

Abstract
Triggered activity in cardiac muscle and intracellular Ca2+ have been linked in the past. However, today not only are there a number of cellular proteins that show clear Ca2+ dependence but also there are a number of arrhythmias whose mechanism appears to be linked to Ca2+-dependent processes. Thus we present a systematic review of the mechanisms of Ca2+ transport (forward excitation-contraction coupling) in the ventricular cell as well as what is known for other cardiac cell types. Second, we review the molecular nature of the proteins that are involved in this process as well as the functional consequences of both normal and abnormal Ca2+ cycling (e.g., Ca2+ waves). Finally, we review what we understand to be the role of Ca2+ cycling in various forms of arrhythmias, that is, those associated with inherited mutations and those that are acquired and resulting from reentrant excitation and/or abnormal impulse generation (e.g., triggered activity). Further solving the nature of these intricate and dynamic interactions promises to be an important area of research for a better recognition and understanding of the nature of Ca2+ and arrhythmias. Our solutions will provide a more complete understanding of the molecular basis for the targeted control of cellular calcium in the treatment and prevention of such.
I. Introduction
Membrane voltage and [Ca2+]i changes have been linked for many decades. However recently, some human ventricular arrhythmias have been associated selectively with mutation of the ryanodine receptor (RyR), the primary release channel of intracellular Ca2+ stores in the cardiac cell (see sect. iiB2). It is the goal of this review to discuss the role of cellular Ca2+ transport in cardiac arrhythmias. We review the basis for Ca2+-dependent arrhythmias by reviewing the building blocks of excitation-contraction coupling. Then, we review what is known to date about the possible role of Ca2+ in different arrhythmias.
A. Overview of Ca2+ Transport in the Cardiac Cell
1. Structural aspects
The cell border is delineated by a glycoprotein layer overlying the sarcolemma, which invaginates the cell near the Z lines of the myofibrils. The resultant transverse tubules (t tubules) are rich in dihydropyridine-sensitive Ca2+ channels (DHPR) and Na+/Ca2+ exchange proteins. The t tubules make contact with a longitudinal network of tubules with lipid membranes called the sarcoplasmic reticulum (SR), which is a prominent Ca2+ storage organelle. Terminal cisternae of the SR abutting the t tubules contain Ca2+ channels with a high affinity for ryanodine (RyRs) that are involved in Ca2+ release from the SR. The RyR are so large that they form ultrastructurally recognizable junctional “foot proteins” in close proximity to the DHPR (Fig. 1). The longitudinal SR envelops the myofibrils and is densely covered by SERCA, SR Ca2+ pump molecules, which drive Ca2+ into the SR where it is buffered in the longitudinal SR by calreticulin and in the junctional SR by calsequestrin, a protein with intermediate affinity for Ca2+. The pump rate of SERCA depends on the [Ca2+]i. The Ca2+ sensitivity of the pump is controlled by the degree of phosphorylation of the regulatory protein, phospholamban, in the SR membrane. The contractile proteins arranged in sarcomeres in the myofibrils occupy 60% of the intracellular space. Mitochondria adjacent to the sarcomeres occupy the remainder of the cell.
Fig. 1.
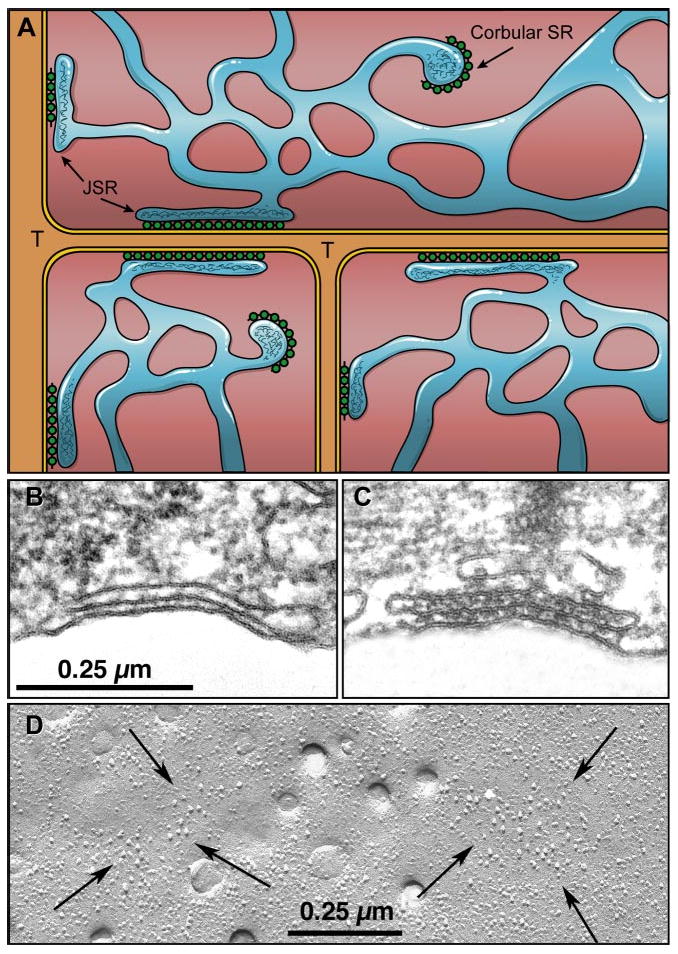
A: Ca2+ release units in cardiac muscle (chick myocardium). Dyads are formed by junctional sarcoplasmic reticulum (SR) with feet on their cytosolic surface and containing calsequestrin (CSQ), associating with the surface membrane or the membrane of t tubules (T). Corbular SR contains the same components but does not associate with the cell membrane. B and C: peripheral couplings; docked, but not yet fully differentiated (embryo 2.5 days). D: freeze fracture of cell membrane arrows surrounding junctional domains containing dihydropyridine receptor (DHPR) particles. [From Franzini-Armstrong et al. (162).]
The ultrastructural elements underlying excitation-contraction coupling (ECC) in the heart are found in the coupling between SR and sarcolemma. Ca2+ are stored in the sarcoplasmic reticulum and can be released from the SR, thus activating the adjacent myofibrils over short diffusion distances (162, 518).
The action potential initiates the release of Ca2+ by mechanisms that are specialized in different cardiac tissues. The most well known specialization of the release units is found in ventricular myocytes, which is penetrated in a highly organized manner by transverse tubules of the cell membrane. The t tubules in these cells make contact with the terminal cisternae of the SR (70, 162, 518). The latter specialized junctional domains of the SR, containing calsequestrin (CASQ) and carrying protein “feet” on their cytoplasmic surface, contact t tubules in the form of dyads. Similar domains of the SR not associated with the cell membrane form so-called corbular SR. The molecular composition of the dyad is now beginning to be revealed. A linking protein junctophyllin is thought to be involved in the docking of t tubules on the dyads (518). While a majority of DHPR and RyR are colocalized in dyads (478), the observation that a significant fraction of RyR is not colocalized with DHPR (40% in adult rat myocytes) suggests that these RyR could be present in the corbular SR. On the luminal side of the junctional domains, the intra SR proteins, CASQ and junctin and triadin, are colocalized with RyR. Studies of the macromolecular protein assemblies have suggested that complete assembly of RyR/DHPR on the one side of the dyad and RyR/TrD (triadin)/JnC (junctin)/CASQ on the luminal side develops during maturation of the animal allowing efficient coupling between surface membrane depolarization and Ca2+ delivery to the myofibrils throughout an adult cardiac myocyte.
The ultrastructure of Purkinje and atrial cells differs from the above structure described for ventricular myocytes in that they have no t tubules and their SR presents in two forms, subsarcolemmal junctional SR and corbular SR located in the core of the cell (505) (Fig. 1). The diameter of a Purkinje cell is 30–40 μm. The diameter of a normal ventricular myocyte is 15–25 μm, whereas an atrial cell is 13–15 μm. Furthermore, the density of the myofibrils is lower in the Purkinje cell compared with that in ventricular myocytes. Accordingly, it has been shown that for both the small atrial myocyte and the large Purkinje cell, ECC relies on a process where surface membrane depolarization and Ca2+ delivery to myofibrils deep inside the myocyte depend on Ca2+ release by the SR, which starts at the cell membrane and which is propagated by chemical transmission to the depth of the cell (513).
2. Model of ECC
A descriptive model of ECC (Fig. 2) has been developed to explain the functional properties of the cardiac cell (476). During the action potential Ca2+ enter the cell through a protein that is sensitive to dihydropyridine, a DHPR or “L-type Ca2+” channels. In ventricular cells the number of Ca2+ channels is estimated to be 15/μm2 and yet only 3% are open at peak currents (317). The amount of Ca2+ entering the cell per second depends on action potential duration and heart rate. Ca2+ influx at t tubules is 2.3× that of the cell surface, but more Ca2+ entry occurs per square micron at the cell surface than at t tubules (69). Ca2+ entering via L-type Ca2+ channels in the t tubules start ECC by triggering release of Ca2+ from the RyR in the terminal cisternae. In the rat (48), the ratio of RyR to L-type Ca2+ channels is 7:1 and would fit spatially with random coupling of the L-type Ca2+ channel to Ca2+ release via RyR (79). The ratio of superficial L-type Ca2+ channels versus t-tubular L-type channels is 1:3 (586), and it is anticipated then that one t-tubular L-type channel faces ∼10 RyRs. Ca2+-induced Ca2+ release (CICR) is proportional to the free intra-SR Ca2+ content and dictates the force of the cardiac contraction. Free Ca2+ in the SR has recently been spatially resolved (484), and data show that intra-SR Ca2+ diffusion is rapid, and local Ca2+ in SR in normal ventricular cells is never less than 50% of the diastolic value. The released Ca2+ activates contraction of the sarcomere. This contraction is short lived due to the rapid elimination of Ca2+ from the cytosol (Fig. 3A). About two-thirds of the Ca2+ is resequestered by the SR (45); the remainder leaves the cell mostly via the low-affinity, high-capacity, Na+/Ca2+ exchanger, while a low-capacity, high-affinity Ca2+ pump lowers the cytosolic Ca2+ level further during the diastolic interval (84). In the steady-state, the sum of the Ca2+ efflux through the membrane balances the influx during the action potential. It follows, then, that the Ca2+ content of the SR depends on the heart rate and on the duration of the action potential. Furthermore, a fraction of the Ca2+ involved in activation of the heartbeat recirculates into the SR and becomes available for activation of the next beat. Thus force of the heartbeat will depend on the force of the previous one. In addition, it takes time for the Ca2+ release process to recover completely from the last release so that sequestered Ca2+ can again be released from the SR. Therefore, the force of the heartbeat will also depend strongly on heart rate, on the duration of the diastolic interval, as well as on the duration of the action potential (594).
Fig. 2.

Diagram of the excitation-contraction coupling system in the cardiac cell. During the action potential Ca2+ enters the cells as a rapid influx followed by a maintained component of the slow inward current. Ca2+ entry does not lead directly to force development as the Ca2+ that enter are rapidly bound to binding sites on the SR that envelops the myofibrils. The rapid influx of Ca2+ via the t tubules is thought to induce release of Ca2+ from a release compartment in the SR, by triggering opening of Ca2+ channels in the terminal cisternae, thus activating the contractile filaments to contract. Relaxation follows because the cytosolic Ca2+ is sequestered again in an uptake compartment of the SR and partly extruded through the cell membrane by the Na+/Ca2+ exchanger and by the low-capacity high-affinity Ca2+ pump. The force of contraction is thus determined by the circulation of Ca2+ from the SR to the myofilaments and back to the SR, and by the amount of Ca2+ that has entered during the preceding action potential. The relaxation rate of the twitch depends on the rate of Ca2+ dissociation from the myofilaments and on the rates of Ca2+ sequestration and extrusion. It is important to note that the process of Na+/Ca2+ exchange is electrogenic so that Ca2+ extrusion through the exchanger leads to a depolarizing current.
Fig. 3.

A, a superimposed tracings are force (thick black line) and intracellular calcium (Cai) transient (thin black line) recordings of the electrically stimulated trabecula. Bottom tracing illustrates the slow change in Cai occurring in normal muscle during the diastolic period (between vertical dotted lines). A, b: force at increased gain and sarcomere length during the twitch and subsequent diastolic pause. Note that no sarcomere length fluctuations (>1.3 nm) occur (535). B: enlarged confocal image depicting the characteristics of line scans during propagation of one microscopic Ca2+ wave (top panel a) and during initiation and propagation of another (bottom panel b) in normal muscle. In Ba, the Ca2+ wave has an asymmetric appearance, as if it encounters a border or failed to propagate in one direction. In Bb, the wave begins as a “V,” indicating equal propagation in both directions; however, this wave stops propagating. The black arrows in both panels mark the same position in the two scans, indicating that the two waves started at the same place. The white arrows indicate the position of sparks at the leading edge of the wave in A. [From Wier et al. (590).]
It is possible to load the SR excessively with Ca2+. This may occur following damage of cardiac cells (115, 386) or after exposure to interventions that increase intracellular Ca2+ levels (digitalis, high [Ca2+]o, high stimulus rate). SR-Ca2+ overload is defined as the condition in which the SR releases Ca2+ spontaneously. Spontaneous uncoordinated Ca2+ release between heartbeats can be observed as spontaneous contractions of small groups of sarcomeres in cells of the myocardium and gives rise to fluctuations of the light-scattering properties of the muscle (287, 303). Spontaneous Ca2+ release increases the diastolic force generated by the contractile filaments and in so doing reduces Ca2+ release during the next heart beat (301, 510). Spontaneous release of Ca2+ is likely to lead to cell depolarization as a result of activation of Ca2+-modulated channels and/or by electrogenic Na+/Ca2+ exchange.
3. ECC coupling in atrial and Purkinje cells from normal hearts
In rabbit atrial cells, immunostaining for either RyR or the L-type Ca2+ channel can be seen near the cell's sarcolemma (84a). Nonjunctional SR elements visualized with RyR staining are in transverse arrays along the Z lines (284, 610). In rat atrial cells, a 0.5- to 1-μm gap exists between junctional and nonjunctional RyR (341). This is in contrast to the unique ultrastructure of the latent atrial pacemaker cell, where subsarcolemmal SR cisternae are prominent and directly opposed to one another in adjacent cells. Differing from normal ventricular cell ultrastructure, where the presence of t tubules ensures reasonably synchronous Ca2+ release throughout the cell, atrial cells exhibit spatial Ca2+ gradients. In response to electrical stimulation, Ca2+ increases first at the cell's periphery and then after a 30- to 50-ms delay, increases in the central regions of the cell (38, 232, 284, 329, 341, 596). Preferential activation of Ca2+ sparks along the cell's periphery and then propagation to the cell's core (487, 596) is consistent with the biphasic nature of the human atrial Ca2+ transient under voltage clamp (198) and the ability of peripheral Ca2+ release to more efficiently regulate Na+/Ca2+ exchanger function (328). Furthermore, in rat atrial cells, a set of “eager” Ca2+ release sites that have a fixed edge location and preset activation pattern are thought to reflect clusters of RyRs that are closely coupled to Ca2+ channels and display a high sensitivity to trigger Ca2+ (341). In voltage-clamped cAMP-stimulated atrial cells, L-type Ca2+ channel evoked peripheral Ca2+ release occurs within 1–4 ms, and this Ca2+ then propagates to the interior of the cell at ∼230 μm/s (596). The efficacy of Ca2+ currents to trigger peripheral Ca2+ release is fivefold greater than that needed for triggering center Ca2+ release. Thus, for rat cells, it is thought that the mechanism of Ca2+ release from junctional SR differs from that in nonjunctional SR. Recent spark data suggest that peripheral atrial Ca2+ sparks are brighter and occur more frequently than central sparks (597). The opposite appears to be the case in feline atrial cells (284) where under permeabilized conditions, Ca2+ spark frequencies in the two regions are similar (488). Thus, in the intact cell, mechanisms that cause Ca2+ release in central SR RyRs are not efficient, most probably because of structural components. An interesting report by MacKenzie et al. (343) states that inherent intracellular calcium buffers in the normal rat atrial cell (for instance, mitochondria and SERCA pumps) prevent global Ca2+ transients under normal-paced conditions. Furthermore, membrane depolarization evoked peripheral Ca2+ release only propagates to the cell's core when the cell is hormonally (e.g., endothelin) stimulated.
As a result of the different structure of the Purkinje cell, coupling of excitation of the cell membrane with Ca2+ release in the core of these (large) cells differs substantially from that of myocytes. Immunostaining experiments in rabbit Purkinje cells show that RyRs are subsarcolemmal as well as within the cell consistent with earlier reports (106, 256, 505, 513). In fact, canine Purkinje cells contain both RyR3 and RyR2 isoforms with the RyR3 protein being located in a subsarcolemmal region. Here the action potential of the Purkinje cell precedes rapid Ca2+ entry into the subsarcolemmal space. The latter induces Ca2+ release which in some species propagates into the core of the Purkinje cells (64). In rabbit Purkinje cells, experiments with ryanodine suggest that Ca2+ changes in the central core of cells are best explained by simple buffered Ca2+ diffusion and not Ca2+ propagation (106). However, in rabbit Purkinje cells, evoked Ca2+ transients and sparks are only seen to originate at peripheral cellular components, suggesting that RyRs in the cell center of this type of cell are “silent” (106). This is consistent with voltage-clamp studies of single rabbit Purkinje cells which show a single-component Ca2+ transient (497). However, in canine Purkinje cells, electrically evoked Ca2+ transients are multiphasic (61, 64). An action potential evokes a sudden increase in Ca2+ particularly along the periphery. In some Purkinje cells from normal hearts, if electrically evoked peripheral release is spatially and temporally inhomogeneous, a local Ca2+ wave is produced and can propagate as a traveling Ca2+ wave the length of the aggregate as well as towards the cell's core (64, 513). Finally, while fundamentals of ECC have been described above, it is important to remember that the time course of the action potential (AP) of the cell can have significant modulatory effects on ECC efficiency. Not only does AP duration affect the time course of the evoked Ca2+ transient (58, 463), but altering the rate of early repolarization can affect both the magnitude and time course of SR Ca2+ release (464).
4. Reversal of ECC
In a ventricular myocyte, the distance between Ca2+ release sites on the terminal cisternae of the SR to the Ca2+ transport molecules at the surface of the t tubules is less than ∼300 nm (i.e., from 40 nm to DHPR to ∼300 nm to Na+/Ca2+ exchangers). It follows that the transport molecules face the largest variation of [Ca2+]i within the cell. This review discusses potential consequences of the SR-related [Ca2+]i changes on function of membrane Ca2+ transport and how this feedback may be involved in modulation of action potential waveform and the development of arrhythmias. We anticipate that the cross-talk between the surface membrane and the SR is strongest in the working myocyte, given their high density of t tubules. In addition, it has become clear that rapid mechanical perturbations of the contracting cell may cause rapid Ca2+ dissociation from troponin C (TnC). Consequently, Ca2+ released from the myofilaments may also trigger Ca2+ release from the SR by CICR. While normal CICR occurs during the action potential, Ca2+ dissociation from the myofilaments may take place when the cell is repolarized. In that case, CICR could elicit a [Ca2+]i transient that in turn affects a different set of membrane channels.
II. Molecular Building Blocks of Excitation-Contraction Coupling
A. Ca2+ Flux Through the Sarcolemma
1. Ca2+ entry through voltage-gated channels
There are several types of ion channels that are Ca2+ permeable. What has been termed as a background Ca2+ channel was originally defined in bilayer experiments (453, 454) (B-type Ca2+ channels). Under these conditions this channel spontaneously opens, has a relatively low conductance, is not blocked by nisoldipine, and is reasonably selective for Ba2+. Further investigations into resting Ca2+ influx into adult cardiac cells have shown the existence of spontaneously active Ca2+- and Ba2+-permeable but Ni2+-insensitive single channels in both cell-attached and inside-out patches (108). These latter channels are activated by phenothiazines such as chlorpromazine, trifluoperazine, and H2O2 but at very negative holding potentials (13, 314). A short report has stated that the voltage-independent B-type Ca2+ channel is regulatory in ceramide-induced rat myocyte apoptosis (201). While whole cell clamp data do not reveal such macroscopic inward currents in myocytes, some have suggested that this Na+-independent Ca2+ channel contributes importantly to tonic Ca2+ entry in the quiescent rat trabecula (307). The molecular nature of these background Ca2+ channels is unknown at this time.
The L-type (L for long lasting, ICaL) and T-type (T for transient, ICaT) Ca2+ currents were initially described in neuronal tissues. Bean et al. (31) first described multiple cardiac Ca2+ channels in canine atrial cells. At that time two types of Ca2+ currents carried by Ba2+ were recognized. Subsequently, ICaL and ICaT have been recorded in cardiac tissues of most species under various conditions. However, within the same species, the density of ICaL and ICaT varies depending on the location of the myocyte within the heart. Hagiwara et al. (189) first described the large density of both the L- and T-type channels in rabbit sinoatrial node (SAN) cells. Zhou and Lipsius (639) described large T-type currents in latent atrial pacemaker cells. Studies of cells dispersed from canine ventricles revealed a large peak T/L current density ratio in Purkinje cells dispersed both from free-running fiber bundles and the subendocardium of the left ventricle (LV) (212, 213, 548). In contrast, myocytes dispersed from mid and epicardial layers have a smaller T/L current ratio (548). Notably, T currents have not been observed in human atrial (366, 556), human ventricular (57, 366), or human Purkinje cells (P. Boyden, unpublished data).
Cardiac L- and T-type Ca2+ channels differ in the following biophysical properties. 1) Voltage range of activation: the T-channel activation occurs at more negative voltages than the L channel, e.g., in 5 mM [Ca2+]o the threshold for activation is −50 and −30 mV for T and L, respectively (548). 2) Voltage range of inactivation: in 5 mM [Ca2+]o, the T channel can be inactivated by membrane depolarization positive to −70 mV. The L channel remains fully available for activation at potentials more negative than −40 mV. 3) Mechanism of inactivation: T channel inactivates solely by membrane depolarization. For the L channel, both membrane depolarization and Ca2+ participate in the inactivation process.
Voltage-dependent inactivation of the L-type Ca2+ current is clearly evident as channels incorporated into lipid bilayers inactivate even when Ca2+ is buffered (453) and as noted from the dependence of the time course of Ba2+ current decay on voltage (14, 187). In fact, inactivation of L-type Ca2+ current can occur at voltage steps where “apparent” activation is absent. The molecular determinants of voltage-dependent inactivation of Ca2+ channels are less well understood than those of K+ or Na+ channels. In studies using Ba2+ as a charge carrier, several critical locations throughout the channel protein have been implicated in the fast (tens to hundreds of milliseconds) voltage-dependent inactivation process. They are the I-II linker, the proximal COOH terminus, the EF hand area in IC, and all four S6 regions (37, 40, 41, 59, 202, 203, 504, 624, 628). One model proposed suggests that a domain of the I-II linker docks to one or all of the S6 segments at the cytoplasmic end (85, 512). Importantly, this mechanism is not involved in the channel's “recovery from inactivation,” only the channel's response to depolarization. Critical of course to these mechanisms is that Ba2+ permeating through these proteins show only voltage-dependent inactivation and no ion-dependent inactivation. However, recent data suggest that inactivation of the L-type Ca2+ channel when Ba2+ is the charge carrier may not be all due to a voltage-dependent process (157, 357).
The molecular basis of the cardiac L-type Ca2+ channel structure is due to the combination of the α1C-subunit [Cav1.2; see Ertel et al. (146) for nomenclature] (four 6-transmembrane segments joined by intracellular linkers with cytoplasmic NH2 and COOH termini) with β2-, α2/δ-, and γ-subunits. Alternative splicings of the α-subunit have been reported (for review, see Ref. 324) and two missense mutations in one exon appear to lead to abnormal Ca2+ current function in cells of patients with Timothy's syndrome (see sect. ivA3). Perhaps in some acquired diseases, alternatively spliced proteins constitute the remodeled Ca2+ channels in arrhythmogenic substrates. The γ-subunit (33 kDa) is also expressed in skeletal muscle and in expression systems can have a modest effect on Ca2+ channel currents (112). Other studies have shown it can modulate Cav3.1, T-type Ca2+ channels (196). Its role in modulation of cardiac Ca2+ channels is minimal.
The Cav1.2 NH2 terminus can act as an inhibitory particle (490) as well as a site for modulation by Ca2+-binding proteins such as CaBP1 (635, 636), Ca2+/calmodulin protein kinase II (CaMKII) (227), calmodulin (CaM) (227, 636), and Cavβ subunits (260). Some have suggested that a reduction in this inhibition can be caused by protein kinase C (PKC), which increases Cav1.2 Ca2+ currents (491); alternatively, phosphorylation of the NH2 terminus by PKC has been proposed to decrease the L-type Ca2+ current (613).
Other major sites of modulation of the α1C-subunit function are within the COOH terminus since it is the target of several kinases that regulate CaV1.2 L-type Ca2+ currents. Both PKA and CaMKII increase L-type Ca2+ current and change channel modal gating (139, 439, 621), and both effects are thought to be due to phosphorylation of the α1C COOH terminus (227, 613). Additionally, Src kinase phosphorylation of the neuronal α1C-isoform at a COOH-terminal residue leads to potentiation of the L-type Ca2+ current (36). However, the mechanisms by which specific kinases modulate L-type Ca2+ currents differ. For example, the major target for PKA in α1C has been identified as Ser1928 (122, 370); however, recent data suggest that phosphorylation of this site may not be required for adrenergic stimulation of L-type Ca2+ currents (166). Although CaMKII activation leads to the same shift in modal gating as that caused by PKA stimulation (139), and CaMKII also phosphorylates the COOH terminus, the specific targets are unknown. One report suggests that Ser1517 may be the target (147), but definitive biochemical evidence is lacking. PKC also phosphorylates Ser1928 (613), although the effects of this phosphorylation on L-type Ca2+ currents are unknown.
The α1C COOH terminus also contains a binding pocket for CaM (278), which mediates Ca2+-dependent inactivation and Ca2+-dependent facilitation of L-type Ca2+ channels (417, 643). The interaction between CaM and α1C is constitutive (145, 278). T-type Ca2+ currents do not show Ca2+-dependent inactivation (136, 213, 508); the α1G- and α1H-subunits lack the determinants for CaM binding in their respective COOH termini.
The voltage sensor for activation is the highly charged S4 segment. Ca2+ binding sites formed by glutamates in the pore loop of each repeat are critical for selectivity of the channel (144). Sections of the α1-subunit pore-forming segments, intracellular loops, and COOH terminus all contribute to Ca2+ channel inactivation. Mutating single amino acids in IIIS6 and IVS6 domains have a significant effect on current decay, suggesting that an area in the inner channel mouth is a key player in channel inactivation. Further point mutations in the intracellular I-II loop and the IVS5-IVS6 linker both affect Ca2+ channel inactivation. Notably, these sections of the protein are also critical sites of β-subunit and/or G protein interactions.
Currently, four potential β-subunits (Cav β1-β4) have been recognized, but in cardiac cells the β2-subunit predominates, providing a rate-limiting step in the expression of Ca2+ channel proteins (102). CaVβ subunits are entirely cytoplasmic, and each subunit includes a variable NH2 terminus, a conserved core that includes an interacting Src homology domain, a guanylate kinase (GK)-like domain (90, 361, 402, 526), and a divergent COOH terminus. Both the NH2 terminus and SH3 domain contribute to isoform-specific regulation of channel inactivation (361). Interestingly, the GK domain contains the binding pocket for the α1 interaction domain (AID) on the α1-subunit. The role of the CaVβ COOH terminus is still largely unknown, but β2 is a target for several kinases known to modulate L-type Ca2+ currents (e.g., the β2a COOH terminus is phosphorylated by PKA on Ser478 and Ser479). This phosphorylation appears to contribute to cAMP-dependent regulation of the channel (72).
The α1C-subunit (Cav1.2) when expressed alone is sufficient for L-type channel activity, but when expressed with a cytosolic β-subunit (52), peak currents increase fourfold, apparently by accelerating the opening of the pore and reducing the rate of channel closure (295, 371, 389, 391, 415). Furthermore, there is a shift in the activation curve, a slowing of activation, and an enhancement of the inactivation process (415). β2a-Subunits slow inactivation (243) due to palmitoylation of the NH2-terminal residues, and subsequent tethering to the membrane (509). A region of I-II intracellular linker the AID of the α-subunit forms the primary binding site for the accessory β-subunits (429). It is thought that this AID region forms an α-helix that becomes buried within a conserved domain common to all β-subunits (90, 402, 555). Interestingly, functional studies have revealed that while the AID region is not necessary for β-subunit modulation of Ca2+ currents, the tethering of β subunits to the AID region optimizes subunit orientation which in turn increases local β-subunit concentration (346, 526). Finally, cardiac L-type Ca2+ current facilitation also occurs when the β2a-subunit is coexpressed with the α1C-subunit (113).
The two-component α2/δ-subunit (170 kDa) remains linked in vitro, with α2-subunit being an extracellular highly glycosylated protein; δ is the short membrane-spanning protein with a fully glycosylated extracellular portion. Several members of this family exist with α2/δ and α2/δ2 being expressed in heart (170, 348). This subunit complex affects both ionic and gating currents of the expressed Ca2+ channels by increasing the number of functional channels at the cell surface (15, 25, 86, 281, 495). In one series of experiments, coexpression of α2/δ with α1C β3-subunits prevented voltage-dependent facilitation (424). In these latter studies, this voltage-dependent facilitation was due to an increase in the number of channels that were able to produce gating current, as well as the number of channels that opened in response to voltage (424). Small molecules like the Ras-related G protein Gem have been shown to bind to β-subunits (33). Such binding apparently interferes with the subunit's ability to traffic the α-subunit to the membrane. Interestingly, gabapentin, a compound that has been shown to bind specifically to the α2/δ-subunit (282), appears to have no consistent effect on expressed L-type Ca2+ currents (348).
Evidence suggests that the cardiac L-type Ca2+ channel protein in rabbit myocytes exists in two forms. One form is full length and comprises ∼20% of all α1C-subunits in rabbit membranes (122, 171, 173), while the other form, truncated at its COOH terminus, comprises ∼80% of all α1-subunits. The truncated form of the channel cannot be directly phosphorylated by PKA (122, 200), but remains in situ near the remaining α-subunit protein (169). Functionally, it has been shown that removal of the COOH terminus from the full-length protein results in an increase in channel activity (280, 582). Several peptides designed to mimic residues of the distal COOH terminus of the Cav1.2 protein also inhibit expressed Ca2+ currents, illustrating that a specific domain of residues 2024–2171 of the subunit functions to inhibit channel conductance (170). It is unclear as to whether the truncated Ca2+ channel and its COOH terminus remain fully functional in the in situ myocyte. However, this mechanism of modulation of L-type Ca2+ current amplitude has the potential of being an important contributor to ECC. For example, activation of N-methyl-d-aspartate (NMDA) receptors and subsequent L-type Ca2+ channel-mediated Ca2+ influx induces such COOH-terminal truncation resulting in sustained changes in Ca2+ channel activity (200).
In addition to α1C (Cav1.2), mRNA and protein from α1D (Cav1.3) subunits have been measured in heart tissues (448, 530, 604). Cav1.3 Ca2+ channel proteins are sparse, but this type of Ca2+ protein may serve a specific functional role. Expression studies have shown that currents mediated by Cav1.3 proteins (plus β2a-subunits) activate more quickly and decay more slowly than those of Cav1.2 proteins (288). Furthermore, steady-state inactivation and activation voltage relations of Ba2+ currents through Cav1.3 channels are shifted in the hyperpolarizing direction. These specific kinetic differences between Cav1.2 and Cav1.3 currents account in part for the decreased sensitivity of expressed Cav1.3 currents to dihydropyridine block (288). Finally, mice lacking α1D-subunit proteins are deaf and exhibit sinoatrial dysfunction and bradycardia, suggesting a role for α1D-proteins in SAN pacemaker activity (347, 425, 633). Interestingly, these mutant mice also show altered atrial Ca2+ currents and have inducible atrial fibrillation but no change in effective refractory period (ERP) (634). While Purkinje cells have not been studied in these mice, canine Purkinje cells express this protein, and two types of L-type Ca2+ currents have been described (548) suggesting that pacemaking in Purkinje cells may also involve Ca2+ currents through Cav1.3 channels.
The molecular basis of neuronal and cardiac T-type Ca2+ channels has been defined (111, 416). In both cases, the low-voltage T-type Ca2+ channel protein (α1H, α1G) (Cav3.3 and Cav3.2) has high sequence identity with the α1C-subunit particularly in the membrane-spanning regions (416). Charged residues of the S4 regions are conserved between α1C and α1H, α1G while a ring of glutamates important in α1C channel selectivity has been partially replaced by aspartates. T-type Ca2+ currents inactivate with voltage but not by Ca2+ (136, 508). Some have suggested that a “ball-and-chain” type mechanism involving the amino side of the COOH terminus contributes to T-type channel inactivation (74, 508). Perhaps more importantly in terms of possible targets for pharmacological modulation, intracellular loop motifs involved in β-subunit binding (306, 429) or Ca2+ binding (124) of the L-type α1C protein are missing in both the α1G and α1H proteins. In canine Purkinje cells, the large T currents most likely are due to Cav3.2 based on their kurtoxin sensitivity (448).
2. Na+/Ca2+ exchange, Ca2+ entry, and Ca2+ efflux
The cardiac Na+/Ca2+ exchanger protein transports Ca2+ across the sarcolemma in exchange for Na+ and is important in maintaining Ca2+ homeostasis in the myocyte. Na+/Ca2+ exchanger activity has been shown to affect various components of normal ECC [i.e., Ca2+ spark frequency, SR Ca2+ release, and SR load (47, 176, 331)]. Normally, Na+/Ca2+ exchange works in the so-called forward mode, i.e., extruding Ca2+ in exchange for extracellular Na+. Reverse-mode operation of Na+/Ca2+ exchanger could provide additional Ca2+ influx into the cell. In the forward mode, Ca2+ are being transported out against their electrochemical gradient, and therefore, this mode of activity requires an expenditure of energy. It is generally accepted that the Na+ transcellular distribution indirectly provides the energy. Stoichiometric determinations have shown that three Na+ are transported for one Ca2+. Thus the exchanger is electrogenic. A recent study suggested that the stoichiometry may be closer to 4:1 Na+/Ca2+ (163, 261), but these data have been challenged (211). Under normal conditions, the reversal potential of the Na+/Ca2+ exchanger is −30 mV (279). Accordingly, negative to this potential Na+ flux is inward and Ca2+ flux is outward generating an inward current. Positive to −30 mV, the Na+/Ca2+ exchanger works in reverse mode, and outward current is generated. For the NCX1 transporter protein, it has been estimated that the turnover rate can be up to 5,000/s (209, 393) with a KD for [Ca2+]i of ∼6 μM (354). Recent data derived from the steady-state voltage and Ca2+ dependence of the Na+/Ca2+ exchanger protein have suggested that within <32 ms of an action potential upstroke, peak Ca2+ in a submembrane space is >3.2 μM (578). Thus Na+/Ca2+ exchanger current influences both the atrial and ventricular action potential (248). Furthermore, a component of observed transmural electrical heterogeneity of the left ventricle has been ascribed to basal differences in INa/Ca currents across the wall (647).
The Na+/Ca2+ exchanger protein is now considered to consist of nine transmembrane segments with a large (∼550 amino acids) intracellular loop (loop f) between segments 5 and 6 (392). The Na+/Ca2+ exchanger gene NCX1 undergoes alternative splicing (NCX1.1, NCX1.3) in the COOH terminus of its large intracellular loop. Splice variants function differently with respect to regulating properties, and expression of NCX1.3 was found to protect against severe Ca2+ overload conditions (230). Distinct regions of the protein have been shown to be involved in the Na+/Ca2+ translocation process (135), while other regions, particularly loop f, are involved in the intrinsic regulation of the Na+/Ca2+ exchanger by Na+ and Ca2+ (355). Ca2+-dependent regulation of exchanger activity is via a high-affinity binding site (0.022–0.4 μM) that is distinct from the Ca2+ transport site (354), is ∼130 amino acids in length, and is located in the center of loop f (316). Ca2+-dependent regulation of Na+/Ca2+ exchanger activity is apparently allosteric in ferret cells such that when [Ca2+]i levels are reduced (approximately <150 nM) Na+/Ca2+ current deactivates (577). A corollary is that when [Ca2+]i is elevated, steady-state activation of Na+/Ca2+ exchanger current increases, by as much as 67% for a doubling of [Ca2+]i. Such activation in normal cells promotes Ca2+ efflux with concomitant production of inward currents. If Ca2+ transients occur as traveling Ca2+ waves between cells, activated Na+/Ca2+ exchanger current would contribute to the occurrence of Ca2+-activated membrane currents.
Binding of the regulator Ca2+ decreases Na+-dependent inactivation of the Na+/Ca2+ exchanger (208). Intrinsic regulation of the Na+/Ca2+ exchanger by Na+ originally observed by Hilgemann (206) was termed Na+-dependent inactivation. This process is enhanced at low intracellular pH and diminished by micromolar Ca2+ (207). Mutagenesis studies suggest that the exchanger inhibitory peptide (XIP) binding site is located on loop f of the protein and is involved in the Na+-dependent inactivation of the exchanger (355). However, recent work with split exchanger proteins suggests that endogenous XIP region is not located between amino acids 265 and 672, since the activity of split exchanger with these loop residues deleted is still blocked (405). Other regulators of exchanger function include free radicals, pH, lipid products, as well as several kinases.
3. Stretch-sensitive Ca2+ flux
Stretch-activated ion channels have been described in both atrial and ventricular cells of several species (34, 226, 626). The channel is permeable to monovalent cations and Ca2+ (275) and thus can provide a source of Ca2+ influx. In single cells and isolated tissues from normal hearts, stretch has been observed to lead to a gradual (10 s) increase in [Ca2+]i (167, 533) as well as increases of inositol trisphosphate (IP3) and inositol tetrakisphosphate (IP4), both of which may modulate [Ca2+]i levels (119) and subsequent force development. In adult guinea pig cells, large stretch-induced [Ca2+]i changes are blocked by streptomycin (34, 168), a blocker of mechanosensitive transduction currents in hair cells (399), are not sensitive to ryanodine or tetrodotoxin (TTX), but sensitive to extracellular Ca2+. Interestingly, streptomycin also blocks stretch-induced atrial tachyarrhythmias in the isolated heart (19a), presumably by inhibiting mechanosensitive cation channels in atrial myocytes (168, 275, 276). In rat cells, stretch produces a slow increase (minutes) in the electrically evoked Ca2+ transient (222). Stretch of either rat myocytes or trabeculae increases both the frequency of SR Ca2+ release (seen as Ca2+ sparks) as well as the level of Akt and endothelial nitric oxide synthase (eNOS) phosphorylation. Thus it has been proposed that in response to stretch, myocytes generate nitric oxide (NO), which acts locally to modify ECC efficiency (419). Interestingly, sensitivity of a myocyte to stretch increases with age and degree of cellular hypertrophy (258).
B. Intracellular Ca2+ Cycling
1. SR Ca2+-ATPase pump
Two Ca2+ can be transported by the cardiac SR Ca2+ pump for each ATP molecule consumed (524), although other stoichiometries have been reported. ATP binds to high-affinity binding sites on the cytoplasmic side of the pump. The terminal phosphate of ATP is transferred to aspartate-351 on the pump protein, and the bound Ca2+ are “occluded.” ATP hydrolysis of the protein alters the structure such that Ca2+ cannot return to the cytoplasmic side. Phosphorylation also reduces the Ca2+ affinity of the pump such that Ca2+ can be released into the lumen of the SR.
The cardiac Ca2+ pump protein is the same as that from slow-twitch muscle (66, 67, 344) and has 10 membrane-spanning regions where each region, M1-M5, has additional α-helical “stalk” regions on the cytoplasmic side. Most of the 96-kDa protein is on the cytoplasmic side of the SR membrane including a β-strand, phosphorylation (aspartate-351) and nucleotide binding sites, stalk domains, and a hinge region. The crucial high-affinity Ca2+ binding sites were initially proposed to reside in the highly anionic stalk region (67); however, more recent data suggest that they are not in the stalk, but within the transmembrane regions M4-M6 and M8 (99, 100). It is likely that in the membrane these transmembrane domains may be arranged in a cylinder to form an ion channel through the SR bilayer (344).
The rate of the cardiac SR Ca2+ pump is highly regulated by phosphorylation of the protein phospholamban (522). In the dephosphorylated state, phospholamban interacts with the SR Ca2+ pump near the phosphorylation site of the pump (246), acting as an inhibitor of the Ca2+ pump activity. Phosphorylation removes the inhibitory effect and increases the pumping rate (205). Phospholamban is a homopentamer; the monomer has 52 amino acids and exhibits one hydrophobic and one hydrophilic domain. A proposed structural model states that the pentamer could have a hydrophilic pore through the SR membrane with phosphorylation sites on the cytoplasmic surface (494). Kovacs et al. (290) have obtained evidence that dephosphorylation of phospholamban can form Ca2+-sensitive channels in lipid bilayers. However, it is not clear whether or how the ionophoretic property might be related to the function of phospholamban in cardiac SR.
Phospholamban is phosphorylated by cAMP-dependent protein kinases (523). Studies from the intact perfused hearts show that β-adrenergic stimulation via PKA reduces the Km for Ca2+ and thus accelerates relaxation of the muscle (362). Ca2+-calmodulin dependent protein kinases and protein kinase C (PKC: Ca2+/phospholipid dependent) (244) also phosphorylate phospholamban at threonine-17 (579). The PKC site on phospholamban is Ser-10, but there is no evidence that this site is ever phosphorylated physiologically. Whether Thr-17 phosphorylation increases Vmax or Ca2+ affinity is controversial. This stimulation can result in an increase in SR content. The cardiac Ca2+ pump has two ATP binding sites: a high-affinity site (Kd ∼1 μM) that is the substrate site and a second lower affinity site (Kd ∼200 μM) that serves as a regulatory role (127, 138). The substrate for the Ca2+ pump is probably MgATP, but other nucleotides can be used (525). Therefore, the ATP level would have to be low to prevent ATP binding to the substrate site. However, decrease of the ATP level during ischemia slows SR Ca2+ pumping and relaxation.
