32] was grown to an OD600 of 0.5. Nucleoprotein complexes were immunoprecipitated (IP) with the anti-FLAG M2 monoclonal antibody (mAb) (Sigma-Aldrich) as previously described [25, 32, 34]. IP DNA was qualitatively detected by PCR and agarose gel electrophoresis using the PCR primers used to generate promoter fragments for EMSA. Real-time quantitative PCR (qPCR) was used to quantitate promoter occupancy by H-NS-FLAG as formerly described [25, 32, 34]. The quantity of IP DNA was calculated as a percentage of the input DNA (10 μL sample taken prior to IP) using the formula IP = 2(CT input -CT IP ), where CT is the fractional threshold cycle of the input and IP DNAs. The relative IP was calculated by normalizing the IP of each sample by the IP of a mock ChIP using the unrelated mouse monoclonal antibody G3A1 IgG1 isotype control (Cell Signaling Technology). To study the effect of HlyU on H-NS occupancy at the hlyA and rtx promoters strain C7258HNS-FLAG containing pBAD-HlyU-His was grown in LB medium containing Amp and Cm at 37°C with agitation to OD600 0.5. At this point the cultures were divided in halves, with one half used as a control and the other induced by addition of L-arabinose to a final concentration of 0.02%. The cultures were incubated for 3 h and the cells collected and processed for ChIP as described previously [25, 32, 34]. For this experiment, a region of the hlyA promoter containing putative H-NS binding sites was amplified with primers HlyA-F9 and HlyA-R7. The rtxCA and rtxBDE promoter regions were amplified with primer combinations 1449-R1/1449-R2 and 1448-F2/1448-R1, respectively. The tcpA promoter, which is not regulated by HlyU, was used as a positive control for H-NS occupancy and amplified with primers TcpA-F1 and TcpA-R2. Negative controls consisted of the VC1922 promoter amplified with primers VC1922-F61 and VC1922-R62 and a region within the rpsM open reading frame (ORF) amplified with primers RpsM-F51 and RpsM-R52.
Cloning and overexpression of the regulator HlyU
The hlyU open reading frame (ORF) was amplified from strain C7258 genomic DNA with primer HlyU-F1 and HlyU-R1 incorporating a C-terminal 6xHis tag. The PCR product was ligated into pCR2.1 and confirmed by DNA sequencing. Then, a 373 bp XbaI-SphI DNA fragment from the resulting plasmid was subcloned into pBAD33 [45] to generate pBAD-HlyU-HIS. Finally, pBAD-HlyU-HIS was transformed into C7258HNS-FLAG by electroporation. Expression of the HlyU-6xHis protein was confirmed by western blot using the histidine tag monoclonal antibody 3D5 (Life Technologies).
Measurement of indole, hemolysin activity and biofilm formation
Indole production was measured as described in [46]. To this end, 0.4 mL of cell-free culture supernatant was treated with the same volume of 20% trichloroacetic acid, mixed, incubated 10 min on ice and cleared by centrifugation. Then 0.4 mL of the treated supernatant was reacted with the same volume of Kovac’s reagent (Sigma-Aldrich). Indole production was estimated using a standard curve of commercial indole (Sigma-Aldrich) and reported as μM indole normalized by the culture OD600. Hemolysin activity on sheep red blood cells was conducted as described in [47] except that a unit of enzyme activity was defined as the amount of enzyme causing an increase in 0.01 optical density units at 570 nm per h at 37°C. Biofilm formation was measured by the crystal violet staining method and expressed as optical density at 570 nM [33, 34].
Enzyme activities
β-Galactosidase activity was measured as described in [48] using the substrate o-nitro phenyl-β-D-galactopyranoside. Specific activities are given in Miller units [1,000 × (OD420/t ×v ×OD600)], where t is the reaction time and v is the volume of enzyme extract per reaction.
Chemotaxis capillary assay
A capillary chemotaxis assay was conducted as described previously [49]. Briefly, overnight cultures of V. cholerae strains in TG medium were diluted 1:30 in fresh medium and incubated with agitation for 6 h at 30°C. The cells were collected by centrifugation and washed with TM buffer consisting of 50 mM Tris-HCl pH 7.4, 5 mM glucose and 5 mM MgCl2. Next, the cells were resuspended in TMN buffer (50 mM Tris-HCl pH 7.4, 5 mM glucose, 100 mM NaCl and 5 mM MgCl2) to OD600 0.1. Cells were pre-incubated for 1 h at 30°C and a capillary containing L-glycine (10 mM), L-serine (10 mM) or water inserted. The capillary was incubated in the cell suspension 1 h at 30°C after which, the number of cells within the capillary was determined by plating serial dilutions on LB agar.
Infant mouse colonization assay
A competitive infant mouse colonization assay was conducted using 3–5 days old CD-1 newborn mice as previously described [33]. The study was conducted in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and the protocol approved by the Morehouse School of Medicine Institutional Animal Care and Use Committee (protocol # 13–27). A competitive index (CI) was calculated by normalizing the mutant to wild type ratio after intestinal colonization (output) by the mutant to wild type ratio in the inoculum (input).
Statistical evaluation
Differences between the means of measured activities obtained for different strains, mutants, transformants and culture conditions were evaluated for statistical significance using an unpaired, one-tailed t test and p values are cited in the text when required.
Results
Transcription profile of a V. cholerae El Tor biotype Δhns mutant
We compared the transcription profile of a wild type V. cholerae strain of the El Tor biotype and its isogenic hns mutant grown in LB medium by RNA-seq. El Tor biotype hns mutants are known to express major virulence factors under this culture condition [27, 28]. A complete list of genes differentially expressed in the hns mutant and their distribution per role category is provided as supporting information in S1 Fig., S1 Table and S2 Table. The raw and processed datasets have been deposited in the Gene Expression Omnibus (GEO) repository (http://www.ncbi.nlm.nih.gov/geo/) and assigned accession number GSE62785.
We identified 701 genes differentially expressed in the Δhns mutant defined as exhibiting a fold change ≥ ±2.0 and q-value < 0.05 (Fig. 1A). As expected, genes known to be transcriptionally silenced by H-NS such as ctxAB, the tcpA-F cluster, tcpPH, toxT [23, 24, 28, 50] and vps genes required for biofilm matrix exopolysaccharide [32] were identified in the dataset. Most H-NS regulated genes fell within the role categories of cellular processes, energy metabolism, regulatory function, and transport and binding proteins (S1 Fig.). The number of differentially regulated genes located in chromosomes I and II exhibited no bias and correlated with chromosome size (Fig. 1B).
Fig 1. Venn diagram representation of the number of genes differentially expressed in an hns mutant.
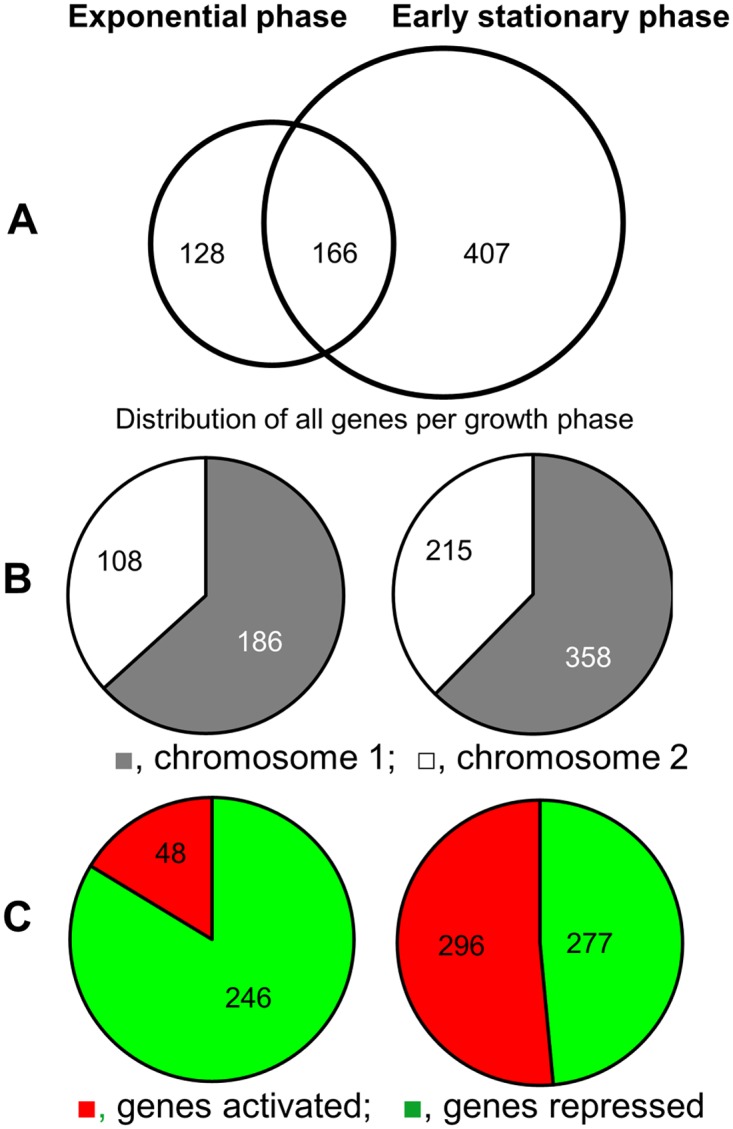
Left side, data from exponential phase; right side, data from early stationary phase. A. Number of H-NS regulated genes in exponential and early stationary phases. B. Distribution of H-NS regulated genes per chromosome in exponential and early stationary phases. C. Distribution of genes positively or negatively regulated by H-NS in exponential and early stationary phases. The data shown resulted from the analysis of two independent cultures of each strain collected in exponential phase and two cultures collected in early stationary phase.
The hns transcriptome differed markedly in exponential and early stationary phases. For instance, the number of genes identified to be regulated by H-NS was higher in the early stationary phase (573) compared to exponential phase (294). Further, H-NS appeared to function mainly as a transcriptional repressor in exponential phase (Fig. 1C), while a larger fraction of genes were found to be positively regulated by H-NS in early stationary phase (Fig. 1C). A selection of genes newly identified to be regulated by H-NS and affecting virulence, stress response, chemotaxis and biofilm development is shown in Table 3.
Table 3. Selected new genes regulated by H-NS affecting V. cholerae virulence, stress response and chemotaxis.
| Loci | Genes | FC * | Loci | Genes | FC * |
|---|---|---|---|---|---|
| Exponential phase | Early stationary phase | ||||
| Virulence | |||||
| VC1447, VC1448 | rtxD, rtxB | 3.1 | VC1447, VC1448 | rtxD, rtxB | 2.5 |
| VC1449, VC1450 | rtxC, rtxA | 4.7 | |||
| VC1651 | vieB | 2.7 | VC1651 | vieB | 1.5 |
| VC1652 | vieA | 2.9 | VC1652 | vieA | 1.8 |
| VC1653 | vieS | 3.1 | VC1653 | vieS | 2.2 |
| VCA0219 | hlyA | 5.2 | VCA0219 | hlyA | 1.7 |
| Cell envelope, transport and binding proteins, stress response | |||||
| VC0156 | tonB | -2.4 | VC0364 | Bfd | 1.2 |
| VC1318 | ompV | 1.2 | VC0365 | Bfr | -1.4 |
| VC1565 | tolC | 2.1 | VC0773 | vibC | -1.0 |
| VC1583 | sodC | 2.9 | VC1565 | tolC | 0.9 |
| VC1585 | katB | 3.7 | VC1854 | ompT | -5.6 |
| VC2464,VC2466, VC2465 | rseA, rseB, rseC | 1.8 | VC2213 | ompA | 1.6 |
| VC2467 | rpoE | 2.1 | VCA0576 | hutA | 2.5 |
| VCA0576 | hutA | 2.6 | VCA0867 | ompW | -1.1 |
| VCA0915 | hutD | 1.4 | VCA1028 | ompS | 1.1 |
| VCA1028 | ompS | 3.4 | VC1583 | sodC | 3.5 |
| VC1585 | katB | 5.7 | |||
* Log2 fold change
** Methyl-accepting chemotaxis protein
H-NS and quorum sensing silence the transcription of the vieSAB regulatory system
The vieSAB operon encodes a three component regulatory system that enhances the expression of ctxAB in vitro and during infection [51–53]. The El Tor biotype hns mutants expressed elevated vieSAB (Table 3). To confirm this result, we constructed vieSAB-lacZ fusions containing the El Tor biotype vieSAB promoter (vieSAB ET) promoter connected to a promoterless lacZ gene. In Fig. 2A we show that the El Tor biotype hns mutant AJB80 grown in LB medium expressed 17-fold higher vieSAB ET -lacZ promoter activity compared to wild type (p < 0.001). We constructed an hns mutant in the classical biotype strain O395ΔlacZ and measured the expression of the vieSAB ET -lacZ promoter fusion in wild type and mutant. As shown in Fig. 2A, the wild type classical biotype strain expressed 54-fold higher vieSAB ET promoter activity compared to the El Tor biotype strain. Further, deletion of hns in the classical biotype resulted in a modest 1.7-fold increase in vieSAB ET-lacZ activity suggesting that H-NS is 10 times less effective in repressing the same promoter when it is introduced in the classical biotype genetic background. A similar experiment was conducted using the vieSAB CL promoter. As shown in Fig. 2B, the vieSAB CL promoter was also silent in the wild type El Tor background and significantly derepressed in the El Tor hns mutant. We note that the vieSAB CL promoter activity was higher in the El Tor hns mutant compared to the vieSAB ET promoter in the same genetic background. The reverse trend was observed for the vieSAB CL promoter in wild type El Tor and classical biotype strains. In spite of these differences, our data shows that both promoters are transcriptionally silenced by H-NS in the El Tor biotype and are expressed in the wild type classical biotype background in which H-NS appears to function as a weaker repressor. The classical biotype strain used in our study is known to lack the quorum sensing regulator HapR. Thus, we considered the possibility of vieSAB being silenced in the El Tor biotype by quorum sensing in addition to H-NS. In Fig. 2C we show that deletion of hapR in strain C7258ΔlacZ resulted in significant derepression of vieSAB and that the ΔhnsΔhapR double mutant exhibited the highest (approximately additive) vieSAB promoter activity. We conclude that both, H-NS and quorum sensing contribute to the transcriptional silencing of vieSAB in El Tor biotype V. cholerae.
Fig 2. Repression of the vieSAB operon by H-NS and quorum sensing.