2. Ryanodine-sensitive SR-Ca2+ release channels (RyR)
Two kinds of Ca2+ release channels found in the SR membrane, a Ca2+-activated channel and an IP3-activated channel, are proteins that form a distinct, highly conserved gene family. It is thought that the major mechanism regulating Ca2+ release in cardiac cells is CICR. CICR requires that Ca2+ provided by the activated L-type Ca2+ channel bind to the SR-Ca2+ release channel and cause opening of a high-conductance channel allowing rapid Ca2+ efflux from the SR. Studies of the SR-Ca2+ release channel have been greatly accelerated by the recognition that ryanodine, a plant alkaloid, is a selective and specific ligand for this channel. The RyR functionally constitutes the Ca2+ release channel of the SR and structurally represents the “foot” structure linking the t tubules to the SR. The recognition of selective ryanodine binding has allowed purification of several isoforms of RyR (RyR1, RyR2, RyR3) from skeletal (236, 239, 298) and cardiac (238, 297) muscle. Most of what is known about RyR comes from electrophysiological experiments on the channels after they have been incorporated into lipid bilayers (298, 456, 457, 502). Such experiments have suggested a biphasic response of the open probability of the channel (Po) to activating [Ca2+]. RyR2 Po increases up to micromolar concentrations of [Ca2+]i and then decreases at higher [Ca2+] (606, 609) (Fig. 4, A and B). Luminal [Ca2+] versus Po of RyR activity slightly differs between control wild-type RyR and mutated RyR (251) (Fig. 4C). Furthermore, the probability that a single RyR will be activated is determined by the amplitude and duration of Ca2+ trigger signals (623). The channel has a high Ca2+ conductance but can also conduct other divalent cations such as Ba2+ and Mg2+ (42, 97, 455) as well as monovalent ions in the absence of Ca2+ (502). Compared with the sarcolemmal Ca2+ channel (Cav1.2) under similar conditions, the SR Ca2+ release channel has lower selectivity for Ca2+ and 10-fold higher conductance (42). The ability of Ca2+ to cause release depends on [Ca2+]i, the rate of rise of [Ca2+]i at its receptor (151), as well as the presence of various nucleotides and Mg2+. RyR channels close rapidly either by deactivation (192) or decrease in trigger Ca2+. An increase of SR luminal [Ca2+] causes a marked increase in the Po of the Ca2+ release channel as well as the cell's resting Ca2+ spark frequency (27, 339, 500). Human atrial RyR share similar biochemical properties compared with ovine or canine ventricular counterparts (107). Refractoriness of SR release may be due in part to SR Ca2+ refilling mediated by the SR Ca2+ pump (521). Ryanodine, at low concentrations (<30 μM), opens the cardiac SR Ca2+ release channel in either vesicles or bilayers to a stable subconducting state and the channel no longer responds to Ca2+, ATP, Mg2+, or ruthenium red (363, 459). This probably is due to the occupation of the high-affinity ryanodine binding sites (Kd ∼10 nM). Very high concentrations of ryanodine (>100 μM) appear to lock the Ca2+ release channel in a closed state.
Fig. 4.

Dependence of ryanodine receptor (RyR) single-channel activities on cytosolic Ca2+ and SR-luminal Ca2+. A: original current traces from cardiac Ca2+ release channels at three differing Ca2+ levels. Upward deflections indicate openings from closed state (small bar at left). B: average single-channel open probability (Po) values determined as in A at +35 mV (closed symbol) and −35 mV (open symbol). See Ref. 606 for more information. C: Po-luminal [Ca2+] relationship of wild-type RyR2 expressed in HEK293 cells compared with the Po-luminal [Ca2+] relationship of RYR2 channels with mutations linked to VT (L433P and R176Q/T2504M). These mutations displayed a leftward shift of the Po-luminal [Ca2+] relationship without a change in the sensitivity to cytosol [Ca2+]. See Ref. 251 for details.
RyR is a homotetramer with a molecular mass of the monomer of ∼320–450 kDa (238, 239, 297). The three-dimensional architecture of RyRs, reconstructed using image processing, matches that of the junctional “feet” observed by electron microscopy in muscle (54, 568). The gene product of cardiac RyR is smaller than, but homologous to, that of skeletal RyR (564,711 Da) (404, 529). The COOH termini of the isoforms are well conserved and contain highly hydrophobic segments probably forming 4 of the ∼10 transmembrane domains (M1-M4), with 2 additional transmembrane sequences near the center of the molecule. Recently, it has been shown (89) that substitution of alanine-3885 for glutamine near the putative transmembrane sequence of the M2 region of RyR3 reduced Ca2+ sensitivity of the channel 10,000-fold. Thus it has been proposed that glutamates of each RyR monomer cooperatively form the Ca2+ sensor of the RyR binding protein. Negatively charged residues within a transmembrane sequence are involved in binding and translocation of cations across the SR membrane (99, 611). This arrangement is attractive because it might confer Ca2+ sensitivity to RyR both at the cytoplasmic as well as the luminal side of the SR membrane.
Novel high-resolution imaging electron microscopic techniques have allowed exciting progress in the structural understanding of SR Ca channels IP3R as well as RyR1, -2, and -3 (468, 481). Future progress will be facilitated by the development of crystallization procedures for these protein complexes (619). These studies have revealed that SR Ca2+ channels are strikingly similar tetrameric structures. We will review here the structure of SR Ca2+ release channels based on data from both cardiac RyR2 (629) and skeletal RyR1 (468).
Three-dimensional reconstruction of RyR1 has revealed a transmembrane domain and a large cytoplasmic assembly (Fig. 5A). The transmembrane domain is shaped as a square prism that is linked by columns in a narrower region to the cytoplasmic assembly (468). The cytoplasmic assembly itself forms a square that is rotated 45 degrees with respect to the prism. The center of the assembly gives access to a trough that connects to the Ca2+ channel in the depth of the transmembrane domain. The corners of the cytoplasmic assembly form the so-called “clamp.” The sides form the “handle.” A series of interconnected tubular structures form a rhomboid structure on the t-tubular surface of the clamp linking four domains (337).
Fig. 5.

A: ultrastructure of RyR1 at 9.5 Å resolution. The receptor is composed of a cytosolic assembly linked to a transmembrane assembly (TMA) through a neck region which conveys columns that form the vestibule of the TMA and the Ca2+ channel in the center of the TMA to the regulatory elements in the clamps and handle domain of the cytosolic assembly (see text for further details). [From Samso et al. (468).] B: schematic diagram of the reported mutation sites of RyR1 and RyR2. NH2 terminus, central domain, and transmembrane (channel) regions are denoted. For more information, see text and http://pc4.fsm.it:81/cardmoc/. [From Yano et al. (617).]
The transmembrane domain is formed of four columns, each of which forms an internal branch and an external branch. The arrangement of the internal and external branches forms a central cavity and four peripheral chambers. The resulting constricted axial structure provides direct continuity between cytoplasmic and transmembrane assemblies. The transmembrane assembly has probably at least six transmembrane α-helices per monomer and closely resembles a closed K+ channel atomic structure (253, 337, 468) and may serve as the single Ca2+ channel formed by the tetramer.
Four columns arise from the external peripheral branches of the transmembrane prism. Each column consists of two connections to the handle; in addition, two adjacent monomers are structurally linked. This creates a connection between each rhomboid structure with a column of the prism via a direct pathway as well as via an external arm of the handle. If the structures in this link exhibit elastic properties, one would expect a torsional force on the prism that should depend on the integrity of the rhomboid structure on the t-tubular surface of the clamp. The twist of the transmembrane prism (as in Fig. 5), observed in the closed state of the channel, is consistent with this notion. Releasing the torque on the molecule, by dissociation of the internal connections in the rhomboid structure, would be accompanied by untwisting the transmembrane prism as has been observed during opening of the channel (480).
The peptide sequences involved in the transmembrane domains are known to some extent, although the exact number of transmembrane sequences (≥6) is still under study. Similarly, the location of the peptide sequences in the cytoplasmic domains is only partially known. The location of the peptide chains in the structure of the Ca2+ channel is still far from complete and even farther from conclusions regarding control mechanisms of the Po of the channel, and therefore, a detailed review of their location (cf. Refs. 332, 334, 350, 466; see also Refs. 35, 191, 333, 334, 465, 467, 629) is beyond the scope of this review. However, the proximity of mutations that affect the channel in skeletal muscle in malignant hyperthermia and central core disease and arrhythmogenic mutations in cardiac muscle suggests that the bridge in the rhomboid structure in the clamp is important to regulation of opening of the channel. The central domain of mutations that is involved in arrhythmias is again found in the bridge within the rhomboid structure of the clamp, suggesting that this structure in the clamp is important in the regulation of the opening probability of the channel.
Similar to what has been hypothesized for RyR1 channel proteins (609), it appears that RyR2 structure involves a critical interdomain interaction that plays a role in modulation of the channel's ability to release Ca2+. In this hypothesis, specific domains of the NH2 terminus interact to “zip” shut regions of the central core region. This zipped conformation has been linked to RyR2 channels with no Ca2+ “leak” (235). In disease and with RyR2 mutations, these regions can become unzipped to “leak” Ca2+ (see sect. ivA1). However, recent data also suggest that highly reactive free radicals destabilize these interdomain interactions and by themselves can cause partial dissociation of the FKBP12.6 binding protein (616).
Several studies have elucidated the sites for modulation of CICR (see reviews in Refs. 44, 88). Smaller modulatory proteins that have been found to copurify with RyR proteins are triadin (68, 183), sorcin, FKBP12.6 (249), PKA catalytic and regulatory subunits, MAKAP anchoring proteins, protein phosphatase (PP) 1 and PP2A (349), and calmodulin/CaMKII (44, 182). Recently, it has become known that RyR2 can be phosphorylated by at least two kinases, PKA and CaMKII, but each has a distinctive effect on RyR2 function. Ca2+ spark frequency of a normal myocyte increases with CaMKII stimulation due to a direct effect of phosphorylation of RyR2 (182). On the other hand, PKA-mediated effects to increase spark frequency appear to be related to an effect on SR load. The role of each of these kinases in abnormal Ca2+ spark frequencies accompanying disease awaits further study. It has been suggested that the FKBP12 protein is required for normal function of RyR2 playing a key role in the efficient so-called coupled gating between neighboring RyR2 channels (401). An immunosuppressant agent, FK506, binds to FKBP12 presumably inhibiting its modulation of RyR1, thereby increasing spontaneous [Ca2+]i transients by increasing the rate of release from the SR (358). FKBP12 null mice have RyR2 channels that exhibit abnormal gating in that there is a high occurrence of subconductance states (492). However, others have reported that removal of FKBP12.6 from RyR2 has no effect on RyR single-channel function (26). In rapamycin- or FK506-treated ventricular cells, presumably the loss of association of FKBP12 from RyR2 underlies the substantial increase in resting Ca2+ spark frequency (358).
A) Potassium and Chloride Channels in the SR Membrane
The presence of large Ca2+ fluxes through the membrane of the SR requires the existence of other channels which allow large countercurrents to protect against electrical instability of the SR membrane. A large-conductance (150–200 pS) K+ channel exists in both ventricular and atrial SR membranes and provides counter ion transport for Ca2+ release (1, 107, 159). Activation kinetics are slow with open times of 100 ms (455). There is no inactivation process. Typical K+ channel blockers (4-aminopyridine, iberiotoxin, amiodarone) are without effect (420). Ca2+ and Mg2+ do not alter the channel's activity (455), but its Po is reduced in low pH. The molecular identity of this protein is unknown at this time.
Additionally, a large-conductance (120 pS) Cl− channel exists in SR membrane and can be also permeable to Ca2+ (516). This Cl− channel's activity is altered with phosphorylation (125, 270, 458), and some have suggested that phospholamban modulates its conductance (125). The molecular identity of this protein remains unknown.
B) Ryanodine Receptors and Calcium Overload of the SR
Spontaneous SR Ca2+ release was first observed by Fabiato and Fabiato (153) in the form of spontaneous oscillatory contractions in skinned fibers. The spontaneous contractions were initiated by loading the SR using low [Ca2+]; the [Ca2+] used for the loading was by itself insufficient to induce Ca2+ release. The observation that skinned myocyte fragments started to contract in an oscillatory fashion led to the concept that a heavily Ca2+-loaded SR is characterized by spontaneous Ca2+ release. The importance of this phenomenon is that spontaneous contractions, caused by cytosolic [Ca2+]i oscillations (403, 588), are accompanied by spontaneous oscillations in current and membrane potential in both single myocytes as well as nondriven multicellular cardiac preparations (80, 264, 287). Agents that reduce Ca2+ load of the SR (e.g., ryanodine, caffeine, EGTA buffer) abolish spontaneous [Ca2+]i oscillations as well as the oscillatory potentials, current, and contractions (4, 353, 519). Therefore, it is thought that spontaneous [Ca2+]i oscillations are not secondary to transmembrane potential changes but, given the correct initiating conditions, may cause depolarization and give rise to nondriven action potentials (61, 64, 81, 353) (Fig. 6).
Fig. 6.
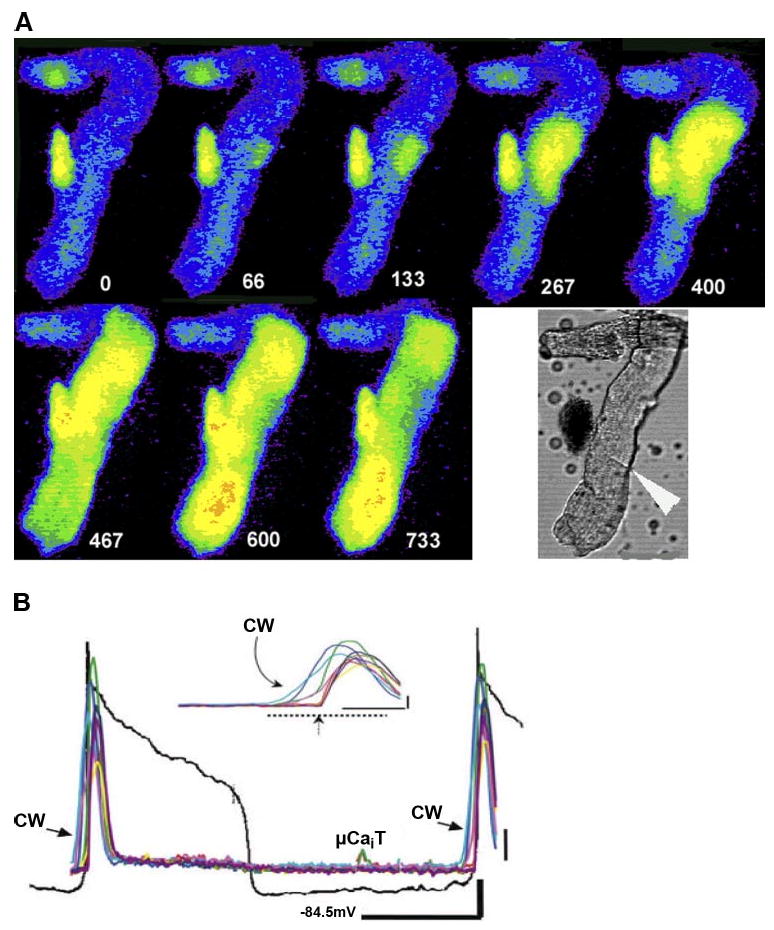
Large cell wide (CW) Ca2+ waves can lead to sufficient membrane depolarization to elicit an action potential (AP). A: selected image frames of Ca2+ from an IZPC (Purkinje cell aggregate from the infarcted heart) during the Ca2+-induced electrical activity. Time relative to t=0 of first frame is depicted by white numbers. Lower white light image is of aggregate during experiment. Large white arrowhead indicates probable cell border. B: transmembrane potential (black line) and Ca2+ (multicolored lines) changes of this aggregate during the CW wave induced electrical activity. μCaiT represents a small micro Ca2+ wavelet that occurred during the recording but that is not shown in these epifluorescent images (see Ref. 61 for more details).
As stated, Fabiato's work on the properties of cardiac SR (152) has provided a potential explanation for spontaneous Ca2+ release in mechanically skinned cells in which the SR was intact. The mechanism for increased probability of opening of the SR Ca2+ channel when the SR is heavily loaded with Ca2+ is still uncertain, but suggests that the channel is directly or indirectly sensitive to the luminal [Ca2+] of the SR. The localization of the Ca2+ sensor in the transmembrane domain of the RyR channel could make it suitable as a sensor of both luminal and cytosolic [Ca2+]. Intact normal cells with a high SR Ca2+ load show similar phenomena (82, 287). Hence, the oscillatory character of a triggered arrhythmia in myocardium with a high cellular Ca2+ load may be due to further increase of Ca2+ entry into the cells during the action potentials of the arrhythmia causing even more Ca2+ loading of the SR. Consequently, as soon as the release process has recovered after an electrically induced Ca2+ release, the overloaded SR again releases a fraction of its Ca2+. The requirement that the Ca2+ release mechanism must recover first would explain the presence of a delay between aftercontractions and afterdepolarizations and the preceding beat.
The released Ca2+ constitutes a “leak” from the SR that tends to reduce the “overload.” This phenomenon has been observed in different forms, which all fall under the general definition of a Ca2+ leak, e.g., increased probability of opening of RyR in lipid bilayer experiments (252), a biochemically detectable loss of Ca2+ from the SR (615), Ca2+ sparks in isolated cells and muscle (485, 590), micro Ca2+ waves in isolated cells and muscle (252, 615) and Purkinje cells after infarction (61), Ca2+ waves that travel inside myocytes but are limited to single cells (396), and multicellular cellular Ca2+ waves (308, 372, 513). The threshold for Ca2+ “leak” appears to be reduced in some arrhythmogenic mutations of the RyR (252) and in the acquired dysfunction of the RyR such as in congestive heart failure (49, 581, 614) and the first days after infarction (61).
3. IP3-dependent Ca2+ release
By immunohistolocalization techniques, the IP3 receptor (IP3R) has been identified in cardiac cells. Its density is less than that of RyR2 but particularly high in Purkinje and atrial cells (178, 342, 513). Most studies show it is located to a region of the intercalated disc (160, 384) with little or no fluorescence in longitudinal SR or mitochondria (274). Three isoforms of IP3R have been identified, with IP3R2 occurring in working cardiac muscle (414) and IP3R2 in the atrial and IP3R1 in the Purkinje fiber system (178, 325). IP3R2 staining in atrial cells is mostly discontinuous, but of a different distribution than that of RyRs. Double-labeling experiments show that RyRs and IP3R2s overlap in subsarcolemmal regions of rat atrial cells (325, 342), and IP3R1 resides mostly with RyR2 in peripheral regions of the Purkinje cell (513). While there are more IP3R2s in atrial versus ventricular cells, binding studies suggest IP3 binding affinities in atrial (Kd = 7.2 nM) and ventricular cells (Kd = 6.8 nM) are similar (325).
IP3R is a tetramer (either homomeric or heteromeric) with a binding site on each subunit. IP3-induced Ca2+ release is regulated by Ca2+ with a biphasic sensitivity (188); that is, IP3-induced opening and subsequent release are augmented with a modest increase in cytosolic Ca2+ (<300 nM) but are inhibited at higher [Ca2+]. However, the predominant cardiac IP3R type 2 is resistant to the inhibitory effects of high Ca2+ (437). IP3R2 has the highest affinity for IP3 (0.10 μM) followed by IP3R1 and then IP3R3 (0.40 μM). ATP modulates IP3R1 and IP3R3 but not IP3R2 (550). For the rat IP3R2, amino acids 1915 to 2175 appear to bind Ca2+ (367). The Ca2+ sensor region is conserved between the various IP3R isoforms [i.e., E2100 is critical for Ca2+-induced changes of IP3R1 (376)]. Isoforms appear to have similar gating and conductance properties and show the bell-shaped sensitivity to Ca2+ (551). These sites are located near the FKBP12, PKA, ATP, and CaM binding sites, all within the regulatory domain of the molecule (71). PKA, while effectively phosphorylating IP3R1, is a weaker modulator of IP3R2 and IP3R3 (595). Residues of the COOH-terminal tail are thought to be a site where ligands bind to transduce activation of the channel (553).
Accessory proteins have been implicated in the Ca2+ regulation of IP3-induced release (627). For example, IP3R2s bind Ca2+/CaM (608), which subsequently inhibits Ca2+ release (2). This interaction is Ca2+ independent, suggesting a role for CaM in tonic inhibition of IP3Rs. One family of Ca2+ binding proteins (CaBPs) are direct ligands of the IP3 channel (612), suggesting that IP3 release channel can become activated by a rise in Ca2+ without the necessity for IP3 and Ca2+ coincidence. Recent studies have shown that the brain IP3R1 complexes with PKA, PP2A, and PP1 (132). PKA increases the sensitivity of IP3R1 to activation by IP3 (595), while PP2A and PP1 would be expected to inhibit channel activity. Other high-affinity, low-capacity calcium binding proteins, such as NCS-1, have been shown to directly increase IP3R1 single-channel activity (473), which subsequently leads to dysregulated intracellular Ca2+ via IP3Rs. Interestingly, this interaction between NCS-1 and IP3R1 is attenuated with lithium.
The role of IP3 Ca2+ release in cardiac ECC is unknown, but IP3R2s from ferret ventricle when incorporated into planar bilayer are Ca2+ selective, IP3 activated, blocked by heparin, and not altered by ryanodine (414). Interestingly, in skinned cardiac fibers, IP3 can induce tension oscillations and enhance submaximal caffeine induced CICR (641) without increasing the Ca2+ sensitivity of Ca2+ release channel (641). Presumably this is because luminal Ca2+ can bind to cytosolic IP3R sites and modulate function. Recent work has linked a highly specialized local Ca2+ pathway between IP3, IP3R, CaM, and CaMKII and nuclear transcription (599). In adult ventricular cells, endothelin-1 increases IP3, which binds to its nuclear membrane receptor. This IP3 receptor is associated with CaM and CaMKII, which then activates type II histone deacetylases (HDACs), leading to the derepression of transcription factor MEF2 (599). Thus IP3Rs appear to play a role in excitation-transcription coupling in the native cell.
In rat atrial cells preincubated in the cell-permeant analog of IP3 (InsP3BM), an IP3 receptor agonist, the number of spontaneous Ca2+ sparks increases significantly, particularly in the subsarcolemmal regions where IP3R2s and RyRs colocalize (325). Furthermore, InsP3BM increases electrically evoked atrial Ca2+ transients suggesting that Ca2+ released from activated IP3Rs activate RyRs mimicking the effects of endothelin in atrial cells (342, 343). IP3-evoked Ca2+ release in ventricular cells is modest compared with that of atrial cells (325). Initial evidence had suggested that IP3 receptor function is critical for the positive inotropic effects of α-adrenergic agonists in guinea pig (378), but these results should be taken with caution since the inhibitor used, xestospongin C, may have other effects. Recent studies using permeabilized atrial cells suggest that IP3 and adenophostin can trigger elementary nonpropagating Ca2+ events that can be prevented by both heparin and 2-aminoethoxy-diphenylborate (2-APB) (642). Furthermore, IP3R2-deficient atrial cells failed to show endothelin-1-induced spontaneous Ca2+ transients (322), suggesting that IP3-dependent Ca2+ release enhances atrial intracellular cell signaling. 2-APB also affects the incidence and frequency of spontaneous Ca2+ events in Purkinje cells from the infarcted heart (62), again suggesting a modulatory role of IP3 in Ca2+ release in Purkinje cells from diseased hearts.
As discussed, Ca2+ waves in cardiac cells depend on the regenerative production of a diffusible molecule that triggers Ca2+ release from adjacent SR stores. Cytosolic Ca2+ may be one such diffusible molecule, but IP3 could also serve as a propagating signal within and between cardiac cells. IP3-dependent Ca2+ waves have been reported in airway epithelial cells (56) and other nonexcitable cells (77, 175). In these latter cells, an endoplasmic reticulum Ca2+ binding protein, calreticulin, clearly inhibits IP3-evoked repetitive Ca2+ waves (77). At this time, no apparent role has been defined for IP3-dependent Ca2+ release in cardiac cell wave propagation.
4. Mitochondria Ca2+ transport
Mitochondria can accumulate a large amount of Ca2+, aided in the presence of inorganic phosphate by the precipitation of insoluble Ca2+-phosphate deposits in the matrix (84). Ca2+ enters via a uniporter pathway down a large electrochemical gradient (∼180 mV) set up by proton extrusion linked to the electron transport system. The uniporter can be blocked competitively by physiological [Mg2+]i and also potently by ruthenium red and lanthanides (45). Ca2+ extrusion occurs mainly via Na+/Ca2+ and Na+/H+ exchangers and thereby is [Na+] dependent (181).
Ca2+ uptake by the mitochondria is too slow to contribute significantly to intracellular Ca2+ transient and myocyte relaxation (46), but may have an important role in the regulation of the [Ca2+]i over periods of many seconds and could, therefore, be expected to contribute to the mechanical restitution of cardiac muscle preparations. This postulate would require that there would be an interaction between the SR and the mitochondria, as observed in skinned rat cardiac trabeculae. In skinned rat fibers, the mitochondria have been observed to decrease the maximally Ca2+-activated force (557). Miyata et al. (377) developed a new approach to measure mitochondrial free [Ca2+] within a living cell by using a fluorescence Mn2+ quenching technique. A dependence of mitochondrial free [Ca2+] on the frequency of electrical stimulation suggests that mitochondria can accumulate Ca2+ under physiological conditions. Also a study by Wendt-Gallitelli et al. (585) on the changes of total mitochondria Ca2+ using electron probe X-ray microanalysis supports the results of Miyata et al. (377). Recent data suggest mitochondrial Ca2+ uptake is apparent only after a progressive Ca2+ load (cytosolic threshold ∼30–500 nM) and is sensitive to the mitochondrial Ca2+ uniporter blocker Ru360 (640). Further direct evidence has been reported for a role of mitochondria in clearing subsarcolemmal Ca2+ near the L-type Ca2+ channels and subsequent inactivation (469).
Instead of mediating cardiac Ca2+ fluxes during the contraction-relaxation cycle, mitochondrial Ca2+ fluxes regulate intramitochondrial processes and thus ATP production. Some matrix enzymes, e.g., pyruvate dehydrogenase, α-oxoglutarate dehydrogenase, and the NAD-dependent isocitrate dehydrogenase, can be activated by Ca2+ in the low micromolar range (128, 129, 359). Therefore, an increase of [Ca2+]i would lead to an increase of [Ca2+] in mitochondria that would increase oxidative metabolism and thereby increase ATP production to meet increased demands caused by high cytosolic [Ca2+], e.g., contractile activation and Ca2+.
Under pathological conditions, mitochondria can also accumulate a large amount of Ca2+. When Ca2+ overload occurs, mitochondria will temporarily compensate for a cellular Ca2+ load by taking up large amounts of Ca2+, which may prevent cell damage. However, Ca2+ accumulation by mitochondria diminishes ATP production and may eventually compromise the mitochondria by inducing the permeability transition. It seems that mitochondrial Ca2+ transport is important in the regulation of intramitochondrial dehydrogenases and in coping with cellular Ca2+ overload. However, beat-to-beat fluctuations in mitochondrial [Ca2+] are small during normal ECC (640).
C. Intracellular Ligands and Buffers
1. Sarcolemmal Ca2+ binding
The interaction between Ca2+ and the sarcolemma is pivotal in the feedback mechanisms described in section iiiC The actual [Ca2+] close to the sarcolemma is determined by the cell's buffering systems, one of which is formed by phospholipids, mostly the phosphatidylserines and phosphatidylinositols of the cell membrane. The density of phosphatidylserine and phosphatidylinositol (427) permits substantial Ca2+ binding (585); the number of sarcolemmal binding sites is estimated at 42 μM (42). The Kd for Ca2+ binding (0.3–1.5 μM) allows these phospholipids to act as a powerful dynamic buffer during the contractile cycle. Hence, feedback of subsarcolemmal [Ca2+] on protein function in the sarcolemma depends critically on this buffer system. Given the low Kd of this buffer, it would be expected that in Ca2+ overloaded cells, the buffer may saturate and cease to buffer [Ca2+] variations near the sarcolemma.
2. Intracellular ligands
Table 1 shows important intracellular Ca2+ ligands in the cardiac cell. It is unlikely that these ligands reflect all intracellular binding sites as was shown by electron microprobe analysis of rapidly stimulated and frozen isolated guinea pig myocytes. Wendt-Gallitelli et al. (585) have shown that the total [Ca2+] rises and falls in the A band of the myofibril from ∼2.6 to ∼5.5 mmol/kg dry wt following a voltage-clamp pulse (from −80 to +5 mV for 180 ms). The rise and fall nearly parallels the free [Ca2+]i transient itself, indicating that binding and dissociation of Ca2+ occur extremely rapidly and that the contractile proteins are responsible for the threefold slower rise of force. These values of total [Ca2+] in the presence of a [Ca2+]i, which ranges between 100 nM at diastole and 1 μM at peak systole, reinforce the notion that the Ca2+ is tightly buffered in the cardiac cell. The concentrations of buffers in the cell indicated in Table 1 are not enough to explain that in excess of 99% of Ca2+ is buffered (585). Hence, Wendt-Gallitelli et al. (585) have postulated an additional 600 μM of rapid Ca2+ binding sites in the cell. Precise knowledge of the properties of these latter buffers is required to assess their modulation of protein function.
Table 1. Ca2+ binding to troponin C and calmodulin in the cardiac cell.
| Parameter | Value | Source |
|---|---|---|
| Troponin Ca2+ specific binding sites | ||
| Concentration (tropT) | 60 μM | Lee and Allen et al. (311) |
| Ca2+ on-rate | 39 μM−1·s−1 | Robertson et al. (442) |
| Ca2+ off-rate | 19.6 s−1 | Robertson et al. (442) |
| Troponin Ca2+ -Mg2+ binding sites | ||
| Concentration (tropT) | 60 μM | Lee and Allen (311) |
| Ca2+ on-rate | 60 μM−1·s−1 | Wang et al. (575) |
| Ca2+ off-rate | 2.4 s−1 | Wang et al. (575) |
| Mg2+ on-rate | 0.04 μM−1·s−1 | Wang et al. (575) |
| Mg2+ off-rate | 20 s−1 | Wang et al. (575) |
| Calmodulin binding sites | ||
| Concentration | 25 μM | Wier and Yue (591) |
| Ca2+ on-rate | 9.2 μM−1·s−1 | Wang et al. (575) |
| Ca2+ off-rate | 7.3 s−1 | Wang et al. (575) |
| Free [Mg2+]i | 0.72 ± 0.06 mM | Gao et al. (172) |
| Diastolic free [Ca2+]i | 0.07–0.25 μM | Stuyvers et al. (515) |
III. Functional Consequences of Calcium Cycling
A. Macroscopic Events
1. The cardiac cycle: cytosolic Ca2+ transients and force development
Figure 3A shows force and the estimated cytosolic [Ca2+] (with a peak of ∼1 μM) as a function of time during a twitch in a muscle loaded by microinjection of fura 2 salt (22). The results are representative of contractions at long and short end-systolic sarcomere length (SL), i.e., at the extremes of the function curve of cardiac muscle. Figure 3A shows a typical behavior of mammalian cardiac muscle, i.e., peak and time course of the Ca2+ transients are remarkably independent of length, albeit that the relaxation phase differs between the short and long muscle. The interpretation of [Ca2+]i transients and their relation to force development requires caution, since it is known that full activation of the contractile system requires saturation of all Ca2+ sites on TnC (requiring ∼60 μM) with simultaneous binding of another 25 μM Ca2+ to calmodulin (43). Hence, even activation of the muscle at only 25% of its maximum, such as in Figure 3A, is accompanied by Ca2+ turnover of ∼30 μM. It is clear that only a small fraction of this Ca2+ is “visible” in the cytosol. With these considerations the cytosolic Ca2+ transient can teach us about a number of important properties of the ECC process. The changes in the kinetics of the transient with stretch are consistent with the hypothesis that force-length relation is determined principally by the length-dependent sensitivity of the contractile system, which resides in the relation between Ca2+ affinity of TnC and stretch (204, 272). This also implies that in the stretched myocardium more Ca2+ is bound (221). The molecular mechanism underlying length dependence of Ca2+ binding to TnC remains unknown, but one current hypothesis is that force exerted on the actin filament deforms the TnC molecule, thus retarding the dissociation of Ca2+ from TnC. This effect is bound to be length dependent since the number of myosin cross-bridges that can attach to actin increases with SL over the operational range of cardiac muscle (1.6 to 2.3 μm). Thus the mechanical load on a sarcomere will influence the dissociation of Ca2+ from TnC. In fact, it has been shown that rapid removal of an external load on a muscle causes a robust additional [Ca2+]i transient (224). This phenomenon can be important when the ECC properties of the myocardium are nonuniform (such as in disease), since nonuniformity of contraction of myocytes may be accompanied by such unloading-related [Ca2+]i transients. Thus the relaxation phase of the [Ca2+]i transient depends on the rates of Ca2+ binding and dissociation to and from TnC, the rate of Ca2+ binding to the sarcolemma Na+/Ca2+ exchanger, and the Ca2+ pump of the SR and on the rate of removal of Ca2+ by these transporters.
B. Microscopic Events
1. Ca2+ sparks in normal cardiac ventricular muscle
Spontaneous release of Ca2+ from the SR is evident in single ventricular cells as spatially localized increases in [Ca2+]i, termed Ca2+ “sparks” (92). Ca2+ sparks are also triggered during voltage-clamp pulses (335) and during action potentials (79), where they have been termed “evoked Ca2+ sparks” or “local [Ca2+] transients.” Local [Ca2+] transients or evoked Ca2+ sparks were originally proposed to be triggered by Ca2+ entering via single L-type Ca2+ channels (78, 336, 574). It is thought now that a cluster of L-type Ca2+ channels is involved in triggering a cluster of RyRs at least in rabbit ventricular cells (237). Even during periods of no electrical activity, Ca2+ released from RyR can occur and is due to the low probability of opening of RyR at diastolic Ca2+ levels (92). Ca2+ sparks may also trigger each other to produce Ca2+ waves, which propagate through the cell (91). It now seems unlikely that a Ca2+ spark arises from a single ryanodine receptor (92, 574) because smaller SR Ca2+ release events can be observed under some conditions (326) and because Ca2+ sparks can sometimes be observed directly to have multiple sites of origin (407). Numerous experiments have suggested that sparks are due to the coherent release of Ca2+ by a cluster of RyR and that Ca2+ sparks evoked by L-type Ca2+ currents summate, spatially and temporally, constituting the electrically evoked whole cell [Ca2+]i transient (78, 79, 335, 336, 589) that couples excitation to contraction.
The relevance of Ca2+ sparks to normal ECC was proven recently by similar observations using confocal microscopy of working ventricular trabeculae under physiological conditions ([Ca2+]i and temperature). Similar to the situation with Ca2+ sparks, Ca2+ waves had been recorded previously only in single isolated cells (511, 528, 588), although waves of sarcomere shortening, limited to single cells, had been reported in multicellular preparations. A possibly related phenomenon, scattered light intensity fluctuations (SLIFs), has been recorded in ventricular muscle but had been related only indirectly to fluctuations in [Ca2+]i. In addition, rapidly propagating Ca2+ waves, accompanied by propagating contractions, have been recorded in trabeculae with focal damage (see sect. iv).
Confocal images of microscopically quiescent trabeculae are illustrated in Figure 3B. Ca2+ sparks are readily visible in the full-frame image as spatially localized bright regions and in the line-scan images as localized transient changes in fluorescence in microscopically quiescent muscle (590). In line-scan images, Ca2+ waves are apparent as regions of elevated [Ca2+]i that move at constant velocity. Ca2+ sparks are common (≤300/s) at body temperature, with ∼10% of these being generated from repeatedly firing single sites. The average spacing between Ca2+ spark sites is 2 μm or intervals of one SL. In addition to single Ca2+ sparks, the line scans showed that Ca2+ sparks larger in extent than 2 μm occurred at ∼10% of the frequency of the single Ca2+ sparks. Ca2+ sparks are similar in time course and spatial spread in unstimulated muscle compared with those recorded in single isolated cardiac cells. The fluorescence ratio can be used to calculate the [Ca2+]i by assuming that the lowest fluorescence at the site of the Ca2+ spark represents fluorescence at the normal “resting” [Ca2+]i (100 nM). The time to rise from 10 to 90% of the peak is 5 ms, and the time to fall from peak to half of peak is 40 ms. The average Ca2+ spark is preceded by a rise of the [Ca2+]i, which starts 10 ms before the Ca2+ spark. Such an event, which suggests the possibility of triggering of some of the Ca2+ sparks by a local rise of [Ca2+]i, has also been described in isolated cells. After the peak of the Ca2+ spark, the width of the region with elevated Ca2+ increases with time consistent with diffusion of Ca2+ away from the Ca2+ spark generating site, as has also been shown in isolated cells (407). Most Ca2+ sparks in muscle reach an amplitude of ∼200 nM with a progressive decrease of the number of larger sparks (>400 nM). A similar distribution has been found in single isolated cells. The peak amplitude Ca2+ sparks is ∼200 nM, which is below the level at which cross-bridges are activated in intact trabeculae. Although the exact estimate of [Ca2+]i on the submicrometer scale is problematic due to both the rapid changes of [Ca2+] and the kinetics of buffering by fluorescent indicators, the observation that Ca2+ sparks occur in microscopically quiescent muscles is not surprising.
A) Calcium Sparks in Atrial and Purkinje Cells
From ultrastructural analysis, peripheral couplings in cat atrial cells are circular and measure 0.2–0.6 μm This is consistent with the size of the individual Ca2+ release events that have been observed in situ (53). In atrial cells from normal hearts, Ca2+ sparks are more frequently seen at the periphery (500 sparks·s−1·pl−1) of the cell compared with the center (100 sparks·s−1·pl−1) (232). Central sparks tend to be larger and longer in spatial spread. In fact, release sparks in atrial cells can occur as doublets, triplets, or compound sparks (53, 597) and can span up to 7 μm with spatial distance between these events being variable within different regions of the cell. In rat atrial cells, electrically evoked Ca2+ transients are comprised of a set of Ca2+ sparks that appear to have a specific spatiotemporal order; that is, at specific peripheral sites, a Ca2+ spark always appears first and the activation order of sites for remaining sparks remains consistent with each subsequent paced beat and is not modified by SR load (38, 341). These data suggest that at least for the rat, there may be L-type Ca2+ channel SR couplings that show a different sensitivity to CICR.
In normal canine Purkinje cells, spontaneous Ca2+ sparks have characteristics similar to those of ventricular cells; however, in the presence of normal [Ca2+]o, there is also a population of wider and larger sparks (513). These compound sparks of Purkinje cells may represent near-synchronous activation of multiple types of Ca2+ release channels. For canine Purkinje cells, this could mean a combination of Ca2+ releases from both IP3R and RyR receptor release channels (513).
2. Microscopic Ca2+ waves in normal muscle
Slowly traveling Ca2+ waves occur rather rarely in trabeculae ([Ca2+]o = 1 mM; 37°C) and appear comparable to those in single cells (511, 528, 588). They occur commonly several seconds after the twitch. Their average frequency is ∼2.5 Hz/cell, and they transmit less than ∼4 SL. More often than not, waves propagate in only one direction. This suggests that if these waves start at a gap junction, their propagation into one or both cells connected to the gap junction would be dictated by chance. It is striking that Ca2+ waves show Ca2+ sparks on their leading edge, with an average distance of Ca2+ sparks along the edge of ∼1 SL (91). These latter authors concluded that in single cells Ca2+ sparks may provide the regenerative mechanism for a Ca2+ propagation wave from one terminal cisternae to another. In this case, the trigger for Ca2+ spark generation during a propagated wave would consist of Ca2+ arriving from an adjacent Ca2+ spark generating site (91). If Ca2+ release from the site is proportional to the rate of rise and the absolute [Ca2+]i reached at the Ca2+ spark site, one would anticipate that waves would propagate at a constant velocity, since the same process would repeat itself at each following site. These results may provide an explanation for spontaneous motion observed previously in the form of SLIF in rat papillary muscles. In this regard, it has been reported recently that the frequency of Ca2+ sparks recovers after stimulation, similar to that observed for SLIFs (286). If single Ca2+ sparks precede the development of Ca2+ waves, it is conceivable that SLIF accompanies Ca2+ waves and that Ca2+ sparks remain undetected by techniques that monitor only sarcomere motion. Nevertheless, small Ca2+ waves, if in only a few myocytes, would be unable to cause a depolarization sufficient to trigger an action potential if the myocytes were well coupled to its neighbors.
3. Ca2+ waves in intact whole hearts
Minamikawa et al. (368) were the first to demonstrate that traveling Ca2+ waves (propagation velocities 60–100 μm/s) occurred in the perfused rat heart. Despite low temporal resolution of their images, they concluded that Ca2+ waves play little if any role in pathology, since they were easily abolished by pacing. Improvement of confocal imaging to more “real” time has allowed further investigations into the types of Ca2+ waves observed in perfused rat hearts. Hama et al. (190) concluded that Ca2+ waves in their normal hearts had limited effects on Ca2+ homeostasis of the myocardium. Kaneko et al. (259) extended these studies (also in rat hearts) by describing three types of Ca2+ waves in perfused hearts. Interestingly, waves of somewhat similar characteristics occurred in both the quiescent and presumably Ca2+-overloaded hearts. The incidence and velocity of in situ Ca2+ waves were related to [Ca2+]o and presumably the Ca2+ load of the SR. More recent studies in normal rat hearts confirm previous work reporting that intercellular Ca2+ wave propagation was mostly confined to two or three cells (18). Furthermore, these authors suggested that spatiotemporal summation of changes in transmembrane potential caused by individual Ca2+ waves may underlie generation of triggered electrical impulses such as those that occur in trabeculae or aggregates of Purkinje cells from the infarcted heart. Nevertheless, despite the numerous technical limitations of these types of isolated heart experiments (see Ref. 514), Ca2+ waves in normal hearts seem of little importance to rhythm in these hearts. On the other hand, Ca2+ waves occurring in the regions of nonuniformity of the ablated rat heart may gain importance (532).
C. Regulation of Ca2+ Transport
The transient elevation of [Ca2+]i following the start of the AP can affect ion channel function, impacting on the time profile of the AP during the plateau and repolarization phases on a beat-to-beat basis. Ca2+ could do so by directly binding to ion channel proteins or by influencing the activity of other proteins, which could then modulate ion channel behavior. In this section we discuss the effect of [Ca2+]i on currents involved in the various phases of the AP.