A. β-galactosidase activity produced by wild type (WT) and mutant strains containing an El Tor biotype vieSAB ET -lacZ promoter fusion. El Tor biotype strain C7258ΔlacZ and classical biotype strain O395ΔlacZ and their isogenic Δhns mutants (AJB80 and AJB608, respectively) were grown to stationary phase in LB medium. B. β-galactosidase activity produced by wild type (WT) and mutant strains containing a classical biotype vieSAB CL -lacZ promoter fusion. Strains C7258ΔlacZ and O395ΔlacZ and their isogenic Δhns mutants were grown to stationary phase in LB medium. C. Activity of the vieSAB ET -lacZ promoter fusion in wild type (C7258ΔlacZ), Δhns (AJB80), ΔhapR (AJB51ΔlacZ) and ΔhnsΔhapR (AJB700) mutants grown to stationary phase in LB medium. β-galactosidase activity (Miller units) was measured as an indicator of promoter activity. Each value represents the mean of six independent experiments. Error bars denote the standard deviation (STDEV).
Since VieA has been suggested to enhance CT production [51–53], we examined if derepression of VieA contributed to the expression of CT genes under ToxR non-permissive conditions in the hns mutant. To this end, we constructed a ctxA-lacZ promoter fusion and compared its activity in El Tor biotype wild type, vieA, vieSAB and hns mutants grown in ToxR non-permissive LB and AKI media. Deletion of vieA and/or vieSAB in the wild type genetic background did not affect ctxAB expression under both culture conditions consistent with vieSAB being transcriptionally silenced by H-NS and quorum sensing (Fig. 3). However, deletion of vieA and/or vieSAB significantly diminished ctxA-lacZ expression in the hns genetic background under both culture conditions (Fig. 3, p < 0.001).
Fig 3. Expression of ctxA in wild type, vieA, vieSAB and hns mutants.

El Tor biotype WT and mutant strains containing a ctxA-lacZ promoter fusion were grown in ToxR-permissive AKI medium and non-permisive LB medium. β-galactosidase activity (Miller units) was measured as an indicator of promoter activity. Each value represents the mean of six independent experiments. Error bars denote the standard deviation (STDEV).
Regulation of the hlyA and rtx promoters by H-NS and HlyU
El Tor biotype strains have been shown to express additional toxic factors such as hemolysin and the RTX toxin [1, 2]. As shown in Table 3, the Δhns mutant overexpressed hlyA encoding hemolysin and genes required for the biosynthesis (rtxCA) and transport (rtxBDE) of the RTX toxin.
Hemolysin expression. Very little hemolysin activity could be detected in exponential phase culture supernatants (OD600 0.6) in the wild type strain (1.5 ± 0.96 units/OD600) compared to the Δhns mutant (11.5 ± 5.4 units/OD600). In both cases, the hemolysin activity declined as the culture progressed to stationary phase (wild type, 0.1 ± 0.05 units/OD600; Δhns 0.54 ± 0.14 units/OD600). Expression of hlyA in V. cholerae is regulated by iron [47] and HlyU, a member of the SmtB/ArsR family of metalloregulators [54]. To determine if this pattern of regulation was disrupted in the Δhns mutant, we constructed an hlyA-lacZ promoter fusion. Wild type and mutant cultures containing the hlyA-lacZ fusion were grown in LB medium to mid-exponential phase and 2, 2,-dipyridyl was added to induce iron-limitation. In Fig. 4A we show that expression the hlyA-lacZ fusion is strongly enhanced in the Δhns mutant (p < 0.01) and diminished in the ΔhlyU genetic background (p < 0.05). We note that deletion of hlyU still diminished hlyA expression in the absence of H-NS (Fig. 4A) (p < 0.05). However, the ΔhnsΔhlyU double mutant still expressed higher activity compared to wild type and hlyU strains. Expression of hlyA was significantly enhanced (p < 0.05) by inducing iron limitation with 2, 2,-dipyridyl in the wild type strain but not in the Δhns mutants (Fig. 4A).
Fig 4. H-NS repression of hemolysin and RTX toxin expression.

A. β-galactosidase activity produced by wild type (WT) and mutant strains containing a hlyA-lacZ promoter fusion. WT and mutants were grown in LB to an OD600 0.5. The cultures were divided in halves, one half treated with 2,2 dipyridyl for 2 h at 37°C and the other half used as a control. B. β-galactosidase activity produced by wild type (WT) and mutant strains containing a divergently transcribed rtxAC- and rtxBDE-lacZ promoter fusions. WT and mutants were grown in LB to stationary phase at 37°C. β-galactosidase activity (Miller units) was measured as an indicator of promoter activity. Each value represents the mean of six independent experiments. Error bars denote the STDEV.
Expression of the RTX toxin and transport system. To confirm the overexpression of RTX toxin and transport genes (Table 3) we constructed rtxCA- and rtxBDE-lacZ promoter fusions. The transcription factor HlyU has been suggested to function as an H-NS anti-repressor of RTX toxin expression in V. vulnificus [55]. In Fig. 4B we show that expression of the divergently transcribed rtxCA and rtxBDE operons are enhanced in the Δhns mutant and significantly diminished in the ΔhlyU genetic background (p < 0.01). Again, HlyU was still required for maximal expression of rtxCA and rtxBDE in the absence of H-NS (Fig. 4B).
H-NS is a repressor of indole biosynthesis
In the cell-to-cell communication catergory, the hns mutant expressed elevated tnaA (encoding tryptophanase) and tryptophan-transport gene VCA0160 in exponential phase (Table 3). Tryptophanase catalyzes the reductive deamination of tryptophan to indole. Indole has been reported to act as a signaling molecule in V. cholerae that enhances biofilm development [46]. We found that the Δhns mutant accumulated 317 ± 26 μM of indole in the medium compared to 160 ± 26 in the wild type precursor (n = 3). We confirmed that growth of the wild type strain C7258 in LB supplemented with exogenous indole (300 μM) resulted in a 4-fold increase in biofilm formation (OD570 in LB 0.62 ± 0.05; OD570 LB + indole 2.45 ± 0.24; n = 3). In a previous microarray study, we showed that expression of tnaA is diminished in strain C7258 lacking CRP [56]. To examine the regulation of tnaA transcription in the V. cholerae Δhns mutant, and the interplay between H-NS and CRP in the control of this promoter, we constructed a tnaA-lacZ promoter fusion. In Fig. 5 we show that tnaA-lacZ activity was enhanced in the Δhns mutant (p < 0.05) and significantly diminished in an isogenic Δcrp mutant (p < 0.01). CRP was still required for full tnaA expression in the mutant lacking H-NS (Fig. 5).
Fig 5. Deletion of hns enhances tryptophanase expression.

WT and mutants containing a tnaA-lacZ promoter fusion were grown in LB to stationary phase at 37°C. β-galactosidase activity (Miller units) was measured as an indicator of promoter activity. Each value represents the mean of at least three independent experiments. Error bars denote the STDEV.
Mutational loss of hns induces an endogenous envelope stress response
The hns mutant exhibited significant dysregulation of genes encoding outer membrane proteins (ompA, ompS, ompT, ompV, ompW and tolC) (Table 3). Some of these omp genes were overexpressed in the mutant (ompA, ompS, ompV, tolC), while ompT and ompW exhibited lower expression (Table 3). The mutant also exhibited aberrant expression of genes involved in haemin and iron binding and transport. For instance, tonB (haemin, vibriobactin and ferrichrome transport), hutA and hutD (haemin transport) and bfd (bacterioferritin-associated ferredoxin) were overexpressed while bfr (bacterioferritin) and vibC (vibriobactin) were down-regulated in the Δhns genetic background (Table 3). We note that genes involved in iron and haemin (bfd, bfr, vibC, hutA) transport, as well as those encoding the major OMPs ompT, ompA, and ompW, were differentially expressed in early stationary phase only. The genes sodC (superoxide dismutase) and katB (catalase), involved in oxidative stress response, were overexpressed in both growth phases studied. In Table 3 we show that the hns mutant expressed elevated rpoE encoding the extra cytoplasmic RNA polymerase alternative sigma factor E (σE) in exponential phase as well as genes rseA, rseB, rseC encoding regulators of rpoE activity (Table 3). Perturbation of the cell envelope has been reported to induce oxidative stress and changes in iron homeostasis [57]. Thus, our results suggested that H-NS plays a critical role in the biogenesis of the cell envelope. We hypothesized that mutational loss of hns could induce an endogenous envelope stress response resulting in the elevated transcription of rpoE in the absence of exogenous stressors. The rpoE gene is transcribed from two promoters; an upstream σ70-dependent promoter (P1) and an σE-dependent promoter (P2) [37]. Upon the occurrence of an envelope stress, σE is released from the cell membrane and activates its own promoter (P2) [58, 59]. To test the hypothesis that loss of hns induces an endogenous envelope stress response, we constructed several promoter fusions containing the P1, P2 and both rpoE promoters fused to a promoterless lacZ gene. Then, the activity of each promoter fusion was tested in the absence and presence of an exogenous envelope stress induced by PolB. As expected, the σ70-dependent P1 promoter was not responsive to PolB (Fig. 6). In contrast, both the P2 and P1P2 lacZ promoter fusions were responsive to PolB and were significantly enhanced (p < 0.01) in the Δhns mutant (Fig. 6). It is noteworthy that mutational loss of H-NS resulted in a much stronger induction of rpoE-lacZ activity compared to treatment with PolB. Treatment of the hns mutant with PolB enhanced rpoE-lacZ activity to a lesser extent compared to the wild type strain (Fig. 6). This increase was nevertheless still statistically significant (p < 0.01).
Fig 6. hns mutants express elevated levels of σE encoded by rpoE.

WT and mutants containing different rpoE-lacZ promoter fusions described in materials and methods were grown in LB to stationary phase at 37°C in the presence and absence of PolB. β-galactosidase activity (Miller units) was measured as an indicator of promoter activity. The P1 fusion contained the rpoE σ70-dependent promoter; P2 contained the σE-dependent promoter and the P1P2 fusion contained both promoters. Symbols: □, control; ■, PolB treated. Each value represents the mean of three independent experiments. Error bars denote the STDEV.
H-NS binds to the vieSAB, tnaA, rtxCA, rtxBDE and hlyA promoters
To determine if H-NS transcriptional repression of the above promoters was direct, we conducted EMSA. As shown in Fig. 7, purified H-NS bound to the vieSAB, tnaA, rtxCA, rtxBDE and hlyA promoters but not to the rpoE promoter. To determine if H-NS could interact with these promoters in the cell, a condition in which physiological levels of H-NS can co-exist with other transcriptional factors, we conducted ChIP. As shown in Fig. 8, significant H-NS occupancy (similar in magnitude to the tcpA promoter used as a positive control) could be demonstrated for all promoters except rpoE. In the ChIP assay, the highest occupancies were observed for the rtx DNA spanning the intergenic region between the rtxC and rtxB ORFs and the hlyA promoter region.
Fig 7. H-NS can bind to the vieSAB, tnaA, rtx and hlyA promoters.

DIG-labeled DNA fragments containing the tcpA, vieSAB, tnaA, rtxBDE, rtxCA, hlyA, rpoE-P1, rpoE-P2 and VC1922 promoters were incubated with 0 (lane1), 10 (lane 2), 20 (lane 3) and 25 (lane 4 in tnaA panel) ng of H-NS protein. The spans of the promoter fragments used relative to the start codon were as follows: tcpA, -306 to-83; vieSAB, -331 to +21; tnaA, -480 to +70; rtxBDE, -188 to +39; rtxCA, -223 to +24; hlyA, -291 to +23; rpoE P1, -414 to-230; rpoE P2, -180 to-12; VC1922, -113 to +43. Lanes 4 in all panels except tnaA, and lane 5 in the tnaA panel contained a 100-fold excess of unlabeled competitor promoter DNA. The tcpA and VC1922 promoters were used as positive and negative controls, respectively.
Fig 8. H-NS associates with vieSAB, tnaA, rtx and hlyA promoters in the cell.

A V. cholerae strain C7258HNS-FLAG was grown to OD600 0.5. Promoter DNA IP with H-NS was determined by agarose gel electrophoresis (top) and quantitated by qPCR (bottom). Relative IP is the amount of promoter IP with anti-FLAG mAb normalized by the quantity of promoter DNA IP with the unrelated IgG1 isotype control mAb. Positive control, tcpA; negatives controls, VC1922, rpsM. Each value represents the mean of three experiments. Error bars denote the STDEV.
We used a ChIP assay to clarify the interaction between H-NS and HlyU at the hlyA, rtxCA, and rtxBDE promoters. To this end, we overexpressed HlyU from the arabinose promoter in plasmid pBAD-HlyU-His and determined H-NS occupancy at the above promoters. As shown in Fig. 9, overexpression of HlyU had a minor effect on H-NS occupancy at the rtx promoters that did not reach statistical significance. Similarly, overexpression of HlyU did not affect H-NS occupancy at the tcpA, promoter, which is not regulated by HlyU. In contrast, overexpression of HlyU diminished H-NS occupancy at the hlyA promoter (p = 0.011) (Fig. 9). It should be noted that the fragment amplified in this ChIP assay exhibited positive H-NS binding in EMSA. These results suggests that HlyU could act as an H-NS anti-repressor for the hlyA gene but not for rtx as reported for the V. vulnificus rtxA1 locus [55].
Fig 9. Effect of HlyU on H-NS occupancy at the rtxCA, rtxBDE and hlyA promoters.

V. cholerae strain C7258HNS-FLAG containing pBAD-HlyU-His was grown to OD600 0.5 and the cultures divided in halves. One half was used as a control and the other half treated with 0.02% L-arabinose to induce HlyU expression. Cultures were incubated as described in materials and methods and the amount of promoter DNA IP with H-NS was quantitated by qPCR. Positive control, tcpA; negative controls, VC1922. Each value represents the mean of three experiments. Error bars denote the STDEV.
H-NS positively regulates chemotaxis
A remarkable characteristic of the Δhns transcriptome is the lower expression of chemotaxis genes in the early stationary phase (Table 3). This included a broad repertoire of loci annotated as methyl-accepting chemotaxis proteins (MCP), response regulators, histidine kinases, coupling proteins, methyl transferases and methyl esterases [60]. Since the function of most of these genes is ill-defined, we conducted a capillary chemotaxis assay to determine if the Δhns mutant has a chemotaxis phenotype. Since serine and glycine have been confirmed to function as V. cholerae attractants [49], we determined if the hns mutant displayed reduced chemotaxis toward these compounds. As shown in Fig. 10, the Δhns mutant displayed reduced taxis toward these amino acids compared to wild type (p < 0.05).
Fig 10. Deletion of hns diminishes chemotaxis.