Well known are the effects of Ca2+ on depolarizing currents during the AP, yet little is known about the influence of [Ca2+]i on sodium current (INa) on a beat-to-beat basis. Recent biochemical evidence suggests that our current thinking that voltage-gated cardiac sodium channels are not directly coupled to Ca2+ signaling events may not be true. These data suggest that the Ca2+ binding sensor protein CaM binds to the α-subunit of the rat brain type II Na+ channel (382). Moreover, COOH-terminal residues bind Ca2+-bound and Ca2+-free CaM, apoCam. The human cardiac sodium channel (Nav1.5) also contains residues within this “IQ” motif that bind Ca2+/CaM (531). In so doing, there is an increase in the proportion of slowly inactivating Na+ channels. A mutation associated with Brugada syndrome in the COOH-terminal region (A1924T) inhibits the slow inactivation induced by Ca2+/CaM (531). More recent data have shown that residues within the EF hand domain of Nav1.5 bind Ca2+ with an affinity within a range known for Ca2+ sensors (593). Such Ca2+ binding increases Na+ channel availability by producing a shift in voltage dependence of channel availability. In a recent report studying various sodium channel isoforms, it was suggested that CaM modulation of function is isoform specific (131), and the effect is to modulate the COOH-terminal inactivation of the III-IV linker of Nav1.5 (277).
[Ca2+]i or current-dependent inactivation of the L-type Ca2+ current has been recognized for many years (312, 364). Hallmarks of this process in native cells include the complex time course of current decay and the presence of a dip in the inactivation curve; that is, in tightly buffered cells, whole cell Ca2+ currents recorded during the final pulse to a fixed test potential of a three-step inactivation protocol show a minimum current level near 0 mV, but current increases again at conditioning steps positive to 0 mV. Finally, Ca2+ current-dependent inactivation is evident in that the decay of ICaL is faster the larger the current amplitude (360) or by Ca2+ buffering or use of Ba or Sr as the charge carrier. Both Ca2+ released from the SR as well as Ca2+ influx through the voltage-activated ICaL contribute to the Ca2+-dependent inactivation (517). In ventricular cells, normal SR Ca2+ release can reduce the calculated integrated Ca2+ influx by 50%. In fact, local SR Ca2+ flux that is sensed by the Ca2+ channel peaks within 5 ms and is independent of the amplitude of the release. The mechanism of how influx and subsequent release of Ca2+ but not other ions, hasten the Ca2+ current decay is unknown. One hypothesis is that Ca2+ directly bind to the α-subunit of the L-type Ca2+ channel protein. An initial hypothesis was that a consensus Ca2+ binding motif (EF hand) is near the inner mouth of the proposed channel and is required for current-induced inactivation of channel activity (124, 390). However, mutagenesis of putative Ca2+ binding sites in this region of the channel does not entirely remove Ca2+-dependent inactivation (418, 637). Other data suggest that 40 amino acids located near the COOH terminus of the α-subunit protein of the L-type Ca2+ channel are implicated in the process (418, 503, 637).
While the COOH terminus of the L-type Ca2+ channel α-subunit contains binding sites for many regulatory proteins, recent data show that CaM can bind to the COOH terminus IQ domain (410, 417, 643) as well as a CaM-binding domain that lies between the EF and IQ regions (385, 410, 447). This suggests that CaM functions as a Ca2+ sensor for calcium-dependent inactivation of the channel. In fact, at Ca2+ levels of 10–100 nM, a portion of the resting COOH-terminal domain binds CaM acting as its tether (423); that is, CaM is preassociated with the channel. In this latter scenario, the two lobes of a single CaM molecule do not tether to the same site but rather bind to multiple noncontiguous sites. Upon the further elevation of Ca2+, one lobe of CaM binds specific IQ residues while the other lobe remains as a secure anchor for CaM. CaM interaction with IQ residues then allows for quick and efficient inactivation of the L-type Ca2+ current. In fact, while one CaM molecule is sufficient for calcium-dependent inactivation of the channel in an expression system, local CaM concentrations near the Ca2+ channel have been estimated to be as great as 2.5 mM (383), much greater than global estimates of CaM (345). Interestingly, residues involved in tethering CaM also appear to be involved in controlling the rate of inactivation of Ba2+ currents, which is assumed to inactivate by a voltage-dependent process (423). While the N lobe of CaM has been implicated in Ca2+-dependent inactivation of other Ca2+ channel isoforms (323), it is unclear at this time as to its role in Cav1.2 Ca2+-dependent inactivation. Recent work by Kim et al. (278) suggests that in the presence of both CaM and Ca2+ which bind and change the conformation of the COOH terminus, certain residues can interact with now nearby residues of the I-II linker. This combined complex then occludes the pore. Interestingly, another Ca2+ sensor, CaBP1, binds to the pore subunit in this way as to prevent Cav1.2 inactivation (636). The role of CaM in this functional effect is not known.
The influx of Ca2+ (estimated to be ∼10 μmol/l cytosol) most likely inactivates L-type Ca2+ channel activity, but under some circumstances, a [Ca2+]I-dependent increase or facilitation of the L-type Ca2+ current has been described (155, 184, 214). Interestingly, facilitation of L-type Ca2+ channels is diminished in the presence of ryanodine and prevented by CaMKII inhibitory agents (11, 605, 620). In fact, dialysis of cells with constitutively active form of CaMKII restores ICaL facilitation (10, 602). Recently Hudmon et al. (227) showed that CaMKII tethers to the Ca2+ channel helping to form the macromolecular complex. When Ca2+ rises via voltage-dependent entry or its subsequent SR release, it activates CaMKII and thus phosphorylates the channel protein (227), inducing modal gating shifts favoring long channel openings (139). This above apparent cross-talk occurs because CaMKII activity in turn reduces the “gain” of ECC (601). The frequency response of CaMKII is modulated by both the amplitude and duration of Ca2+ spikes (123). On the other hand, CaM and its binding to the pore-forming unit of the channel appears to control both inactivation and facilitation of P/Q-type Ca2+ channels (126). In this latter scenario, Ca2+ binding to the NH2-terminal lobe of CaM initiates inactivation while the Ca2+ sensing portion of the NH2-terminal lobe initiates Ca2+ current facilitation. Some have described a scenario where the time course of Ca-CaM, CaMKII, and two of its phosphorylation sites is one where the time course of Ca-CaM largely follows that of the global Ca2+ transient, while that of CaM bound to CaMKII declines more slowly. Thus with repetitive stimuli, CaMKII accumulates in its active state (345).
Once an L-type Ca2+ channel is activated and inactivated, it follows a predictable time course as it recovers from inactivation, repriming itself for the next stimulus. This recovery process is voltage and Cai dependent. Voltage-dependent recovery has a reasonably fast phase and slow (477) or very slow (65, 263) phase. Importantly, recovery from [Ca2+]i-induced inactivation may occur at positive plateau potentials and depends on both SR and Na+/Ca2+ exchanger function (364, 496, 498). Presumably recovery from [Ca2+]i-dependent inactivation allows the L-type Ca2+ channels to reopen and provide Ca2+ influx during early afterdepolarizations (see sect. ivB3a).
Current generated by the Na+/Ca2+ exchanger protein (INa/Ca) depends on [Ca2+]i (578), due to the contribution of [Ca2+]i to the diffusion gradient for Ca2+ (51) as well as its regulatory role on Na+/Ca2+ exchanger function. Regulatory Ca2+ is not transported by the exchanger protein. Instead, it serves to “activate” the exchanger. In excised patches Ca2+ affinity for the regulatory site on the cytoplasmic loop of the Na+/Ca2+ protein is 0.1–0.3 μM (163, 206). It is thought that [Ca2+]i augments peak outward INa/Ca by promoting the exchanger's recovery from Na+-independent inactivation. If the low-affinity Ca2+ binding sites remain so in intact cells, then Na+/Ca2+ currents should be modulated via this Ca2+ binding on a beat-to-beat basis as Ca2+ influx and SR release ensue.
In normal myocytes the time course of decline of inward INa/Ca that occurs upon membrane repolarization is related to the time course of the spatially averaged [Ca2+]i transient (16, 141, 578). In myocytes from diseased hearts showing abnormal [Ca2+]i cycling, INa/Ca could contribute substantially to both altered outward and inward currents. Therefore, in myocytes from hypertrophied/failed hearts where the relaxation phase of [Ca2+]i transients may be slowed (e.g., Ref. 50), slowly decaying inward Na+/Ca2+ exchanger currents would exist during diastole. Under conditions of disease where the L-type Ca2+ channel function is downregulated [e.g., postcoronary artery occlusion (3, 63)] and perhaps the Na+/Ca2+ exchanger is upregulated (220, 330, 426, 499, 630), a large Ca2+ influx seen upon depolarization could be carried by the Na+/Ca2+ exchanger. Currents generated by Na+/Ca2+ exchanger could be both sustained and oscillatory during a maintained depolarization. These currents have been termed the transient inward current, Iti (310).
Currents favoring cardiac repolarization can also be modified by changes in [Ca2+]i. The Po of the native cardiac delayed rectifier channel (IKs) is increased with an increase in [Ca2+]i (472, 542), thereby producing enhanced outward currents with increased [Ca2+]i. Elevation of [Ca2+]i above 10 nM enhances IKs without an effect on the current-voltage relationship (265, 572). Noise analysis has shown that [Ca2+]i increases the Po of the native IKs channels without changing their unit amplitude (542). What remains unclear is whether this K+ channel modulation is due to Ca2+ binding directly to a site on the channel protein or due to activation of a Ca2+-dependent signaling molecule. However, recent evidence suggests that CaM-dependent NOS3 activation confers the Ca2+ sensitivity on IKs (24). Furthermore, there is a Ca2+ interaction between CaM and a CaM binding pocket on the KCNQ1 COOH terminus that appears critical for IKs channel assembly (174, 483). Previous studies evaluating the role of Ca2+/CaM in KCNQ function had revealed conflicting results (165, 174, 584, 622). Some long QT mutations in KCNQ1 channels disrupt this CaM interaction preventing functional assembly of channels (174, 483). Finally, CaM also regulates KCNQ1 gating, relieving inactivation in a Ca2+-dependent fashion (174). Thus it appears that Ca2+/CaM affects activation, inactivation of the channel, and interaction with its accessory protein, KCNE1.
The rapid component of the native delayed rectifier current (IKr) has distinct negatively charged channel residues near a voltage sensor that bind extracellular Ca2+ affecting the alignment of the S3-S4 segments (254). Recently reexamined by Fernandez et al. (156), it was shown that specific residues in a pocket between the S2 and S3 segments contribute to these low-affinity binding sites for Ca2+. With Ca2+ binding, the process of voltage-dependent activation is modified. Thus modest external Ca2+ concentrations have been shown to significantly affect the fraction of HERG channels participating in action potential repolarization (255). Intracellular Ca2+ changes would not be expected to cause such effects.
Ca2+ influx may also contribute to a “dynamic” rectification of the inward rectifying K+ current (IK1) since both the probability of opening of the channel in subconductance states and rectification of IK1 appear to be [Ca2+]i dependent (356). An increase in [Ca2+]i is expected to increase rectification of IK1 (234, 352) but not nearly as much as [Mg2+]i or polyamines. Interestingly, cytochalasin but not colchicine removes this Ca2+-dependent effect, suggesting a role for cytoskeletal actin filaments in rectification of this channel (356). Recent data obtained in intact voltage-clamped cells suggest that [Ca2+]i-dependent IK1 rectification contributes to <2% of total rectification (625) and that Ca2+-dependent reduction in IK1 contributes to remodeled IK1 in cells from failing hearts (154).
Transient outward currents, described in early Purkinje fiber (161, 493) and canine myocyte studies (549), reflect the sum of a K+ current through a voltage-dependent, [Ca2+]i-independent channel (Itol) and one through a [Ca2+]i-dependent Cl− conducting channel [Ito2 also called ICl(Ca)] (161, 216, 645, 646).
Kv4 α-subunit proteins are thought to underlie the voltage-dependent transient outward currents in animal as well as human ventricular cells. The integral components of the native Kv4 channel complex are a group of Ca2+ binding proteins called KChIPs (9). KChIPs have some similarities to other calcium-binding proteins (e.g., recoverin, DREAM, calsenilin, GCAPS, and NCS-1) and KChIP2 is predominately expressed in heart. Expression of KChIP2 with Kv4.2 proteins produces increased density of Kv4.2 currents as well as changes in activation and inactivation properties of these currents (hyperpolarizing direction). The effects of KChIP2s on Kv4.3 proteins are specific, since no effect was seen when KChIPs were coexpressed with Kv1.4 or Kv2.1 proteins (9). Interestingly, KChIP1 has similar effects on Kv4.2 and Kv4.3 subunits but opposite effects on Kv4.1 subunits (387). Recent studies using splice variants of human KChIP2 have shown that KChIP effects on Kv4.3 current decay is significantly reduced when intracellular Ca2+ is buffered. On the other hand, the effects of KChIP to increase current density and speed recovery are Ca2+ independent (130). A minimal KChIP isoform accelerates recovery and slows inactivation kinetics of Kv4.3, and Ca2+ binding to KChIP2d relieves KChIP-induced slowing of Kv4.3 inactivation (411). The modulatory effects of KChIP1 on Kv4 currents is eliminated when EF-hand motifs are mutated, suggesting that modulation is Ca2+ dependent (9). Recent data suggest that KChIP2 also affects Kv1.5 currents in a Ca2+-dependent way (320).
More important to the voltage-dependent transient outward current in atrial cells may be the proper functioning of the Kv1.4 K+ channel (8). Unlike Kv4.3 channels, Kv1.4 channels are dephosphorylated by Ca2+-regulated calcineurin and phosphorylated by CaMKII (445). Phosphorylation via CaMKII results in a slowing of current inactivation and acceleration in recovery from inactivation. This is entirely consistent with findings in human atrial cells where CaMKII inhibitors reduce sustained atrial currents restoring native peak Ito currents (538).
In normal canine and feline myocytes, the amplitude of Ito2 is small relative to the voltage-dependent, 4-aminopyridine-sensitive Ito1 (164, 497, 549). For this reason, Ito2 is studied in the presence of catecholamines in some studies (549, 645), or high concentrations of strophanthidin (493) (presumably resulting in increased [Ca2+]i). Conversely, Ca2+ depletion of the SR, e.g., by ryanodine, and Cl− transport blockers, e.g., DIDS, have been shown to block Ito2 in cardiac cells (269, 549, 645). Despite the importance of this [Ca2+]i-dependent Cl− channel for normal cardiac repolarization as well as its potential involvement in arrhythmogenesis (193, 217, 268, 559, 644), little is known about its physiology and specific pharmacology. Recently, a low-conductance (1.0–1.3 pS) Ca2+-activated Cl− channel of high membrane density (3/μm2) has been described in canine myocytes (103). Despite the relatively low Ca2+ sensitivity of this channel, they can conduct significant current transiently or in a sustained manner depending on [Ca2+]i and time course of the subplasmalemmal [Ca2+]i transient (103, 269, 292, 340), suggesting an apparent voltage-dependent Ca2+ sensitivity of the channel. This Cl− current is activated upon depolarization after ICaL-induced Ca2+ release from the SR as well as upon caffeine-induced Ca2+ release (406). Characteristically Ito2 decays before the global cellular Ca2+ transient has reached its peak (497). However, under strict voltage-clamp conditions with different “clamped” levels of intracellular Ca2+, Ito2 shows little or no voltage-induced inactivation (269, 644). The specific protein giving rise to the small-conductance Ca2+-activated Cl− channels (CLCA) in native cardiac cells remain unknown. Tissue expression levels of cloned CLCA subunits (bCLCA1, mCLCA1, hCLCA1, hCLCA2) reveal none is highly expressed in heart. The presence of a [Ca2+]i-dependent Cl− current in normal human electrophysiology remains controversial (149, 319). For instance, it appears that a caffeine-evoked [Ca2+]i-dependent current is not Cl− sensitive in human atrial myocytes (289, 319). Rather, human atrial cells express a [Ca2+]i-dependent nonspecific cation channel (168). Finally, the role of this current in beat-to-beat AP will depend on the status of the L-type Ca2+ channel and SR load. In acquired disease, the Ca2+ currents are often remodeled (for review, see Ref. 422).
Two apparently different Ca2+-dependent cation channels have been identified in adult and neonatal ventricular cells. Ca2+ is needed for activation of both channels, and each channel appears to be equally permeable to Na+, K+, Li+, and Cs+. Activation of the first type of cation channel in neonatal (104) and adult cardiac cells requires [Ca2+]i to exceed 0.3 μM (142). The Po of the channel increases with [Ca2+]i and is insensitive to voltage. The native channel is activated with an intracellular injection of Ca2+ (351) or by caffeine-induced Ca2+ release from the SR (245). Thus the kinetics of the channel in ventricular (104, 142, 600) and human atrial cells (289) are determined by changes in [Ca2+]i and not by voltage. In adult guinea pig myocytes, the estimated channel density is 0.04–0.4/μm2.
A second Ca2+-activated cation channel, observed in bilayer experiments of canine ventricular sarcolemma vesicles, is sensitive to both [Ca2+]i and voltage (210). In the presence of resting [Ca2+]i levels (100 nM), a membrane potential more positive than −60 mV causes activation. The probability of opening increases to a maximum with depolarization to 0 mV. It has been estimated that this channel may be expressed at a high density in Purkinje fibers where transient inward currents associated with Ca2+ overload conditions (39, 80, 264) are as large as 100 nA.
An alternatively spliced form of the TRP3 protein (transient receptor potential, like protein 3) has been identified in heart (398). When expressed in oocytes, this variant, TRP3sv, encodes a cation-selective channel that is Ca2+ activated, but appears unrelated to the family of “capacitative Ca2+ entry currents.” Such currents were partially blocked by trivalents like GdCl3 or LaCl3. TRPC4 and TRPC5 channels have been identified in neonatal cardiac cells (482), while in embryonic and neonatal mouse cells, a store-operated Ca2+ influx channel has been proposed and persists in RyR2 knockout mice (554). In neonatal rat cells, Ca2+ store depletion activates inward currents perhaps due to TRP channels (229). In adult canine ventricle and Purkinje cells, TRPC3, -6, and -7 have been identified (137). A Mg2+-inhibited TRPM6/7-like current has been described biophysically in both voltage-clamped pig and rat cells (185). Activation with diacylglycerol (DAG) appears to activate a Ca2+-activated nonselective channel in rat (180). These authors suggest it is due to TRPM4b channels, similar to findings in human atrial cells (179). Interestingly, TRPV4 channels complex with RyR channels in smooth muscle cells (140), and TRPC6 channels associate with RyR2 proteins in cardiac cells (137).
At high [Ca2+]i, inactivation of the L-type Ca2+ current has been described (496) and is presumably due to a Ca2+-induced reduction of the Po of the channel(124). The site of this inactivation may be Ca2+ binding sites on the COOH terminus of the α-subunit of the L-type Ca2+ channel protein (see above). Factors influencing the intracellular [Ca2+]i as well as changes in [Ca2+]i itself can alter the size of T-type Ca2+ currents, especially in canine Purkinje cells (7, 309, 547, 552).
IV. Calcium and Arrhythmogenesis
A. Inherited Mutations That Cause Ca2+-Dependent Arrhythmias
1. RyR2 mutations and arrhythmias
Coumel et al. (109), Swan et al. (520), and Leenhardt et al. (313) recognized catecholaminergic polymorphic ventricular tachycardia (CPVT) as a malignant clinical entity that causes stress-related syncope and sudden death in children, occurs without structural heart disease and can be treated by suitable β-blockers. CPVT is similar to familial polymorphic ventricular tachycardia (FPVT) (299) but occurs without a long QT interval and is not accompanied by the pattern of torsade des pointes arrhythmia. The electrocardiogram (ECG) pattern seen during exercise with induced bidirectional arrhythmias preceding VT is reminiscent of triggered arrhythmias occurring with digitalis intoxication. Genetic analysis by Swan et al. (520), Priori et al. (431), and Laitinen et al. (299) have now shown that these patients exhibit mutations of RyR2 (human ryanodine receptor type 2) that map to chromosome 1q42-q43. More recent genotype-phenotype analysis of patients with CPVT has shown that there are two groups of patients: the predominately male patient with RyR2 CPVT has early symptoms and is at high risk of a cardiac event, and the other group is the nongenotyped CPVT female who shows symptoms later in life (430). The variable expressivities of RyR2 mutations are further illustrated by the fact that 17% of the gene carriers had no phenotype.
Tiso et al. (541) reported that one of the (at least 6) forms of arrhythmogenic right ventricular dysplasia, ARVD type 2, a catecholamine-sensitive autosomal dominant cardiomyopathy, is also caused by mutations in the hRyR2 (human ryanodine receptor type 2) (29, 438, 520).
A) Functional Implications
The 60 missense mutations (Fig. 5B) reported so far (see updated information at http://www.fsm.it/cardmoc/) occur in domains of RyR2 that may be involved in regulation of the Po of RyR channels by the phosphorylation-dependent binding protein FKBP12.6 or direct alteration in gating (23, 30, 95, 197, 299, 300, 430, 431, 539, 541). In fact, the properties of the hRyR2 mutation R4497C as it is expressed in HEK cells are altered compared with wild-type; there is an enhanced basal activity of the RyR channel that is accompanied by augmented spontaneous Ca2+ release (250). In other studies (315), only PKA activation of mutant RyR2 proteins produces a significant gain-of-function defect consistent with enhanced Ca2+ leak. These findings suggest that abnormal Ca2+ cycling based on dysfunction of SR Ca2+ release channels are involved in potentially lethal and genetically transmitted human cardiac arrhythmias.
Most studies have characterized the properties of mutant RyR2 channels in expressed cell systems but recently mice with knock-in RyR2 mutations have been produced (87, 262). Cells from both knock-in mice show a gain-in-function represented by presence of delayed after-depolarizations (DADs) and/or [Ca2+]i oscillations. Functional characteristics of these mutants compared with wild-type have ranged from enhanced sensitivity to Ca2+ (251, 252) to increased “leak.” The mechanisms of the increased leak under these circumstances appear to fall into a few general categories. First is the interdomain hypothesis (235, 609), which states that under normal conditions, the NH2-terminal of the RyR channel folds over in such a way as to interact with certain residues of the central domain of the same channel protein. With this interdomain interaction, the channel is in a stable closed state. “Leak” arises in mutant channels when this interdomain interaction is weakened. A second hypothesis is that the “leak” is due to faulty protein-protein interactions owing to PKA-mediated hyperphosphorylation of RyR2. The latter is assumed to cause dissociation of the FKBP12.6 protein to result in abnormal Ca2+ channel activity (580). Recent data suggest that the R2474S mutation of RyR2, which causes the “unzipping” of the inter-domains, is still able to interact with FKBP12.6 but, now, PKA phosphorylation of RyR2 has a greater effect on its dissociation (397). Finally, it has been proposed that RyR2 mutations associated with CPVT/ARVD2 (251) exhibit a lower threshold for store-overload-induced Ca2+ release. Whether these mechanisms of RyR dysfunction occur in native cells and lead to spontaneous Ca2+ release and arrhythmias is not as yet known.
2. CASQ mutations and arrhythmias
In four families CPVT has been associated with homozygous and heterozygous missense mutations in the calsquesterin2 gene (CASQ2) (296, 428).
A) Functional Implications
Calsequestrin is the most abundant Ca2+ binding protein in the cardiac SR. It contains up to 50 Ca2+ binding sites. Original work on cardiac CASQ showed a Kd of 400–600 μM (369), while recent data using a Ca2+ overlay procedure (133) suggest that the Kd of wild-type CASQ2 is ∼2.2 mM (133). The folding and stabilization of CASQ depends on the concentration of Ca2+ (576). COOH-terminal residues of CASQ and triadin interact within the SR lumen (489), while the NH2-terminal residues appear neither to interact with triadin nor junctin (576). It is not entirely clear how these conformational changes in CASQ affect the response of RyR to luminal Ca2+. However, the addition of CASQ to RyRs with triadin and junction inhibits RyR channel activity even at low luminal Ca2+ levels (186). In fact, CASQ remains associated with the junctional membrane when Ca2+ is 1–2 mM but is removed when Ca2+ luminal is >4 mM. As such, CASQ increases relative Po of RyRs when Ca2+ luminal concentration is >1.5 mM (32).
The identified missense mutation in patients with CPVT changes the charge of the CASQ protein in such a way as to alter Ca2+ binding and thus the conformational changes. Because CASQ plays an important role in SR Ca2+ release (537), cells with mutated CASQ would have reduced SR storage capacity. In fact, Viatchenko-Karpinski et al. (562) showed that when rat cells were infected with the CASQ mutant D307H, the Ca2+ storage capacity of the SR is reduced (and/or the CASQ/RyR2 interaction is disrupted). This in turn reduces Ca2+ transients and spark amplitudes. By reducing the effective buffering of the SR, cells become more prone to abnormal Ca2+ release in the presence of adrenergic stimulation presumably by promoting the generation of Ca2+ waves (291). One study has shown that a point mutation of CASQ altered Ca2+-dependent binding of the mutant CASQ to both triadin-1 and junctin (536). Recent data on new CASQ2 mutations in patients with CPVT suggest that while some mutations can lead to altered binding to Ca2+ to CASQ2, others lead directly to altered regulation of RyR2 (133). Finally, as yet, no atrial arrhythmias have been genetically linked to RyR2 or CASQ2 genes.
3. Ca2+ channel mutations and arrhythmias
Timothy's syndrome is a multiorgan disorder that can cause atrioventricular (AV) block, T-wave alternans, and ventricular tachyarrhythmias. In one large family where the inheritance pattern was clear, affected individuals had prolonged QT intervals and spontaneous arrhythmias (507). No mutations were found in known long QT syndrome genes. However, an analysis of one Cav1.2 splice variant (exon 8a) revealed a missense mutation in residues of the COOH terminus end of the S6 segment of Domain 1. Another two individuals also had severe polymorphic ventricular tachycardias and a severe variant of this syndrome (506). Again de novo mutations were found in exon 8 of Cav1.2. Biophysical studies revealed that these mutations produce sustained Ca2+ currents that lacked voltage-dependent inactivation. One mutation, G436R, in the rabbit Cav1.2 gene produces spontaneous mode 2 gating that depends on CaMKII-dependent protein phosphorylation (147). As such, it is predicted that these mutated Cav1.2 Ca2+ currents will persist during the action potentials of cardiac cells. Simulations showed that such a change in Ca2+ influx leads to abnormal Ca2+ dynamics and DADs. Presumably then, the arrhythmias in these individuals would be sensitive to Ca2+ channel blockers.
4. Ankyrin B mutations and arrhythmias
Congenital long QT syndrome has been associated with mutations in ion channels and recently in a large French family with mutations in the adaptor ankyrin B protein (381, 475). Affected family members show severe bradycardia and atrial fibrillation (475). Ankyrin B proteins are critical for anchoring other proteins to a specific cell location. For example, ankyrin B places Na+-K+ pumps, Na+/Ca2+ proteins, and IP3R proteins at t-tubule SR sites (379, 381). In mice where ankyrin B has been knocked down, some cardiac ion channels remain normal, but altered Ca2+ transients occur and with adrenergic stress action potentials with either early afterdepolarizations (EADs) or DADs occur (381). The human mutation of ankyrin B blocks the interaction of ankyrin B to its target effectors and would most likely lead to altered Ca2+ homeostasis and Ca2+-dependent arrhythmias.
Mutations of ankyrin G are associated with one form of the Brugada syndrome (380); however, arrhythmias have not as yet been linked to dysfunction of Ca2+.
5. Long QT syndromes and Ca2+-dependent arrhythmias
Long QT syndrome is a cardiac syndrome that is characterized by prolongation of the QT interval of the ECG. This prolongation is presumably due to APD prolongation of at least some of the ventricular cells. Patients with long QT can have specific ventricular arrhythmias such as torsade de pointes and ventricular fibrillation (VF). Genes associated with long QT syndrome in patients are KCNQ1, KCNH2, SCN5A, ankyrin B, KCNE1, and KCNE2. For the genes that encode ion channels, both gain-in-function and loss-in-function mutations have been described (444). It is thought that “triggers” that lead to the arrhythmias in these patients are due to disturbances of impulse initiation/conduction or both. Clearly from basic electrophysiological studies, it is known that just by prolonging the APD of a cardiac cell, one could set up conditions for reentry as well as increase the likelihood of EADs or DADs (110). But, are abnormal Ca2+ releases critically involved in the triggers of long QT-associated arrhythmias? Unfortunately, there are no Ca2+ data on cells from patients with these gene-based arrhythmias. Information so far has been from mouse models of long QT.
DeltaKPQ SCN5A causes a gain in Na+ channel function, and cells isolated from DeltaKPQ SCN5A knock-in mice generate EADs and cardiac arrhythmias particularly at rapid rates (394). No DADs were reported, and no Ca2+ imaging was performed to determine the role of intracellular Ca2+ in these EAD-generating cells. A more severe phenotype has been described in mice with a N1325S mutation of SCN5A (540). Again, these mice show the expected long QT, arrhythmias, and persistent Na+ currents. Cells from these mice showed EADs and late EADs (phase 3 EADs which could be due to Ca2+ waves) particularly at short pacing cycle lengths. Again, no DADs were observed in these cells, and Ca2+ was not imaged. However, if no other adaptive mechanisms exist in these mutant mice, then the prolonged APD secondary to persistent Na+ entry could in fact lead to increased Ca2+ influx, which in turn would increase SR stores and increase the likelihood of spontaneous intracellular Ca2+ releases, Ca2+ waves, and DADs.
B. Automaticity
1. Normal automaticity
Phasic nondriven rhythmic electrical activity occurs in various regions of the normal heart. The term normal automaticity refers to nondriven electrical activity of the sinoatrial nodal (SAN) cells, latent atrial pacemaker cells, and Purkinje fibers. Recent data combining voltage clamp with Ca2+-imaging techniques have implicated a role for Ca2+ in modulating the slope of phase 4 depolarization and thus automatic firing rates of the following cell types.
A) SAN Cells
In SAN cells, Ca2+ release channels are distributed in the cytoplasm as regular bands, localized to Z lines with some near to the sarcolemma (440). A highly localized subsarcolemmal Ca2+ release from the SR occurs concurrently with phase 4 depolarization (55, 231) and spreads in a wavelike manner by CICR (55). Ryanodine, which reduces conductance of the Ca2+ release channel in cardiac cells (459), slows the final phase of depolarization and thus pacemaker activity of cat, guinea pig, and rabbit SAN cells (55, 321, 440, 441). Furthermore, a compound used to chelate intracellular [Ca2+]i (BAPTA-AM) reduces Ca2+ transients and slows the firing rate of the SAN cell (321). Ryanodine also significantly reduces the positive chronotropic effect of β-adrenergic stimulation (304, 440) consistent with the idea that β-adrenergic stimulation modulates RyR Ca2+ release to augment sinus node firing rates (564), and that high basal cAMP levels of SAN cells are linked to this SR Ca2+ cycling (563). However, this finding is controversial (223), and fundamental work to show how the local Ca2+ releases lead to the global SAN Ca2+ transient has not been done. In fact, there is no information at this time on the fundamental basis of the SAN local Ca2+ release. Whether ryanodine reduces the contribution of Ca2+-dependent Na+/Ca2+ exchanger current (55), Ca2+-dependent T-type Ca2+ currents, or Ca2+-dependent IKs to phase 4 depolarization in SAN cells remains controversial at this time. Nevertheless, such evidence suggests that intracellular Ca2+ at least modulates SAN activity.
B) Atrial Pacemaker Cells
In cells from the pacemaker region of normal cat right atria, a late diastolic component of nondriven rhythmic activity depends on SR Ca2+ release (231, 461). In one study, a slow SR leak of Ca2+ during diastole provides persistent Ca2+ extrusion via the Na+/Ca2+ exchanger, which in turn generates inward current and atrial cell depolarization (461, 638). In normal cat atrial and ventricular myocytes, the rate of spontaneous Ca2+ leak from the SR is very low and thus no diastolic depolarization occurs (28). Interestingly, SAN cells do not show this diastolic Ca2+ efflux (28). In a combined confocal and voltage-clamp study of latent atrial pacemaker cells, the local release of Ca2+ from the SR occurring during late-phase diastolic depolarization is nickel sensitive, suggesting a role for the voltage-activated T-type Ca2+ channels in modulating latent pacemaker function (231). Agonists such as endothelin-1 increase the rate of spontaneous Ca2+ release as well as nondriven electrical events (342), suggesting that endothelin increases IP3 production, which subsequently sensitizes RyRs to release Ca2+ spontaneously. Spatially altered properties of the subcellular Ca2+ release also underlie arrhythmogenic events, such as Ca2+ waves, in cat atrial cells (283).
C) AV Nodal Cells
Morphologically, normal spontaneously active AV nodal cells show action potentials preceding large Ca2+ transients. After it peaks, the Ca2+ declines slowly, occurring even during the pacemaker depolarization. Ryanodine completely blocks AV node Ca2+ transients and presumably also abolishes the pacemaker activity (194). Thus the slow decline of Ca2+ during depolarization and subsequent pacemaker depolarization may activate Na+/Ca2+ exchanger in these cells (194).
D) Normal Purkinje Fibers
Data from several laboratories (76, 443) have clearly shown that the normally polarized individual canine Purkinje cell is quiescent and lacks normal automaticity in the absence as well as in the presence of catecholamines (443) despite the fact that If has been identified in this cell type (76). This is quite unlike adult SAN cells where the individual cell shows normal automaticity, and under voltage-clamp, If is prominent (240). Moreover, it appears that the minimal element needed for automaticity is a “dense pack” aggregate of Purkinje cells. In fact, in Purkinje cell aggregates from normal canine hearts, focally arising Ca2+ waves occur in the absence of electrical stimulation (64). These spontaneous Ca2+ waves appear to originate at cell borders similar to those in ventricular trabeculae (see below), can propagate the full extent of an aggregate, and often initiate membrane depolarization. In some cases, these depolarizations are accompanied by nondriven electrical activity of the well-polarized Purkinje cell (61). Thus spontaneous Ca2+ release clearly modulates normal Purkinje cell pacemaker function.
2. Abnormal automaticity
Fibers that become chronically depolarized show nondriven electrical activity that does not depend on an initiating beat. Diseased Purkinje as well as diseased human atrial fibers both show such abnormal automaticity. In one study, rates of firing of the abnormal foci were strongly modulated by agents that affect SR function (60, 148). Recent studies in atrial cells from both patients with and without atrial fibrillation (AF) as well as in cells from the rapid pacing model of AF have an increase in Ca2+ spark and Ca2+ wave frequency (225), as well as RyR2 channel dysfunction (561). Dysfunction of RyR2 in AF in the absence of adrenergic stimulation leads to an increase in the probability of opening of release channels (561). It is not clear whether such dysfunction in Ca2+ is accompanied by DADs and/or abnormal atrial cell automaticity. On the other hand, recent fluorescence data provide conclusive evidence that an increased frequency of micro Ca2+ transients in diseased Purkinje cell aggregates contribute to the abnormally automatic arrhythmias originating in the Purkinje network of the postmyocardial infarction heart (Fig. 6) (61). These frequent micro Ca2+ transients are sensitive to ryanodine, suggesting that in this model of acquired disease, spontaneous Ca2+ release is the fundamental abnormality leading to arrhythmias.
3. Triggered activity
A) Role of Calcium in EADS
While the role of propagating Ca2+ waves in membrane DADs is reasonably well accepted (see below), EADs in ventricular myocytes appear not to be due to a spontaneous regional increase in [Ca2+]i or propagating Ca2+ waves. Rather, during EADs, fluorescence transients show synchronous changes of [Ca2+]i throughout the myocyte lacking distinct high peaks (121, 373, 374, 567). These findings are consistent with the idea that a change in membrane potential primarily causes the observed increases in [Ca2+]i during an EAD. Evidence supporting a role for the L-type Ca2+ window current in the BAY K 8644-induced EADs in sheep Purkinje cells has been the demonstration of the appropriate voltage- and time-dependent properties of the whole cell L-type Ca2+ current as well as of its single-channel events (215, 247). In rabbit Purkinje cells, isoproterenol induces large EADs (20–30 mV) during which spatially uniform, circumferential rise in subsarcolemmal Ca2+ is observed (105). β-Adrenergic stimulation of canine ventricular cells produces EADs that are thought to be accompanied by spontaneous Ca2+ release (566). Early Ca2+ “aftertransients” or aftercontractions rise earlier than upstroke of change in membrane voltage of the EAD, suggesting that spontaneous Ca2+ release and nickel-sensitive inward Na+/Ca2+ exchanger current underlie the EADs (565). Simultaneous voltage and Ca2+ mapping data of rabbit hearts with experimental long QT syndrome suggest that during EADs in the epicardium, a rise of [Ca2+]i precedes the voltage rise by ∼20 ms in one focus while within millimeters the signals are synchronous (94). The actual amplitude of the Ca2+ signal that precedes the voltage signal is not known, and therefore, it is difficult to assess whether this “initiating” Ca2+ pulse is secondary to spontaneous Ca2+ release.
In other models, EADs have been shown to depend on Ca2+ loading and Na+/Ca2+ exchange current (412, 413) and/or CaMKII activity (12, 603). In the latter studies, action potential prolongation preceding EAD generation increases both L-type Ca2+ current and [Ca2+]i transient, while transient inward currents were associated with elevated [Ca2+]i. Although it is known that elevated [Ca2+]i can inactivate L-type Ca2+ current, its predominant effect is to further enhance Ca2+ currents through activation of CaMKII (12). In fact, recent experimental data suggest that CaMKII inhibitors suppress clofilium-induced EADs in the isolated heart and the appearance of Iti in rabbit myocytes (12, 603).
EADs occurring at potentials more negative than that of activation of L-type Ca2+ current have been called high membrane potential or phase 3 EADs (114, 413) and can be elicited after spontaneous termination of rapid pacing in isolated atrial preparations (73). Subcellular Ca2+ dynamics most probably underlie these depolarizations since they are augmented by an increased SR Ca2+ load. In fact, in two-dimensional simulations of LQT2 with increased sympathetic tone, spontaneous Ca2+ release in a region of the tissue generated an EAD which propagated, and due to the inhomogeneity of the substrate, initiated more nondriven electrical activity (228). Whether these Ca2+ abnormalities are in the form of spontaneous Ca2+ release and traveling Ca2+ waves is unknown at this time.
B) Arrhythmias, DADS, and Calcium Waves in Myocardium
Like in any arrhythmia, triggered arrhythmias both result from the previous impulse and lead to subsequent impulse generation. It has been shown that DADs are based on a spontaneous increase in [Ca2+]i leading to a transient inward current on the one hand and to activation of the contractile filaments on the other hand (158, 264). Kass et al. (264) proposed that a small [Ca2+]i transient, assumed to be due to “spontaneous” Ca2+ release from the SR, leads to a transient inward current. Hence, a sufficiently large Ca2+ load of the SR would create an unstable state where the spontaneous Ca2+ release could become so large that the resulting transient inward current would depolarize the cells sufficiently to trigger a new action potential, which would perpetuate itself as a triggered arrhythmia. Spontaneous Ca2+ release in individual cells in tissue has been shown to lead to DADs which are determined in amplitude by the amplitude of the local [Ca2+]i transient and by the spatial extent of this transient. Large and extensive Ca2+ transients are likely to generate a large Iti which may overcome the electrotonic drain of adjacent cells and generate a DAD of sufficient amplitude to trigger an ectopic beat.
I) Myocytes
Spontaneous Ca2+ release from the SR at submicrometer scale has been well-documented in both isolated dispersed cells and cardiac trabeculae using confocal microscopy. Regional Ca2+ waves occur after an action potential-induced synchronous Ca2+ transient in a myocyte accompanied by an aftercontraction and a DAD. Typically, the interval between the last stimulation and the onset of the first Ca2+ wave shortens and the probability of multiple foci of Ca2+ waves increases when the stimulus frequency or [Ca2+]o is increased (82). These observations are consistent with the concept that the increase in [Ca2+]i causes a transient net inward current and resulting in a DAD. Any of the [Ca2+]i-dependent currents described above might be involved in the generation of net inward current and most probably depends on the cell type.
Ca2+ waves usually start at one end of a myocyte, where one might envisage gap junctions. When a Ca2+ wave begins in a focus within a myocyte, it spreads at equal velocity in all directions (528). Sometimes Ca2+ waves emerge, as a domelike region of spontaneously elevated [Ca2+]i (300 nM) ∼20 μm in diameter, and propagate as a localized 10-μm-wide band of elevated [Ca2+]i (528, 592). Amplitude and width of Ca2+ waves are fairly constant during propagation (242, 528), and their velocity of propagation is typically ∼100 μm/s in quiescent cells (242, 302, 327, 510, 528, 587, 588, 592).
Ca2+ waves in isolated myocytes occur randomly with a frequency that may vary from <0.1 to ∼5 Hz, although remarkably stable intervals between spontaneous Ca2+ waves can be observed (241). In an individual cell, the frequency of Ca2+ waves increases monotonically with increased SR Ca2+ loading as does the number of foci (82). When Ca2+ waves start from two or more foci within a myocyte, the waves appear to collide without augmentation of [Ca2+]i. After the collision, [Ca2+]i declines without evidence of further wave propagation, demonstrating refractoriness of the propagation mechanism (39, 242). Thus Ca2+ waves are the consequence of a process with a “refractory period.” If an action potential is elicited during the propagation of a Ca2+ wave, the amplitude of the global Ca2+ transient and the accompanying twitch induced by that action potential are reduced by the preceding Ca2+ wave. The decrease of the Ca2+ transient of the action potential is more pronounced if the interval with the preceding Ca2+ wave transient is short (375). It appears that resultant twitch of the myocyte recovers with a time course similar to that of the mechanical restitution curve. This is indirect evidence that the spontaneous transient and twitch generation share the same mechanisms involved in intracellular Ca2+ cycling (83).