A chemotaxis capillary assay was used to measure WT and mutant taxis toward amino acids as described in materials and methods. A non-motile (motY) mutant and water were used as negative controls. Each value represents the mean of three experiments. The error bars denote the STDEV.
Diminished chemotaxis has been shown to enhance V. cholerae infectivity and provide a competitive advantage for colonization of the suckling mouse intestine [61]. We conducted a suckling mouse competitive colonization assay to determine if the diminished chemotaxis exhibited by the hns mutant had a positive effect on intestinal colonization. However, despite the hns mutant exhibiting diminished chemotaxis, the El Tor biotype mutant showed a 2-fold reduction in infant mouse colonization capacity compared to wild type (CI = 0.45, median of 6 mice). An identical competition experiment in LB medium yielded a CI of 0.48 (n = 4). These results suggest that the diminished intestinal colonization exhibited by the hns mutant is a general consequence of its reduced growth rate.
Discussion and Conclusions
Transcription profiling using RNA-seq showed that loss of H-NS has a significant impact on the transcriptome affecting the expression of 18% of all predicted genes. Here we identify new genes involved in chemotaxis, cell envelope biogenesis, biofilm formation and virulence that are regulated by H-NS. In some cases, the effect of H-NS was found to be growth phase-dependent. H-NS is one of the most abundant proteins in the cell and it is expressed at comparable levels at low cell density, high cell density and early stationary phase [32]. Therefore, the growth phase differences in the number and kind of H-NS responsive genes is not due to differences in H-NS expression. Instead, these differences most likely reflect the expression of other nucleoid-associated proteins that could counteract H-NS repression. For instance, we have shown that induction of IHF in the stationary phase results in lower H-NS occupancy at the flrA and rpoN promoters [25]. FIS, which has been reported to counteract H-NS repression at several promoters [12, 13], declines sharply at high cell density in V. cholerae [62]. DPS (DNA protection during starvation), which is expressed in the stationary phase, could indirectly affect H-NS repression due to its effect on DNA topology which changes from negatively supercoiled in log phase cells to more relaxed in stationary phase cells [63]. The activity of CRP known to counteract H-NS repression at a number of promoters [12,13] is regulated by carbon source availability and could increase in stationary phase due to nutrient limitation [64]. Further, RNA polymerase containing σS, expressed in the stationary phase, has been reported to be more resistant to inhibition of transcription initiation by H-NS [65].
The vieSAB regulatory system is a regulator of CT biosynthesis, the expression of which differs between V. cholerae biotypes [3]. This operon is expressed at a higher level in the classical biotype and 20% of the genes that are differentially expressed in the classical versus El Tor biotype are controlled by VieA [3]. In fact, VieA was found to regulate the expression of 10% of the classical biotype genome [3] suggesting that this regulator is a major contributor to the phenotypic differences observed between V. cholerae biotypes. Here we show that H-NS represses the vieSAB operon by binding to its promoter in vitro and in vivo. Overexpression of the vieSAB operon in the El Tor Δhns mutant revealed a new layer of H-NS transcriptional silencing of virulence gene expression. In agreement with previous studies, deletion of vieA and vieSAB resulted in identical phenotypes suggesting that VieA is the component of this regulatory system that enhances toxin expression [53]. We note, however, that deletion of vieA and vieSAB diminished ctxAB expression only in the hns mutant. Overexpression of VieA from a heterologous promoter to bypass H-NS repression had no effect on ctxAB expression in wild type V. cholerae (data not shown). This result could be explained by H-NS transcriptional silencing of the ToxR regulatory cascade at multiple steps [28]. Since vieA mutants express diminished toxT mRNA [52], our data are consistent with VieA acting at the level of ToxT to enhance ctxAB expression when H-NS is absent. Despite V. cholerae of the El Tor biotype expressing lower levels of vieSAB compared to the classical biotype [3], the regions preceding the vieS ORF in the two biotypes aligned with ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/) are 99% identical. Furthermore, the virtual footprint software (http://www.prodoric.de/vfp) [66] identified three identically spaced H-NS putative binding sites upstream of vieS in both biotypes suggesting that H-NS can potentially bind to the vieSAB promoter in both biotypes. In spite of the promoter similarity, the same vieSAB-lacZ constructs exhibited much higher activities in the classical versus the El Tor biotype. The classical biotype strain used in this study differs from the El Tor strain in that the former does not express the quorum sensing regulator HapR. Here we show that the vieSAB operon is also repressed by quorum sensing. We hypothesize that HapR could bind the vieSAB promoter to diminish transcription activity together with H-NS in a roughly additive manner. Binding of H-NS and HapR to negatively regulate transcription has been reported for the vpsT promoter [32, 67]. A DNA sequence closely matching the HapR binding motif-1 described in [68] is located upstream the vieSAB ORF. Alternatively, HapR could act indirectly by inhibiting the expression and/or activity of a positive factor that counteracts H-NS repression.
The El Tor biotype Δhns mutant over expressed additional toxic factors with demonstrated activities in cell culture. This is the case of hemolysin exhibiting pore-forming and vacuolating activities [69] and the RTX toxin causing actin depolymerization [2, 70]. The genes encoding these toxins were expressed at a low level in the wild type strain but were significantly over expressed in the Δhns mutant. DNA binding data, and ChIP showed that repression of hlyA and rtx genes by H-NS is direct. Repression of hlyA by H-NS has also been reported in V. anguillarum [71]. Interestingly, regulation of hlyA expression by iron is lost in the Δhns mutant which, together with the RNA-seq data, suggests that loss of H-NS is accompanied by significant disruption of iron homeostasis. Expressions of hlyA in V. cholerae and rtx genes in V. vulnificus are positively regulated by the transcriptional factor HlyU [54, 55]. We conducted a ChIP assay to examine the effect of HlyU on H-NS occupancy at the hlyA and rtx promoters. For the hlyA gene, the virtual footprint predicts the occurrence of two potential H-NS binding sites separated by 115 bp, one located downstream from the start codon. Such an arrangement suggests the formation of the reported DNA: H-NS: DNA bridge structure trapping the RNA polymerase at promoters [21]. Overexpression of HlyU diminished H-NS occupancy at a hlyA promoter fragment spanning the putative H-NS binding site. Since binding of HlyU to the hlyA promoter has not been demonstrated [55], we suggest that HlyU diminishes H-NS occupancy indirectly through a hitherto unknown regulator.
Our data are not consistent with HlyU acting as an H-NS antirepressor at the V. cholerae rtx promoters as proposed for the V. vulnificus rtxA1 promoter [55]. The HlyU proteins of V. vulnificus and V. cholerae are closely homologous (93% similarity, 82% identity). However, alignment of the region protected by HlyU at the V. vulnificus rtxA1 promoter to the DNA located between the rtxCA and rtxBDE operons in V. cholerae using the ClustalW2 software did not reveal significant homology. Furthermore, we expressed and purified V. cholerae HlyU and could not demonstrate binding of this protein to the rtxCA and rtxBDE promoters by EMSA (data not shown).