Traveling Ca2+ waves and membrane depolarizations occur in normal canine Purkinje cell aggregates (64) as well as in Purkinje cells that have survived in the border zone of an infarcted heart (61). In this latter study, the spatial extent, number, and duration of the spontaneously occurring Ca2+ waves present in the aggregate at any one time dictate the amplitude and duration of the membrane depolarization that occurs and thus the likelihood of triggering a nondriven electrical event (61). In rabbit Purkinje cells, transient inward current (Iti) underlying DADs was recorded both in the absence of spontaneous Ca2+ release and in the presence of variable spontaneous release (105). In an experimental model of disordered Ca2+ dynamics in left atrial preparations, Chou et al. (96) have shown that after rapid rates of pacing of the pulmonary vein (PV) sleeve area, spontaneous Ca2+ release, presumably from myocardial cells, precedes the depolarization of the PV cells. This leads to nondriven triggered activity in these cells which perpetuates the arrhythmic period (96). The mechanism of this focal spontaneous Ca2+ release is unknown, but presumably it is related to the rapid pacing-induced Ca2+ accumulation in the SR of PV cells. With the use of high-resolution calcium and voltage mapping (219), DADs and spontaneous Ca2+ release have now been mapped to the origin of the autonomically enhanced PV ectopy of the canine pulmonary veins.
To control SR Ca2+ release, Schlotthauer and Bers (474) applied a rapid caffeine superfusion to normal rabbit ventricle cells. The amplitude of the caffeine-induced membrane depolarization (cDADs) doubled for every 88 nM change in [Ca2+]i. Furthermore, if a cDAD voltage change of 12.5 mV occurred (∼424 nM Ca2+ change), an action potential was elicited. The cDAD-induced action potential threshold was altered if the membrane voltage change occurred faster (474). DADs are relatively easily induced in ventricular hypertrophy secondary to renal hypertension (17), to isoproterenol infusion (365), and in failing ventricular trabeculae (120, 560). In some cases, a role for Iti has been shown as the ionic mechanism and elevated [Ca2+]i has been implicated (365, 558). But traveling Ca2+ waves have only been illustrated in isoproterenol-stimulated cells (121) and trabeculae from the failing rat heart (120).
II) Cardiac muscle
ter Keurs and collaborators (115) discovered that only when cardiac muscle is damaged locally, such as by microelectrode impalement or dissection procedures, Ca2+ waves start near the damaged region and propagate in a coordinated fashion into adjacent tissue. These aftercontractions in multicellular preparations occur as the combined result of the mechanical effects and elevated cellular Ca2+ levels owing to the regional damage and thus may give rise to premature beats as well as triggered arrhythmias. These aftercontractions appear to be initiated by stretch and release of the damaged region during the regular twitch, and they propagate into neighboring myocardium, hence, the term triggered propagated contractions (TPCs). Damage-induced TPCs may, therefore, serve as the mechanism that couples regional damage with the initiation of premature beats and arrhythmias in the adjacent myocardium. The displacement of the TPC, or the Ca2+ wave that causes the TPC, occurs at a velocity of propagation (Vprop) along the long axis of the muscle which varies at room temperature from 0.1 to 15 mm/s (372, 386) and is correlated tightly with the amplitude of the twitch preceding the TPC, suggesting that the Ca2+ load of the SR dictates Vprop. In contrast, sarcomere stretch, which increases twitch force for any level of loading of the SR, does not increase Vprop of the TPC (118). Studies of the effects of interventions such as varied [Ca2+]o, Ca2+ channel agonists, and antagonists also support the idea that the Ca2+ load of the SR is the main determinant of Vprop (117). On the other hand, interventions that cause a leak of Ca2+ from the SR (caffeine and ryanodine) increase Vprop, suggesting that Vprop also depends on the diastolic cytosolic Ca2+ level (372). Finally, the rate of initiation of TPCs is tightly correlated with Vprop when the Ca2+ load of the SR is modulated, suggesting that the triggering process and the propagation process share closely related mechanisms.
III) Mechanisms underlying propagated Ca2+ waves
A) Initiation of Ca2+ waves in myocytes
Fabiato's work (152) on the properties of cardiac SR has provided a potential explanation for spontaneous Ca2+ release in isolated myocytes. He observed that in mechanically skinned cells in which the SR was intact, excessive Ca2+ loading of the SR caused spontaneous Ca2+ release (152). The mechanism for increased probability of opening of the SR-Ca2+ channel when the SR is heavily loaded with Ca2+ is still uncertain but suggests that the channel is directly or indirectly sensitive to the luminal [Ca2+] of the SR. The localization of a Ca2+ sensor in the transmembrane domain of the RyR channel would make it suitable as a sensor of both luminal and cytosolic [Ca2+]. Intact cells with a high SR-Ca2+ load show similar phenomena (82, 287). Hence, the oscillatory character of a triggered arrhythmia in myocardium with a high cellular Ca2+ load may be due to further increase of Ca2+ entry into the cells during the action potentials of the arrhythmia causing even more Ca2+ loading of the SR. Consequently, as soon as the release process has recovered after an electrically induced Ca2+ release, the overloaded SR again releases a fraction of its Ca2+. The requirement that the Ca2+ release mechanism must recover first would explain the presence of a delay between aftercontractions and afterdepolarizations to the preceding beat.
B) Initiation of TPCs in multicellular preparations
TPCs arise invariably in damaged regions of cardiac muscle. Spontaneous activity in the damaged zone is usually random; hence, the accompanying Ca2+ transients are small and do not propagate through the muscle, but can cause Ca2+ overload and spontaneous activity in the border zone. This process continues until the [Ca2+] gradient between cells is minimal or until gap junctions close (302). The existence of spontaneous SR Ca2+ release activity and contractions increases resting tension and decreases twitch force (287, 511). Thus twitch force of the damaged cells and cells of the border zone is less than that of the central region of the trabeculae. During an electrically evoked twitch, contraction of the central region of the normal trabeculae stretches the damaged region. During the rapid relaxation phase of the twitch, the stretched damaged region shortens suddenly. Stretch or quick release of damaged ends of trabeculae during the electrically driven twitch trigger TPCs (118) and may provide an explanation for the triggering mechanism. TPCs always start shortly after rapid shortening of damaged areas, suggesting that it is actually the shortening during relaxation that initiates a TPC. The observation (5, 6, 21, 224) that rapid shortening of a contracting muscle causes release of Ca2+ from the myofilaments provides a candidate mechanism for initiation of TPCs. In fact, Ca2+ that dissociate from the contractile filaments due to the quick release of the damaged areas during relaxation accelerate the initiation of a TPC and Ca2+ wave if CICR has recovered sufficiently to allow amplification of the initial Ca2+ transient in the damaged region and/or the border zone (571). Reduction of the afterload to <20% of twitch amplitude eliminates TPCs completely; this effect can be immediately reversed by forcing the muscle to contract again against a high afterload. At the same time, it is clear that as long as a TPC is observed, Vprop is not influenced by the manipulation of the afterload. So, apparently the probability of triggering a TPC depends on the force supported by the damaged area and the border zone and hence on the degree of stretch of these areas during the twitch. The TPC appeared to be initiated when force declined rapidly while the stretched areas shortened during the relaxation phase of the twitch. Studies of the contributing factors (nonuniform ECC vs. force decline in the stretched area vs. stretch of that area) suggest that rapid decline of force in the stretched area is responsible for rapid dissociation of Ca2+ from the contractile filaments, which initiates the Ca2+ waves (534, 570). These authors suggest that the rate of Ca2+ dissociation depends on force development and is significantly accelerated during a rapid reduction of force; elimination of force by externally unloading the muscle would eliminate accelerated Ca2+ dissociation from the myofilaments and hence eliminate the initiating event of the Ca2+ waves and TPCs (534, 570).
C) Propagation of Ca2+ waves
The fact that Ca2+ waves travel at a constant velocity and with constant amplitude through an isolated myocyte and/or a multicellular preparation provides an important clue about the mechanism of wave propagation. Diffusion of Ca2+ alone would clearly be too slow and would be accompanied by a decline of the observed wave amplitude. Propagation of electrical activity is much faster (1 m/s for the action potential in ventricular myocardium), and thus electrotonic conduction is too fast (∼0.1 m/s) to be compatible with the observed values of Vprop in trabeculae. A mechanism of Ca2+ wave propagation in cells has been proposed that consists of diffusion of Ca2+ due to the local increase of [Ca2+]i and subsequent CICR from adjacent SR, similar to the waves propagated by Ca2+ sparks. The transition from nonpropagating sparks to propagating sparks and a Ca2+ wave is possibly caused by an increase in Ca2+ sensitivity of the SR Ca2+ release channel as a consequence of greater SR Ca2+ loading (92).
Propagation with a constant velocity is consistent with a model of CICR propagated by Ca2+ diffusion along its concentration gradient to adjacent sarcomeres and adjacent cells (82, 118, 285, 386) and is supported by work on saponin-skinned muscle fibers which also exhibit propagating local contractions, suggesting the cell membrane is not essential for the phenomenon (115). The observation that neither initiation of TPCs nor their propagation is affected by gadolinium ions suggests that stretch-activated channels play little or no role in the initiation or propagation of damage-induced TPCs (632). As was shown by the lack of effects of varied afterload and varied sarcomere length on Vprop, it is unlikely that stretch of the myofibrils is essential to the propagation process.
The characteristics of Ca2+ waves and TPCs in trabeculae are quite similar. In addition, neither spontaneous activity in single myocytes nor TPCs in trabeculae require an intact sarcolemma, and both are abolished by agents that interfere with SR Ca2+ loading or release. On the other hand, at first glance, a striking difference between them is the propagation velocity. The velocity of Ca2+ waves in unstimulated cells is about 10 times lower than Vprop. However, TPCs are generated in cardiac muscle preparations at short intervals after the twitch such that their properties are affected by residual binding of Ca2+ to intracellular ligands (see sect. iiC). This is in contrast to the situation in myocytes where the moment of appearance of a Ca2+ wave following the twitch is both random and usually later. Hence, elimination of Ca2+ from the ligands during late diastole, after the Ca2+ extrusion processes have done their work, should reduce Vprop in muscle (386).
Vprop has been shown to vary experimentally from 0.1 to 6 mm/s, and maximal values of 15 mm/s have been observed. This speed is stunning if one realizes that only movement of a Ca2+ transient at speed lower than 0.03 mm/s can be explained by diffusion of Ca2+ based on the concentration gradient and the diffusion constant in an aqueous medium. The speed should be lower in the cytosol, since the effective diffusion constant is lower than in water owing to Ca2+ binding to, and dissociation from, ligands. Backx et al. (20) investigated which parameters of Ca2+ diffusion and CICR are required for the high Vprop in muscles by modeling the behavior of a myofibril accompanied by its SR during a sudden focal Ca2+ release. From the model we learned that Ca2+ transients propagate through the cytosol at a rate modified by binding to troponin and calmodulin and sequestration by the SR, as well as by the rate of Ca2+ release from adjacent release sites of the SR. Vprop increased indeed in the model from 0.1 to 15 mm/s if the “model cell” was loaded with Ca2+, which resulted in both a rise of the diastolic [Ca2+]i and the Ca2+ load of the SR. The former led to increased occupancy of the intracellular ligands with Ca2+ and, hence, an increase in the effective diffusion constant for Ca2+; the latter led to an increase of the amount of Ca2+ released by the SR and, assumedly, an increase of the rate of Ca2+ release (20). This combination would be expected to result from loading of cardiac cells with Ca2+ during repetitive stimulation as well as due to exposure to high [Ca2+]o or Ca2+ agonists (372, 386). An important conclusion on the basis of the model is that the observed range of Vprop could be achieved without postulating conduction of electrical or mechanical signals (20).
Although these observations have provided a reasonable framework for explanation of propagated Ca2+ waves, the model is still only a working model and many questions remain unanswered. For example, the mechanism of propagation of a Ca2+ wave from one cardiac cell to another has received little attention. It has been reported that there is apparently continuous propagation of a Ca2+ wave from one cell to another with no delay or change of velocity at the cell-to-cell junction (527). On the other hand, it has been noted that in myocytes without Ca2+ overload a local increase in [Ca2+]i using caged Ca2+ does not propagate (395) and that a Ca2+ wave induced by local application of caffeine decreases in both amplitude and velocity as it propagates along the cell (545). The high Vprop suggests that the barrier for Ca2+ diffusion imposed by gap junctions between cells is minor compared with the other parameters in the model such as Ca2+ binding to ligands in the cell and Ca2+ extrusion and sequestration processes. Zhang et al. (631) tested the importance of gap junctions to the properties of the TPCs in experiments where the trabeculae were exposed to the gap junction blockers heptanol and octanol. Although these compounds like many drugs have probably numerous side effects, their main effect is assumed to be a reduction of the open frequency of gap junctions. Exposure of the muscles to these alcohols decreased both the rate of initiation and Vprop dramatically with only a small decrease in twitch force (631). This suggests that closure of gap junctions reduces the rate of initiation and Vprop by reducing the effective rate of Ca2+ diffusion from cell to cell.
Second, since mitochondria are located in close proximity to RyR (436), it has been suggested that Ca2+ uptake via the mitochondrial uniporter may contribute to a local control of Ca2+ wave propagation (479).
IV) Propagated Ca2+ release elicits DADs
Whenever a TPC arises, it is accompanied by a depolarization similar to a DAD. It appears that the duration of the depolarization correlates exactly with the time during which the TPC travels through the trabeculae. The amplitude of the afterdepolarization also correlates exactly with the amplitude of the TPC (115). The tight correspondence between the time course of TPCs and those of the depolarizations suggests that the depolarization is elicited by a Ca2+-dependent current, which exists as long as the [Ca2+]i transient (wave) persists, as has been proposed by Kass et al. (264). In the small trabeculae used for TPC studies, this depolarization can be recorded over a distance of a few millimeters without much decrement due to electrotonic conduction. This assumption was verified experimentally by interrupting the propagation process of the TPC by locally heating the muscle. Local heating of the muscle caused the TPC to stop at the site of heating. In contrast, the concomitant depolarization could still be measured at a distance of ∼1 mm distal of the heating site (115) again as a result of electrotonic conduction of the DAD for which the current generators are located in the region with elevated [Ca2+]i. This observation clearly indicates that the depolarization cannot be the source of the TPC but must be induced by the TPC. The effect of local heating makes it also unlikely that TPCs are induced as a result of a linear gradient of Ca2+ overload along the muscle from a maximum in the damaged region to a minimum at the other end of the muscle. Such a gradient could potentially cause apparent propagation of a contraction if Ca2+ overload-induced Ca2+ release would occur along the muscle at a latency that is small in the damaged region and increases linearly toward the other end of the muscle.
A TPC accompanied by a DAD can become sufficiently large to elicit an action potential with twitch. The action potential triggered by the first TPC may add so much Ca2+ to the cell that a triggered arrhythmia starts. Triggered arrhythmias indeed occur in the damaged muscle when the Ca2+ load of the SR is large. At room temperature triggered arrhythmias occur during the first hour after damage to the muscle has occurred. In such a case, the full-blown arrhythmia is usually preceded by the repeated occurrence of single premature beats. At 37°C, the time span over which these damage-related events occur in human trabecula is much shorter, and the TPCs, which cause the premature beats, disappear in 10 min or less (116). Under those conditions it is likely that their occurrence is limited by rapid closure of gap junctions as a result of persistently elevated Ca2+ levels in the damaged cells. In addition, the pH in these cells may be low due to the enormous metabolic load resulting from intense ion movement across their membranes or across membranes of adjacent cells. The lowered pH may promote gap junction closure.
In the intact canine wedge preparation under conditions of enhanced Ca2+ influx, simultaneous voltage/Ca2+ imaging studies have shown that multiple, simultaneous spontaneous Ca2+ release events can occur, and when they occur, the calcium release starts from a group of cells and then propagates outward but within a 3- to 4-mm region (267) (Fig. 7). The amplitude and occurrence of these spontaneous Ca2+ release events are related to the region of the ventricle (endocardial cells more prone than epicardium) as well as to the region with the greatest accumulation of diastolic Ca2+ in this preparation. Presumably this is due to the slower uptake (less SERCA2a) of Ca2+ in endocardial cells (573). In an ischemia/reperfusion model in the isolated guinea pig heart, monomorphic VTs and VF have been associated with spontaneous Ca2+ release. In 17% of the cases, the onset of spontaneous Ca2+ release preceded epicardial depolarizations by 2–15 ms (305), implying reverse ECC and arrhythmogenesis. Such Ca2+ changes appeared confined to a local region (1.8 × 1.8 mm2) of these hearts.
Fig. 7.

Multiple simultaneous spontaneous Ca2+ release events in the isolated canine wedge preparation under Ca2+-loaded conditions. A: representative transmural map (endocardium to the left, epicardium to the right) depicting activity of imaged Ca2+. Imaged area is 14 mm × 14 mm. SCRamp means spontaneous calcium release amplitude as determined from recordings such as those shown in B. Relative fluorescent ratio units (ORU) are depicted by the various shades of gray in the bar. Note this map reveals that there are two “hot” spots of relatively high-amplitude SCR (denoted by a, c). Corresponding local Ca2+ transients during this time are seen in B. Note that while there is SCR near endocardium (site a) and midmyocardium (site c), there is no release at site d (epicardium). The SCRs are depicted as delayed after Ca2+ transients in B. [From Katra and Laurita (267).] C: one local Ca2+ transient in another preparation with selected images (0 to +660 ms) above showing the isochrones of Ca2+ levels during the inscription of the after Ca2+ transient. Note that SCR starts at one focus (∼1.2 mm2) and then during the course of the global transient propagates outward at ∼26 mm/s. At its maximum this SCR covers ∼35 mm2. (From Laurita laboratory, unpublished data.)
Additional studies have suggested a similar coupling between the mechanical events during the twitch calcium and membrane electrophysiology. Studies of both skeletal and cardiac muscle have shown that a quick release of the muscle during contraction causes rapid release of Ca2+ from contractile filaments (224). Lab et al. (293) provided a link between these observations by showing that quick releases can induce a [Ca2+]i transient accompanied by a DAD. Cardiac disease leads invariably to mechanical nonuniformity of myocardium. While the role of electrical nonuniformity of the myocardium in re-entry arrhythmias is well established, it is less well known to what extent nonuniform myocardial stress and strain distributions and nonuniform ECC may play a role in the initiation of extrasystoles that start arrhythmias. It is well known that tens of micromoles (per liter cell volume) of Ca2+ shuttle during the cardiac cycle between the SR and the cytosol where TnC is the dominant ligand. Hence, it is conceivable that nonuniformity of myocardium may lead to extrasystoles by several mechanisms including both abnormal SR Ca 2+ transport following damage and abnormal mechanical events in nonuniform myocardium, which cause dissociation of Ca2+ from TnC. It has been discussed how “spontaneous” SR Ca2+ release causes both transient inward currents and arrhythmogenic DADs as well as aftercontractions (see sect. ivB3). A sufficiently large SR Ca2+ load in cells at the rim of a damaged region could create an unstable state where spontaneous SR Ca2+ release may become so large that the resulting Iti depolarizes the cells enough to trigger a new action potential, which perpetuates itself as a triggered arrhythmia (110). Alternatively, events that result from the tug of war between normal myocardium and weak cells in the ischemic zone could trigger the Ca2+ release and lead to arrhythmias. This tug of war may play a role in Ca2+ release, triggered in damaged regions of isolated rat ventricular and human atrial trabeculae, resulting in Ca2+ release that appears to be initiated after stretch of the damaged region during the regular twitch and propagates into neighboring myocardium by the combination of Ca2+ diffusion and Ca2+-induced SR Ca2+ release.
Experimentally, a mechanical discontinuity along the trabeculae has been created by exposing the preparation to a small constant flow of solution with a composition that reduces ECC in myocytes only within that segment (570) (Fig. 8). Force, sarcomere length, as well as [Ca2+]i were measured regionally. When the jet contained caffeine, butane-dione-monoxime (BDM), or low [Ca2+], muscle-twitch force decreased and the sarcomeres in the exposed segment were stretched by the shortening of the normal regions outside the jet. During relaxation the sarcomeres in the exposed segment shortened rapidly. Short trains of stimulation at 2.5 Hz reproducibly caused Ca2+ waves to rise from the borders exposed to the jet. Interestingly, these Ca2+ waves started during force relaxation of the last stimulated twitch and propagated into segments both inside and outside of the jet. Arrhythmias, in the form of nondriven rhythmic activity, were triggered when the amplitude of the Ca2+ wave increased by raising [Ca2+]o. The arrhythmias disappeared when the muscle uniformity was restored by turning the jet off (570). The authors have used the four-state model of the cardiac cross-bridge (Xb) with feedback of force development to Ca2+ binding by TnC and observed that the force-Ca2+ relationship as well as the force-sarcomere length relationship and the time course of the force and Ca2+ transients in cardiac muscle can be reproduced faithfully by a single effect of force on deformation of the TnC-Ca2+ complex and thereby on the dissociation rate of Ca2+. Importantly, this feedback predicts that rapid decline of force in the activated sarcomere causes release of Ca2+ from TnC-Ca2+, which is sufficient to initiate arrhythmogenic Ca2+ release from the SR. These results show that nonuniform contraction can cause Ca2+ waves underlying TPCs and suggest that Ca2+ dissociated from myofilaments plays an important role in the initiation of arrhythmogenic Ca2+ waves (534, 570).
Fig. 8.

Initiation of Ca2+ waves in experimental multicellular preparation of nonuniform excitation-contraction coupling. A: Ca2+ waves induced by a local BDM exposure at various Ca2+ concentrations (1, 2, and 4 mM). BDM is delivered to the trabecula via a jet system that superfuses the region denoted by the dotted lines. Note that increasing Ca2+ led to the initiation of Ca2+ waves that propagate into the segment inside the jet and into the normal muscle. Both the amplitude of the initial and propagating Ca2+ transient as well as propagation velocity increased with increase in Ca2+. Arrows indicate initiation sites. B: collision of Ca2+ waves in the jet region (white arrow). C: Ca2+ traces, F records, and SL (sarcomere records) from average profiles indicated by the square bracket in B. Note that the onset of the initial Ca2+ rise of the wave in B (denoted by the black arrow in B) corresponds with the time at which the twitch had relaxed to 10% (F onset) and late during relaxation. [From Wakayama et al. (570).]
Arrhythmogenic Ca2+ waves underlying triggered propagated contractions arise from Ca2+-overloaded regions near damaged areas in the cardiac muscle. Ca2+ waves can also be induced in undamaged muscle, in regions with nonuniform ECC by the cycle of stretch and release in the border zone (BZ) between the damaged and intact regions. The same authors studied the hypothesis that rapid shortening of sarcomeres in BZ during relaxation causes Ca2+ release from TnC on thin filaments and initiates Ca2+ waves and went on to test whether elimination of this shortening will inhibit the initiation of Ca2+ waves, while SR Ca2+ overload will enhance the waves. In this study, increase of [Ca2+]i during TPCs was observed only after quick release of the muscle that followed a short (∼200 ms) stretch during twitch. This observation confirmed our hypothesis that Ca2+ dissociated from TnC plays an important role in the acceleration of Ca2+ waves underlying TPCs (571). When a small jet of HEPES solution with 10 mM [Ca2+], or [Ca2+]o containing BDM, was used to weaken a small muscle segment (10% of muscle length), the high [Ca2+]o jet induced spontaneous diastolic sarcomere contractions in the jet region while attenuating the twitch sarcomere shortening outside the jet-stretched region. Stimulus trains induced Ca2+ waves inside the high [Ca2+]o jet region upon twitch relaxation by 60%; Ca2+ waves started in the border zone of the BDM jet. The initial local [Ca2+]i rise of the waves by high [Ca2+]o was twice that by BDM. And arrhythmias occurred frequently (40%) in trabeculae after exposure to the high-[Ca2+]o jet. Exponential stretches (10% muscle length) early during relaxation reduced sarcomere shortening in the weakened segment of the muscles and decreased force of a triggered propagating contraction (FTPC) by 50%, and both delayed and reduced Vprop commensurate with the reduction FTPC. The results are consistent with the hypothesis that Ca2+ release from TnC initiates arrhythmogenic propagating Ca2+ release in mechanically nonuniform myocardium. Prevention of sarcomere shortening reduces the Ca2+ dissociation from TnC, and initiation and propagation of these Ca2+ waves would be potentiated by high SR-Ca2+ overload.
C. Re-entrant Excitation
1. Ca2+ and impulse propagation
Important components of reentrant excitation are impulse propagation and unidirectional block. Both theoretical and experimental models (257, 338, 446, 486) suggest that under some circumstances the L-type Ca2+ current and [Ca2+]i can affect cardiac impulse propagation. In neuronal tissues activity-dependent modulation of action potentials occurs as Ca2+ accumulates in the cell. Action potential propagation is slowed with a sudden flash-induced increase of [Ca2+]i (338). Ca2+ accumulation during action potential propagation impedes propagation probably due to [Ca2+]i-dependent inactivation of the L-type Ca2+ current in settings where the Ca2+ currents of neuronal action potentials are reduced and improves conduction at sites of impedance mismatch (338). Similar studies have now been completed in patterned growth cultures of neonatal myocytes (446) and adult cell pair preparations (257). Importantly, these latter studies emphasize that the L-type Ca2+ current of the myocyte at the region of a current-to-load mismatch can become essential for impulse propagation (or block) (446). Reversal of such block can be accomplished by using BAY K 8644, an agonist of the L-type Ca2+ current. In an experimental cell model of action potential conduction, discontinuous action potential propagation increases the peak Ca2+ current of cells while also causing a brief increase in the cellular Ca2+ transient (569).
The critical relationship between altered [Ca2+]i to discontinuities of conduction in arrhythmogenic substrates such as those of acute ischemia or those during the healing/healed phase postmyocardial infarction is not clear at this time. However, the mechanism of “pseudo-block” during reentrant arrhythmias in the healing infarcted heart may depend not only on the cell surface redistribution of gap junction proteins, but also on the loss of function of the L-type Ca2+ channels and its subsequent impact of intracellular Ca2+ cycling (19). In mapping studies of reentrant excitation in ventricles postmyocardial infarction, an L-type Ca2+ current agonist is antiarrhythmic (75), presumably due to its effect to increase the amplitude of the remodeled L-type Ca2+ currents and [Ca2+]i in the border zone myocytes that form the substrate of these reentrant arrhythmias (433).
If altered Ca2+ can initiate arrhythmias, how does elevated [Ca2+]i contribute to perpetuation of a re-entrant arrhythmia? Two types of studies have been completed in an attempt to address this question. First, simulations of spiral waves have shown that they destabilize when [Ca2+]i transient nonuniformities are allowed to occur (98). The result is a fibrillatory pattern of excitation. However, when both [Ca2+]i and voltage were measured experimentally during VF in the nonremodeled heart, there appears to be a loss of association of phasic [Ca2+]i transients with fibrillatory waves (400, 598), suggesting that [Ca2+]i is not essential for VF maintenance.
2. Ca2+ and gap junction permeability
Intercellular electrical transmission occurs via a set of ion channel proteins and specialized membrane structures called gap junctions. Each channel is formed by close apposition of two hemichannels each of which is in an opposing cell (618). Gap junctions can provide passage of many molecules (cAMP, Ca2+, IP3, ATP) up to 1 kDa in size (56, 462, 470). In cardiac cells, gap junctional conductance can be regulated acutely by pH, Ca2+, cAMP, and cGMP (449). Therefore, Ca2+ can be both flowing through gap junctions as well inhibiting their conductance. In the first case, Ca2+ released upon RyR activation can travel as a wave across a cell (631) and propagate to adjoining cells via gap junctions (513). In cells transfected with both connexin43 and RyR receptors propagation of Ca2+ waves between cells is sensitive to octanol (544). Furthermore, in this experimental cell model, both Ca2+ wave propagation and gap junctional conductance between a pair of cardiac cells are related to the state of tyrosine phosphorylation of connexin43 (543). How the Ca2+ wave crosses the gap junction is unknown, but extracellular disulfide bonds of the connexin43 proteins between the adjoining cells appear critical for wave propagation (544).
D. Nonuniform ECC and Electromechanical Alternans
T-wave alternans as well as QT alternans are ECG descriptors that are characterized by beat-to-beat changes in T-wave morphology as well as the QT interval. Both measures are positive prognostic indicators of arrhythmic events (451, 452, 501, 546). Although both observations have been linked to spatial and temporal heterogeneity of repolarization of action potentials in the heart (408, 409), spatial and temporal changes in intracellular Ca2+ within myocytes have also been implicated (432). Several recent reviews discuss the nomenclature and current understanding of the relationship between Ca2+ and voltage in alternans (143, 150, 450, 471, 583). In fact, in the nonremodeled guinea pig heart, alternans of both APD and [Ca2+]i occur preferentially at the base of the heart (432). These latter data imply that the normal spatial heterogeneity of Ca2+ transients (a base to apex gradient) is potentially a cellular mechanism of Ca2+ alternans (93, 266, 573). Mechanical alternans accompanies [Ca2+]i transient alternans (218, 273, 294, 460), and both are sensitive to agents that affect SR function (e.g., ryanodine). In atrial cells Ca2+ transient alternans can be seen where there was failure of Ca2+ wave propagation from the periphery to the cell's center every other stimulated beat (233). In fact, cellular Ca2+ alternans has been related to the amount of Ca2+ released from the SR, which is determined by the trigger (L-type Ca2+ current), the SR load, and the metabolic and phosphorylation state of the RyR. Some have also suggested that diastolic fluctuations of SR Ca2+ occur during alternans, and this leads to Ca2+ transient alternans. This has recently been examined directly (421), and these results suggest that factors other than SR load are critical for frequency-related Ca2+ alternans in a single cell.
Mechanical alternans would be expected on the basis of APD alternans in a model of ECC as described in Reference 195. The model predicts two types of alternans, i.e., the alternans in which APD and force increase and decrease together, i.e., “in-phase” alternans, or the changes in APD occur “out of phase” with those of force. The amount of Ca2+ released during any beat depends on the Ca2+ influx during the preceding action potential. This is true for myocytes with a robust SR function; in cells with a diminished SR Ca2+ storage ability, APD determines concomitant Ca2+ release directly. Acute changes in [Ca2+]i affect the membrane currents (see above) and thus provide a feedback mechanism that controls APD of the concurrent beat. Furthermore, at high heart rates (at which alternans is usually found), relaxation may be incomplete. As a result of the elevated [Ca2+]i, ligands in the cytosol including the SR Ca2+ pumps would be occupied and the effect of variation of Ca2+ influx would be more pronounced. Alternans also occurs in metabolically compromised muscle. Hence, the force of each contraction may influence the cytosolic phosphate level; the latter will reduce force of the next contraction (271). Force of contraction increases the sensitivity of TnC for Ca2+. As a consequence, elevated cytosolic phosphate levels will reduce the binding of released Ca2+ to the contractile filaments. Perhaps this is related to the Ca2+ transient alternans of ischemia (434).
Triggered propagated contractions could be also involved in the generation of a mechanical alternans. Here a strong contraction would trigger a substantial Ca2+ release that propagates as TPC. The strong TPC would reduce the Ca2+ release during the next electrically driven beat. This beat would be weak and followed by a small TPC. The subsequent beat would be strong again. If the [Ca2+]i feedback mechanism to membrane currents described above is operational, one would expect an “out of phase” alternans with evidence of DADs of varying amplitude. Elimination of SR function would eliminate this alternans. Recent data regarding alternans produced by small depolarizing steps may reflect this, since alternans in this case was due to large Ca2+ transients resulting from intracellular wave propagation (134).
In a recent study discordant electrical alternans was described when membrane repolarization alternated with the opposite phase between groups of neighboring cells (408). This type of alternans was directly linked to the formation of unidirectional lines of block and reentrant ventricular fibrillation. Whether mechanical alternans accompanies or causes these arrhythmogenic beats remains unknown at this time. While some believe that heterogeneities in electrical restitution properties are central to the basis for discordant alternans, we still underappreciate the role of nonuniform ECC and/or the intrinsic variability of the Ca2+ transient across the ventricular wall (266). In fact, when normal cells which show APD/Ca2+ transient alternans are voltage clamped with an action potential waveform of fixed duration, Ca2+ transient alternans persist (98, 177). Studies combining optical voltage and Ca2+ fluorescence measures indicate that the shapes of the Ca2+ transients recorded from cells within 1.1 mm in the intact heart are similar but differ significantly from alternans observed with acute ischemia (101, 434, 435). Furthermore, recent evidence points to a role of Ca2+ cycling rather than electrical restitution in T-wave alternans in normal hearts (93, 432).
V. Summary
Ca2+ play a key role in both the short- and long-term properties of cardiac cells and are thus involved in the development of arrhythmias. The nature of the many mechanisms via which Ca2+ exerts its effects is by no means fully understood. The notion that these ions do play a key role in the development of arrhythmias is not surprising. The structure of cardiac cells enables rapid electrical conduction as well as rapid activation of the contractile system even though diffusion of Ca2+ is slow. Nature has, therefore, provided amplification stations between the sarcolemma and the myofibrils so that both the delivery and the removal of Ca2+ is accelerated. Simultaneously, the Ca2+ sensitivity of many proteins in the cardiac cell is so high such that activation of contractile proteins occurs at [Ca2+]i only slightly above the diastolic level. Furthermore, [Ca2+]i affects the electrical processes at the surface membrane profoundly. It is therefore plausible that instability of the Ca2+ transport systems is involved in the mechanisms that lead to overall instability of the tissue during arrhythmias. In addition, Ca2+ plays an important role in the long-term life of the cardiac cell by affecting both the composition and cellular distribution of proteins which dictate the phenotype of the cell. Turnover of these proteins is so fast that it is likely that the very factors which determine the initiation of an arrhythmia may themselves change the cardiac cell phenotype and thus alter the cell's future response to the same factors. Solving the nature of these intricate and dynamic interactions promises to be an important area of research for a better recognition and understanding of the nature of Ca2+ and arrhythmias. In so doing, our solutions will provide a more complete understanding of the molecular basis for the targeted control of cellular calcium in the treatment and prevention of such.
Acknowledgments
We thank Dr. Wen Dun for helping with this paper.
Address for reprint requests and other correspondence: P. A. Boyden, Dept of Pharmacology, Columbia College of Physicians and Surgeons, 630 West 168th St., New York, NY 10032 (pab4@columbia.edu).
GRANTS: This work was supported by National Heart, Lung, and Blood Institute Grants HL-58860 and HL-66140, Canadian Institutes for Health Research, Alberta Heritage Foundation for Medical Research, and a NATO Collaborative Research Grant.
References
- 1.Abramcheck CW, Best PM. Physiological role and selectivity of the in situ potassium channel of the sarcoplasmic reticulum in skinned frog skeletal muscle fibers. J Gen Physiol. 1989;93:1–21. doi: 10.1085/jgp.93.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adkins CE, Morris SA, De Smedt H, Sienaert I, Torok K, Taylor CW. Ca2+-calmodulin inhibits Ca2+ release mediated by type-1, -2 and -3 inositol trisphosphate receptors. Biochem J. 2000;345:357–363. [PMC free article] [PubMed] [Google Scholar]
- 3.Aggarwal R, Boyden PA. Diminished calcium and barium currents in myocytes surviving in the epicardial border zone of the 5 day infarcted canine heart. Circ Res. 1995;77:1180–1191. doi: 10.1161/01.res.77.6.1180. [DOI] [PubMed] [Google Scholar]
- 4.Allen DG, Eisner DA, Orchard CH. Characterization of oscillations of intracellular calcium concentration in ferret ventricular muscle. J Physiol. 1984;352:113–128. doi: 10.1113/jphysiol.1984.sp015281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allen DG, Kentish JC. Calcium concentration in the myoplasm of skinned ferret ventricular muscle following changes in muscle length. J Physiol. 1988;407:489–503. doi: 10.1113/jphysiol.1988.sp017427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allen DG, Kurihara S. The effects of muscle length on intracellular calcium transients in mammalian cardiac muscle. J Physiol. 1982;327:79–94. doi: 10.1113/jphysiol.1982.sp014221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alvarez JL, Vassort G. Properties of the low threshold Ca2+ current in single frog atrial cardiomyocytes. A comparison with the high threshold Ca2+ current. J Gen Physiol. 1992;100:519–545. doi: 10.1085/jgp.100.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amos GJ, Wettwer E, Metzger F, Li Q, Himmel HM, Ravens U. Differences between outward currents of human atrial and subepicardial ventricular myocytes. J Physiol. 1996;491:31–50. doi: 10.1113/jphysiol.1996.sp021194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.An WF, Bowlby MR, Betty M, Cao J, Ling H, Mendoza G, Hinson JW, Mattson KI, Strassle BW, Trimmer JS, Rhodes KJ. Modulation of A type potassium channels by a family of calcium sensors. Nature. 2000;403:553–556. doi: 10.1038/35000592. [DOI] [PubMed] [Google Scholar]
- 10.Anderson ME. Ca2+ dependent regulation of cardiac L type Ca2+ channels: is a unifying mechanism at hand? J Mol Cell Cardiol. 2001;33:639–650. doi: 10.1006/jmcc.2000.1354. [DOI] [PubMed] [Google Scholar]
- 11.Anderson ME, Braun AP, Schulman H, Premack BA. Multifunctional Ca2+/calmodulin dependent protein kinase mediates Ca2+-induced enhancement of the L-type Ca2+ current in rabbit ventricular myocytes. Circ Res. 1994;75:854–861. doi: 10.1161/01.res.75.5.854. [DOI] [PubMed] [Google Scholar]
- 12.Anderson ME, Braun AP, Wu Y, Lu T, Wu Y, Schulman H, Sung RJ. KN-93, an inhibitor of multifunctional Ca2+/calmodulin-dependent protein kinase, decreases early afterdepolarizations in rabbit heart. J Pharmacol Exp Ther. 1998;287:996–1006. [PubMed] [Google Scholar]
- 13.Antoine S, Lefevre T, Coraboeuf E, Nottin R, Coulombe A. B-type Ca2+ channels activated by chlorpromazine and free radicals in membrane of human atrial myocytes. J Mol Cell Cardiol. 1998;30:2623–2636. doi: 10.1006/jmcc.1998.0818. [DOI] [PubMed] [Google Scholar]
- 14.Argibay JA, Fischmeister R, Hartzell HC. Inactivation, reactivation and pacing dependence of calcium current in frog cardiocytes: correlation with current density. J Physiol. 1988;401:201–226. doi: 10.1113/jphysiol.1988.sp017158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arikkath J, Campbell KP. Auxiliary subunits: essential components of the voltage gated calcium channel complex. Curr Opin Neurobiol. 2003;13:298–307. doi: 10.1016/s0959-4388(03)00066-7. [DOI] [PubMed] [Google Scholar]
- 16.Armoundas AA, Hobai IA, Tomaselli GF, Winslow RL, O'Rourke B. Role of sodium-calcium exchange in modulating the action potential of ventricular myocytes from normal and failing hearts. Circ Res. 2003;93:46–53. doi: 10.1161/01.RES.0000080932.98903.D8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aronson RS. Afterpotentials and triggered activity in hypertrophied myocardium from rats with renal hypertension. Circ Res. 1981;48:720–727. doi: 10.1161/01.res.48.5.720. [DOI] [PubMed] [Google Scholar]
- 18.Baader AP, Buchler L, Bircher-Lehmann L, Kleber AG. Real time, confocal imaging of Ca2+ waves in arterially perfused rat hearts. Cardiovasc Res. 2002;53:105–115. doi: 10.1016/s0008-6363(01)00423-0. [DOI] [PubMed] [Google Scholar]
- 19.Baba S, Dun W, Cabo C, Boyden PA. Remodeling in cells from different regions of the reentrant circuit during ventricular tachycardia. Circulation. 2005;112:2386–2396. doi: 10.1161/CIRCULATIONAHA.105.534784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19a.Babuty D, Lab MJ. Heterogeneous changes of monophasic action potential induced by sustained stretch in atrium. J Cardiovasc Electrophysiol. 2001;12:329. doi: 10.1046/j.1540-8167.2001.00323.x. [DOI] [PubMed] [Google Scholar]
- 20.Backx PH, de Tombe PP, van Deen JHK, Mulder BJM, ter Keurs HEDJ. A model of propagating calcium-induced calcium release mediated by calcium diffusion. J Gen Physiol. 1989;93:963–977. doi: 10.1085/jgp.93.5.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Backx PH, Gao WD, Azan-Backx MD, Marban E. The relationship between contractile force and intracellular Ca2+ in intact rat cardiac trabeculae. J Gen Physiol. 1995;105:1–19. doi: 10.1085/jgp.105.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Backx PH, ter Keurs HEDJ. Fluorescent properties of rat cardiac trabeculae microinjected with fura-2 salt. Am J Physiol Heart Circ Physiol. 1993;264:H1098–H1110. doi: 10.1152/ajpheart.1993.264.4.H1098. [DOI] [PubMed] [Google Scholar]
- 23.Bagattin A, Veronese C, Bauce B, Wuyts W, Settino L, Nava A, Rampazzo A, Danieli GA. Denaturing HPLC based approach for detecting RYR2 mutations involved in malignant arrhythmias. Clin Chem. 2004;50:1148–1155. doi: 10.1373/clinchem.2003.030734. [DOI] [PubMed] [Google Scholar]
- 24.Bai CX, Namekata I, Kurokawa J, Tanaka H, Shigenobu K, Furukawa T. Role of nitric oxide in Ca2+ sensitivity of the slowly activating delayed rectifier K current in cardiac myocytes. Circ Res. 2005;96:64–72. doi: 10.1161/01.RES.0000151846.19788.E0. [DOI] [PubMed] [Google Scholar]
- 25.Bangalore R, Mehrke G, Gingrich K, Hofmann F, Kass RS. Influence of L-type Ca channel Alpha2/Delta subunit on ionic and gating current in transiently transfected HEK293 cells. Am J Physiol Heart Circ Physiol. 1996;270:H1521–H1528. doi: 10.1152/ajpheart.1996.270.5.H1521. [DOI] [PubMed] [Google Scholar]
- 26.Barg S, Copello JA, Flesicher S. Different interactions of cardiac and skeletal muscle ryanodine receptors with FK-506 binding protein isoforms. Am J Physiol Cell Physiol. 1997;272:C1726–C1733. doi: 10.1152/ajpcell.1997.272.5.C1726. [DOI] [PubMed] [Google Scholar]
- 27.Bassani JW, Yuan W, Bers D. Fractional SR Ca2+ release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am J Physiol Cell Physiol. 1995;268:C1313–C1319. doi: 10.1152/ajpcell.1995.268.5.C1313. [DOI] [PubMed] [Google Scholar]
- 28.Bassani RA, Bassani JWM, Lipsius SL, Bers DM. Diastolic SR Ca efflux in atrial pacemaker cells and Ca-overloaded myocytes. Am J Physiol Heart Circ Physiol. 1997;273:H886–H892. doi: 10.1152/ajpheart.1997.273.2.H886. [DOI] [PubMed] [Google Scholar]
- 29.Bauce B, Nava A, Rampazzo A, Daliento L, Muriago M, Basso C, Thiene G, Danieli GA. Familial effort polymorphic ventricular arrhythmias in arrhythmogenic right ventricular cardiomyopathy map to chromosome 1q42–43. Am J Cardiol. 2000;85:573–579. doi: 10.1016/s0002-9149(99)00814-0. [DOI] [PubMed] [Google Scholar]
- 30.Bauce B, Rampazzo A, Basso C, Bagattin A, Daliento L, Tiso N, Turrini P, Thiene P, Danieli GA, Nava A. Screening for ryanodine receptor type 2 mutations in families with effort induced polymorphic ventricular arrhythmias and sudden death: early diagnosis of asymptomatic carriers. J Am Coll Cardiol. 2002;40:341–349. doi: 10.1016/s0735-1097(02)01946-0. [DOI] [PubMed] [Google Scholar]
- 31.Bean BP. Two kinds of calcium channels in canine atrial cells: differences in kinetics, selectivity, pharmacology. J Gen Physiol. 1985;86:1–30. doi: 10.1085/jgp.86.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beard NA, Casarotto MG, Wei L, Varsanyi M, Laver DR, Dulhunty AF. Regulation of ryanodine receptors by calsequestrin: effect of high luminal Ca2+ and phosphorylation. Biophys J. 2005;88:3444–3454. doi: 10.1529/biophysj.104.051441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beguin P, Nagashima K, Gonol T, Shibasaki T, Takahashi K, Kashima Y, Ozaki N, Geering K, Iwanaga T, Seino S. Regulation of Ca2+ channel expression at the cell surface by the small G-protein Kir/Gem. Nature. 2001;411:701–706. doi: 10.1038/35079621. [DOI] [PubMed] [Google Scholar]
- 34.Belus A, White E. Streptomycin and intracellular calcium modulate the response of single guinea-pig ventricular myocytes to axial stretch. J Physiol. 2003;546:501–509. doi: 10.1113/jphysiol.2002.027573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benacquista BL, Sharma MR, Samso M, Zorzato F, Treves S, Wagenknecht T. Amino acid residues 4425–4621 localized on the three-dimensional structure of the skeletal muscle ryanodine receptor. Biophys J. 2000;78:1349–1358. doi: 10.1016/S0006-3495(00)76689-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bence-Hanulec KK, Marshall J, Blair LA. Potentiation of neuronal L calcium channels by IGF-1 requires phosphorylation of the alpha1 subunit on a specific tyrosine residue. Neuron. 2000;27:121–131. doi: 10.1016/s0896-6273(00)00014-3. [DOI] [PubMed] [Google Scholar]
- 37.Berjukow S, Marksteiner R, Sokolov S, Weiss R, Margreiter E, Hering S. Amino acids in segment IVS6 and beta subunit interaction support distinct conformational changes during Cav2.1 inactivation. J Biol Chem. 2001;276:17076–17082. doi: 10.1074/jbc.M010491200. [DOI] [PubMed] [Google Scholar]
- 38.Berlin JR. Spatiotemporal changes of Ca2+ during electrically evoked contractions in atrial and ventricular cells. Am J Physiol Heart Circ Physiol. 1995;269:H1165–H1170. doi: 10.1152/ajpheart.1995.269.3.H1165. [DOI] [PubMed] [Google Scholar]
- 39.Berlin JR, Cannell MB, Lederer WJ. Cellular origins of the transient inward current in cardiac myocytes. Role of fluctuations and waves of elevated intracellular calcium. Circ Res. 1989;65:115–126. doi: 10.1161/01.res.65.1.115. [DOI] [PubMed] [Google Scholar]
- 40.Bernatchez G, Talwar D, Parent L. Mutations in the EF-hand motif impair the inactivation of barium currents of the cardiac alpha1C channel. Biophys J. 1998;75:1727–1739. doi: 10.1016/S0006-3495(98)77614-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berrou L, Bernatchez G, Parent L. Molecular determinants of inactivation within the I-II linker of alpha1E (CaV2.3) calcium channels. Biophys J. 2001;80:215–228. doi: 10.1016/S0006-3495(01)76008-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bers D. Sarcoplasmic reticulum calcium uptake, content and release. In: Bers D, editor. Excitation-Contraction Coupling and Cardiac Contractile Force. London: Kluwer Academic; 2002. pp. 203–244. [Google Scholar]
- 43.Bers DM. Calcium fluxes involved in control of cardiac myocyte contraction. Circ Res. 2000;87:275–281. doi: 10.1161/01.res.87.4.275. [DOI] [PubMed] [Google Scholar]
- 44.Bers DM. Macromolecular complexes regulating cardiac ryanodine receptor function. J Mol Cell Cardiol. 2004;37:417–429. doi: 10.1016/j.yjmcc.2004.05.026. [DOI] [PubMed] [Google Scholar]
- 45.Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. Dordrecht, The Netherlands: Kluwer Academic; 1991. [Google Scholar]
- 46.Bers DM, Bridge JHB. Relaxation of rabbit ventricular muscle by Na-Ca exchange and sarcoplasmic reticulum Ca-pump: ryanodine and voltage sensitivity. Circ Res. 1989;65:334–342. doi: 10.1161/01.res.65.2.334. [DOI] [PubMed] [Google Scholar]
- 47.Bers DM, Bridge JHB, Spitzer KW. Intracellular calcium transients during rapid cooling contractures in guinea pig myocytes. J Physiol. 1989;417:537–553. doi: 10.1113/jphysiol.1989.sp017817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bers DM, Stiffel VM. Ratio of ryanodine to dihydropyridine receptors in cardiac and skeletal muscle and implications for EC coupling. Am J Physiol Cell Physiol. 1993;264:C1587–C1593. doi: 10.1152/ajpcell.1993.264.6.C1587. [DOI] [PubMed] [Google Scholar]
- 49.Bers DM, Eisner DA, Valdivia HH. Sarcoplasmic reticulum Ca2+ and heart failure: roles of diastolic leak and Ca2+ transport. Circ Res. 2003;93:487–490. doi: 10.1161/01.RES.0000091871.54907.6B. [DOI] [PubMed] [Google Scholar]
- 50.Beuckelmann DJ, Nabauer M, Erdmann E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation. 1992;85:1046–1055. doi: 10.1161/01.cir.85.3.1046. [DOI] [PubMed] [Google Scholar]
- 51.Beuckelmann DJ, Wier WG. Sodium-calcium exchange in guinea pig cardiac cells: exchange current and changes in intracellular Ca2+ J Physiol. 1989;414:499–520. doi: 10.1113/jphysiol.1989.sp017700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Birnbaumer L, Qin N, Olcese R, Tareilus E, Platano D, Costantin J, Stefani E. Structure and functions of calcium channel β subunits. J Bioenerg Biomembr. 1998;30:357–375. doi: 10.1023/a:1021989622656. [DOI] [PubMed] [Google Scholar]
- 53.Blatter LA, Huser J, Rios E. Sarcoplasmic reticulum Ca2+ release flux underlying Ca2+ sparks in cardiac muscle. Proc Natl Acad Sci USA. 1997;94:4176–4181. doi: 10.1073/pnas.94.8.4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Block BA, Imagawa T, Campbell KP, Franzini-Armstrong C. Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J Cell Biol. 1988;107:2587–2600. doi: 10.1083/jcb.107.6.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bogdanov KY, Vinogradova TM, Lakatta EG. Sinoatrial nodal cell ryanodine receptor and Na+-Ca2+ exchanger: molecular partners in pacemaker regulation. Circ Res. 2001;88:1254–1258. doi: 10.1161/hh1201.092095. [DOI] [PubMed] [Google Scholar]
- 56.Boitano S, Dirksen ER, Sanderson MJ. Intercellular propagation of calcium waves mediated by inositol triphosphate. Science. 1992;258:292–295. doi: 10.1126/science.1411526. [DOI] [PubMed] [Google Scholar]
- 57.Bosch RF, Zeng X, Grammer JB, Popovic K, Mewis C, Kuhl-kamp V. Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovasc Res. 1999;44:121–131. doi: 10.1016/s0008-6363(99)00178-9. [DOI] [PubMed] [Google Scholar]
- 58.Bouchard RA, Clark RB, Giles WR. Effects of action potential duration on excitation-contraction coupling in rat ventricular myocytes. Action potential voltage-clamp measurements. Circ Res. 1995;76:790–801. doi: 10.1161/01.res.76.5.790. [DOI] [PubMed] [Google Scholar]
- 59.Bourinet E, Zamponi GW, Stea A, Soong TW, Lewis BA, Jones LP, Yue DT, Snutch TP. The alpha 1E calcium channel exhibits permeation properties similar to low-voltage-activated calcium channels. J Neurosci. 1996;16:4983–4993. doi: 10.1523/JNEUROSCI.16-16-04983.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boutjdir M, El-Sherif N, Gough WB. Effects of caffeine and ryanodine on delayed afterdepolarizations and sustained rhythmic activity in the 1-day old myocardial infarction in the dog. Circulation. 1990;81:1393–1400. doi: 10.1161/01.cir.81.4.1393. [DOI] [PubMed] [Google Scholar]
- 61.Boyden PA, Barbhaiya C, Lee T, ter Keurs HEDJ. Nonuniform Ca2+ transients in arrhythmogenic purkinje cells that survive in the infarcted canine heart. Cardiovasc Res. 2003;57:681–693. doi: 10.1016/s0008-6363(02)00725-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boyden PA, Dun W, Barbhaiya C, ter Keurs HEDJ. 2APB- and JTV519(K201) sensitive micro Ca2+ waves in arrhythmogenic Purkinje cells that survive in infarcted canine heart. Heart Rhythm. 2004;1:218–226. doi: 10.1016/j.hrthm.2004.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boyden PA, Pinto JMB. Reduced calcium currents in subendocardial Purkinje myocytes that survive in the 24 and 48 hour infarcted heart. Circulation. 1994;89:2747–2759. doi: 10.1161/01.cir.89.6.2747. [DOI] [PubMed] [Google Scholar]
- 64.Boyden PA, Pu J, Pinto JMB, ter Keurs HEDJ. Ca2+ transients and Ca2+ waves in Purkinje cells. Role in action potential initiation. Circ Res. 2000;86:448–455. doi: 10.1161/01.res.86.4.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Boyett MR, Honjo H, Harrison SM, Zang WJ, Kirby MS. Ultra-slow voltage dependent inactivation of the calcium current in guinea pig and ferret ventricular myocytes. Pflügers Arch. 1994;428:39–50. doi: 10.1007/BF00374750. [DOI] [PubMed] [Google Scholar]
- 66.Brandl CJ, deLeon S, Martin DR, MacLennan DH. Adult forms of the Ca2+ ATPase of sarcoplasmic reticulum. J Biol Chem. 1987;262:3768–3774. [PubMed] [Google Scholar]
- 67.Brandl CJ, Green NM, Korczak B, MacLennan DH. Two Ca2+ ATPase genes: homologies and mechanistic implications of deduced amino acid sequences. Cell. 1986;44:597–607. doi: 10.1016/0092-8674(86)90269-2. [DOI] [PubMed] [Google Scholar]
- 68.Brandt NR, Caswell AH, Carl SA, Ferguson DG, Brandt T, Brunschwig JP, Bassett AL. Detection and localization of traidin in rat ventricular muscle. J Membr Biol. 1993;131:219–228. doi: 10.1007/BF02260110. [DOI] [PubMed] [Google Scholar]
- 69.Brette F, Salle L, Orchard CH. Quantification of calcium entry at the t-tubules and surface membrane in rat ventricular myocytes. Biophys J. 2006;90:381–389. doi: 10.1529/biophysj.105.069013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brette F, Orchard C. T-tubule function in mammalian cardiac myocytes. Circ Res. 2003;92:1182–1192. doi: 10.1161/01.RES.0000074908.17214.FD. [DOI] [PubMed] [Google Scholar]
- 71.Bultynck G, de Smet P, Rossi D, Callewaert G, Missiaen L, Sorrentino V, De Smedt H, Parys JB. Characterization and mapping of the 12kDa FK506-binding (FKBP12) binding site on different isoforms of the ryanodine receptor and of the inositol 1,4,5 trisphosphate receptor. Biochem J. 2001;354:413–422. doi: 10.1042/0264-6021:3540413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bunemann M, Gerhardstein BL, Gao T, Hosey MM. Functional regulation of L-type calcium channels via protein kinase A-mediated phosphorylation of the beta 2 subunit. J Biol Chem. 1999;274:33851–33854. doi: 10.1074/jbc.274.48.33851. [DOI] [PubMed] [Google Scholar]
- 73.Burashnikov A, Antzelevitch C. Reinduction of atrial fibrillation immediately after termination of the arrhythmia is mediated by late phase 3 early afterdepolarization-induced triggered activity. Circulation. 2003;107:2355–2360. doi: 10.1161/01.CIR.0000065578.00869.7C. [DOI] [PubMed] [Google Scholar]
- 74.Burgess DE, Crawford O, Delisle BP, Satin J. Mechanism of inactivation gating of human T-type (low-voltage activated) calcium channels. Biophys J. 2002;82:1894–1906. doi: 10.1016/S0006-3495(02)75539-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cabo C, Schmitt H, Wit AL. New mechanism of antiarrhythmic drug action: increasing L type calcium current prevents reentrant ventricular tachycardia. Circulation. 2000;102:2417–2425. doi: 10.1161/01.cir.102.19.2417. [DOI] [PubMed] [Google Scholar]
- 76.Callewaert G, Carmeliet EE, Vereecke J. Single cardiac Purkinje cells: general electrophysiology and voltage-clamp analysis of the pace-maker current. J Physiol. 1984;349:643–661. doi: 10.1113/jphysiol.1984.sp015179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Camacho P, Lechleiter JD. Calreticulin inhibits repetitive intracellular Ca2+ waves. Cell. 1995;82:765–771. doi: 10.1016/0092-8674(95)90473-5. [DOI] [PubMed] [Google Scholar]
- 78.Cannell MB, Cheng H, Lederer WJ. The control of calcium release in heart muscle. Science. 1995;268:1045–1049. doi: 10.1126/science.7754384. [DOI] [PubMed] [Google Scholar]
- 79.Cannell MB, Cheng H, Lederer WJ. Spatial nonuniformities in Cai during excitation contraction coupling in cardiac myocytes. Biophys J. 1994;67:1942–1956. doi: 10.1016/S0006-3495(94)80677-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cannell MB, Lederer WJ. The arrhythmogenic current ITI in the absence of electrogenic sodium-calcium exchange in sheep cardiac Purkinje fibres. J Physiol. 1986;374:201–219. doi: 10.1113/jphysiol.1986.sp016075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Capogrossi MC, Houser SR, Bahinski A, Lakatta EG. Synchronous occurrence of spontaneous localized calcium release from the sarcoplasmic reticulum generates action potentials in rat cardiac ventricular myocytes at normal resting membrane potential. Circ Res. 1987;61:489–503. doi: 10.1161/01.res.61.4.498. [DOI] [PubMed] [Google Scholar]
- 82.Capogrossi MC, Lakatta EG. Frequency modulation and synchronization of spontaneous oscillations in cardiac cells. Am J Physiol Heart Circ Physiol. 1985;248:H412–H418. doi: 10.1152/ajpheart.1985.248.3.H412. [DOI] [PubMed] [Google Scholar]
- 83.Capogrossi MC, Suarez-Isla BA, Lakatta EG. The interaction between electrically stimulated twitches and spontaneous contractile waves in single cardiac myocytes. J Gen Physiol. 1986;88:615–633. doi: 10.1085/jgp.88.5.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Carafoli E. Intracellular calcium homeostasis. Annu Rev Biochem. 1987;56:395–433. doi: 10.1146/annurev.bi.56.070187.002143. [DOI] [PubMed] [Google Scholar]
- 84a.Carl S, Felix K, Caswell AH, Brandt NR, Ball WJ, Vaghy PL, Meissner G, Ferguson DG. Immunolocalization of sarcolemmal dihydropyridine receptor and sarcoplasmic reticular triadin and ryanodine receptor in rabbit atrium and ventricle. J Cell Biol. 1995;129:672–682. doi: 10.1083/jcb.129.3.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cens T, Restituito S, Galas S, Charnet P. Voltage and calcium use the same molecular determinants to inactivate calcium channels. J Biol Chem. 1999;274:5483–5490. doi: 10.1074/jbc.274.9.5483. [DOI] [PubMed] [Google Scholar]
- 86.Cens T, Restituito S, Vallentin A, Charnet P. Promotion and inhibition of L-type Ca2+ channel facilitation by distinct domains of the subunit. J Biol Chem. 1998;273:18308–18315. doi: 10.1074/jbc.273.29.18308. [DOI] [PubMed] [Google Scholar]
- 87.Cerrone M, Colombi B, Santoro M, di Barletta MR, Scelsi M, Villani L, Napolitano C, Priori SG. Bidirectional ventricular tachycardia and fibrillation elicited in a knock-in mouse model carrier of a mutation in the cardiac ryanodine receptor. Circ Res. 2005;96:e77–e82. doi: 10.1161/01.RES.0000169067.51055.72. [DOI] [PubMed] [Google Scholar]
- 88.Chelu MG, Danila CI, Gilman CP, Hamilton SL. Regulation of ryanodine receptors by FK506 binding proteins. Trends Cardiovasc Med. 2004;14:227–234. doi: 10.1016/j.tcm.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 89.Chen SR, Ebisawa K, Li X, Zhang L. Molecular identification of the ryanodine receptor Ca2+ sensor. J Biol Chem. 1998;273:14675–14678. doi: 10.1074/jbc.273.24.14675. [DOI] [PubMed] [Google Scholar]
- 90.Chen YH, Li MH, Zhang Y, He L, Yamada Y, Fitzmaurice A, Shen Y, Zhang H, Tong L, Yang J. Structural basis of the α1-β subunit interaction of voltage-gated Ca2+ channels. Nature. 2004;429:675–680. doi: 10.1038/nature02641. [DOI] [PubMed] [Google Scholar]
- 91.Cheng H, Lederer MR, Lederer WJ, Cannell MB. Calcium sparks and [Ca2+]i waves in cardiac myocytes. Am J Physiol Cell Physiol. 1996;270:C148–C159. doi: 10.1152/ajpcell.1996.270.1.C148. [DOI] [PubMed] [Google Scholar]
- 92.Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- 93.Choi BR, Salama G. Simultaneous maps of optical action potentials and calcium transients in guinea pig hearts: mechanisms underlying concordant alternans. J Physiol. 2000;529:171–188. doi: 10.1111/j.1469-7793.2000.00171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Choi BR, Burton F, Salama G. Cytosolic Ca2+ triggers early afterdepolarizations and torsade de pointes in rabbit hearts with long QT syndrome, LQT2. J Physiol. 2002;543:615–631. doi: 10.1113/jphysiol.2002.024570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Choi G, Kopplin LJ, Tester DJ, Will ML, Haglund CM, Ackerman MJ. Spectrum and frequency of cardiac channel defects in swimming triggered arrhythmia syndromes. Circulation. 2004;110:2119–2124. doi: 10.1161/01.CIR.0000144471.98080.CA. [DOI] [PubMed] [Google Scholar]
- 96.Chou C, Nihei M, Zhou S, Tan A, Kawase A, Macias ES, Fishbein MC, Lin SF, Chen PS. Intracellular calcium dynamics and anisotropic reentry in isolated canine pulmonary veins and left atrium. Circulation. 2005;111:2889–2897. doi: 10.1161/CIRCULATIONAHA.104.498758. [DOI] [PubMed] [Google Scholar]
- 97.Chu A, Fill M, Stefani E, Entman ML. Cytoplasmic Ca2+ does not inhibit the cardiac sarcoplasmic reticulum ryanodine receptor Ca2+ channel, although Ca2+-induced Ca2+ inactivation of Ca2+ release is observed in native vesicles. J Membr Biol. 1993;135:49–59. doi: 10.1007/BF00234651. [DOI] [PubMed] [Google Scholar]
- 98.Chudin E, Goldhaber J, Garfinkel A, Weiss J, Kogan B. Intracellular Ca2+ dynamics and the stability of ventricular tachycardia. Biophys J. 1999;77:2930–2941. doi: 10.1016/S0006-3495(99)77126-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Clarke DM, Loo TW, Inesi G, MacLennan DH. Location of high affinity Ca2+ binding sites within the predicted transmembrane domain of the sarcoplasmic reticulum Ca2+-ATPase. Nature. 1989;339:476–478. doi: 10.1038/339476a0. [DOI] [PubMed] [Google Scholar]
- 100.Clarke DM, Maruyama M, Loo TW, Leberer E, Inesi G, MacLennan DH. Functional consequences of glutamate, aspartate, glutamine and asparagine mutations in the stalk sector of the Ca2+-ATPase of sarcoplasmic reticulum. J Biol Chem. 1989;264:11246–11251. [PubMed] [Google Scholar]
- 101.Clusin WT. Calcium and cardiac arrhythmias: DADs, EADs and Alternans. Crit Rev Clin Lab Sci. 2003;40:337–375. doi: 10.1080/713609356. [DOI] [PubMed] [Google Scholar]
- 102.Colecraft HM, Alseikhan B, Takahashi SX, Chaudhuri D, Mitt-man S, Yegnasubramanian V, Alvania RS, John DC, Marban E, Yue DT. Novel functional properties of Ca2+ channel beta subunits revealed by their expression in adult rat heat cells. J Physiol. 2002;541:435–452. doi: 10.1113/jphysiol.2002.018515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Collier ML, Levesque PC, Kenyon JL, Hume JR. Unitary Cl− channels activated by cytoplasmic Ca2+ in canine ventricular myocytes. Circ Res. 1996;78:936–944. doi: 10.1161/01.res.78.5.936. [DOI] [PubMed] [Google Scholar]
- 104.Colquhoun D, Neher E, Reuter H, Stevens C. Inward current channels activated by intracellular Ca in cultured cardiac cells. Nature. 1981;294:752–754. doi: 10.1038/294752a0. [DOI] [PubMed] [Google Scholar]
- 105.Cordeiro JM, Bridge JHB, Spitzer KW. Early and delayed afterdepolarizations in rabbit heart Purkinje cells viewed by confocal microscopy. Cell Calcium. 2001;29:289–297. doi: 10.1054/ceca.2000.0192. [DOI] [PubMed] [Google Scholar]
- 106.Cordeiro JM, Spitzer KW, Giles W, Ershler PR, Cannell MB, Bridge JHB. Location of the initiation of calcium transients and sparks in rabbit Purkinje cells. J Physiol. 2001;531:301–314. doi: 10.1111/j.1469-7793.2001.0301i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cote K, Proteau S, Teijeira J, Rousseau E. Characterization of the sarcoplasmic reticulum K and Ca2+ release channel, ryanodine receptor, in human atrial cells. J Mol Cell Cardiol. 2000;32:2051–2063. doi: 10.1006/jmcc.2000.1236. [DOI] [PubMed] [Google Scholar]
- 108.Coulombe A, Lefevre IA, Baro I, Coraboeuf E. Barium- and calcium-permeable channels open at negative membrane potentials in rat ventricular myocytes. J Membr Biol. 1989;111:57–67. doi: 10.1007/BF01869209. [DOI] [PubMed] [Google Scholar]
- 109.Coumel P, Fidelle J, Lucet V, Attuel P, Bouvrain Y. Catecholaminergic-induced severe ventricular arrhythmias with Adams-Stokes syndrome in children: report of four cases. Br Heart J. 1978;40(Suppl):28–37. [Google Scholar]
- 110.Cranefield PF, Aronson RS. Cardiac Arrhythmias: The Role of Triggered Activity. Mount Kisco, NY: Futura; 1988. [Google Scholar]
- 111.Cribbs LL, Lee JH, Yang J, Satin J, Zhang Y, Daud A, Barclay J, Williamson MP, Fox M, Rees M, Perez-Reyes E. Cloning and characterization of alpha1H from human heart, a member of the T-type Ca2+ channel gene family. Circ Res. 1998;83:103–109. doi: 10.1161/01.res.83.1.103. [DOI] [PubMed] [Google Scholar]
- 112.Curtis BM, Catterall WA. Purification of the calcium antagonist receptor of the voltage sensitive calcium channel from skeletal muscle transverse tubules. Biochemistry. 1984;23:2113–2118. doi: 10.1021/bi00305a001. [DOI] [PubMed] [Google Scholar]
- 113.Dai S, Klugbauer N, Zong X, Seisenberger C, Hofmann F. The role of subunit composition on prepulse facilitation of the cardiac L-type calcium channel. FEBS Lett. 1999;442:70–74. doi: 10.1016/s0014-5793(98)01632-9. [DOI] [PubMed] [Google Scholar]
- 114.Damiano BP, Rosen MR. Effects of pacing on triggered activity induced by early afterdepolarizations. Circulation. 1984;69:1013–1025. doi: 10.1161/01.cir.69.5.1013. [DOI] [PubMed] [Google Scholar]
- 115.Daniels MC, Fedida D, Lamont C, ter Keurs HEDJ. Role of the sarcolemma in triggered propagated contractions in rat cardiac trabeculae. Circ Res. 1991;68:1408–1421. doi: 10.1161/01.res.68.5.1408. [DOI] [PubMed] [Google Scholar]
- 116.Daniels MC, Kieser T, ter Keurs HEDJ. Triggered propagated contractions in human atrial trabeculae. Cardiovasc Res. 1993;27:1831–1835. doi: 10.1093/cvr/27.10.1831. [DOI] [PubMed] [Google Scholar]
- 117.Daniels MC, ter Keurs HEDJ. Propagated contractions in rat cardiac trabeculae: effects of caffeine, ryanodine BayK8644 and D-600. Biophys J. 1990;57:170a. [Google Scholar]
- 118.Daniels MC, ter Keurs HEDJ. Spontaneous contractions in rat cardiac trabeculae: trigger mechanism and propagation velocity. J Gen Physiol. 1990;95:1123–1137. doi: 10.1085/jgp.95.6.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Dassouli A, Sulpice JC, Roux S, Crozatier B. Stretch induced inositol triphosphate and tetrakisphosphate production in rat cardiocytes. J Mol Cell Cardiol. 1993;25:973–982. doi: 10.1006/jmcc.1993.1109. [DOI] [PubMed] [Google Scholar]
- 120.Davidoff AW, Boyden PA, Schwartz K, Michel JB, Zhang YM, Obayashi M, Crabbe D, ter Keurs HEDJ. Congestive heart failure after myocardial infarction in the rat: cardiac force and spontaneous sarcomere activity. Ann NY Acad Sci. 2004;1015:84–95. doi: 10.1196/annals.1302.007. [DOI] [PubMed] [Google Scholar]
- 121.De Ferrari GM, Viola M, D'Amato E, Antolini R, Forti S. Distinct patterns of calcium transients during early and delayed afterdepolarizations induced by isoproterenol in ventricular myocytes. Circulation. 1995;91:2510–2515. doi: 10.1161/01.cir.91.10.2510. [DOI] [PubMed] [Google Scholar]
- 122.De Jongh KS, Murphy BJ, Colvin AA, Hell JW, Takahashi M, Catterall WA. Specific phosphorylation of a site in the full-length form of the Alpha1 subunit of the cardiac L-type calcium channel by adenosine 3′,5′-cyclic monophosphate-dependent protein kinase. Biochem. 1996;35:10392–10402. doi: 10.1021/bi953023c. [DOI] [PubMed] [Google Scholar]
- 123.De Koninck P, Schulman H. Sensitivity of CaM kinase II to the frequency of Ca2+ oscillations. Science. 1998;279:227–230. doi: 10.1126/science.279.5348.227. [DOI] [PubMed] [Google Scholar]
- 124.De Leon M, Wang Y, Jones L, Perez-Reyes E, Wei X, Soong TW, Snutch TP, Yue DT. Essential Ca2+-binding motif for Ca2+-sensitive inactivation of L-type Ca2+ channels. Science. 1995;270:1502–1506. doi: 10.1126/science.270.5241.1502. [DOI] [PubMed] [Google Scholar]
- 125.Decrouy A, Juteau M, Rousseau E. Examination of the role of phosphorylation and phospholamban in the regulation of the cardiac sarcoplasmic reticulum Cl− channel. J Membr Biol. 1995;146:315–326. doi: 10.1007/BF00233951. [DOI] [PubMed] [Google Scholar]
- 126.DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, Yue DT. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature. 2001;411:484–489. doi: 10.1038/35078091. [DOI] [PubMed] [Google Scholar]
- 127.De Meis L, Vianna AL. Energy interconversion by the Ca-dependent ATPase of the sarcoplasmic reticulum. Annu Rev Biochem. 1979;48:275–292. doi: 10.1146/annurev.bi.48.070179.001423. [DOI] [PubMed] [Google Scholar]
- 128.Denton RM, Randle PJ, Martin BR. Stimulation by calcium ions of pyruvate dehydrogenase phosphate phosphatase. Biochem J. 1972;128:161–163. doi: 10.1042/bj1280161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Denton RM, Richards DA, Chin JG. Calcium ions and the regulation of NAD+ linked isocitrate dehydrogenase from the mitochondria of rat heart and other tissues. Biochem J. 1978;176:899–906. doi: 10.1042/bj1760899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Deschenes I, DiSilvestre D, Juang GJ, Wu RC, An WF, Tomaselli GF. Regulation of Kv4.3 current by KChIP2 splice variants: a component of native cardiac Ito? Circulation. 2002;106:423–429. doi: 10.1161/01.cir.0000025417.65658.b6. [DOI] [PubMed] [Google Scholar]
- 131.Deschenes I, Neyroud N, DiSilvestre D, Marban E, Yue DT, Tomaselli GF. Isoform-specific modulation of voltage-gated Na+ channels by calmodulin. Circ Res. 2002;90:49e–57. doi: 10.1161/01.res.0000012502.92751.e6. [DOI] [PubMed] [Google Scholar]
- 132.DeSouza N, Reiken S, Ondrias K, Yang YM, Matkovich SJ, Marks AR. Protein kinase A and two phosphatases are components of the inositol 1,4,5-trisphosphate receptor macromolecular signaling complex. J Biol Chem. 2002;277:39397–39400. doi: 10.1074/jbc.M207059200. [DOI] [PubMed] [Google Scholar]
- 133.Di Barletta MR, Viatchenko-Karpinski S, Nori A, Memmi M, Terentyev D, Turcato F, Valle G, Rizzi N, Napolitano C, Gyorke S, Volpe P, Priori SG. Clinical phenotype and functional characterization of CASQ2 mutations associated with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2006;114:1012–1019. doi: 10.1161/CIRCULATIONAHA.106.623793. [DOI] [PubMed] [Google Scholar]
- 134.Diaz ME, O'Neill SC, Eisner DA. Sarcoplasmic reticulum calcium content fluctuation is the key to cardiac alternans. Circ Res. 2004;94:650–656. doi: 10.1161/01.RES.0000119923.64774.72. [DOI] [PubMed] [Google Scholar]
- 135.Doering AE, Nicoll DA, Lu Y, Lu L, Weiss JN, Philipson KD. Topology of a functionally important region of the cardiac Na/Ca exchanger. J Biol Chem. 1998;273:778–783. doi: 10.1074/jbc.273.2.778. [DOI] [PubMed] [Google Scholar]
- 136.Droogmans G, Nilius B. Kinetic properties of the cardiac T-type calcium channel in the guinea-pig. J Physiol. 1989;419:627–650. doi: 10.1113/jphysiol.1989.sp017890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dun W, Hirose M, Gor Y, Venkatraman Y, Feinmark SJ, Boyden PA. Identification and localization of TRPC6 channel protein in canine ventricular myocytes and Purkinje cells (Abstract) Biophys J. 2006;90:414a. [Google Scholar]
- 138.Dupont Y. Kinetics and regulation of sarcoplasmic reticulum ATPase. Eur J Biochem. 1977;72:185–190. doi: 10.1111/j.1432-1033.1977.tb11238.x. [DOI] [PubMed] [Google Scholar]
- 139.Dzhura I, Wu Y, Colbran RJ, Balser JR, Anderson ME. Calmodulin kinase determines calcium-dependent facilitation of L-type calcium channels. Nat Cell Biol. 2000;2:173–177. doi: 10.1038/35004052. [DOI] [PubMed] [Google Scholar]
- 140.Earley S, Heppner TJ, Nelson MT, Brayden JE. TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BK Ca channels. Circ Res. 2005;97:1270–1279. doi: 10.1161/01.RES.0000194321.60300.d6. [DOI] [PubMed] [Google Scholar]
- 141.Egan TM, Noble D, Noble SJ, Powell T, Spindler AJ, Twist VW. Sodium-calcium exchange during the action potential in guinea pig ventricular cells. J Physiol. 1989;411:639–661. doi: 10.1113/jphysiol.1989.sp017596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ehara T, Noma A, Ono K. Calcium-activated non-selective cation channel in ventricular cells isolated from adult guinea-pig hearts. J Physiol. 1988;403:117–133. doi: 10.1113/jphysiol.1988.sp017242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Eisner DA, Li Y, O'Neill SC. Alternans of intracellular calcium: mechanism and significance. Heart Rhythm. 2006;3:743–745. doi: 10.1016/j.hrthm.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 144.Ellinor PT, Yang J, Sather WA, Zhang JF, Tsien RW. Ca2+ channel selectivity at a single locus for high affinity Ca2+ interactions. Neuron. 1995;15:1121–1132. doi: 10.1016/0896-6273(95)90100-0. [DOI] [PubMed] [Google Scholar]
- 145.Erickson MG, Alseikhan BA, Peterson BZ, Yue DT. Preassociation of calmodulin with voltage-gated Ca2+ channels revealed by FRET in single living cells. Neuron. 2001;31:973–985. doi: 10.1016/s0896-6273(01)00438-x. [DOI] [PubMed] [Google Scholar]
- 146.Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E, Schwartz A, Snutch TP, Tanabe T, Birnbaumer L, Tsien RW, Catterall W. Nomenclature of voltage gated calcium channels. Neuron. 2000;25:533–535. doi: 10.1016/s0896-6273(00)81057-0. [DOI] [PubMed] [Google Scholar]
- 147.Erxleben C, Liao Y, Gentile S, Chin D, Gomez-Alegria C, Mori Y, Birnbaumer L, Armstrong DL. Cyclosporin and Timothy syndrome increase mode 2 gating of CaV1.2 calcium channels through aberrant phosphorylation of S6 helices. Proc Natl Acad Sci USA. 2006;103:3932–3937. doi: 10.1073/pnas.0511322103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Escande D, Coraboeuf E, Planche C. Abnormal pacemaking is modulated by sarcoplasmic reticulum in partially depolarized myocardium from dilated right atria in humans. J Mol Cell Cardiol. 1987;19:231–241. doi: 10.1016/s0022-2828(87)80590-4. [DOI] [PubMed] [Google Scholar]
- 149.Escande D, Coulombe A, Faivre JF, Deroubaix E, Coraboeuf E. Two types of transient outward currents in adult human atrial cells. Am J Physiol Heart Circ Physiol. 1987;252:H142–H148. doi: 10.1152/ajpheart.1987.252.1.H142. [DOI] [PubMed] [Google Scholar]
- 150.Euler DE. Cardiac alternans: mechanisms and pathophysiological significance. Cardiovasc Res. 1999;42:583–590. doi: 10.1016/s0008-6363(99)00011-5. [DOI] [PubMed] [Google Scholar]
- 151.Fabiato A. Time and calcium dependence of activation and inactivation of calcium induced release of calcium from the sarcoplasmic reticulum of a skinned cardiac Purkinje cell. J Gen Physiol. 1985;85:247–289. doi: 10.1085/jgp.85.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Fabiato A. Spontaneous versus triggered contractions of calcium tolerant cardiac cells from the adult rat ventricle. Basic Res Cardiol. 1985;80(2):83–87. [PubMed] [Google Scholar]