Here we show that H-NS binds to the tnaA promoter to repress tryptophanase and indole production, a positive regulator of biofilm formation [46]. It has been proposed that indole enhances the expression of vps genes required for biofilm matrix polysaccharide biosynthesis [46]. Thus, our studies indicate that H-NS represses biofilm development at two levels: direct repression of vps genes [32] and lowering of indole production. Consistent, with a previous transcription profiling of a Δcrp mutant [56], we found that the tnaA gene is activated by CRP. In E. coli, expression of tnaA is subject to complex regulation that involves CRP transcriptional activation and tryptophan-induced translation anti-termination [72]. The organization of the tnaA region in V. cholerae chromosome II resembles that of E. coli where the tnaA ORF is preceded by a large mRNA leader that includes a sequence encoding the TnaC leader peptide (annotated VCA0161.1 in V. cholerae). In E. coli, transcription is initiated upstream of tnaC upon activation by the CRP-cAMP complex [72]. Inspection of the V. cholerae tnaA region with the virtual footprint software showed the presence of a CRP binding site upstream of the leader peptide encoding sequence consistent with the regulation of our tnaA-lacZ fusion. In contrast, H-NS binding sites predicted by this software were only found downstream of the leader peptide. On this basis, it is unlikely that CRP acts at this promoter by counteracting H-NS repression. This interpretation is consistent with our finding that CRP was still required for maximal tnaA expression in the absence of H-NS.
Transcriptional profiling of the Δhns mutant indicated that loss of H-NS results in significant changes in OMP transcription, genes involved in iron binding and transport and oxidative stress. Perturbation of the cell envelope has been reported to result in alterations of iron homeostasis and oxidative stress [57]. Our results are consistent with previous observations that an hns mutant expressed elevated catalase and peroxidase encoding genes [27]. Thus, our data suggest that mutational loss of H-NS has a significant impact on the cell envelope. Accordingly, the Δhns mutant overexpressed rpoE encoding σE, which is required for virulence in the cholera bacterium [37]. We propose that altered OMP transcription in the Δhns mutant induces an endogenous envelope stress response resulting in σE release from the inner membrane and activation of its own promoter. In agreement with this interpretation, we did not observe a direct interaction between H-NS and the rpoE promoter. Similar to E. coli, the genes encoding σE, the negative regulators of its activity RseA and RseB, as well as RseC are organized in an operon [58, 59]. Consistent with this organization, genes rseA, rseB and rseC were also overexpressed in the Δhns mutant. However, analogous to E. coli, it is likely that RseA is proteolytically degraded under conditions of envelope stress [58, 59].
A remarkable feature of the Δhns transcriptome is the down-regulation of numerous genes predicted to affect chemotaxis. This result explains the diminished motility of El Tor biotype hns mutants in the swarm agar assay in spite of being flagellated [25, 27]. It has been suggested that H-NS affects gene expression indirectly or by post-transcriptional mechanisms when acting as a positive regulator [12, 13]. Chemotaxis is critical for V. cholerae to adapt to environmental changes and compete with other bacteria by sensing and responding to chemical gradients. In particular, the MCP family of proteins down-regulated in the hns mutant are predicted to sense environmental cues and transduce this information to the flagellar motor through the participation of additional che genes, allowing the bacterium to swim toward favorable substrata or away from toxic environments. Down-regulation of these genes in the hns mutant could severely hamper its survival capacity in nature. We note that of the several che genes down-regulated in the Δhns mutant, only cheZ is located in the cluster II reported to be essential for controlling the direction of flagellar rotation [60]. One MCP (VCA1056) down-regulated in the Δhns mutant was reported to be induced upon human infection [49]. Although the function of the remaining MCP and che genes down-regulated in the hns mutant are not defined, we confirmed that the hns mutant exhibits reduced chemotaxis toward glycine and serine.
In the suckling mouse, diminished chemotaxis would be expected to confer a competitive advantage to the hns mutant [61]. Nevertheless, the hns mutant colonized less compared to wild type, suggesting that loss of other H-NS functions outweighs the potential advantage of being less chemotactic in this model. Loss of H-NS in the El Tor biotype mutant, however, had a less severe effect on intestinal colonization compared to the classical biotype [73].
From the studies described in this article we conclude that:
H-NS regulates the expression of a significant fraction of the cholera bacterium’s genome in a growth phase-dependent manner. Salient features of the V. cholerae hns transcriptome are the down-regulation of multiple genes annotated as methyl-accepting chemotaxis proteins resulting in diminished chemotaxis, and the aberrant expression of porins with induction of an endogenous envelope stress response.
H-NS, in conjunction with quorum sensing, silences the transcription of the VieSAB three-component regulatory system, a major contributor to biotype-specific differences in gene expression.
H-NS directly repressed the transcription of hemolysin, the RTX toxin and the RTX toxin transport system and indole biosynthesis. Transcriptional silencing of hemolysin (hlyA) could be partially counteracted by the transcriptional activator HlyU.
The identification of new genes silenced by H-NS encoding virulence regulators and known cytotoxic factors that are differentially expressed in V. cholerae biotypes could facilitate the discovery of additional transcription factors that may contribute to the emergence of new pathogenic variants of the cholera bacterium by anti-silencing.
Supporting Information
(TIF)
(XLSX)
(XLSX)
Acknowledgments
We are grateful to Michael Crowley and David Crossman (University of Alabama at Birmingham Heflin Center for Genomic Sciences) for assistance in RNA-seq performance and data analysis.
Data Availability
The raw and processed datasets have been deposited in the Gene Expression Omnibus (GEO) repository (http://www.ncbi.nlm.nih.gov/geo/) and assigned accession number GSE62785.
Funding Statement