- 153.Fabiato A, Fabiato F. Calcium-induced release of calcium from the sarcoplasmic reticulum of skinned cells from adult human, dog, cat, rabbit, rat, frog hearts and from fetal and new-born rat ventricles. Ann NY Acad Sci. 1978;307:491–522. doi: 10.1111/j.1749-6632.1978.tb41979.x. [DOI] [PubMed] [Google Scholar]
- 154.Fauconnier J, Lacampagne A, Rauzier JM, Vassort G, Richard S. Ca2+ dependent reduction of IK1 in rat ventricular cells: a novel paradigm for arrhythmia in heart failure. Cardiovasc Res. 2005;68:204–212. doi: 10.1016/j.cardiores.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 155.Fedida D, Noble D, Spindler AJ. Use-dependent reduction and facilitation of Ca2+ current in guinea-pig myocytes. J Physiol. 1988;405:439–460. doi: 10.1113/jphysiol.1988.sp017341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Fernandez D, Ghanta A, Kinard KI, Sanguinetti MC. Molecular mapping of a site for Cd2+-induced modification of human ether-a-go-go-related gene (hERG) channel activation. J Physiol. 2005;567:737–755. doi: 10.1113/jphysiol.2005.089094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ferreira G, Yi J, Rios E, Shirokov R. Ion-dependent inactivation of barium current through L-type calcium channels. J Gen Physiol. 1997;109:449–461. doi: 10.1085/jgp.109.4.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ferrier GR. The effects of tension on acetylstrophanthidin-induced transient depolarizations and aftercontractions in canine myocardial and Purkinje tissues. Circ Res. 1976;38:156–162. doi: 10.1161/01.res.38.3.156. [DOI] [PubMed] [Google Scholar]
- 159.Fink RHA, Stephenson DG. Ca2+-movements in muscle modulated by the state of K+-channels in the sarcoplasmic reticulum membranes. Pflügers Arch. 1987;409:374–380. doi: 10.1007/BF00583791. [DOI] [PubMed] [Google Scholar]
- 160.Fitzgerald M, Anderson KE, Woodcock EA. Inositol-1,4,5 triphopshate IP3 and IP3 receptor concentrations in heart tissues. Clin Exp Pharmacol Physiol. 1994;21:257–260. doi: 10.1111/j.1440-1681.1994.tb02509.x. [DOI] [PubMed] [Google Scholar]
- 161.Fozzard HA, Hiraoka M. The positive dynamic current and its inactivation properties in cardiac Purkinje fibers. J Physiol. 1973;234:569–586. doi: 10.1113/jphysiol.1973.sp010361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Franzini-Armstrong C, Protasi F, Tijskens P. The assembly of calcium release units in cardiac muscle. Ann NY Acad Sci. 2005;1047:76–85. doi: 10.1196/annals.1341.007. [DOI] [PubMed] [Google Scholar]
- 163.Fujioka Y, Hiroe K, Matsuoka S. Regulation kinetics of NaCa exchange current in guinea pig ventricular myocytes. J Physiol. 2000;529:611–623. [PubMed] [Google Scholar]
- 164.Furukawa T, Myerburg RJ, Furukawa N, Bassett AL, Kimura S. Differences in transient outward currents of feline endocardial and epicardial myocytes. Circ Res. 1990;67:1287–1291. doi: 10.1161/01.res.67.5.1287. [DOI] [PubMed] [Google Scholar]
- 165.Gamper N, Li Y, Shapiro MS. Structural requirements for differential sensitivity of KCNQ K+ channels to modulation by Ca2+/calmodulin. Mol Biol Cell. 2005;16:3538–3551. doi: 10.1091/mbc.E04-09-0849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ganesan AN, Maack C, Johns DC, Sidor A, O'Rourke B. β-Adrenergic stimulation of L-type Ca2+ channels in cardiac myocytes requires the distal carboxyl terminus of α1C but not serine 1928. Circ Res. 2006;98:e11–e18. doi: 10.1161/01.RES.0000202692.23001.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gannier F, White E, Garnier D, Le Guennec JY. A possible mechanism for large stretch induced increase in Cai in isolated guinea pig ventricular myocytes. Cardiovasc Res. 1996;32:158–167. [PubMed] [Google Scholar]
- 168.Gannier F, White E, Lacampagne A, Garnier D, Le Guennec JY. Streptomycin reverses a large stretch induced increase in intracellular calcium in isolated guinea pig ventricular myocytes. Cardiovasc Res. 1994;28:1193–1198. doi: 10.1093/cvr/28.8.1193. [DOI] [PubMed] [Google Scholar]
- 169.Gao T, Bunemann M, Gerhardstein BL, Ma H, Hosey MM. Role of the C terminus of the Alpha1C (Cav1.2) subunit in membrane targeting of cardiac L-type calcium channels. J Biol Chem. 2000;275:25436–25444. doi: 10.1074/jbc.M003465200. [DOI] [PubMed] [Google Scholar]
- 170.Gao T, Cuadra AE, Ma H, Bunemann M, Gerhardstein BL, Cheng T, Eick RT, Hosey MM. C-terminal fragments of the Alpha1C (Cav1.2) subunit associate with and regulate L-type calcium channels containing C-terminal-truncated Alpha1C subunits. J Biol Chem. 2001;276:21089–21097. doi: 10.1074/jbc.M008000200. [DOI] [PubMed] [Google Scholar]
- 171.Gao T, Puri TS, Gerhardstein BL, Chien AJ, Green RD, Hosey MM. Identification and subcellular localization of the subunits of L-type calcium channels and adenylyl cyclase in cardiac myocytes. J Biol Chem. 1997;272:19401–19407. doi: 10.1074/jbc.272.31.19401. [DOI] [PubMed] [Google Scholar]
- 172.Gao WD, Backx PH, Azan-Backx MD, Marban E. Myofilament Ca2+ sensitivity in intact versus skinned rat ventricular muscle. Circ Res. 1994;74:408–415. doi: 10.1161/01.res.74.3.408. [DOI] [PubMed] [Google Scholar]
- 173.Gerhardstein BL, Gao T, Bunemann M, Puri TS, Adair A, Ma H, Hosey MM. Proteolytic processing of the C terminus of the Alpha1C subunit of L-type calcium channels and the role of a proline-rich domain in membrane tethering of proteolytic fragments. J Biol Chem. 2000;275:8556–8563. doi: 10.1074/jbc.275.12.8556. [DOI] [PubMed] [Google Scholar]
- 174.Ghosh S, Nunziato DA, Pitt GS. KCNQ1 assembly and function is blocked by LQTS mutations that disrupt interaction with calmodulin. Circ Res. 2006;98:1048–1054. doi: 10.1161/01.RES.0000218863.44140.f2. [DOI] [PubMed] [Google Scholar]
- 175.Girard S, Clapham D. Acceleration of intracellular calcium waves in Xenopus oocytes by calcium influx. Science. 1993;260:229–232. doi: 10.1126/science.8385801. [DOI] [PubMed] [Google Scholar]
- 176.Goldhaber JI, Lamp ST, Walter DO, Garfinkel A, Fukumoto GH, Weiss JN. Local regulation of the threshold for calcium sparks in rat ventricular myocytes: role of sodium-calcium exchange. J Physiol. 1999;520:431–438. doi: 10.1111/j.1469-7793.1999.00431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Goldhaber JI, Xie LH, Duong T, Motter C, Khuu K, Weiss JN. Action potential duration restitution and alternans in rabbit ventricular myocytes: the key role of intracellular calcium cycling. Circ Res. 2005;96:459–466. doi: 10.1161/01.RES.0000156891.66893.83. [DOI] [PubMed] [Google Scholar]
- 178.Gorza L, Schiaffino S, Volpe P. Inositol 1,4,5-triphosphate receptor in heart: evidence for its concentration in Purkinje myocytes of the conduction system. J Cell Biol. 1993;121:345–353. doi: 10.1083/jcb.121.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Guinamard R, Chatelier A, Demion M, Potreau D, Patri S, Rahmati M, Bois P. Functional characteristics of a Ca2+ activated nonselective cation channel in human atrial cardiomyocytes. J Physiol. 2004;558:75–83. doi: 10.1113/jphysiol.2004.063974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Guinamard R, Chatelier A, Lenfant J, Bois P. Activation of the Ca2+ activated nonselective cation channel by diacylglycerol analogues in rat cardiomyocytes. J Cardiovasc Electrophysiol. 2004;15:342–348. doi: 10.1046/j.1540-8167.2004.03477.x. [DOI] [PubMed] [Google Scholar]
- 181.Gunter TE, Pfeifer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol Cell Physiol. 1990;258:C755–C786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- 182.Guo T, Zhang T, Mistral R, Bers DM. Ca2+/calmodulin-dependent protein kinase II phosphorylation of ryanodine receptor does affect calcium sparks in mouse ventricular myocytes. Circ Res. 2006;99:398–406. doi: 10.1161/01.RES.0000236756.06252.13. [DOI] [PubMed] [Google Scholar]
- 183.Guo W, Jorgensen AO, Jones LR, Campbell KP. Biochemical characterization and molecular cloning of cardiac triadin. J Biol Chem. 1996;271:458–465. doi: 10.1074/jbc.271.1.458. [DOI] [PubMed] [Google Scholar]
- 184.Gurney AM, Charnet P, Pye JM, Nargeot J. Augmentation of cardiac calcium current by flash photolysis of intracellular caged-Ca2+ molecules. Nature. 1989;341:65–68. doi: 10.1038/341065a0. [DOI] [PubMed] [Google Scholar]
- 185.Gwanyanya A, Amuzescu B, Zakharov S, Macianskiene R, Sipido KR, Bolotina VM, Vereecke J, Mubagwa K. Magnesium-inhibited TRPM6/7 like channel in cardiac myocytes: permeation of divalent cations and pH mediated regulation. J Physiol. 2004;559:761–776. doi: 10.1113/jphysiol.2004.067637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Gyorke I, Hester N, Jones LR, Gyorke S. The role of calsequestrin, triadin, junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J. 2004;86:2121–2128. doi: 10.1016/S0006-3495(04)74271-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Hadley RW, Lederer WJ. Ca2+ and voltage inactivate Ca2+ channels in guinea pig ventricular myocytes through independent mechanisms. J Physiol. 1991;444:257–268. doi: 10.1113/jphysiol.1991.sp018876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Hagar RE, Ehrlich BE. Regulation of the type III InsP3 receptor by InsP3 and ATP. Biophys J. 2000;79:271–278. doi: 10.1016/S0006-3495(00)76289-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hagiwara N, Irisawa H, Kameyama M. Contribution of two types of calcium currents to the pacemaker potentials of rabbit sinoatrial node cells. J Physiol. 1988;395:233–253. doi: 10.1113/jphysiol.1988.sp016916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hama T, Takahashi A, Ichihara A, Takamatsu T. Real time in situ confocal imaging of calcium wave in the perfused whole heart of the rat. Cell Signal. 1998;10:331–337. doi: 10.1016/s0898-6568(97)00136-8. [DOI] [PubMed] [Google Scholar]
- 191.Hamilton SL, Serysheva I, Strasburg GM. Calmodulin and excitation-contraction coupling. News Physiol Sci. 2000;15:281–284. doi: 10.1152/physiologyonline.2000.15.6.281. [DOI] [PubMed] [Google Scholar]
- 192.Han S, Schiefer A, Isenberg G. Ca2+ load of guinea pig ventricular myocytes determines efficacy of brief Ca2+ currents as trigger for Ca2+ release. J Physiol. 1994;480:411–421. doi: 10.1113/jphysiol.1994.sp020371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Han X, Ferrier GR. Ionic mechanisms of transient inward current in the absence of Na-Ca exchange in rabbit Purkinje fibers. J Physiol. 1992;456:19–38. doi: 10.1113/jphysiol.1992.sp019324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Hancox JC, Levi AJ, Brooksby P. Intracellular calcium transients recorded with Fura-2 in spontaneously active myocytes isolated from the atrioventricular node of the rabbit heart. Proc Biol Sci. 1994;255:99–105. doi: 10.1098/rspb.1994.0014. [DOI] [PubMed] [Google Scholar]
- 195.Hanley PJ, ter Keurs HE, Cannell MB. Excitation-contraction coupling in the heart and the negative inotropic action of volatile anesthetics. Anesthesiology. 2004;101:999–1014. doi: 10.1097/00000542-200410000-00027. [DOI] [PubMed] [Google Scholar]
- 196.Hansen JP, Chen RS, Larsen JK, Chu PJ, Janes DM, Weis KE, Best PM. Calcium channel γ6 subunits are unique modulators of low voltage-activated (Cav3.1) calcium current. J Mol Cell Cardiol. 2004;37:1147–1158. doi: 10.1016/j.yjmcc.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 197.Hasdemir C, Priori SG, Overholt ED, Lazzara R. Catecholaminergic polymorphic ventricular tachycardia, recurrent syncope, implantable loop recorder. J Cardiovasc Electrophysiol. 2004;15:729. doi: 10.1046/j.1540-8167.2004.03408.x. [DOI] [PubMed] [Google Scholar]
- 198.Hatem SN, Benardeau A, Rucker-Martin C, Marty I, de Chamisso P, Villaz M, Mercadier JJ. Different compartments of sarcoplasmic reticulum participate in the excitation-contraction coupling process in human atrial myocytes. Circ Res. 1997;80:345–353. doi: 10.1161/01.res.80.3.345. [DOI] [PubMed] [Google Scholar]
- 199.Heinemann SH, Terlau H, Stuhmer W, Imoto K, Numa S. Calcium channel characteristics conferred on the sodium channel by single mutations. Nature. 1992;356:441–443. doi: 10.1038/356441a0. [DOI] [PubMed] [Google Scholar]
- 200.Hell JW, Westenbroek RE, Breeze LJ, Wang KK, Chavkin C, Catterall WA. N-Methyl-d-aspartate receptor-induced proteolytic conversion of postsynaptic class C L-type calcium channels in hippocampal neurons. Proc Natl Acad Sci USA. 1996;93:3362–3367. doi: 10.1073/pnas.93.8.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Henaff M, Antoine S, Mercadier JJ, Coulombe A, Hatem S. The voltage independent B type Ca2+ channel modulates apoptosis of cardiac myocytes. FASEB J. 2002;16:99–101. doi: 10.1096/fj.01-038fje. [DOI] [PubMed] [Google Scholar]
- 202.Hering S, Aczel S, Grabner M, Doring F, Berjukow S, Mitterdorfer J, Sinnegger MJ, Striessnig J, Degtiar VE, Wang Z, Glossmann H. Transfer of high sensitivity for benzothiazepines from L-type to class A (BI) calcium channels. J Biol Chem. 1996;271:24471–24475. doi: 10.1074/jbc.271.40.24471. [DOI] [PubMed] [Google Scholar]
- 203.Hering S, Aczel S, Kraus RL, Berjukow S, Striessnig J, Timin EN. Molecular mechanism of use-dependent calcium channel block by phenylalkylamines: role of inactivation. Proc Natl Acad Sci USA. 1997;94:13323–13328. doi: 10.1073/pnas.94.24.13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Hibberd MG, Jewell BR. Calcium and length dependent force production in rat ventricular muscle. J Physiol. 1982;329:527–540. doi: 10.1113/jphysiol.1982.sp014317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Hicks MJ, Shigekawa M, Katz AM. Mechanism by which cyclic adenosine 3′:5′-monophosphate-dependent protein kinase stimulates calcium transport in cardiac sarcoplasmic reticulum. Circ Res. 1979;44:384–391. doi: 10.1161/01.res.44.3.384. [DOI] [PubMed] [Google Scholar]
- 206.Hilgemann DW. Regulation and deregulation of cardiac Na/Ca exchange in giant excised sarcolemmal membrane patches. Nature. 1990;344:242–245. doi: 10.1038/344242a0. [DOI] [PubMed] [Google Scholar]
- 207.Hilgemann DW, Collins A, Matsuoka S. Steady-state and dynamic properties of cardiac sodium-calcium exchange. Secondary modulation by cytoplasmic calcium and ATP. J Gen Physiol. 1992;100:933–961. doi: 10.1085/jgp.100.6.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hilgemann DW, Matuoska S, Nagel GA, Collins A. Steady state and dynamic properties of cardiac sodium-calcium exchange; sodium dependent inactivation. J Gen Physiol. 1992;100:905–932. doi: 10.1085/jgp.100.6.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Hilgemann DW, Nicoll DA, Philipson KD. Charge movement during Na+ translocation by native and cloned cardiac Na+/Ca2+ exchanger. Nature. 1991;352:715–718. doi: 10.1038/352715a0. [DOI] [PubMed] [Google Scholar]
- 210.Hill JA, Coronado R, Strauss HC. Reconstitution and characterization of a calcium-activated channel from heart. Circ Res. 1988;62:411–415. doi: 10.1161/01.res.62.2.411. [DOI] [PubMed] [Google Scholar]
- 211.Hinata M, Yamamura H, Li L, Watanabe Y, Watano T, Imaizumi Y, Kimura J. Stoichiometry of Na/Ca exchange is 3:1 in guinea pig ventricular myocytes. J Physiol. 2002;545:453–461. doi: 10.1113/jphysiol.2002.025866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Hirano Y, Fozzard HA, January CT. Characteristics of L- and T-type Ca2+ currents in canine cardiac Purkinje cells. Am J Physiol Heart Circ Physiol. 1989;256:H1478–H1492. doi: 10.1152/ajpheart.1989.256.5.H1478. [DOI] [PubMed] [Google Scholar]
- 213.Hirano Y, Fozzard HA, January CT. Inactivation properties of T-type calcium current in canine cardiac Purkinje cells. Biophys J. 1989;56:1007–1016. doi: 10.1016/S0006-3495(89)82745-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Hirano Y, Hiraoka M. Dual modulation of unitary L-type Ca2+ channel currents by [Ca2+]i in fura-2-loaded guinea-pig ventricular myocytes. J Physiol. 1994;480:449–463. doi: 10.1113/jphysiol.1994.sp020374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Hirano Y, Moscucci A, January CT. Direct measurement of L type Ca window current in heart cells. Circ Res. 1992;70:445–455. doi: 10.1161/01.res.70.3.445. [DOI] [PubMed] [Google Scholar]
- 216.Hiraoka M. The role of the positive dynamic current on the action potential of cardiac Purkinje fibers. Jpn J Physiol. 1975;25:705–717. doi: 10.2170/jjphysiol.25.705. [DOI] [PubMed] [Google Scholar]
- 217.Hiraoka M, Kawano S. Calcium-sensitive and insensitive transient outward current in rabbit ventricular myocytes. J Physiol. 1989;410:187–212. doi: 10.1113/jphysiol.1989.sp017528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Hirayama Y, Saitoh H, Atarashi H, Hayakawa H. Electrical and mechanical alternans in canine myocardium in vivo, dependence on intracellular calcium cycling. Circulation. 1993;88:2894–2902. doi: 10.1161/01.cir.88.6.2894. [DOI] [PubMed] [Google Scholar]
- 219.Hirose M, Laurita KR. Calcium mediated triggered activity is an underlying cellular mechanism of ectopy originating from pulmonary vein in canine. Am J Physiol Heart Circ Physiol. doi: 10.1152/ajpheart.00826.2006. In press. [DOI] [PubMed] [Google Scholar]
- 220.Hobai IA, O'Rourke B. Enhanced Ca2+ activated Na+ Ca2+ exchange activity in canine pacing-induced heart failure. Circ Res. 2000;87:690–698. doi: 10.1161/01.res.87.8.690. [DOI] [PubMed] [Google Scholar]
- 221.Hofmann PA, Fuchs F. Bound calcium and force development in skinned cardiac muscle bundles: effect of sarcomere length. J Mol Cell Cardiol. 1988;20:667–677. doi: 10.1016/s0022-2828(88)80012-9. [DOI] [PubMed] [Google Scholar]
- 222.Hongo K, White E, Le Guennec JY, Orchard CH. Changes in [Ca2+]i, [Na+]i and Ca2+ current in isolated rat ventricular myocytes following an increase in cell length. J Physiol. 1996;491:609–619. doi: 10.1113/jphysiol.1996.sp021243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Honjo H, Inada M, Lancaster MK, Yamamoto M, Niwa R, Jones SA, Shibata N, Mitsui K, Horiuchi T, Kamiya K, Kodama I, Boyett MR. Sarcoplasmic reticulum Ca2+ release is not a dominating factor in sinoatrial node pacemaker activity. Circ Res. 2003;92:e41–e44. doi: 10.1161/01.res.0000055904.21974.be. [DOI] [PubMed] [Google Scholar]
- 224.Housmans PR, Lee NKM, Blinks JR. Active shortening retards the decline of the intracellular calcium transient in mammalian heart muscle. Science. 1983;221:159–161. doi: 10.1126/science.6857274. [DOI] [PubMed] [Google Scholar]
- 225.Hove-Madsen L, Llach A, Bayes-Genis A, Roura S, Rodriguez Font E, Aris A, Cinca J. Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation. 2004;110:1358–1363. doi: 10.1161/01.CIR.0000141296.59876.87. [DOI] [PubMed] [Google Scholar]
- 226.Hu H, Sachs F. Stretch-activated ion channels in the heart. J Mol Cell Cardiol. 1997;29:1511–1523. doi: 10.1006/jmcc.1997.0392. [DOI] [PubMed] [Google Scholar]
- 227.Hudmon A, Schulman H, Kim J, Maltez JM, Tsien RW, Pitt GS. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J Cell Biol. 2005;171:537–547. doi: 10.1083/jcb.200505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Huffaker R, Lamp ST, Weiss JN, Kogan B. Intracellular calcium cycling, early afterdepolarizations, reentry in simulated long QT syndrome. Heart Rhythm. 2004;1:441–448. doi: 10.1016/j.hrthm.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 229.Hunton DL, Zou L, Pang Y, Marchase RB. Adult rat cardiomyocytes exhibit capacitative calcium entry. Am J Physiol Heart Circ Physiol. 2004;286:H1124–H1132. doi: 10.1152/ajpheart.00162.2003. [DOI] [PubMed] [Google Scholar]
- 230.Hurtado C, Prociuk M, Maddaford TG, Dibrov E, Mesaeli N, Hryshko LV, Pierce GN. Cells expressing unique Na-Ca exchange (NCX1) splice variants exhibit different susceptibilities to Ca overload. Am J Physiol Heart Circ Physiol. 2006;290:H2155–H2162. doi: 10.1152/ajpheart.00958.2005. [DOI] [PubMed] [Google Scholar]
- 231.Huser J, Blatter LA, Lipsius S. Intracellular Ca2+ release contributes to automaticity in cat atrial pacemaker cells. J Physiol. 2000;542:415–422. doi: 10.1111/j.1469-7793.2000.00415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Huser J, Lipsius SL, Blatter LA. Calcium gradients during excitation-contraction coupling in cat atrial myocytes. J Physiol. 1996;494:641–651. doi: 10.1113/jphysiol.1996.sp021521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Huser J, Wang YG, Sheehan KA, Cifuentes F, Lipsius SL, Blatter LA. Functional coupling between glycolysis and excitation-contraction coupling underlies alternans in cat heart cells. J Physiol. 2000;524:795–806. doi: 10.1111/j.1469-7793.2000.00795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Ibarra J, Morley GE, Delmar M. Dynamics of the inward rectifier potassium current during the action potential of guinea pig ventricular myocytes. Biophys J. 1991;60:1534–1539. doi: 10.1016/S0006-3495(91)82187-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Ikemoto N, Yamamoto T. Postulated role of inter-domain interaction within the ryanodine receptor in Ca2+ channel regulation. Trends Cardiovasc Med. 2000;10:310–316. doi: 10.1016/s1050-1738(01)00067-6. [DOI] [PubMed] [Google Scholar]
- 236.Imagawa T, Smith JS, Coronado R, Campbell KP. Purified ryanodine receptor from skeletal muscle sarcoplasmic reticulum is the calcium permeable pore of the calcium release channel. J Biol Chem. 1987;262:16636–16643. [PubMed] [Google Scholar]
- 237.Inoue M, Bridge JH. Ca2+ sparks in rabbit ventricular myocytes evoked by action potentials. Involvement of clusters of L type Ca2+ channels. Circ Res. 2003;92:532–538. doi: 10.1161/01.RES.0000064175.70693.EC. [DOI] [PubMed] [Google Scholar]
- 238.Inui M, Saito A, Fleischer S. Isolation of the ryanodine receptor from cardiac sarcoplasmic reticulum and identity with the feet structures. J Biol Chem. 1987;262:15637–15642. [PubMed] [Google Scholar]
- 239.Inui M, Saito A, Fleischer S. Purification of the ryanodine receptor and identity with feet structures of junctional terminal cisternae of sarcoplasmic reticulum from fast skeletal muscle. J Biol Chem. 1987;262:1740–1747. [PubMed] [Google Scholar]
- 240.Irisawa H, Brown HF, Giles W. Cardiac pacemaking in the sinoatrial node. Physiol Rev. 1993;73:197–227. doi: 10.1152/physrev.1993.73.1.197. [DOI] [PubMed] [Google Scholar]
- 241.Ishide N, Miura M, Sakurai M, Takishima T. Initiation and development of calcium waves in rat myocytes. Am J Physiol Heart Circ Physiol. 1992;263:H327–H332. doi: 10.1152/ajpheart.1992.263.2.H327. [DOI] [PubMed] [Google Scholar]
- 242.Ishide N, Urayama T, Inoue K, Komaru T, Takishima T. Propagation and collision characteristics of calcium waves in rat myocytes. Am J Physiol Heart Circ Physiol. 1990;259:H940–H950. doi: 10.1152/ajpheart.1990.259.3.H940. [DOI] [PubMed] [Google Scholar]
- 243.Isom LL, DeJongh KS, Catterall WA. Auxiliary subunits of voltage gated ion channels. Neuron. 1994;12:1183–1194. doi: 10.1016/0896-6273(94)90436-7. [DOI] [PubMed] [Google Scholar]
- 244.Iwasa Y, Hosey MM. Phosphorylation of cardiac sarcolemma proteins by the calcium-activated phospholipid-dependent protein kinase. J Biol Chem. 1984;259:534–540. [PubMed] [Google Scholar]
- 245.Jabr RI, Cole WC. Oxygen-derived free radical stress activates nonselective cation current in guinea pig ventricular myocytes. Role of sulfhydrl groups. Circ Res. 1995;76:812–824. doi: 10.1161/01.res.76.5.812. [DOI] [PubMed] [Google Scholar]
- 246.James P, Inui M, Tada M, Chiesi M, Carafoli E. Nature and site of phospholamban regulation of the Ca2+ pump of sarcoplasmic reticulum. Nature. 1989;342:90–92. doi: 10.1038/342090a0. [DOI] [PubMed] [Google Scholar]
- 247.January C, Riddle JM. Early afterdepolarizations: mechanisms of induction and block. A role for the L type Ca current. Circ Res. 1989;64:977–990. doi: 10.1161/01.res.64.5.977. [DOI] [PubMed] [Google Scholar]
- 248.Janvier NC, Boyett MR. The role of NaCa exchange current in the cardiac action potential. Cardiovasc Res. 1996;32:69–84. [PubMed] [Google Scholar]
- 249.Jayaraman T, Brillantes AM, Timerman AP, Erdjument-Bromage H, Fleischer S, Tempst P, Marks AR. FK506 binding protein associated with the calcium release channel (ryanodine receptor) J Biol Chem. 1992;267:9474–9477. [PubMed] [Google Scholar]
- 250.Jiang D, Xiao B, Zhang L, Chen W. Enhanced basal activity of a cardiac Ca2+ release channel (ryanodine receptor) mutant associated with ventricular tachycardia and sudden death. Circ Res. 2002;91:218–225. doi: 10.1161/01.res.0000028455.36940.5e. [DOI] [PubMed] [Google Scholar]
- 251.Jiang D, Wang R, Xiao B, Kong H, Hunt DJ, Choi P, Zhang L, Chen SR. Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ Res. 2005;97:1173–1181. doi: 10.1161/01.RES.0000192146.85173.4b. [DOI] [PubMed] [Google Scholar]
- 252.Jiang D, Xiao B, Yang D, Wang R, Choi P, Zhang L, Cheng H, Chen SR. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR) Proc Natl Acad Sci USA. 2004;101:13062–13067. doi: 10.1073/pnas.0402388101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Jiang QX, Wang DN, MacKinnon R. Electron microscopic analysis of KvAP voltage-dependent K+ channels in an open conformation. Nature. 2004;430:806–810. doi: 10.1038/nature02735. [DOI] [PubMed] [Google Scholar]
- 254.Johnson JP, Jr, Balser JR, Bennett PB. A novel extracellular calcium sensing mechanism in voltage-gated potassium ion channels. J Neurosci. 2001;21:4143–4153. doi: 10.1523/JNEUROSCI.21-12-04143.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Johnson JP, Mullins FM, Bennett PB. Human ether a go go related gene K channel gating probed with extracellular Ca2+ J Gen Physiol. 1999;113:565–580. doi: 10.1085/jgp.113.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Jorgensen AO, Shen ACY, Arnold W, McPherson P, Campbell KP. The Ca2+ release channel/ryanodine receptor is localized in junctional and corbular sarcoplasmic reticulum in cardiac muscle. J Cell Biol. 1993;120:969–980. doi: 10.1083/jcb.120.4.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Joyner RW, Kumar R, Wilders R, Jongsma HJ, Verheijek EE, Golod DA, vanGinneken ACG, Wagner MB, Goolsby WN. Modulating L type calcium current affects discontinuous cardiac action potential conduction. Biophys J. 1996;71:237–245. doi: 10.1016/S0006-3495(96)79220-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Kamkin A, Kiseleva I, Isenberg G. Stretch activated currents in ventricular myocytes: amplitude and arrhythmogenic effects increase with hypertrophy. Cardiovasc Res. 2000;48:409–420. doi: 10.1016/s0008-6363(00)00208-x. [DOI] [PubMed] [Google Scholar]
- 259.Kaneko T, Tanaka H, Oyamada M, Kawata S, Takamatsu T. Three distinct types of Ca2+ waves in Langendorff-perfused rat heart revealed by real-time confocal microcopy. Circ Res. 2000;86:1093–1099. doi: 10.1161/01.res.86.10.1093. [DOI] [PubMed] [Google Scholar]
- 260.Kanevsky N, Dascal N. Regulation of maximal open probability is a separable function of Cavβ subunit in L-type Ca2+ channel, dependent on NH2 terminus of α1C (Cav1.2{alpha}) J Gen Physiol. 2006;128:15–36. doi: 10.1085/jgp.200609485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Kang TM, Hilgemann DW. Multiple transport modes of the cardiac Na+/Ca2+ exchanger. Nature. 2004;427:544–548. doi: 10.1038/nature02271. [DOI] [PubMed] [Google Scholar]
- 262.Kannankeril PJ, Mitchell BM, Goonasekera SA, Chelu MG, Zhang W, Sood S, Kearney DL, Danila CI, De Biasi M, Wehrens XHT, Pautler RG, Roden DM, Taffet GE, Dirksen RT, Anderson ME, Hamilton SL. Mice with the R176Q cardiac ryanodine receptor mutation exhibit catecholamine-induced ventricular tachycardia and cardiomyopathy. Proc Natl Acad Sci USA. 2006;103:12179–12184. doi: 10.1073/pnas.0600268103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Kass RS, Scheuer T. Slow inactivation of calcium channels in the cardiac Purkinje fiber. J Mol Cell Cardiol. 1982;14:615–618. doi: 10.1016/0022-2828(82)90148-1. [DOI] [PubMed] [Google Scholar]
- 264.Kass RS, Tsien RW, Weingart R. Ionic basis of transient inward current induced by strophanthidin in cardiac purkinje fibres. J Physiol. 1978;281:209–226. doi: 10.1113/jphysiol.1978.sp012417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Kass RS, Wiegers SE. The ionic basis of concentration-related effects of noradrenaline on the action potential of calf cardiac Purkinje fibres. J Physiol. 1982;322:541–558. doi: 10.1113/jphysiol.1982.sp014054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Katra RP, Pruvot E, Laurita KR. Intracellular calcium handling heterogeneities in intact guinea pig heart. Am J Physiol Heart Circ Physiol. 2004;286:H648–H656. doi: 10.1152/ajpheart.00374.2003. [DOI] [PubMed] [Google Scholar]
- 267.Katra RP, Laurita KR. Cellular mechanism of calcium-mediated triggered activity in the heart. Circ Res. 2005;96:535–542. doi: 10.1161/01.RES.0000159387.00749.3c. [DOI] [PubMed] [Google Scholar]
- 268.Kawano S, Hiraoka M. Transient outward currents and action potential alterations in rabbit ventricular myocytes. J Mol Cell Cardiol. 1991;23:681–693. doi: 10.1016/0022-2828(91)90978-u. [DOI] [PubMed] [Google Scholar]
- 269.Kawano S, Hirayama Y, Hiraoka M. Activation mechanism of Ca sensitive transient outward current in rabbit ventricular myocytes. J Physiol. 1995;486:593–604. doi: 10.1113/jphysiol.1995.sp020837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Kawano S, Nakamura F, Tanaka T, Hiraoka M. Cardiac sarcoplasmic reticulum chloride channels regulated by PKA. Circ Res. 1992;71:585–589. doi: 10.1161/01.res.71.3.585. [DOI] [PubMed] [Google Scholar]
- 271.Kentish JC. The effects of inorganic phosphate and creatine phosphate on force production in skinned muscles from rat ventricle. J Physiol. 1986;370:585–604. doi: 10.1113/jphysiol.1986.sp015952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Kentish JC, ter Keurs HEDJ, Ricciardi L, Bucx JJ, Noble MIM. Comparison between the sarcomere length force relations of intact and skinned trabeculae from rat right ventricle. Circ Res. 1986;58:755–768. doi: 10.1161/01.res.58.6.755. [DOI] [PubMed] [Google Scholar]
- 273.Kihara Y, Morgan JP. Abnormal Cai handling is the primary cause of mechanical alternans: study in ferret ventricular muscles. Am J Physiol Heart Circ Physiol. 1991;261:H1746–H1755. doi: 10.1152/ajpheart.1991.261.6.H1746. [DOI] [PubMed] [Google Scholar]
- 274.Kijima Y, Saito A, Jetton TL, Magnuson MA, Fleischer S. Different intracellular localization of inositol 1,4,5-triphosphate and ryanodine receptors in cardiomyocytes. J Biol Chem. 1993;268:3499–3506. [PubMed] [Google Scholar]
- 275.Kim D. A mechanosensitive K+ channel in heart cells. Activation by arachidonic acid. J Gen Physiol. 1992;100:1021–1040. doi: 10.1085/jgp.100.6.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Kim D, Fu C. Activation of a nonselective cation channel by swelling in atrial cells. J Membr Biol. 1993;135:27–37. doi: 10.1007/BF00234649. [DOI] [PubMed] [Google Scholar]
- 277.Kim J, Ghosh S, Liu H, Tateyama M, Kass RS, Pitt GS. Calmodulin mediates Ca2+ sensitivity of sodium channels. J Biol Chem. 2004;279:45004–45012. doi: 10.1074/jbc.M407286200. [DOI] [PubMed] [Google Scholar]
- 278.Kim J, Ghosh S, Nunziato DA, Pitt GS. Identification of the components controlling inactivation of voltage-gated Ca2+ channels. Neuron. 2004;41:745–754. doi: 10.1016/s0896-6273(04)00081-9. [DOI] [PubMed] [Google Scholar]
- 279.Kimura J, Miyamae S, Noma A. Identification of sodium-calcium exchange current in single ventricular cells of guinea pig. J Physiol. 1987;384:199–222. doi: 10.1113/jphysiol.1987.sp016450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Klockner U, Mikala G, Varadi M, Varadi G, Schwartz A. Involvement of the carboxyl-terminal region of the alpha 1 subunit in voltage-dependent inactivation of cardiac calcium channels. J Biol Chem. 1995;270:17306–17310. doi: 10.1074/jbc.270.29.17306. [DOI] [PubMed] [Google Scholar]
- 281.Klugbauer N, Lacinova L, Marais E, Hobum M, Hofmann F. Molecular diversity of the calcium channel Alpha2Delta subunit. J Neurosci. 1999;19:684–691. doi: 10.1523/JNEUROSCI.19-02-00684.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Klugbauer N, Marais E, Hofmann F. Calcium channel alpha2/delta subunits: differential expression function and drug binding. J Bioenerg Biomembr. 2003;35:639–647. doi: 10.1023/b:jobb.0000008028.41056.58. [DOI] [PubMed] [Google Scholar]
- 283.Kockskamper J, Blatter LA. Subcellular Ca2+ alternans represents a novel mechanism for the generation of arrhythmogenic Ca2+ waves in cat atrial myocytes. J Physiol. 2002;545:65–79. doi: 10.1113/jphysiol.2002.025502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Kockskamper J, Sheehan KA, Bare DJ, Lipsius S, Mignery GA, Blatter LA. Activation and propagation of Ca2+ release during excitation contraction coupling in atrial myocytes. Biophys J. 2001;81:2590–2605. doi: 10.1016/S0006-3495(01)75903-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Kort AA, Capogrossi MC, Lakatta EG. Frequency, amplitude, propagation velocity of spontaneous calcium-dependent contractile waves in intact adult rat cardiac muscle and isolated myocytes. Circ Res. 1985;57:844–855. doi: 10.1161/01.res.57.6.844. [DOI] [PubMed] [Google Scholar]
- 286.Kort AA, Lakatta EG. Bimodal effect of stimulation on light fluctuation transients monitoring spontaneous sarcoplasmic reticulum Ca2+ release in rat cardiac muscle. Circ Res. 1988;63:960–968. doi: 10.1161/01.res.63.5.960. [DOI] [PubMed] [Google Scholar]
- 287.Kort AA, Lakatta EG. Calcium-dependent mechanical oscillations occur spontaneously in unstimulated mammalian cardiac tissues. Circ Res. 1984;54:396–404. doi: 10.1161/01.res.54.4.396. [DOI] [PubMed] [Google Scholar]
- 288.Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Engel J, Striessnig J. Alpha 1D (Cav1.3) subunits can form L-type Ca2+ channels activating at negative voltages. J Biol Chem. 2001;276:22100–22106. doi: 10.1074/jbc.M101469200. [DOI] [PubMed] [Google Scholar]
- 289.Koster OF, Szigeti GP, Beuckelmann DJ. Characterization of a Cai dependent current in human atrial and ventricular cardiomyocytes in the absence of Na+ and K+ Cardiovasc Res. 1999;41:175–187. doi: 10.1016/s0008-6363(98)00202-8. [DOI] [PubMed] [Google Scholar]
- 290.Kovacs RJ, Nelson MT, Simmerman HK, Jones LR. Phospholamban forms Ca2+-sensitive channels in lipid bilayers. J Biol Chem. 1988;263:18364–18368. [PubMed] [Google Scholar]
- 291.Kubalova Z, Gyorke I, Terentyeva R, Viatchenko-Karpinski S, Terentyev D, Williams SC, Gyorke S. Modulation of cytosolic and intra-sarcoplasmic reticulum calcium waves by calsequestrin in rat cardiac myocytes. J Physiol. 2004;561:515–524. doi: 10.1113/jphysiol.2004.073940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Kuruma A, Hartzell HC. Bimodal control of a Ca2+ activated Cl− channel by different Ca2+ signals. J Gen Physiol. 2000;115:59–80. doi: 10.1085/jgp.115.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Lab M, Allen DG, Orchard CH. The effects of shortening on myoplasmic calcium concentration and on the action potential in mammalian ventricular muscle. Circ Res. 1984;55:825–829. doi: 10.1161/01.res.55.6.825. [DOI] [PubMed] [Google Scholar]
- 294.Lab MJ, Lee JA. Changes in intracellular calcium during mechanical alternans in isolated ferret ventricular muscle. Circ Res. 1990;66:585–595. doi: 10.1161/01.res.66.3.585. [DOI] [PubMed] [Google Scholar]
- 295.Lacerda AE, Kim HS, Ruth P, Perez Reyes E, Fockerzi V, Hofmann F, Birnbaumer L, Brown AM. Normalization of current kinetics by interaction between alpha 1 and beta subunits of the skeletal muscle dihydropyridine sensitive Ca2+ channel. Nature. 1991;352:527–530. doi: 10.1038/352527a0. [DOI] [PubMed] [Google Scholar]
- 296.Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O, Levy-Nissenbaum E, Khoury A, Lorber A, Goldman B, Lancet D, Eldar MA. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet. 2001;69:1378–1384. doi: 10.1086/324565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Lai FA, Anderson K, Rousseau E, Liu Q, Meissner G. Evidence for a Ca2+ channel within the ryanodine receptor complex from cardiac sarcoplasmic reticulum. Biochem Biophys Res Commun. 1988;151:441–449. doi: 10.1016/0006-291x(88)90613-4. [DOI] [PubMed] [Google Scholar]
- 298.Lai FA, Erickson HP, Rousseau E, Liu Q, Meissner G. Purification and reconstitution of the calcium release channel from skeletal muscle. Nature. 1988;331:315–319. doi: 10.1038/331315a0. [DOI] [PubMed] [Google Scholar]
- 299.Laitinen PJ, Brown KM, Piip OK, Swan H, Devaney JM, Brahmbhatt B, Donarum EA, Marino M, Tiso N, Viitasalo M, Toivonen L, Stephan DA, Kontula K. Mutations of the cardiac ryanodine receptor (RYR2) gene in familial polymorphic ventricular tachycardia. Circulation. 2001;103:485–490. doi: 10.1161/01.cir.103.4.485. [DOI] [PubMed] [Google Scholar]
- 300.Laitinen PJ, Swan H, Kontula K. Molecular genetics of exercise induced polymorphic ventricular tachycardia: identification of three novel cardiac ryanodine receptor mutations and two common calsequestrin 2 amino acid polymorphisms. Eur J Hum Genet. 2003;11:888–891. doi: 10.1038/sj.ejhg.5201061. [DOI] [PubMed] [Google Scholar]
- 301.Lakatta EG, Jewell BR. Length dependent activation. Its effect on the length tension relation in cat ventricular muscle. Circ Res. 1977;40:251–257. doi: 10.1161/01.res.40.3.251. [DOI] [PubMed] [Google Scholar]
- 302.Lakatta EG, Capogrossi MC, Kort AA, Stern MD. Spontaneous myocardial calcium oscillations: overview with emphasis on ryanodine and caffeine. Fed Proc. 1985;44:2977–2983. [PubMed] [Google Scholar]
- 303.Lakatta EG, Lappe DL. Diastolic scattered light fluctuation, resting force and twitch force in mammalian cardiac muscle. J Physiol. 1981;315:369–394. doi: 10.1113/jphysiol.1981.sp013753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Lakatta EG, Maltsev VA, Bogdanov KY, Stern MD, Vinogradova TM. Cyclic variation of intracellular calcium. A critical factor for cardiac pacemaker cell dominance. Circ Res. 2003;92:e45–e50. doi: 10.1161/01.res.0000055920.64384.fb. [DOI] [PubMed] [Google Scholar]
- 305.Lakkireddy V, Bub G, Baweja P, Syed A, Boutjdir M, El-Sherif N. The kinetics of spontaneous calcium oscillations and arrhythmogenesis in the in vivo heart during ischemia/reperfusion. Heart Rhythm. 2006;3:58–66. doi: 10.1016/j.hrthm.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 306.Lambert RC, Maulet Y, Mouton J, Beattie R, Volsen S, De Waard M, Feltz A. T-type Ca2+ current properties are not modified by Ca2+ channel β subunit depletion in nodosus ganglion neurons. J Neurosci. 1997;17:6621–6628. doi: 10.1523/JNEUROSCI.17-17-06621.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Lamont C, Eisner DA. The sarcolemmal mechanisms involved in the control of diastolic intracellular calcium in isolated rat cardiac trabeculae. Pflügers Arch. 1996;432:961–969. doi: 10.1007/s004240050223. [DOI] [PubMed] [Google Scholar]