National Institutes of Health AI104993 AJS, National Institutes of Health AI103693 JAB. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Kaper JB, Morris G Jr, Levine MM. Cholera. Clin Microbiol Rev. 1995;8: 48–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lin W, Fullner KJ, Clayton R, Sexton JA, Rogers MB, Calia KE, et al. Identification of a Vibrio cholerae RTX toxin gene cluster that is tightly linked to the cholera toxin prophage. Proc Natl Acad Sci USA. 1999;96: 1071–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beyhan S, Tischler AD, Camilli A, Yildiz FH. Differences in gene expression between the classical and El Tor biotypes of Vibrio cholerae O1. Infect Immun. 2006;74: 3633–3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Herrington DA, Hall RH, Losonsky GA, Mekalanos JJ, Taylor RK, Levine MM. Toxin, the toxin co-regulated pili and the toxR regulon are essential for Vibrio cholerae pathogenesis in humans. J Exp Med. 1988;168: 1487–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Matz C, McDougald D, Moreno AM, Yung PY, Yildiz FH, et al. Biofilm formation and phenotypic variation enhance predation-driven persistence of Vibrio cholerae. Proc Natl Acad Sci USA. 2005;102: 16819–16824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jensen MA, Faruque SM, Mekalanos JJ, Levin BR. Modeling the role of bacteriophage in the control of cholera outbreaks. Proc Natl Acad Sci USA. 2006;103: 4652–4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Faruque SM, Naser IB, Islam MJ, Faruque AS, Ghosh AN, Nair GB, et al. Seasonal epidemics of cholera inversely correlate with the prevalence of environmental cholera phages. Proc Natl Acad Sci USA. 2005;102: 1702–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Faruque SM, Islam MJ, Ahmad QS, Faruque AS, Sack DA, Nair GB, et al. Self-limiting nature of seasonal cholera epidemics: role host-mediated amplification of phage. Proc Natl Acad Sci USA. 2005;102: 6119–6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Faruque SM, Biswas K, Nashir Udden SM, Ahmad QS, Sack DA, Nair GB, et al. Mekalanos JJ. Transmissibility of cholera: in vivo formed biofilms and their relationship to infectivity and persistence in the environment. Proc Natl Acad Sci USA. 2006;103: 6350–6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schoolnik GK, Yildiz FH. The complete genome sequence of Vibrio cholerae: a tale of two chromosomes and two lifestyles. Genome Biol. 2000;1: reviews 1016.1–1016.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tamayo R, Patimalla B, Camilli A. Growth in a biofilm induces a hyperinfective phenotype in Vibrio cholerae. Infect Immun. 2010;78: 3560–3569. 10.1128/IAI.00048-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Atlung T, Ingmer H. H-NS: a modulator of environmentally regulated gene expression. Mol Microbiol. 1997;24: 7–17. [DOI] [PubMed] [Google Scholar]
- 13. Dorman CJ. H-NS: a universal regulator for a dynamic genome. Nature Rev Microbiol 2004;2: 391–400. [DOI] [PubMed] [Google Scholar]
- 14. Cerdan R, Bloch V, Yang Y, Bertin P, Dumas C, Rimsky S, et al. Crystal structure of the N-terminal dimerization domain of VicH, the H-NS-like protein of Vibrio cholerae. J Mol Biol. 2003;334: 179–185. [DOI] [PubMed] [Google Scholar]
- 15. Nye MB, Taylor RK. Vibrio cholerae H-NS domain structure and function with respect to transcriptional repression of ToxR regulon genes reveals differences among H-NS family members. Mol Microbiol. 2003;50: 427–444. [DOI] [PubMed] [Google Scholar]
- 16. Spurio R, Falconi M, Brandi A, Pon CL, Gualerzi CO. The oligomeric structure of the nucleoid protein H-NS is necessary for recognition of intrinsically curved DNA and for DNA binding. EMBO J. 1997;16: 1795–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dame RT, Wyman C, Goosen N. Structural basis for preferential binding of H-NS to curved DNA. Biochimie 2001;83: 231–234. [DOI] [PubMed] [Google Scholar]
- 18. Lang B, Blot N, Bouffartigues E, Buckle M, Geertz M, Gualerzi CO, et al. High-affinity DNA binding sites for H-NS provide a molecular basis for selective silencing within proteobacterial genomes. Nucleic Acids Res. 2007;35: 6330–6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Owen-Hughes TA, Pavitt GD, Santos DS, Sidebotham JM, Hulton CSJ, Hinton JCD. et al. The chromatin-associated protein H-NS interacts with curved DNA to influence DNA topology and gene expression. Cell. 1992;71: 255–265. [DOI] [PubMed] [Google Scholar]
- 20. Ueguchi C, Mizuno T. The Escherichia coli nucleoid protein H-NS functions directly as a transcriptional repressor. EMBO J. 1993;12: 1039–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dorman CJ, Kane KA. Bridging and anti-bridging: a role for bacterial nucleoid-associated proteins in regulating the expression of laterally acquired genes. FEMS Microbiol Rev. 2009;33: 587–592. 10.1111/j.1574-6976.2008.00155.x [DOI] [PubMed] [Google Scholar]
- 22. Yu RR, DiRita VJ. Regulation of gene expression in Vibrio cholerae by toxT involves both antirepression and RNA polymerase stimulation. Mol Microbiol. 2002;43: 119–134. [DOI] [PubMed] [Google Scholar]
- 23. Stonehouse EA, Kovacikova G, Taylor RK, Skorupski K. Integration host factor positively regulates virulence gene expression in Vibrio cholerae. J Bacteriol. 2008;190: 4736–4748. 10.1128/JB.00089-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stonehouse EA, Hulbert RR, Nye MB, Skorupski K, Taylor RK. H-NS binding and repression of the ctx promoter in Vibrio cholerae. J Bacteriol. 2011;193: 979–988. 10.1128/JB.01343-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang H, Ayala JC, Benitez JA, Silva, AJ. Interaction of the histone-like nucleoid structuring protein and the general stress response regulator RpoS at Vibrio cholerae promoters that regulate motility and hemagglutinin/protease expression. J Bacteriol. 2012;194: 1205–1215. 10.1128/JB.05900-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tending C, Badaut C, Krin E, Gounon P, Ngo S, Danchin A, et al. Isolation and characterization of vicH, encoding a new pleiotropic regulator in Vibrio cholerae. J Bacteriol. 2000;182: 2006–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Silva AJ, Sultan SZ, Liang W, Benitez JA. Role of the histone-like nucleoid structuring protein (H-NS) in the regulation of RpoS and RpoS-dependent genes in Vibrio cholerae. J Bacteriol. 2008;190: 7335–7345. 10.1128/JB.00360-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nye MB, Pfau JD, Skorupski K, Taylor RK. Vibrio cholerae H-NS silences virulence gene expression at multiple steps in the ToxR regulatory cascade. J Bacteriol. 2000;182: 4295–4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. De Lorenzo V, Eltis L, Kessler B, Timmis KN. Analysis of the Pseudomonas gene products using lacIq/Ptrp-lac plasmids and transposons that confer conditional phenotypes. Gene. 1993;123: 17–24. [DOI] [PubMed] [Google Scholar]
- 30. Iwanaga M, Yamamoto K, Higa N, Ichinose Y, Nakasone N, Tanabe M. Culture conditions for stimulating cholera toxin production by Vibrio cholerae O1 El Tor. Microbiol Immunol. 1986;30: 1075–1083. [DOI] [PubMed] [Google Scholar]
- 31. Donnenberg MS, Kaper JB. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive selection suicide vector. Infect Immun. 1991;59: 4310–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang H, Ayala JC, Silva AJ, Benitez JA. The histone-like nucleoid structuring protein (H-NS) is a repressor of Vibrio cholerae exopolysaccharide biosynthesis (vps) genes. Appl Environ Microbiol. 2012;78: 2482–2488. 10.1128/AEM.07629-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Silva AJ, Leitch GJ, Camilli A, Benitez JA. Contribution of hemagglutinin/protease and motility to the pathogenesis of El Tor biotype cholera. Infect Immun. 2006;74: 2072–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang H, Ayala JC, Benitez JA, Silva AJ. The LuxR-type regulator VpsT negatively controls the transcription of rpoS encoding the general stress response regulator in Vibrio cholerae biofilms. J Bacteriol. 2014;196: 1020–1030. 10.1128/JB.00993-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang H, Zhang L, Silva AJ, Benitez JA. A quinazoline-2,4-diamino analog suppresses Vibrio cholerae flagellar motility by interacting with motor protein PomB and induces envelope stress. Antimicrob Agents Chemother. 2013;57: 3950–3959. 10.1128/AAC.00473-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boardman BK, Meehan BM, Fullner-Satchell KJ. Growth Phase Regulation of Vibrio cholerae RTX Toxin Export. J Bacteriol. 2007;189: 1827–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kovacikova G, Skorupski K. The alternative sigma factor sigma (E) plays an important role in intestinal survival and virulence in Vibrio cholerae. Infect Immun. 2002;70: 5355–5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Silva AJ, Pham K, Benitez JA. Haemagglutinin/protease expression and mucin gel penetration in El Tor biotype Vibrio cholerae. Microbiology. 2003;149: 1883–891. [DOI] [PubMed] [Google Scholar]