- 308.Lamont C, Luther PW, Balke CW, Wier WG. Intercellular Ca2+ waves in rat heart muscle. J Physiol. 1998;512:669–676. doi: 10.1111/j.1469-7793.1998.669bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Le Grande B, Deroubaix E, Coulombe A, Coraboeuf E. Stimulatory effect of ouabain on T- and L-type calcium currents in guinea pig cardiac myocytes. Am J Physiol Heart Circ Physiol. 1990;258:H1620–H1623. doi: 10.1152/ajpheart.1990.258.5.H1620. [DOI] [PubMed] [Google Scholar]
- 310.Lederer WJ, Tsien RW. Transient inward current underlying arrhythmogenic effects of cardiotonic steroids in Purkinje fibers. J Physiol. 1976;263:73–100. doi: 10.1113/jphysiol.1976.sp011622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Lee JA, Allen DG. EMD 53998 sensitizes the contractile proteins to calcium in intact ferret ventricular muscle. Circ Res. 1991;69:927–936. doi: 10.1161/01.res.69.4.927. [DOI] [PubMed] [Google Scholar]
- 312.Lee KS, Marban E, Tsien RW. Inactivation of calcium channels in mammalian heart cells: joint dependence on membrane potential and intracellular calcium. J Physiol. 1985;364:395–411. doi: 10.1113/jphysiol.1985.sp015752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children. Circulation. 1995;91:1512–1519. doi: 10.1161/01.cir.91.5.1512. [DOI] [PubMed] [Google Scholar]
- 314.Lefevre T, Coraboeuf E, Ghazi A, Coulombe A. Divalent cation channels activated by phenothiazines in membrane of rat ventricular myocytes. J Membr Biol. 1995;147:147–158. doi: 10.1007/BF00233543. [DOI] [PubMed] [Google Scholar]
- 315.Lehnart SE, Wehrens XH, Laitinen PJ, Reiken S, Deng SX, Cheng Z, Landry DW, Kontula K, Swan H, Marks AR. Sudden death in familial polymorphic ventricular tachycardia associated with calcium release channel (ryanodine receptor) leak. Circulation. 2004;109:3208–3214. doi: 10.1161/01.CIR.0000132472.98675.EC. [DOI] [PubMed] [Google Scholar]
- 316.Levitsky DO, Nicoll DA, Philipson KD. Identification of the high affinity Ca2+-binding domain of the cardiac Na+-Ca2+ exchanger. J Biol Chem. 1994;269:22847–22852. [PubMed] [Google Scholar]
- 317.Lew WY, Hryshko LV, Bers DM. Dihydropyridine receptors are primarily functional L type calcium channels in rabbit ventricular myocytes. Circ Res. 1991;69:1139–1145. doi: 10.1161/01.res.69.4.1139. [DOI] [PubMed] [Google Scholar]
- 319.Li GR, Feng J, Wang Z, Fermini B, Nattel S. Comparative mechanisms of 4 aminopyridine resistant Ito in human and rabbit atrial myocytes. Am J Physiol Heart Circ Physiol. 1995;269:H463–H472. doi: 10.1152/ajpheart.1995.269.2.H463. [DOI] [PubMed] [Google Scholar]
- 320.Li H, Guo W, Mellor RL, Nerbonne JM. KChIP2 modulates the cell surface expression of Kv1.5-encoded K+ channels. J Mol Cell Cardiol. 2005;39:121–132. doi: 10.1016/j.yjmcc.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 321.Li J, Qu J, Nathan RD. Ionic basis of ryanodine's negative chronotropic effect on pacemaker cells isolated from the sinoatrial node. Am J Physiol Heart Circ Physiol. 1997;273:H2481–H2489. doi: 10.1152/ajpheart.1997.273.5.H2481. [DOI] [PubMed] [Google Scholar]
- 322.Li X, Zima AV, Sheikh F, Blatter LA, Chen J. Endothelin 1 induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol 1,4,5 trisphosphate (IP3) receptor type 2-deficient mice. Circ Res. 2005;96:1274–1281. doi: 10.1161/01.RES.0000172556.05576.4c. [DOI] [PubMed] [Google Scholar]
- 323.Liang H, DeMaria CD, Erickson MG, Mori M, Alseikhan B, Yue DT. Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron. 2003;39:951–960. doi: 10.1016/s0896-6273(03)00560-9. [DOI] [PubMed] [Google Scholar]
- 324.Liao P, Yong TF, Liang MC, Yue DT, Soong TW. Splicing for alternative structures of Cav1.2 Ca2+ channels in cardiac and smooth muscles. Cardiovasc Res. 2005;68:197–203. doi: 10.1016/j.cardiores.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 325.Lipp P, Laine M, Tovey SC, Burrell KM, Berridge MJ, Li W, Bootman MD. Functional InsP3 receptors that may modulate excitation-contraction coupling in the heart. Curr Biol. 2000;10:939–942. doi: 10.1016/s0960-9822(00)00624-2. [DOI] [PubMed] [Google Scholar]
- 326.Lipp P, Niggli E. Submicroscopic calcium signals as fundamental events of excitation contraction coupling in guinea pig cardiac myocytes. J Physiol. 1996;492:31–38. doi: 10.1113/jphysiol.1996.sp021286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Lipp P, Niggli E. Microscopic spiral waves reveal positive feedback in subcellular calcium signaling. Biophys J. 1993;65:2272–2276. doi: 10.1016/S0006-3495(93)81316-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Lipp P, Pott L, Callewaert G, Carmeliet E. Simultaneous recordings of Indo-1 fluorescence and Na/Ca exchange current reveals two components of Ca2+ release from sarcoplasmic reticulum of cardiac atrial myocytes. FEBS Lett. 1990;275:181–184. doi: 10.1016/0014-5793(90)81467-3. [DOI] [PubMed] [Google Scholar]
- 329.Lipp P, Pott L, Callewaert G, Carmeliet EE. Calcium transients caused by calcium entry are influenced by the sarcoplasmic reticulum in guinea-pig atrial myocytes. J Physiol. 1992;454:321–338. doi: 10.1113/jphysiol.1992.sp019266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Litwin SE, Bridge JH. Enhanced NaCa exchange in the infarcted heart. Implications for excitation contraction coupling. Circ Res. 1997;81:1083–1093. doi: 10.1161/01.res.81.6.1083. [DOI] [PubMed] [Google Scholar]
- 331.Litwin SE, Li J, Bridge JH. Na-Ca exchange and the trigger for sarcoplasmic reticulum Ca release: studies in adult rabbit ventricular myocytes. Biophys J. 1998;75:359–371. doi: 10.1016/S0006-3495(98)77520-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Liu Z, Zhang J, Sharma MR, Li P, Chen SR, Wagenknecht T. Three-dimensional reconstruction of the recombinant type 3 ryanodine receptor and localization of its amino terminus. Proc Natl Acad Sci USA. 2001;98:6104–6109. doi: 10.1073/pnas.111382798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Liu Z, Wang R, Zhang J, Chen SRW, Wagenknecht T. Localization of a disease-associated mutation site in the three-dimensional structure of the cardiac muscle ryanodine receptor. J Biol Chem. 2005;280:37941–37947. doi: 10.1074/jbc.M505714200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Liu Z, Zhang J, Li P, Chen SRW, Wagenknecht T. Three-dimensional reconstruction of the recombinant type 2 ryanodine receptor and localization of its divergent region 1. J Biol Chem. 2002;277:46712–46719. doi: 10.1074/jbc.M208124200. [DOI] [PubMed] [Google Scholar]
- 335.Lopez-Lopez JR, Shacklock PS, Balke CW, Wier WG. Local stochastic release of Ca2+ in voltage clamped rat heart cells: visualization with confocal microscopy. J Physiol. 1994;480:21–29. doi: 10.1113/jphysiol.1994.sp020337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Lopez-Lopez JR, Shacklock PS, Balke CW, Wier WG. Local Ca2+ transients triggered by single L type Ca2+ channel currents in cardiac cells. Science. 1995;268:1042–1045. doi: 10.1126/science.7754383. [DOI] [PubMed] [Google Scholar]
- 337.Ludtke SJ, Serysheva II, Hamilton SL, Chiu W. The pore structure of the closed RyR1 channel. Structure. 2005;13:1203–1211. doi: 10.1016/j.str.2005.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Luscher C, Lipp P, Luscher HR, Niggli E. Control of action potential propagation by intracellular Ca2+ in cultured rat dorsal root ganglion cells. J Physiol. 1996;490:319–324. doi: 10.1113/jphysiol.1996.sp021146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Luyanenko V, Gyorke I, Gyorke S. Regulation of calcium release by calcium inside the sarcoplasmic reticulum in ventricular myocytes. Pflügers Arch. 1996;432:1047–1054. doi: 10.1007/s004240050233. [DOI] [PubMed] [Google Scholar]
- 340.Machaca K, Hartzell HC. Reversible Ca2+ gradients between the subsarcolemma and cytosol differentially activate Ca-dependent Cl currents. J Gen Physiol. 1999;113:249–266. doi: 10.1085/jgp.113.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Mackenzie L, Bootman MD, Berridge MJ, Lipp P. Predetermined recruitment of calcium release sites underlies excitation-contraction coupling in rat atrial myocytes. J Physiol. 2001;530:417–429. doi: 10.1111/j.1469-7793.2001.0417k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Mackenzie L, Bootman MD, Laine M, Berridge MJ, Thuring J, Holmes A, Li WH, Lipp P. The role of inositol 1,4,5-trisphosphate receptors in Ca(2+) signaling and the generation of arrhythmias in rat atrial myocytes. J Physiol. 2002;541:395–409. doi: 10.1113/jphysiol.2001.013411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Mackenzie L, Roderick HL, Berridge MJ, Conway SJ, Bootman MD. The spatial pattern of atrial cardiomyocyte calcium signaling modulates contraction. J Cell Sci. 2004;117:6327–6337. doi: 10.1242/jcs.01559. [DOI] [PubMed] [Google Scholar]
- 344.MacLennan DH, Brandl CJ, Korczak B, Green NM. Calcium ATPases: contribution of molecular genetics to our understanding of structure and function. In: Hille B, Frambrough DM, editors. Proteins of Excitable Membranes. New York: Wiley; 1987. pp. 287–300. [PubMed] [Google Scholar]
- 345.Maier LS, Bers DM. Calcium, calmodulin, calcium-calmodulin kinase II: heartbeat to heartbeat and beyond. J Mol Cell Cardiol. 2002;34:919–939. doi: 10.1006/jmcc.2002.2038. [DOI] [PubMed] [Google Scholar]
- 346.Maltez JM, Nunziato DA, Kim J, Pitt GS. Essential Cavβ modulatory properties are AID-independent. Nat Struct Mol Biol. 2005;12:372–377. doi: 10.1038/nsmb909. [DOI] [PubMed] [Google Scholar]
- 347.Mangoni ME, Couette B, Bourinet E, Platzer J, Reimer D, Striessnig J, Nargeot J. Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc Natl Acad Sci USA. 2003;100:5543–5548. doi: 10.1073/pnas.0935295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Marais E, Klugbauer N, Hofmann F. Calcium channel alpha(2)delta subunits-structure and Gabapentin binding. Mol Pharm. 2001;59:1243–1248. doi: 10.1124/mol.59.5.1243. [DOI] [PubMed] [Google Scholar]
- 349.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 350.Masumiya H, Wang R, Zhang J, Xiao B, Chen SRW. Localization of the 12.6-kDa FK506-binding protein (FKBP126) binding site to the NH2-terminal domain of the cardiac Ca2+ release channel (ryanodine receptor) J Biol Chem. 2003;278:3786–3792. doi: 10.1074/jbc.M210962200. [DOI] [PubMed] [Google Scholar]
- 351.Matsuda H. Effects of intracellular calcium injection on steady state membrane currents in isolated single ventricular cells. Pflügers Arch. 1983;397:81–83. doi: 10.1007/BF00585176. [DOI] [PubMed] [Google Scholar]
- 352.Matsuda H, Cruz J dos S. Voltage dependent block of internal Ca2+ ions of inwardly rectifying K channels in guinea pig ventricular cells. J Physiol. 1993;470:295–311. doi: 10.1113/jphysiol.1993.sp019859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Matsuda H, Noma A, Kurachi Y, Irisawa H. Transient depolarization and spontaneous voltage fluctuations in isolated single cells from guinea-pig ventricles. Circ Res. 1982;51:142–151. doi: 10.1161/01.res.51.2.142. [DOI] [PubMed] [Google Scholar]
- 354.Matsuoka S, Nicoll DA, Hryshko LV, Levitsky DO, Weiss JN, Philipson KD. Regulation of the cardiac NaCa exchanger by Ca2+. Mutational analysis of the Ca2+ binding domain. J Gen Physiol. 1995;105:403–420. doi: 10.1085/jgp.105.3.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Matuoska S, Nicoll DA, He Z, Philipson KD. Regulation of cardiac NaCa exchanger by endogenous XIP region. J Gen Physiol. 1997;109:273–286. doi: 10.1085/jgp.109.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Mazzanti M, Assandri R, Ferroni A, DiFranesco D. Cytoskeletal control of rectification and expression of four substates in cardiac inward rectifier K channels. FASEB J. 1996;10:357–361. doi: 10.1096/fasebj.10.2.8641571. [DOI] [PubMed] [Google Scholar]
- 357.Mazzanti M, DeFelice LJ, Liu YM. Gating of L-type Ca2+ channels in embryonic chick ventricle cells: dependence on voltage, current and channel density. J Physiol. 1991;443:307–344. doi: 10.1113/jphysiol.1991.sp018835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.McCall E, Li L, Satoh H, Shannon TR, Blatter LA, Bers DM. Effects of FK-506 on contraction and Ca2+ transients in rat cardiac myocytes. Circ Res. 1996;79:1110–1121. doi: 10.1161/01.res.79.6.1110. [DOI] [PubMed] [Google Scholar]
- 359.McCormack JG, Denton RM. The effects of calcium ions and adenine nucleotides on the activity of pig heart 2-oxoglutarate dehydrogenase complex. Biochem J. 1979;180:533–544. doi: 10.1042/bj1800533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.McDonald TF, Pelzer S, Trautwein W, Pelzer D. Regulation and modulation of calcium channels in cardiac, skeletal and smooth muscle cells. Physiol Rev. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- 361.McGee AW, Nunziato DA, Maltez JM, Prehoda KE, Pitt GS, Bredt DS. Calcium channel function regulated by the SH3-GK module in β subunits. Neuron. 2004;42:89–99. doi: 10.1016/s0896-6273(04)00149-7. [DOI] [PubMed] [Google Scholar]
- 362.McIvor ME, Orchard CH, Lakatta EG. Dissociation of changes in apparent myofibrillar Ca2+ sensitivity and twitch relaxation induced by adrenergic and cholinergic stimulation in isolated ferret cardiac muscle. J Gen Physiol. 1988;92:509–529. doi: 10.1085/jgp.92.4.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Meissner G. Ryanodine activation and inhibition of the Ca release channel of sarcoplasmic reticulum. J Biol Chem. 1986;261:6300–6306. [PubMed] [Google Scholar]
- 364.Mentrard D, Vassort G, Fischmeister R. Calcium-mediated inactivation of the calcium conductance in cesium loaded frog heart cells. J Gen Physiol. 1984;83:105–131. doi: 10.1085/jgp.83.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Meszaros J, Khananshvili D, Hart G. Mechanisms underlying delayed afterdepolarizations in hypertrophied left ventricular myocytes of rats. Am J Physiol Heart Circ Physiol. 2001;281:H903–H914. doi: 10.1152/ajpheart.2001.281.2.H903. [DOI] [PubMed] [Google Scholar]
- 366.Mewes T, Ravens U. L-type calcium currents of human myocytes from ventricle of non-failing and failing hearts and from atrium. J Mol Cell Cardiol. 1994;26:1307–1320. doi: 10.1006/jmcc.1994.1149. [DOI] [PubMed] [Google Scholar]
- 367.Mignery GA, Johnston PA, Sudhof TC. Mechanism of Ca2+ inhibition of inositol 1,4,5-trisphosphate (InsP3) binding to the cerebellar InsP3 receptor. J Biol Chem. 1992;267:7450–7455. [PubMed] [Google Scholar]
- 368.Minamikawa T, Cody SH, Williams DA. In situ visualization of spontaneous calcium waves within perfused whole rat heart by confocal imaging. Am J Physiol Heart Circ Physiol. 1997;272:H236–H243. doi: 10.1152/ajpheart.1997.272.1.H236. [DOI] [PubMed] [Google Scholar]
- 369.Mitchell RD, Simmerman HK, Jones LR. Ca2+ binding effects on protein conformation and protein interactions of canine cardiac calsequestrin. J Biol Chem. 1988;263:1376–1381. [PubMed] [Google Scholar]
- 370.Mitterdorfer J, Froschmayr M, Grabner M, Moebius FF, Glossmann H, Streissing J. Identification of PKA phosphorylation sites in the carboxyl terminus of the L type calcium channel alpha1 subunit. Biochemistry. 1996;35:9400–9406. doi: 10.1021/bi960683o. [DOI] [PubMed] [Google Scholar]
- 371.Mitterdorfer J, Froschmayr M, Grabner M, Striessnig J, Glossmann H. Calcium channels: the beta subunit increases the affinity of dihydropyridine and Ca2+ binding sites of the alpha 1-subnit. FEBS Lett. 1994;352:141–145. doi: 10.1016/0014-5793(94)00938-4. [DOI] [PubMed] [Google Scholar]
- 372.Miura M, Boyden PA, ter Keurs HEDJ. Ca2+ waves during triggered propagated contractions in intact trabeculae: determinants of the velocity of propagation. Circ Res. 1999;84:1459–1468. doi: 10.1161/01.res.84.12.1459. [DOI] [PubMed] [Google Scholar]
- 373.Miura M, Ishide N, Numaguchi H, Takishima T. Diversity of early afterdepolarizations in guinea pig myocytes; Spatial characteristics of intracellular Ca2+ concentration. Heart Vessels. 1995;10:266–274. doi: 10.1007/BF01744906. [DOI] [PubMed] [Google Scholar]
- 374.Miura M, Ishide N, Oda H, Sakurai M, Shinozaki T, Takishima T. Spatial features of calcium transients during early and delayed afterdepolarizations. Am J Physiol Heart Circ Physiol. 1993;265:H439–H444. doi: 10.1152/ajpheart.1993.265.2.H439. [DOI] [PubMed] [Google Scholar]
- 375.Miura M, Ishide N, Sakurai M, Shinozaki T, Takishima T. Interactions between calcium waves and action potential-induced calcium transients in guinea pig myocytes. Heart Vessels. 1994;9:79–86. [Google Scholar]
- 376.Miyakawa T, Mizushima A, Hirose K, Yamazawa T, Bezprozvanny I, Kurosaki T, Iino M. Ca2+-sensor region of IP3 receptor controls intracellular Ca2+ signaling. EMBO J. 2001;20:1674–1680. doi: 10.1093/emboj/20.7.1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Miyata H, Silverman HS, Sollott SJ, Lakatta EG, Stern MD, Hansford RG. Measurement of mitochondrial free Ca2+ concentration in living single rat cardiac myocytes. Am J Physiol Heart Circ Physiol. 1991;261:H1123–H1134. doi: 10.1152/ajpheart.1991.261.4.H1123. [DOI] [PubMed] [Google Scholar]
- 378.Miyamoto S, Izumi M, Hori M, Kobayashi M, Ozaki H, Karaki H. Xestospongin C, a selective and membrane-permeable inhibitor of IP3 receptor, attenuates the positive inotropic effect of alpha-adrenergic stimulation in guinea-pig papillary muscle. Br J Pharmacol. 2000;130:650–654. doi: 10.1038/sj.bjp.0703358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Mohler PJ, Davis JQ, Bennett V. Anykrin B coordinates the NaK ATPase, NaCa exchanger and the IP3 receptor in a cardiac t-tubule/SR microdomain. PLos Biology. 2005;3:2158–2167. doi: 10.1371/journal.pbio.0030423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Mohler PJ, Rivolta I, Napolitano C, LeMaillet G, Lambert S, Priori SG, Bennett V. Nav1.5.E1053K mutation causing Brugada syndrome blocks binding to anykrinG and expression of NAv1.5 on the surface of cardiomyocytes. Proc Natl Acad Sci. 2004;101:17533–17538. doi: 10.1073/pnas.0403711101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Mohler PJ, Schott JJ, Gramolini AO, Dilly K, Guatimosim S, DuBell WH, Song LS, Haurogne K, Kyndt F, Ali ME, Rogers TB, Lederer WJ, Escande D, Le Marec H, Bennett V. AnykrinB mutation causes type4 longQT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421:634–639. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- 382.Mori M, Konno T, Ozawa T, Murata M, Imoto K, Nagayama K. Novel interaction of the voltage-dependent sodium channel (VDSC) with calmodulin: does VDSC acquire calmodulin-mediated Ca2+ sensitivity? Biochemistry. 2000;39:1316–1323. doi: 10.1021/bi9912600. [DOI] [PubMed] [Google Scholar]
- 383.Mori MX, Erickson MG, Yue DT. Functional stoichiometry and local enrichment of calmodulin interacting with Ca2+ channels. Science. 2004;304:432–435. doi: 10.1126/science.1093490. [DOI] [PubMed] [Google Scholar]
- 384.Moschella MC, Marks AR. Inositol 1,4,5-triphosphate receptor expression in cardiac myocytes. J Cell Biol. 1993;120:1137–1146. doi: 10.1083/jcb.120.5.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Mouton J, Feltz A, Maulet Y. Interactions of calmodulin with two peptides derived from the C-terminal cytoplasmic domain of the Ca(v)1.2 Ca2+ channel provide evidence for a molecular switch involved in Ca2+-induced inactivation. J Biol Chem. 2001;276:22359–22367. doi: 10.1074/jbc.M100755200. [DOI] [PubMed] [Google Scholar]
- 386.Mulder BJM, de Tombe PP, ter Keurs HEDJ. Spontaneous and propagated contractions in rat cardiac trabeculae. J Gen Physiol. 1989;93:943–961. doi: 10.1085/jgp.93.5.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Nakamura TY, Nandi S, Pountney DJ, Artman M, Rudy B, Coetzee WA. Different effects of the Ca(2+)-binding protein, KChIP1, on two Kv4 subfamily members, Kv4.1 and Kv4.2. FEBS Lett. 2001;499:205–209. doi: 10.1016/s0014-5793(01)02560-1. [DOI] [PubMed] [Google Scholar]
- 389.Neely A, Olcese R, Baldelli P, Wei X, Birnbaumer L, Stefani E. Dual activation of the cardiac Ca2+ channel alpha 1C subunit and its modulation by the beta subunit. Am J Physiol Cell Physiol. 1995;268:C732–C740. doi: 10.1152/ajpcell.1995.268.3.C732. [DOI] [PubMed] [Google Scholar]
- 390.Neely A, Olcese R, Wei X, Birnbaumer L, Stefani E. Calcium-dependent inactivation of a cloned cardiac calcium channel alpha(1) subunit (alpha 1c) expressed in Xenopus oocytes. Biophys J. 1994;66:1895–1903. doi: 10.1016/S0006-3495(94)80983-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Neely A, Wei X, Olcese R, Birnbaumer L, Stefani E. Potentiation by the beta subunit of the ratio of the ionic current to the charge movement in the cardiac calcium channel. Science. 1993;262:575–578. doi: 10.1126/science.8211185. [DOI] [PubMed] [Google Scholar]
- 392.Nicoll DA, Ottolia M, Lu L, Lu Y, Philipson KD. A new topological model of the cardiac sarcolemmal Na+-Ca2+ exchanger. J Biol Chem. 1999;274:910–917. doi: 10.1074/jbc.274.2.910. [DOI] [PubMed] [Google Scholar]
- 393.Niggli E, Lederer WJ. Molecular operations of the sodium-calcium exchanger revealed by conformation currents. Nature. 1991;349:621–624. doi: 10.1038/349621a0. [DOI] [PubMed] [Google Scholar]
- 394.Nuyens D, Stengl M, Dugarmaa S, Rossenbacker T, Compernolle V, Rudy Y, Smits JF, Flameng W, Clancy CE, Moons L, Vos MA, Dewerchin M, Benndorf K, Collen D, Carmeliet E, Carmeliet P. Abrupt rate accelerations or premature beats cause life-threatening arrhythmias in mice with long-QT3 syndrome. Nat Med. 2001;7:1021–1027. doi: 10.1038/nm0901-1021. [DOI] [PubMed] [Google Scholar]
- 395.O'Neill SC, Mill JG, Eisner DA. Local activation of contraction in isolated rat ventricular myocytes. Am J Physiol Cell Physiol. 1990;258:C1165–C1168. doi: 10.1152/ajpcell.1990.258.6.C1165. [DOI] [PubMed] [Google Scholar]
- 396.Obayashi M, Xiao B, Stuyvers BD, Davidoff AW, Mei J, Chen SRW, ter Keurs HEDJ. Spontaneous diastolic contractions and phosphorylation of the cardiac ryanodine receptor at serine-2808 in congestive heart failure in rat. Cardiovasc Res. 2006;69:140–151. doi: 10.1016/j.cardiores.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 397.Oda T, Yano M, Yamamoto T, Tokuhisa T, Okuda S, Doi M, Ohkusa T, Ikeda Y, Kobayashi S, Ikemoto N, Matsuzaki M. Defective regulation of interdomain interactions within the ryanodine receptor plays a key role in the pathogenesis of heart failure. Circulation. 2005;111:3400–3410. doi: 10.1161/CIRCULATIONAHA.104.507921. [DOI] [PubMed] [Google Scholar]
- 398.Ohki G, Miyoshi T, Murata M, Ishibashi K, Imai M, Suzuki M. A calcium-activated cation current by an alternatively spliced form of Trp3 in the heart. J Biol Chem. 2000;275:39055–39060. doi: 10.1074/jbc.M003606200. [DOI] [PubMed] [Google Scholar]
- 399.Ohmori H. Mechano-electrical transduction currents in isolated vestibular hair cells of the chick. J Physiol. 1985;359:189–217. doi: 10.1113/jphysiol.1985.sp015581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Omichi C, Lamp ST, Lin SF, Yang J, Baher A, Zhou S, Attin M, Lee MH, Karagueuzian HS, Kogan B, Qu Z, Garfinkel A, Chen PS, Weiss JN. Intracellular Ca dynamics in ventricular fibrillation. Am J Physiol Heart Circ Physiol. 2004;286:H1836–H1844. doi: 10.1152/ajpheart.00123.2003. [DOI] [PubMed] [Google Scholar]
- 401.Ondrias K, Marx SO, Gaburjakova M, Marks AR. FKBP12 modulates gating of the ryanodine receptor/calcium release channel. Anal NY Acad Sci. 1998;853:149–156. doi: 10.1111/j.1749-6632.1998.tb08263.x. [DOI] [PubMed] [Google Scholar]
- 402.Opatowsky Y, Chen CC, Campbell KP, Hirsch JA. Structural analysis of the voltage-dependent calcium channel β subunit functional core and its complex with the α1 interaction domain. Neuron. 2004;42:387–399. doi: 10.1016/s0896-6273(04)00250-8. [DOI] [PubMed] [Google Scholar]
- 403.Orchard CH, Eisner DA, Allen DG. Oscillations of intracellular Ca2+ in mammalian cardiac muscle. Nature. 1983;304:735–738. doi: 10.1038/304735a0. [DOI] [PubMed] [Google Scholar]
- 404.Otsu K, Willard HF, Khanna VK, Zorzato F, Green NM, Mac-Lennan DH. Molecular cloning of cDNA encoding the Ca2+ release channel (ryanodine receptor) of rabbit cardiac muscle sarcoplasmic reticulum. J Biol Chem. 1990;265:13472–13483. [PubMed] [Google Scholar]
- 405.Ottolia M, John S, Qiu Z, Philipson KD. Split Na+-Ca2+ exchangers. Implications for function and expression. J Biol Chem. 2001;276:19603–19609. doi: 10.1074/jbc.M101489200. [DOI] [PubMed] [Google Scholar]
- 406.Papp Z, Sipido KR, Callewaert G, Carmeliet EE. Two components of Cai activated Cl current during large Cai transients in single rabbit heart Purkinje cells. J Physiol. 1995;483:319–330. doi: 10.1113/jphysiol.1995.sp020588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Parker I, Zang WJ, Wier WG. Ca2+ sparks involving multiple Ca2+ release sites along Z lines in rat heart cells. J Physiol. 1996;497:31–38. doi: 10.1113/jphysiol.1996.sp021747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Pastore JM, Girouard SD, Laurita KR, Akar FG, Rosenbaum DS. Mechanism linking T wave alternans to the genesis of cardiac fibrillation. Circulation. 1999;99:1385–1394. doi: 10.1161/01.cir.99.10.1385. [DOI] [PubMed] [Google Scholar]
- 409.Pastore JM, Rosenbaum DS. Role of structural barriers in the mechanism of alternans-induced reentry. Circ Res. 2000;87:1157–1163. doi: 10.1161/01.res.87.12.1157. [DOI] [PubMed] [Google Scholar]
- 410.Pate P, Mochca-Morales J, Wu Y, Zhang JZ, Rodney GG, Serysheva II, Williams BY, Anderson ME, Hamilton SL. Determinants for calmodulin binding on voltage-dependent Ca2+ channels. J Biol Chem. 2000;275:39786–39792. doi: 10.1074/jbc.M007158200. [DOI] [PubMed] [Google Scholar]
- 411.Patel SP, Campbell DL, Morales MJ, Strauss HC. Heterogeneous expression of KChIP2 isoforms in the ferret heart. J Physiol. 2002;539:649–656. doi: 10.1113/jphysiol.2001.015156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Patterson E, Scherlag BJ, Lazzara R. Early afterdepolarizations produced by d,l sotalol and clofilium. J Cardiovasc Electrophysiol. 1997;8:667–678. doi: 10.1111/j.1540-8167.1997.tb01830.x. [DOI] [PubMed] [Google Scholar]
- 413.Patterson E, Lazzara R, Szabo B, Liu H, Tang D, Li YH, Scherlag BJ, Po SS. Sodium-calcium exchange initiated by the Ca2+ transient: an arrhythmia trigger within pulmonary veins. J Am Coll Cardiol. 2006;47:1196–1206. doi: 10.1016/j.jacc.2005.12.023. [DOI] [PubMed] [Google Scholar]
- 414.Perez PJ, Ramos-Franco J, Fill M, Mignery GA. Identification and functional reconstitution of the type2 inositol 1,4,5-trisphosphate receptor from ventricular cardiac myocytes. J Biol Chem. 1997;272:23961–23969. doi: 10.1074/jbc.272.38.23961. [DOI] [PubMed] [Google Scholar]
- 415.Perez Garcia MT, Kamp TJ, Marban E. Functional properties of cardiac L type calcium channels transiently expressed in HEK293 cells. Role of alpha 1 and beta subunit. J Gen Physiol. 1995;105:289–305. doi: 10.1085/jgp.105.2.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Perez Reyes E, Cribbs LL, Daud A, Lacerda AE, Barclay J, Williamson MP, Fox M, Rees M, Lee JH. Molecular characterization of a neuronal low voltage activated T type calcium channel. Nature. 1998;391:896–900. doi: 10.1038/36110. [DOI] [PubMed] [Google Scholar]
- 417.Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for the Ca2+ dependent inactivation of the L type Calcium channel. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 418.Peterson BZ, Lee JS, Mulle JG, Wang Y, de Leon M, Yue DT. Critical determinants of Ca(2+)-dependent inactivation within an EF-hand motif of L-type Ca(2+) channels. Biophys J. 2000;78:1906–1920. doi: 10.1016/S0006-3495(00)76739-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Petroff MG, Kim SH, Pepe S, Dessy C, Marban E, Balligand JL, Sollott SJ. Endogenous nitric oxide mechanisms mediate the stretch dependence of Ca2+ release in cardiomyocytes. Nat Cell Biol. 2001;3:867–873. doi: 10.1038/ncb1001-867. [DOI] [PubMed] [Google Scholar]
- 420.Picard L, Cote K, Teijeira J, Greentree D, Rousseau E. Sarcoplasmic reticulum K+ channels from human and sheep atrial cells display a specific electro-pharmacological profile. J Mol Cell Cardiol. 2002;34:1163–1172. doi: 10.1006/jmcc.2002.2041. [DOI] [PubMed] [Google Scholar]
- 421.Picht E, DeSantiago J, Blatter LA, Bers DM. Cardiac alternans do not rely on diastolic sarcoplasmic reticulum calcium content fluctuations. Circ Res. 2006;99:740–748. doi: 10.1161/01.RES.0000244002.88813.91. [DOI] [PubMed] [Google Scholar]
- 422.Pitt GS, Dun W, Boyden PA. Remodeled cardiac calcium channels. JMCC. 2006;41:373–388. doi: 10.1016/j.yjmcc.2006.06.071. [DOI] [PubMed] [Google Scholar]
- 423.Pitt GS, Zuhlke RD, Hudmon A, Schulman H, Reuter H, Tsien RW. Molecular basis of calmodulin tethering and Ca2+-dependent inactivation of L-type ca2+ channels. J Biol Chem. 2001;276:30794–30802. doi: 10.1074/jbc.M104959200. [DOI] [PubMed] [Google Scholar]
- 424.Platano D, Qin N, Noceti F, Birnbaumer L, Stefani E, Olcese R. Expression of the α2 · δ subunit interferes with prepulse facilitation in cardiac L-type calcium channels. Biophys J. 2000;78:2959–2972. doi: 10.1016/S0006-3495(00)76835-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, Zheng H, Streissing J. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L type Ca2+ channels. Cell. 2000;102:89–97. doi: 10.1016/s0092-8674(00)00013-1. [DOI] [PubMed] [Google Scholar]
- 426.Pogwizd SM, Qi M, Yuan W, Samarel AM, Bers DM. Upregulation of NaCa exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ Res. 1999;85:1009–1019. doi: 10.1161/01.res.85.11.1009. [DOI] [PubMed] [Google Scholar]
- 427.Post JA, Langer GA, Opdenkamp JAF, Verkleij AJ. Phospholipid asymmetry in cardiac sarcolemma. Analysis of intact cells and “gas dissected” membranes. Biochim Biophys Acta. 1988;943:255–266. doi: 10.1016/0005-2736(88)90557-3. [DOI] [PubMed] [Google Scholar]
- 428.Postma AV, Denjoy I, Hoorntje TM, Lupoglazoff JM, DaCosta A, Sebillon P, Mannens M, Wilde AAM, Guicheney P. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2002;91:e21–e26. doi: 10.1161/01.res.0000038886.18992.6b. [DOI] [PubMed] [Google Scholar]
- 429.Pragnell M, De Waard M, Mori Y, Tanabe T, Snutch TP, Campbell KP. Calcium channel beta-subunit binds to a conserved motif in the I-II cytoplasmic linker of the alpha 1-subunit. Nature. 1994;368:67–70. doi: 10.1038/368067a0. [DOI] [PubMed] [Google Scholar]
- 430.Priori SG, Napolitano C, Memmi M, Colombi B, Drago F, Gasparini M, DeSimone L, Coltorti F, Bloise R, Keegan R, Cruz Filho FES, Vignati G, Benatar A, DeLogu A. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69–74. doi: 10.1161/01.cir.0000020013.73106.d8. [DOI] [PubMed] [Google Scholar]
- 431.Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli GA. Mutations in the cardiac ryanodine receptor gene (hRYR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- 432.Pruvot E, Katra RP, Rosenbaum DS, Laurita KR. Role of calcium cycling versus restitution in the mechanism of repolarization alternans. Circ Res. 2004;94:1083–1090. doi: 10.1161/01.RES.0000125629.72053.95. [DOI] [PubMed] [Google Scholar]
- 433.Pu J, Ruffy F, Boyden PA. Effects of Bay Y5959 on Ca2+ currents and intracellular Ca2+ in cells that have survived in the epicardial border zone of the infarcted canine heart. J Cardiovasc Pharmacol. 1999;33:929–937. doi: 10.1097/00005344-199906000-00014. [DOI] [PubMed] [Google Scholar]
- 434.Qian YW, Clusin WT, Lin SF, Han J, Sung RJ. Spatial heterogeneity of calcium transient alternans during the early phase of myocardial ischemia in the blood-perfused rabbit heart. Circulation. 2001;104:2082–2087. doi: 10.1161/hc4201.097136. [DOI] [PubMed] [Google Scholar]
- 435.Qian YW, Sung RJ, Lin SF, Province R, Clusin WT. Spatial heterogeneity of action potential alternans during global ischemia in the rabbit heart. Am J Physiol Heart Circ Physiol. 2003;285:H2722–H2733. doi: 10.1152/ajpheart.00369.2003. [DOI] [PubMed] [Google Scholar]
- 436.Ramesh V, Sharma VK, Sheu SS, Franzini-Armstrong C. Structural proximity of mitochondria to calcium release units in rat ventricular myocardium may suggest a role in Ca2+ sequestration. Ann NY Acad Sci. 1998;853:341–344. doi: 10.1111/j.1749-6632.1998.tb08295.x. [DOI] [PubMed] [Google Scholar]
- 437.Ramos-Franco J, Fill M, Mignery GA. Isoform-specific function of single inositol 1,4,5-trisphosphate receptor channels. Biochem J. 1998;75:834–839. doi: 10.1016/S0006-3495(98)77572-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Rampazzo A, Nava A, Miorin M, Fonderico P, Pope B, Tiso N, Livolsi B, Zinbello R, Thiene G, Danieli GA. ARVD4, a new locus for arrhythmogenic right ventricular cardiomyopathy, maps to chromosome 2 long arm. Genomics. 1997;45:259–263. doi: 10.1006/geno.1997.4927. [DOI] [PubMed] [Google Scholar]
- 439.Reuter H, Scholz H. The regulation of Ca conductance of cardiac muscle by adrenaline. J Physiol. 1977;246:49–62. doi: 10.1113/jphysiol.1977.sp011657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Rigg L, Heath BM, Cui Y, Terrar DA. Localisation and functional significance of ryanodine receptors during Beta adrenergic stimulation in the guinea pig sino-atrial node. Cardiovasc Res. 2000;48:254–264. doi: 10.1016/s0008-6363(00)00153-x. [DOI] [PubMed] [Google Scholar]
- 441.Rigg L, Terrar DA. Possible role of calcium release from the SR in pacemaking in guinea pig sinoatrial node. Exp Physiol. 1996;81:877–880. doi: 10.1113/expphysiol.1996.sp003983. [DOI] [PubMed] [Google Scholar]
- 442.Robertson SP, Johnson P, Potter JD. The time course of calcium exchange with calmodulin, troponin para albumin and myosin in response to transient increases of calcium. Biophys J. 1981;34:559–569. doi: 10.1016/S0006-3495(81)84868-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Robinson RB, Boyden PA, Hoffman BF, Hewett KW. The electrical restitution process in dispersed canine cardiac Purkinje and ventricular cells. Am J Physiol Heart Circ Physiol. 1987;253:H1018–H1025. doi: 10.1152/ajpheart.1987.253.5.H1018. [DOI] [PubMed] [Google Scholar]
- 444.Roden DM, Viswanathan PC. Genetics of acquired long QT syndrome. J Clin Invest. 2005;115:2025–2032. doi: 10.1172/JCI25539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445.Roeper J, Lorra C, Pongs O. Frequency dependent inactivation of mammalian A type K channel Kv1.4 regulated by Ca2+/Calmodulin dependent protein kinase. J Neurosci. 1997;17:3379–3391. doi: 10.1523/JNEUROSCI.17-10-03379.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Rohr S, Kucera JP. Involvement of the calcium inward current in cardiac impulse propagation: induction of unidirectional conduction block by nifedipine and reversal by Bay K 8644. Biophys J. 1997;72:754–766. doi: 10.1016/s0006-3495(97)78710-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Romanin C, Gamsjaeger R, Kahr H, Schaufler D, Carlson O, Abernethy DR, Soldatov NM. Ca2+ sensors of L-type Ca2+ channel. FEBS Lett. 2000;487:301–306. doi: 10.1016/s0014-5793(00)02361-9. [DOI] [PubMed] [Google Scholar]
- 448.Rosati B, Dun W, Hirose M, Boyden PA, McKinnon D. Molecular basis of the T and L type Ca2+ currents in canine Purkinje fibers. J Physiol. doi: 10.1113/jphysiol.2006.127480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Rosen MR, Boyden PA. Is there a pharmacology of discontinuous conduction? In: Spooner PM, Joyner RW, Jalife J, editors. Discontinuous Conduction in the Heart. Armonk, NY: Futura; 1997. pp. 471–482. [Google Scholar]
- 450.Rosenbaum DS. T wave alternans: a mechanism of arrhythmogenesis comes of age after 100 years. J Cardiovasc Electrophysiol. 2001;12:207–209. doi: 10.1046/j.1540-8167.2001.00207.x. [DOI] [PubMed] [Google Scholar]
- 451.Rosenbaum DS, Jackson LE, Smith JM, Garna H, Ruskin JN, Cohen RJ. Electrical alternans and vulnerability to ventricular arrhythmias. N Engl J Med. 1994;330:235–241. doi: 10.1056/NEJM199401273300402. [DOI] [PubMed] [Google Scholar]
- 452.Rosenbaum MB, Acunzo RS. Pseudo 2:1 AV block and T wave alternans in the long QT syndromes. J Am Coll Cardiol. 1991;18:1363–1366. doi: 10.1016/0735-1097(91)90560-v. [DOI] [PubMed] [Google Scholar]
- 453.Rosenberg RL, Hess P, Reeves JP, Smilowitz H, Tsien RW. Calcium channels in planar lipid bilayers: insights into mechanism of ion permeation and gating. Science. 1986;231:1564–1566. doi: 10.1126/science.2420007. [DOI] [PubMed] [Google Scholar]
- 454.Rosenberg RL, Hess P, Tsien RW. Cardiac calcium channels in planar lipid bilayers: L-type channels and calcium-permeable channels open at negative membrane potentials. J Gen Physiol. 1988;92:27–54. doi: 10.1085/jgp.92.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Rousseau E, Chabot H, Beaudry C, Muller B. Reconstitution and regulation of cation selective channels from cardiac sarcoplasmic reticulum. Mol Cell Biochem. 1992;114:109–117. doi: 10.1007/BF00240305. [DOI] [PubMed] [Google Scholar]
- 456.Rousseau E, Meissner G. Single cardiac sarcoplasmic reticulum Ca2+ release channel: activation by caffeine. Am J Physiol Heart Circ Physiol. 1989;256:H328–H333. doi: 10.1152/ajpheart.1989.256.2.H328. [DOI] [PubMed] [Google Scholar]
- 457.Rousseau E, Pinkos J. pH modulates conducting and gating behaviour of single calcium release channels. Pflügers Arch. 1990;415:645–647. doi: 10.1007/BF02583520. [DOI] [PubMed] [Google Scholar]
- 458.Rousseau E, Roberson M, Meissner G. Properties of single chloride selective channel from sarcoplasmic reticulum. Eur Biophys J. 1988;16:143–151. doi: 10.1007/BF00261900. [DOI] [PubMed] [Google Scholar]
- 459.Rousseau E, Smith JS, Meissner G. Ryanodine modifies conductance and gating behavior of single Ca2+ release channels. Am J Physiol Cell Physiol. 1987;253:C364–C368. doi: 10.1152/ajpcell.1987.253.3.C364. [DOI] [PubMed] [Google Scholar]
- 460.Rubenstein DS, Lipsius SL. Premature beats elicit a phase reversal of mechanoelectrical alternans in cat ventricular myocytes: a possible mechanism for reentrant arrhythmias. Circulation. 1995;91:201–214. doi: 10.1161/01.cir.91.1.201. [DOI] [PubMed] [Google Scholar]
- 461.Rubenstein DS, Lipsius SL. Mechanisms of automaticity in subsidiary pacemakers from cat right atrium. Circ Res. 1989;64:648–657. doi: 10.1161/01.res.64.4.648. [DOI] [PubMed] [Google Scholar]
- 462.Saez JC, Connor JA, Spray DC, Bennett MV. Hepatocyte gap junctions are permeable to the second messenger, inositiol 1,4,5-trisphosphate and to calcium ions. Proc Natl Acad Sci USA. 1989;86:2708–2712. doi: 10.1073/pnas.86.8.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Sah R, Ramirez RJ, Kaprielian R, Backx PH. Alterations in action potential profile enhance excitation-contraction coupling in rat cardiac myocytes. J Physiol. 2001;533:201–214. doi: 10.1111/j.1469-7793.2001.0201b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Sah R, Ramirez RJ, Backx PH. Modulation of Ca2+ release in cardiac myocytes by changes in repolarization rate. Role of phase-1 action potential repolarization in excitation-contraction coupling. Circ Res. 2002;90:165–173. doi: 10.1161/hh0202.103315. [DOI] [PubMed] [Google Scholar]