- 39. Rothmel RD, Shinabarger D, Parsek M, Aldrich T, Chakrabarty AM. Functional analysis of the Pseudomonas putida regulatory protein CatR: transcriptional studies and determination of the CatR DNA binding site by hydroxyl-radical foot printing. J Bacteriol. 1991;173: 4717–4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stewart V, Yanofsky C. Role of leader peptide synthesis in tryptophanase operon expression in Escherichia coli K-12. J Bacteriol. 1986;167: 383–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Giannoukos G, Ciulla DM, Huang K, Haas BJ, Izard J, Levin JZ, et al. Efficient and robust RNA-seq process for cultured bacteria and complex community transcriptomes. Genome Biol. 2012;13: R23 10.1186/gb-2012-13-3-r23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-seq. Bioinformatics. 2009;25: 1105–1111. 10.1093/bioinformatics/btp120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols. 2012; 7: 562–578. 10.1038/nprot.2012.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10: R25 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose pBAD promoter. J Bacteriol. 1995;177: 4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mueller RS, Beyhan S, Saini SG, Yildiz FH, Bartlett DH. Indole acts as an extracellular cue regulating gene expression in Vibrio cholerae . J Bacteriol. 2009;191: 3504–3516. 10.1128/JB.01240-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stroebner JA, Payne SM. Iron-regulated hemolysin production and utilization of heme and hemoglobin by Vibrio cholerae. Infect Immun. 1998;56: 2891–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Miller JH. Experiments in molecular genetics. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 1972. [Google Scholar]
- 49. Nishiyama S, Suzuki D, Itoh Y, Suzuki K, Tajima H, Hyakutake A, et al. Mlp24 (McpX) of Vibrio cholerae implicated in pathogenicity functions as a chemoreceptor for multiple amino acids. Infect Immun. 2012;80: 3170–3178. 10.1128/IAI.00039-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kovacikova G, Skorupski K. Overlapping binding sites for the virulence gene regulators AphA, AphB and cAMP-CRP at the Vibrio cholerae tcpPH promoter. Mol Microbiol. 2001;41: 393–407. [DOI] [PubMed] [Google Scholar]
- 51. Lee SH, Angelichio MJ, Mekalanos JJ, Camilli A. Nucleotide sequence and spatiotemporal expression of the Vibrio cholerae vieSAB genes during infection. Infect Immun. 1998;180: 2298–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tischler AD, Lee SH, Camilli A. The Vibrio cholerae vieSAB locus encodes a pathway contributing to cholera toxin production. J Bacteriol. 2002;184: 4104–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tischler AD, Camilli A. Cyclic diguanylate regulates Vibrio cholerae virulence gene expression. Infect Immun. 2005;73: 5873–5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Williams SG, Manning PA. Transcription of the Vibrio cholerae haemolysin gene, hlyA, and cloning of a positive regulator locus, hlyU. Mol Microbiol. 1991;5: 2031–2038. [DOI] [PubMed] [Google Scholar]
- 55. Liu M, Crosa JH. The regulator HlyU, the repeat-in-toxin gene rtxA1, and their roles in the pathogenesis of Vibrio vulnificus infections. MicrobiologyOpen. 2012;4: 502–513. 10.1002/mbo3.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Liang W, Pascual-Montano A, Silva AJ, Benitez JA. The cyclic AMP receptor protein modulates quorum sensing, motility and multiple genes that affect intestinal colonization in Vibrio cholerae. Microbiology. 2007;153: 2964–2975. [DOI] [PubMed] [Google Scholar]
- 57. Sikora AE, Beyhan S, Bagdasarian M, Yildiz F, Sandkvist M. Cell Envelope Perturbation Induces Oxidative Stress and Changes in Iron Homeostasis in Vibrio cholerae. J Bacteriol. 2009;191: 5398–5408. 10.1128/JB.00092-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Raivio TL, Silhavy TJ. Periplasmic stress and ECF Sigma factors. Ann Rev Microbiol. 2001;55: 591–624. [DOI] [PubMed] [Google Scholar]
- 59. Raivio TL. Envelope stress responses and Gram-negative bacterial pathogenesis. Mol Microbiol. 2005;56: 1119–1128. [DOI] [PubMed] [Google Scholar]
- 60. Boin MA, Austin MJ, Hase CC. Chemotaxis in Vibrio cholerae. FEMS Microbiol Lett. 2004;239: 1–8. [DOI] [PubMed] [Google Scholar]
- 61. Butler SM, Camilli A. Both chemotaxis and net motility greatly influence the infectivity of Vibrio cholerae. Proc Natl Acad Sci USA. 2004;101: 5018–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lenz DH, Bassler BL. The small nucleoid protein Fis is involved in Vibrio cholerae quorum sensing Mol Microbiol. 2007;63: 859–871. [DOI] [PubMed] [Google Scholar]
- 63. Dorman CJ. Genome architecture and global gene regulation in bacteria: making progress towards a unified model. Nature Rev Microbiol. 2013;11: 349–355. 10.1038/nrmicro3007 [DOI] [PubMed] [Google Scholar]
- 64. Stulke J, Hillen W. Carbon catabolite repression in bacteria. Curr Opin Microbiol. 1999;2: 195–201. [DOI] [PubMed] [Google Scholar]
- 65. Shin M, Song M, Rhee JH, Hong Y, Kim YJ, Seok YJ, et al. DNA looping-mediated repression by histone-like protein H-NS: specific requirement of σ70 as a cofactor for looping. Genes Dev. 2005;19: 2388–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Münch R, Hiller K, Grote A, Scheer M, Klein J, Schobert M, et al. Virtual Footprint and PRODORIC: an integrative framework for regulon prediction in prokaryotes. Bioinformatics. 2005; 21: 4187–4189. [DOI] [PubMed] [Google Scholar]
- 67. Srivastava D, Harris RC, Waters CM. Integration of cyclic di-GMP and quorum sensing in the control of vpsT and aphA in Vibrio cholerae . J Bacteriol. 2011;193: 6331–5341. 10.1128/JB.05167-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tsou AM, Cai T, Liu Z, Zhu J, Kulkarni RV. Regulatory targets of quorum sensing in Vibrio cholerae: evidence for two distinct HapR-binding motifs. Nucl Acids Res. 2009;37: 2747–2756. 10.1093/nar/gkp121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Figueroa-Arredondo P, Heuser JE, Akopyants NS, Hiroshi Morisaki J, Giono-Cerezo S, Enriquez-Rincon J et al. Cell vacuolation caused by Vibrio cholerae hemolysin. Infect Immun. 2001;69: 1613–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cordero CL, Kudryashov DS, Reisler E, Fullner Satchell KJ. The actin cross-linking domain of the Vibrio cholerae RTX toxin directly catalyzes the covalent cross-linking of actin. J Biol Chem. 2006;281: 32366–32374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mou X, Spinard EJ, Driscoll MV, Zhao W, Nelson DR. H-NS is a negative regulator of the two hemolysin/cytotoxin gene clusters in Vibrio anguillarum. Infect Immun. 2013;81: 3566–3576. 10.1128/IAI.00506-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yanofsky C. RNA-based regulation of genes of tryptophan synthesis and degradation in bacteria. RNA. 2007;13: 1141–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ghosh A, Paul K, Chowdhury R. Role of the histone-like nucleoid structuring protein in colonization, motility, and bile-dependent repression of virulence gene expression in Vibrio cholerae. Infect Immun. 2006;74: 3060–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
(XLSX)
(XLSX)
Data Availability Statement
The raw and processed datasets have been deposited in the Gene Expression Omnibus (GEO) repository (http://www.ncbi.nlm.nih.gov/geo/) and assigned accession number GSE62785.