- 465.Samso M, Shen X, Allen PD. Structural characterization of the RyR1-FKBP12 interaction. J Mol Biol. 2006;356:917–927. doi: 10.1016/j.jmb.2005.12.023. [DOI] [PubMed] [Google Scholar]
- 466.Samso M, Trujillo R, Gurrola GB, Valdivia HH, Wagenknecht T. Three-dimensional location of the imperatoxin A binding site on the ryanodine receptor. J Cell Biol. 1999;146:493–500. doi: 10.1083/jcb.146.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Samso M, Wagenknecht T. Apocalmodulin and Ca2+-calmodulin bind to neighboring locations on the ryanodine receptor. J Biol Chem. 2002;277:1349–1353. doi: 10.1074/jbc.M109196200. [DOI] [PubMed] [Google Scholar]
- 468.Samso M, Wagenknecht T, Allen PD. Internal structure and visualization of transmembrane domains of the RyR1 calcium release channel by cryo-EM. Nat Struct Mol Biol. 2005;12:539–544. doi: 10.1038/nsmb938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Sanchez JA, Garcia MC, Sharma VK, Young KC, Matlib MA, Sheu SS. Mitochondria regulate inactivation of L-type Ca2+ channels in rat heart. J Physiol. 2001;536:387–396. doi: 10.1111/j.1469-7793.2001.0387c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Sandberg K, Ji H, Iida Y, Catt KJ. Intercellular communication between follicular angiotensin receptors and Xenopus laevis oocytes: mediation by an inositol 1,4,5-trisphosphate-dependent mechanism. J Cell Biol. 1992;117:157–167. doi: 10.1083/jcb.117.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 471.Sato D, Shiferaw Y, Garfinkel A, Weiss JN, Qu Z, Karma A. Spatially discordant alternans in cardiac tissue. Role of calcium cycling. Circ Res. 2006;99:520–527. doi: 10.1161/01.RES.0000240542.03986.e7. [DOI] [PubMed] [Google Scholar]
- 472.Scamps F, Carmeliet E. Delayed K current and external K in single Purkinje cells. Am J Physiol Cell Physiol. 1989;257:C1086–C1092. doi: 10.1152/ajpcell.1989.257.6.C1086. [DOI] [PubMed] [Google Scholar]
- 473.Schlecker C, Boehmerle W, Jeromin A, DeGray B, Varshney A, Sharma Y, Szigeti-Buck K, Ehrlich BE. Neuronal calcium sensor-1 enhancement of InsP3 receptor activity is inhibited by therapeutic levels of lithium. J Clin Invest. 2006;116:1668–1674. doi: 10.1172/JCI22466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Schlotthauer K, Bers DM. Sarcoplasmic reticulum Ca2+ release causes myocyte depolarization. Underlying mechanism and threshold for triggered action potentials. Circ Res. 2000;87:774–780. doi: 10.1161/01.res.87.9.774. [DOI] [PubMed] [Google Scholar]
- 475.Schott JJ, Charpentier F, Peltier S, Foley P, Drouin E, Bouhour JB, Donnelly P, Vergnaud G, Bachner L, Moisan JP. Mapping of a gene for long QT syndrome to chromosome 4q25–27. Am J Hum Genet. 1995;57:1114–1122. [PMC free article] [PubMed] [Google Scholar]
- 476.Schouten VJA, Deen JK, deTombe PP, Verveen AA. Force interval relationship in heart muscle of mammals. A calcium compartment model. Biophys J. 1987;51:13–26. doi: 10.1016/S0006-3495(87)83307-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Schouten VJA, Morad M. Regulation of Ca current in frog ventricular myocytes by the holding potential, cAMP and frequency. Pflügers Arch. 1989;415:1–11. doi: 10.1007/BF00373135. [DOI] [PubMed] [Google Scholar]
- 478.Sedarat F, Lin E, Moore EDW, Tibbits GF. Deconvolution of confocal images of dihydropyridine and ryanodine receptors in developing cardiomyocytes. J Appl Physiol. 2004;97:1098–1103. doi: 10.1152/japplphysiol.00089.2004. [DOI] [PubMed] [Google Scholar]
- 479.Seguchi H, Ritter M, Shizukuishi M, Ishida H, Chokoh G, Nakazawa H, Spitzer KW, Barry WH. Propagation of Ca2+ release in cardiac myocytes: role of mitochondria. Cell Calcium. 2005;38:1–9. doi: 10.1016/j.ceca.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 480.Serysheva II, Schatz M, van Heel M, Chiu W, Hamilton SL. Structure of the skeletal muscle calcium release channel activated with Ca2+ and AMP-PCP. Biophys J. 1999;77:1936–1944. doi: 10.1016/S0006-3495(99)77035-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Serysheva II, Hamilton SL, Chiu W, Ludtke SJ. Structure of Ca2+ release channel at 14 A resolution. J Mol Biol. 2005;345:427–431. doi: 10.1016/j.jmb.2004.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Seth M, Sumbilla C, Mullen S, Lewis D, Klein MG, Hussain A, Soboloff J, Gill DL, Inesi G. Sarcoplasmic reticulum Ca ATPase-(SERCA) gene silencing and remodeling of the Ca2+ signaling mechanism in cardiac myocytes. Proc Natl Acad Sci USA. 2004;101:16683–16688. doi: 10.1073/pnas.0407537101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Shamgar L, Ma L, Schmitt N, Haitin Y, Peretz A, Wiener R, Hirsch J, Pongs O, Attali B. Calmodulin is essential for cardiac IKS channel gating and assembly. Impaired function in long-QT mutations. Circ Res. 2006;98:1055–1063. doi: 10.1161/01.RES.0000218979.40770.69. [DOI] [PubMed] [Google Scholar]
- 484.Shannon TR, Guo T, Bers DM. Ca2+ scraps: local depletions of free Ca2+ in cardiac sarcoplasmic reticulum during contractions leave substantial Ca2+ reserve. Circ Res. 2003;93:40–45. doi: 10.1161/01.RES.0000079967.11815.19. [DOI] [PubMed] [Google Scholar]
- 485.Shannon TR, Pogwizd SM, Bers DM. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ Res. 2003;93:592–594. doi: 10.1161/01.RES.0000093399.11734.B3. [DOI] [PubMed] [Google Scholar]
- 486.Shaw RM, Rudy Y. Ionic mechanisms of propagation in cardiac tissue. Roles of the sodium and L-type calcium currents during reduced excitability and decreased gap junction coupling. Circ Res. 1997;81:727–741. doi: 10.1161/01.res.81.5.727. [DOI] [PubMed] [Google Scholar]
- 487.Sheehan KA, Blatter LA. Regulation of junctional and nonjunctional sarcoplasmic reticulum calcium release in excitation contraction coupling in cat atrial myocytes. J Physiol. 2003;546:119–135. doi: 10.1113/jphysiol.2002.026963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Sheehan KA, Zima AV, Blatter LA. Regional differences in spontaneous Ca2+ spark activity and regulation in cat atrial myocytes. J Physiol. 2006;572:799–809. doi: 10.1113/jphysiol.2005.103267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Shin DW, Ma J, Kim DH. The Asp-rich region at the carboxyl-terminus of calsequestrin binds to Ca2+ and interacts with triadin. FEBS Lett. 2000;486:178–182. doi: 10.1016/s0014-5793(00)02246-8. [DOI] [PubMed] [Google Scholar]
- 490.Shistik E, Ivanina T, Blumenstein Y, Dascal N. Crucial role of N terminus in function of cardiac L-type Ca2+ channel and its modulation by protein kinase C. J Biol Chem. 1998;273:17901–17909. doi: 10.1074/jbc.273.28.17901. [DOI] [PubMed] [Google Scholar]
- 491.Shistik E, Keren-Raifman T, Idelson GH, Blumenstein Y, Dascal N, Ivanina T. The N terminus of the cardiac L-type Ca2+ channel alpha 1C subunit. The initial segment is ubiquitous and crucial for protein kinase C modulation, but is not directly phosphorylated. J Biol Chem. 1999;274:31145–31149. doi: 10.1074/jbc.274.44.31145. [DOI] [PubMed] [Google Scholar]
- 492.Shou W, Aghdasi B, Armstrong DL, Guo Q, Bao S, Charng MJ, Mathews LM, Schneider MD, Hamilton SL, Matzuk MM. Cardiac defects and altered ryanodine receptor function in mice lacking FKBP12. Nature. 1998;391:489–492. doi: 10.1038/35146. [DOI] [PubMed] [Google Scholar]
- 493.Siegelbaum S, Tsien RW. Calcium-activated transient outward current in calf cardiac Purkinje fibres. J Physiol. 1980;299:485–506. doi: 10.1113/jphysiol.1980.sp013138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Simmerman HKB, Collins JA, Theiber JL, Weger AD, Jones LR. Sequence analysis of phospholamban. Identification of phosphorylation sites and two major structural domains. J Biol Chem. 1986;261:13333–13341. [PubMed] [Google Scholar]
- 495.Singer D, Biel M, Lotan I, Flockerzi V, Hofmann F, Dascal N. The roles of the subunits in the function of the calcium channel. Science. 1991;253:1553–1557. doi: 10.1126/science.1716787. [DOI] [PubMed] [Google Scholar]
- 496.Sipido KR, Callewaert G, Carmeliet E. Inhibition and rapid recovery of Ca2+ current during Ca2+ release from sarcoplasmic reticulum in guinea pig ventricular myocytes. Circ Res. 1995;76:102–109. doi: 10.1161/01.res.76.1.102. [DOI] [PubMed] [Google Scholar]
- 497.Sipido KR, Callewaert G, Carmeliet E. [Ca2+]i transients and [Ca2+]i-dependent chloride current in single purkinje cells from rabbit heart. J Physiol. 1993;468:641–667. doi: 10.1113/jphysiol.1993.sp019793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Sipido KR, Callewaert G, Porciatti F, Vereecke J, Carmeliet E. Cai dependent membrane currents in guinea pig cells in the absence of Na/Ca exchange. Pflügers Arch. 1995;430:871–878. doi: 10.1007/BF00386189. [DOI] [PubMed] [Google Scholar]
- 499.Sipido KR, Volders PG, de Groot M, Verdonck F, Van de Werf F, Wellens HJJ, Vos MA. Enhanced Ca2+ release and NaCa exchange activity in hypertrophied canine ventricular myocytes. Potential link between contractile adaptation and arrhythmogenesis. Circulation. 2000;102:2137–2144. doi: 10.1161/01.cir.102.17.2137. [DOI] [PubMed] [Google Scholar]
- 500.Sitsapesan R, Williams AJ. Regulation of current flow through ryanodine receptors by luminal Ca2+ J Membr Biol. 1997;159:179–185. doi: 10.1007/s002329900281. [DOI] [PubMed] [Google Scholar]
- 501.Smith JM, Clancy EA, Valeri CR, Ruskin JN, Cohen RJ. Electrical alternans and cardiac electrical instability. Circulation. 1988;77:110–121. doi: 10.1161/01.cir.77.1.110. [DOI] [PubMed] [Google Scholar]
- 502.Smith JS, Imagawa T, Ma J, Fill M, Campbell KP, Coronado R. Purified ryanodine receptor from rabbit skeletal muscle is the calcium-release channel of sarcoplasmic reticulum. J Gen Physiol. 1988;92:1–26. doi: 10.1085/jgp.92.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Soldatov NM, Oz M, O'Brien KA, Abernathy DR, Morad M. Molecular determinants of L type Ca2+ channel inactivation. J Biol Chem. 1998;273:957–963. doi: 10.1074/jbc.273.2.957. [DOI] [PubMed] [Google Scholar]
- 504.Soldatov NM, Zuhlke RD, Bouron A, Reuter H. Molecular structures involved in L-type calcium channel inactivation. Role of the carboxyl-terminal region encoded by exons 40–42 in alpha1C subunit in the kinetics and Ca2+ dependence of inactivation. J Biol Chem. 1997;272:3560–3566. doi: 10.1074/jbc.272.6.3560. [DOI] [PubMed] [Google Scholar]
- 505.Sommer JR, Johnson EA. Cardiac muscle. A comparative study of Purkinje fibers and ventricular fibers. J Cell Biol. 1968;36:497–526. doi: 10.1083/jcb.36.3.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, Sanguinetti MC, Keating MT. Inaugural article: severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc Natl Acad Sci USA. 2005;102:8089–8096. doi: 10.1073/pnas.0502506102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. CaV1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 508.Staes M, Talavera K, Klugbauer N, Prenen J, Lacinova L, Droogmans G, Hofmann F, Nilius B. The amino side of the C-terminus determines fast inactivation of the T-type calcium channel Alpha1G. J Physiol. 2001;530:35–45. doi: 10.1111/j.1469-7793.2001.0035m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Stephens GJ, Page KM, Bogdanov KY, Dolphin A. The alpha1 B Ca2+ channel amino terminus contributes determinants for beta subunit mediated voltage dependent inactivation properties. J Physiol. 2000;525:377–390. doi: 10.1111/j.1469-7793.2000.t01-1-00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Stern MD, Capogrossi MC, Lakatta EG. Spontaneous calcium release from the sarcoplasmic reticulum in myocardial cells: mechanisms and consequences. Cell Calcium. 1988;9:247. doi: 10.1016/0143-4160(88)90005-x. [DOI] [PubMed] [Google Scholar]
- 511.Stern MD, Kort AA, Bhatnagar GM, Lakatta EG. Scattered light intensity fluctuations in diastolic rat cardiac muscle caused by spontaneous calcium dependent cellular mechanical oscillations. J Gen Physiol. 1983;82:119–153. doi: 10.1085/jgp.82.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512.Stotz SC, Zamponi GW. Identification of inactivation determinants in the domain IIS6 region of high voltage-activated calcium channels. J Biol Chem. 2001;276:33001–33010. doi: 10.1074/jbc.M104387200. [DOI] [PubMed] [Google Scholar]
- 513.Stuyvers BD, Dun W, Matkovich SJ, Sorrentino V, Boyden PA, ter Keurs HEDJ. Ca2+ sparks and Ca2+ waves in Purkinje cells: a triple layered system of activation. Circ Res. 2005;97:35–43. doi: 10.1161/01.RES.0000173375.26489.fe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Stuyvers BDMY, Boyden PA, ter Keurs HEDJ. Calcium waves physiological relevance in cardiac function. Circ Res. 2000;86:1016–1018. doi: 10.1161/01.res.86.10.1016. [DOI] [PubMed] [Google Scholar]
- 515.Stuyvers BDMY, Miura M, ter Keurs HEDJ. Dynamics of viscoelastic properties of rat cardiac sarcomeres during the diastolic interval: involvement of Ca2+ J Physiol. 1997;502:661–677. doi: 10.1111/j.1469-7793.1997.661bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Sukhareva M, Morrisette J, Coronado R. Mechanism of chloride dependent release of Ca2+ in the sarcoplasmic reticulum of rabbit skeletal muscle. Biophys J. 1994;76:751–765. doi: 10.1016/S0006-3495(94)80536-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Sun H, LeBlanc N, Nattel S. Mechanisms of inactivation of L-type calcium channels in human atrial myocytes. Am J Physiol Heart Circ Physiol. 1997;272:H1625–H1635. doi: 10.1152/ajpheart.1997.272.4.H1625. [DOI] [PubMed] [Google Scholar]
- 518.Sun XH, Protasi F, Takahashi M, Takeshima H, Ferguson DG, Franzini-Armstrong C. Molecular architecture of membranes involved in excitation-contraction coupling of cardiac muscle. J Cell Biol. 1995;129:659–671. doi: 10.1083/jcb.129.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Sutko JL, Kenyon JL. Ryanodine modification of cardiac muscle responses to potassium-free solutions. J Gen Physiol. 1983;82:385–404. doi: 10.1085/jgp.82.3.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Swan H, Piippo K, Viitasalo M, Heikkila P, Paavonen T, Kainulainen K, Kere J, Keto P, Kontula K, Toivonen L. Arrhythmic disorder mapped to chromosome 1q42-q43 causes malignant polymorphic ventricular tachycardia in structurally normal hearts. J Am Coll Cardiol. 1999;34:2035–2042. doi: 10.1016/s0735-1097(99)00461-1. [DOI] [PubMed] [Google Scholar]
- 521.Szentesi P, Pignier C, Egger M, Kranias EG, Niggli E. Sarcoplasmic reticulum Ca2+ refilling controls recovery from Ca2+ induced Ca2+ release refractoriness in heart muscle. Circ Res. 2004;95:807–813. doi: 10.1161/01.RES.0000146029.80463.7d. [DOI] [PubMed] [Google Scholar]
- 522.Tada M, Katz AM. Phosphorylation of the sarcoplasmic reticulum and sarcolemma. Annu Rev Physiol. 1982;44:401–423. doi: 10.1146/annurev.ph.44.030182.002153. [DOI] [PubMed] [Google Scholar]
- 523.Tada M, Kirchberger M, Repke DI, Katz AM. The stimulation of calcium transport in cardiac sarcoplasmic reticulum by adenosine 3′:5′-monophosphate-dependent protein kinase. J Biol Chem. 1974;249:6174–6180. [PubMed] [Google Scholar]
- 524.Tada M, Yamada M, Kadoma M, Inui M, Ohmori F. Calcium transport by cardiac sarcoplasmic reticulum and phosphorylation of phospholamban. Mol Cell Biochem. 1982;46:74–95. doi: 10.1007/BF00236776. [DOI] [PubMed] [Google Scholar]
- 525.Tada M, Yamamoto T, Tonomura Y. Molecular mechanism of active calcium transport by sarcoplasmic reticulum. Physiol Rev. 1978;58:1–79. doi: 10.1152/physrev.1978.58.1.1. [DOI] [PubMed] [Google Scholar]
- 526.Takahashi SX, Miriyala J, Tay LH, Yue DT, Colecraft HM. A CaVβ SH3/guanylate kinase domain interaction regulates multiple properties of voltage-gated Ca2+ channels. J Gen Physiol. 2005;126:365–377. doi: 10.1085/jgp.200509354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Takamatsu T, Minamikawa T, Kawachi H, Fujita S. Imaging of calcium wave propagation in guinea pig ventricular cell pairs by confocal laser scanning microscopy. Cell Struct Funct. 1991;16:341–346. doi: 10.1247/csf.16.341. [DOI] [PubMed] [Google Scholar]
- 528.Takamatsu T, Wier WG. Calcium waves in mammalian heart: quantification of origin magnitude waveform and velocity. FASEB J. 1990;4:1519–1525. doi: 10.1096/fasebj.4.5.2307330. [DOI] [PubMed] [Google Scholar]
- 529.Takeshima H, Nishimura S, Matsumoto T, Ishida H, Kangawa K, Minamino N, Matsuo H, Ueda M, Hanaoka M, Hirose T. Primary structure and expression from complementary DNA of skeletal muscle ryanodine receptor. Nature. 1989;339:439–445. doi: 10.1038/339439a0. [DOI] [PubMed] [Google Scholar]
- 530.Takimoto K, Li D, Nerbonne JM, Levitan ES. Distribution, splicing and glucocorticoid-induced expression of cardiac alpha 1C and alpha 1D voltage-gated Ca2+ channel mRNAs. J Mol Cell Cardiol. 1997;29:3035–3042. doi: 10.1006/jmcc.1997.0532. [DOI] [PubMed] [Google Scholar]
- 531.Tan HL, Kupershmidt S, Zhang R, Stepanovic S, Roden DM, Wilde AAM, Anderson ME, Balser JR. A calcium sensor in the sodium channel modulates cardiac excitability. Nature. 2002;415:442–447. doi: 10.1038/415442a. [DOI] [PubMed] [Google Scholar]
- 532.Tanaka H, Oyamada M, Tsujii E, Nakajo T, Takamatsu T. Excitation-dependent intracellular Ca2+ waves at the border zone of the cryo-injured rat heart revealed by ral time confocal microscopy. J Mol Cell Cardiol. 2002;34:1501–1512. doi: 10.1006/jmcc.2002.2096. [DOI] [PubMed] [Google Scholar]
- 533.Tavi P, Han C, Weckstrom M. Mechanisms of stretch induced changes in Cai in rat atrial myocytes. Role of increased troponin C affinity and stretch activated ion channels. Circ Res. 1998;83:1165–1177. doi: 10.1161/01.res.83.11.1165. [DOI] [PubMed] [Google Scholar]
- 534.Ter Keurs HEDJ, Wakayama Y, Miura M, Shinozaki T, Stuyvers BD, Boyden PA, Landesberg A. Arrhythmogenic Ca2+ release from cardiac myofilaments. Prog Biophys Mol Biol. 2006;90:151–171. doi: 10.1016/j.pbiomolbio.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 535.Ter Keurs HEDJ. Regulation of cardiac contraction in the normal and failing heart: cellular aspects. Can J Cardiol. 1993;9:1F–11F. [Google Scholar]
- 536.Terentyev D, Cala SE, Houle TD, Viatchenko-Karpinski S, Gyorke I, Terentyeva R, Williams SC, Gyorke S. Triadin over-expression stimulates excitation-contraction coupling and increases predisposition to cellular arrhythmia in cardiac myocytes. Circ Res. 2005;96:651–658. doi: 10.1161/01.RES.0000160609.98948.25. [DOI] [PubMed] [Google Scholar]
- 537.Terentyev D, Viatchenko-Karpinski S, Gyorke I, Volpe P, Williams SC, Gyorke S. Calsequestrin determines the functional size and stability of cardiac intracellular calcium stores: mechanism for hereditary arrhythmia. Proc Natl Acad Sci USA. 2003;100:11759–11764. doi: 10.1073/pnas.1932318100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538.Tessier S, Karczewski P, Krause EG, Pansard Y, Acar C, Lang-Lazdunski M, Mercadier JJ, Hatem S. Regulation of the transient outward K+ current by Ca2+/calmodulin-dependent protein kinase II in human atrial myocytes. Circ Res. 1999;85:810–819. doi: 10.1161/01.res.85.9.810. [DOI] [PubMed] [Google Scholar]
- 539.Tester DJ, Spoon DB, Valdivia HH, Makielski JC, Ackerman MJ. Targeted mutational analysis of the RYR2-encoded cardiac ryanodine receptor in sudden unexplained death: a molecular autopsy of 49 medical examiner/coroner's cases. Mayo Clin Proc. 2004;79:1380–1384. doi: 10.4065/79.11.1380. [DOI] [PubMed] [Google Scholar]
- 540.Tian XL, Yong SL, Wan X, Wu L, Chung MK, Tchou PJ, Rosenbaum DS, Van Wagoner DR, Kirsch GE, Wang Q. Mechanisms by which SCN5A mutation N1325S causes cardiac arrhythmias and sudden death in vivo. Cardiovasc Res. 2004;61:256–267. doi: 10.1016/j.cardiores.2003.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Tiso N, Stephan DA, Nava A, Bagattin A, Devaney JM, Stanchi F, Larderet G, Brahmbhatt B, Brown K, Bauce B, Muriago M, Basso C, Thiene G, Danieli GA, Rampazzo A. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2) Hum Mol Gen. 2001;10:189–194. doi: 10.1093/hmg/10.3.189. [DOI] [PubMed] [Google Scholar]
- 542.Tohse N, Kameyama M, Irisawa H. Intracellular Ca and protein kinase C modulate K current in guinea pig heart cells. Am J Physiol Heart Circ Physiol. 1987;253:H1321–H1324. doi: 10.1152/ajpheart.1987.253.5.H1321. [DOI] [PubMed] [Google Scholar]
- 543.Toyofuku T, Yabuki M, Otsu K, Kuzuya T, Tada M, Hori M. Functional role of c-Src in gap junctions of the cardiomyopathic heart. Circ Res. 1999;85:672–681. doi: 10.1161/01.res.85.8.672. [DOI] [PubMed] [Google Scholar]
- 544.Toyofukyu T, Yabuki M, Otsu K, Kuzuya T, Hori M, Tada M. Intercellular calcium signaling via gap junction in connexin43 transfected cells. J Biol Chem. 1998;273:1519–1528. doi: 10.1074/jbc.273.3.1519. [DOI] [PubMed] [Google Scholar]
- 545.Trafford AW, Lipp P, O'Neill SC, Niggli E, Eisner DA. Propagating calcium waves initiated by local caffeine application in rat ventricular myocytes. J Physiol. 1995;489:319–326. doi: 10.1113/jphysiol.1995.sp021053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546.Tsai J, Cao JM, Zhou S, Swissa M, Cates AW, KenKnight BH, Chen LS, Karagueuzian HS, Chen PS. T wave alternans as a predictor of spontaneous ventricular tachycardia in a canine model of sudden cardiac death. J Cardiovasc Electrophysiol. 2002;13:51–55. doi: 10.1046/j.1540-8167.2002.00051.x. [DOI] [PubMed] [Google Scholar]
- 547.Tseng GN, Boyden PA. Different effects of intracellular Ca2+ and a phorbol ester on the T and L types Ca2+ currents in ventricular and Purkinje cells. Am J Physiol Heart Circ Physiol. 1991;261:H364–H379. doi: 10.1152/ajpheart.1991.261.2.H364. [DOI] [PubMed] [Google Scholar]
- 548.Tseng GN, Boyden PA. Multiple types of Ca currents in single canine Purkinje myocytes. Circ Res. 1989;65:1735–1750. doi: 10.1161/01.res.65.6.1735. [DOI] [PubMed] [Google Scholar]
- 549.Tseng GN, Hoffman BF. Two components of transient outward current in canine ventricular myocytes. Circ Res. 1989;64:633–647. doi: 10.1161/01.res.64.4.633. [DOI] [PubMed] [Google Scholar]
- 550.Tu H, Wang Z, Nosyreva E, DeSmedt H, Bezprozvanny I. Functional characterization of mammalian inositol 1,4,5-trisphosphate receptor isoforms. Biophys J. 2005;88:1046–1055. doi: 10.1529/biophysj.104.049593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551.Tu H, Wang Z, Bezprozvanny I. Modulation of mammalian inositol 1,4,5-trisphosphate receptor isoforms by calcium: a role of calcium sensor region. Biophys J. 2005;88:1056–1069. doi: 10.1529/biophysj.104.049601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.Tytgat J, Vereecke J, Carmeliet E. Differential effects of verapamil and flunarizine on cardiac L type and T type Ca channels. Naunyn-Schmeideberg's Arch Pharmacol. 1988;337:690–692. doi: 10.1007/BF00175798. [DOI] [PubMed] [Google Scholar]
- 553.Uchida K, Miyauchi H, Furuichi T, Michikawa T, Mikoshiba K. Critical regions for activation gating of inositol 1,4,5-trisphoshate receptor. J Biol Chem. 2003;278:16551–16560. doi: 10.1074/jbc.M300646200. [DOI] [PubMed] [Google Scholar]
- 554.Uehara A, Yasukochi M, Imanaga I, Nishi M, Takeshima H. Store-operated Ca2+ entry uncoupled with ryanodine receptor and junctional membrane complex in heart muscle cells. Cell Calcium. 2002;31:89–96. doi: 10.1054/ceca.2001.0257. [DOI] [PubMed] [Google Scholar]
- 555.Van Petegem F, Clark KA, Chatelain FC, Minor DL., Jr Structure of a complex between a voltage-gated calcium channel β-subunit and an α-subunit domain. Nature. 2004;429:671–675. doi: 10.1038/nature02588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556.Van Wagoner DR, Pond AL, Lamorgese M, Rossie SS, McCarthy PM, Nerbonne JM. Atrial L-type Ca2+ currents and human atrial fibrillation. Circ Res. 1999;85:428–436. doi: 10.1161/01.res.85.5.428. [DOI] [PubMed] [Google Scholar]
- 557.Veksler VI, Levitskaya EI, Kuznetkov AV, Sak VA, Kapelko VI. Mitochondrial function and calcium activated tension in saponin treated rat myocardial fibers. J Muscle Res Cell Motil. 1987;8:61. [Google Scholar]
- 558.Verkerk AO, Veldkamp MW, Baartscheer A, Schumacher CA, Klopping C, van Ginneken AC, Ravesloot JH. Ionic mechanism of delayed afterdepolarizations in ventricular cells isolated from human end-stage failing hearts. Circulation. 2001;104:2728–2733. doi: 10.1161/hc4701.099577. [DOI] [PubMed] [Google Scholar]
- 559.Verkerk AO, Veldkamp MW, Bouman LN, Van Ginneken AC. Calcium-activated Cl− current contributes to delayed afterdepolarizations in single purkinje and ventricular myocytes. Circulation. 2000;101:2639–2644. doi: 10.1161/01.cir.101.22.2639. [DOI] [PubMed] [Google Scholar]
- 560.Vermeulen JT, McGuire MA, Opthof T, Coronel R, de Bakker JM, Klopping C, Janse MJ. Triggered activity and automaticity in ventricular trabeculae of failing human and rabbit hearts. Cardiovasc Res. 1994;28:1547–1554. doi: 10.1093/cvr/28.10.1547. [DOI] [PubMed] [Google Scholar]
- 561.Vest JA, Wehrens XH, Reiken S, Lehnart SE, Dobrev D, Chandra P, Danilo P, Jr, Ravens U, Rosen MR, Marks AR. Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation. 2005;111:2025–2032. doi: 10.1161/01.CIR.0000162461.67140.4C. [DOI] [PubMed] [Google Scholar]
- 562.Viatchenko-Karpinski S, Terentyev D, Gyorke I, Terentyeva R, Volpe P, Priori SG, Napolitano C, Nori A, Williams SC, Gyorke S. Abnormal calcium signaling and sudden cardiac death associated with mutation of calsequestrin. Circ Res. 2004;94:471–477. doi: 10.1161/01.RES.0000115944.10681.EB. [DOI] [PubMed] [Google Scholar]
- 563.Vinogradova TM, Lyashkov AE, Zhu W, Ruknudin A, Sirenko S, Yang D, Deo S, Barlow M, Johnson S, Caffrey JL, Zhou YY, Xiao RP, Cheng H, Stern MD, Maltsev V, Lakatta EG. High basal protein kinase A dependent phosphorylation drives rhythmic internal Ca2+ store oscillations and spontaneous beating of cardiac pacemaker cells. Circ Res. 2006;98:505–514. doi: 10.1161/01.RES.0000204575.94040.d1. [DOI] [PubMed] [Google Scholar]
- 564.Vinogradova TM, Bogdanov KY, Lakatta EG. Beta-adrenergic stimulation modulates ryanodine receptor Ca2+ release during diastolic depolarization to accelerate pacemaker activity in rabbit sinoatrial nodal cells. Circ Res. 2002;90:73–79. doi: 10.1161/hh0102.102271. [DOI] [PubMed] [Google Scholar]
- 565.Volders PG, Kulcsar A, Vos MA, Sipido KR, Wellens HJ, Lazzara R, Szabo B. Similarities between early and delayed afterdepolarizations induced by isoproterenol in canine ventricular myocytes. Cardiovasc Res. 1997;34:348–359. doi: 10.1016/s0008-6363(96)00270-2. [DOI] [PubMed] [Google Scholar]
- 566.Volders PG, Vos MA, Szabo B, Sipido KR, de Groot SH, Gorgels APM, Wellens HJJ, Lazzara R. Progress in the understanding of cardiac early afterdepolarizations and torsades de pointes: time to revise current concepts. Cardiovasc Res. 2000;46:376–392. doi: 10.1016/s0008-6363(00)00022-5. [DOI] [PubMed] [Google Scholar]
- 567.Volders PGA, Sipido KR, Vos MA, Kulcsar A, Verduyn SC, Wellens HJJ. Cellular basis of biventricular hypertrophy and arrhythmogenesis in dogs with chronic complete atrioventricular block and acquired torsade de pointes. Circulation. 1998;98:1136–1147. doi: 10.1161/01.cir.98.11.1136. [DOI] [PubMed] [Google Scholar]
- 568.Wagenknecht T, Grassucci R, Frank J, Saito A, Inui M, Fleischer S. Three-dimensional architecture of the calcium channel/foot structure of sarcoplasmic reticulum. Nature. 1989;338:167–170. doi: 10.1038/338167a0. [DOI] [PubMed] [Google Scholar]
- 569.Wagner MB, Wang YG, Kumar R, Golod DA, Goolsby WN, Joyner RW. Measurements of calcium transients in ventricular cells during discontinuous action potential conduction. Am J Physiol Heart Circ Physiol. 2000;278:H444–H451. doi: 10.1152/ajpheart.2000.278.2.H444. [DOI] [PubMed] [Google Scholar]
- 570.Wakayama Y, Miura M, Stuyvers BD, Boyden PA, ter Keurs HEDJ. Spatial nonuniformity of excitation contraction coupling causes arrhythmogenic Ca2+ waves in rat cardiac muscle. Circ Res. 2005;96:1266–1273. doi: 10.1161/01.RES.0000172544.56818.54. [DOI] [PubMed] [Google Scholar]
- 571.Wakayama Y, Miura M, Sugai Y, Kagaya Y, Watanabe J, ter Keurs HEDJ, Shirato K. Stretch and quick release of rat cardiac trabeculae accelerates Ca2+ waves and triggered propagated contractions. Am J Physiol Heart Circ Physiol. 2001;281:H2133–H2142. doi: 10.1152/ajpheart.2001.281.5.H2133. [DOI] [PubMed] [Google Scholar]
- 572.Walsh KB, Arena JP, Kwok WM, Freeman L, Kass RS. Delayed-rectifier potassium channel activity in isolated membrane patches of guinea pig ventricular myocytes. Am J Physiol Heart Circ Physiol. 1991;260:H1390–H1393. doi: 10.1152/ajpheart.1991.260.4.H1390. [DOI] [PubMed] [Google Scholar]
- 573.Wan X, Laurita KR, Pruvot E, Rosenbaum DS. Molecular correlates of repolarization alternans in cardiac myocytes. J Mol Cell Cardiol. 2005;39:419–428. doi: 10.1016/j.yjmcc.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 574.Wang S, Song LS, Lakatta EG, Cheng H. Ca2+ signaling between single L type Ca2+ channels and ryanodine receptors in heart cells. Nature. 2001;410:592–596. doi: 10.1038/35069083. [DOI] [PubMed] [Google Scholar]
- 575.Wang S, George SE, Davis JP, Johnson JD. Structural determinants of Ca2+ exchange and affinity in the C terminal of cardiac troponin C. Biochemistry. 1998;37:14539–14544. doi: 10.1021/bi9814641. [DOI] [PubMed] [Google Scholar]
- 576.Wang S, Trumble WR, Liao H, Wesson CR, Dunker AK, Kang CH. Crystal structure of calsequestrin from rabbit skeletal muscle sarcoplasmic reticulum. Nature Struct Biol. 1998;5:476–483. doi: 10.1038/nsb0698-476. [DOI] [PubMed] [Google Scholar]
- 577.Weber CR, Ginsburg KS, Philipson KD, Shannon TR, Bers DM. Allosteric regulation of Na/Ca exchange current by cytosolic Ca in intact cardiac myocytes. J Gen Physiol. 2001;117:119–131. doi: 10.1085/jgp.117.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578.Weber CR, Piacentino IIIV, Ginsburg KS, Houser SR, Bers DM. Na+-Ca2+ exchange current and submembrane [Ca2+] during the cardiac action potential. Circ Res. 2002;90:182–189. doi: 10.1161/hh0202.103940. [DOI] [PubMed] [Google Scholar]
- 579.Wegener AD, Simmerman HKB, Lindemann JP, Jones LR. Phospholamban phosphorylation in intact ventricles. Phosphorylation of serine 16 and threonine 17 in response to β-adrenergic stimulation. J Biol Chem. 1989;264:11468–11474. [PubMed] [Google Scholar]
- 580.Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken S, Mohler PJ, Sun J, Guatimosim S, Song LS, Rosemblit N, D'Armiento J, Napolitano C, Memmi M, Priori S, Lederer WJ, Marks AR. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise induced sudden cardiac death. Cell. 2003;113:829–840. doi: 10.1016/s0092-8674(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 581.Wehrens XHT, Lehnart SE, Reiken S, van der Nagel R, Morales R, Sun J, Cheng Z, Deng SX, de Windt LJ, Landry DW, Marks AR. Enhancing calstabin binding to ryanodine receptors improves cardiac and skeletal muscle function in heart failure. Proc Natl Acad Sci USA. 2005;102:9607–9612. doi: 10.1073/pnas.0500353102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.Wei SK, Colecraft HM, DeMaria CD, Peterson BZ, Zhang R, Kohout TA, Rogers TB, Yue DT. Ca2+ channel modulation by recombinant auxiliary B subunits expressed in young adult heart cells. Circ Res. 2000;86:175–184. doi: 10.1161/01.res.86.2.175. [DOI] [PubMed] [Google Scholar]
- 583.Weiss JN, Karma A, Shiferaw Y, Chen PS, Garfinkel A, Qu Z. From pulsus to pulseless: the saga of cardiac alternans. Circ Res. 2006;98:1244–1253. doi: 10.1161/01.RES.0000224540.97431.f0. [DOI] [PubMed] [Google Scholar]
- 584.Wen H, Levitan IB. Calmodulin is an auxiliary subunit of KCNQ2/3 potassium channels. J Neurosci. 2002;22:7991–8001. doi: 10.1523/JNEUROSCI.22-18-07991.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585.Wendt-Gallitelli MF, Isenberg G. Total and free myoplasmic calcium during a contraction cycle: X ray microanalysis in guinea pig ventricular myocytes. J Physiol. 1991;435:349–372. doi: 10.1113/jphysiol.1991.sp018514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 586.Wibo M, Bravo G, Godfraind T. Postnatal maturation of excitation contraction coupling in rat ventricle in relation to the subcellular localization and surface density of 1,4 dihydropyridine and ryanodine receptors. Circ Res. 1991;68:662–673. doi: 10.1161/01.res.68.3.662. [DOI] [PubMed] [Google Scholar]
- 587.Wier WG, Blatter LA. Ca2+ oscillations and Ca2+ waves in mammalian cardiac and vascular smooth muscle cells. Cell Calcium. 1991;12:241–254. doi: 10.1016/0143-4160(91)90024-9. [DOI] [PubMed] [Google Scholar]
- 588.Wier WG, Cannell MB, Berlin JR, Marban E, Lederer WJ. Cellular and subcellular heterogeneity of intracellular calcium concentration in single heart cells revealed by fura-2. Science. 1987;235:325–328. doi: 10.1126/science.3798114. [DOI] [PubMed] [Google Scholar]
- 589.Wier WG, Egan TM, Lopez-Lopez JR, Balke CW. Local control of excitation contraction coupling in rat heart cells. J Physiol. 1994;474:463–471. doi: 10.1113/jphysiol.1994.sp020037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590.Wier WG, ter Keurs HE, Marban E, Gao WD, Balke CW. Ca2+ “sparks” and waves in intact ventricular muscle resolved by confocal imaging. Circ Res. 1997;81:462–469. doi: 10.1161/01.res.81.4.462. [DOI] [PubMed] [Google Scholar]
- 591.Wier WG, Yue DT. Intracellular calcium transients underlying the short term force interval relationship in ferret ventricular myocardium. J Physiol. 1986;376:507–530. doi: 10.1113/jphysiol.1986.sp016167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 592.Williams DA, Delbridge LM, Cody SH, Harris PJ, Morgan TO. Spontaneous and propagated calcium release in isolated cardiac myocytes viewed by confocal microscopy. Am J Physiol Cell Physiol. 1992;262:C731–C742. doi: 10.1152/ajpcell.1992.262.3.C731. [DOI] [PubMed] [Google Scholar]
- 593.Wingo TL, Shah VN, Anderson ME, Lybrand TP, Chazin WJ, Balser JR. An EF hand in the sodium channel couples intracellular calcium to cardiac excitability. Nature Mol Biol. 2004;11:219–225. doi: 10.1038/nsmb737. [DOI] [PubMed] [Google Scholar]
- 594.Wohlfart B. Relationships between peak force, action potential duration and stimulus interval in rabbit myocardium. Acta Physiol Scand. 1979;106:395–409. doi: 10.1111/j.1748-1716.1979.tb06419.x. [DOI] [PubMed] [Google Scholar]
- 595.Wojcikiewicz RJ, Luo SG. Differences among type I, II, III inositol-1,4,5-trisphosphate receptors in ligand-binding affinity influence the sensitivity of calcium stores to inositol-1,4,5-trisphosphate. Mol Pharmacol. 1998;53:656–662. doi: 10.1124/mol.53.4.656. [DOI] [PubMed] [Google Scholar]
- 596.Woo SH, Cleemann L, Morad M. Ca2+ current-gated focal and local Ca2+ release in rat atrial myocytes: evidence from rapid 2-D confocal imaging. J Physiol. 2002;543:439–453. doi: 10.1113/jphysiol.2002.024190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 597.Woo SH, Cleemann L, Morad M. Spatiotemporal characteristics of junctional and nonjunctional focal Ca2+ release in rat atrial myocytes. Circ Res. 2003;92:e1–e11. doi: 10.1161/01.res.0000051887.97625.07. [DOI] [PubMed] [Google Scholar]
- 598.Wu S, Weiss JN, Chou C, Attin M, Hayashi H, Lin SF. Dissociation of membrane potential and intracellular calcium during ventricular fibrillation. J Cardiovasc Electrophysiol. 2005;16:186–192. doi: 10.1046/j.1540-8167.2005.40334.x. [DOI] [PubMed] [Google Scholar]
- 599.Wu X, Zhang T, Bossuyt J, Li X, McKinsey TA, Dedman JR, Olson EN, Chen J, Brown JH, Bers DM. Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling. J Clin Invest. 2006;116:675–682. doi: 10.1172/JCI27374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600.Wu Y, Anderson ME. Ca2+-activated non-selective cation current in rabbit ventricular myocytes. J Physiol. 2000;522:51–57. doi: 10.1111/j.1469-7793.2000.0051m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601.Wu Y, Colbran RJ, Anderson ME. Calmodulin kinase is a molecular switch for cardiac excitation-contraction coupling. Proc Natl Acad Sci USA. 2001;98:2877–2881. doi: 10.1073/pnas.051449198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602.Wu Y, MacMillan LB, McNeill RB, Colbran RJ, Anderson ME. CaM kinase augments cardiac L type Ca2+ current: a cellular mechanism for long QT arrhythmias. Am J Physiol Heart Circ Physiol. 1999;276:H2168–H2178. doi: 10.1152/ajpheart.1999.276.6.H2168. [DOI] [PubMed] [Google Scholar]
- 603.Wu Y, Roden DM, Anderson ME. Calmodulin kinase inhibition prevents development of the arrhythmogenic transient inward current. Circ Res. 1999;84:906–912. doi: 10.1161/01.res.84.8.906. [DOI] [PubMed] [Google Scholar]
- 604.Wyatt CN, Campbell V, Brodbeck J, Brice NL, Page KM, Berrow NS, Brickley K, Terracciano CM, Naqvi RV, MacLeod KT, Dolphin AC. Voltage dependent binding and calcium channel current inhibition by an anti-alpha1D subunit antibody in rat dorsal root ganglion neurons and guinea pig myocytes. J Physiol. 1997;502:307–319. doi: 10.1111/j.1469-7793.1997.307bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605.Xiao RP, Cheng H, Lederer WJ, Suzuki T, Lakatta EG. Dual regulation of Ca2+/calmodulin dependent kinase II activity by membrane voltage and by calcium influx. Proc Natl Acad Sci USA. 1994;91:9659–9663. doi: 10.1073/pnas.91.20.9659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 606.Xu L, Mann G, Meissner G. Regulation of cardiac Ca2+ release channel (ryanodine receptor) by Ca2+,H+, Mg2+ and adenine nucleotides under normal and ischemic conditions. Circ Res. 1996;79:1100–1109. doi: 10.1161/01.res.79.6.1100. [DOI] [PubMed] [Google Scholar]
- 608.Yamada M, Miyawaki A, Saito K, Nakajima T, Yamamoto-Hino M, Ryo Y, Furuichi T, Mikoshiba K. The calmodulin-binding domain in the mouse type 1 inositol 1,4,5-trisphosphate receptor. Biochem J. 1995;308:83–88. doi: 10.1042/bj3080083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 609.Yamamoto T, Ikemoto N. Spectroscopic monitoring of local conformational changes during the intramolecular domain-domain interaction of the ryanodine receptor. Biochemistry. 2002;41:1492–1501. doi: 10.1021/bi015581z. [DOI] [PubMed] [Google Scholar]
- 610.Yamasaki Y, Furuya Y, Araki K, Matsuura K, Kobayashi M, Ogata T. Ultra-high-resolution scanning electron microscopy of the sarcoplasmic reticulum of the rat atrial myocardial cells. Anat Rec. 1997;248:70–75. doi: 10.1002/(SICI)1097-0185(199705)248:1<70::AID-AR8>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 611.Yang J, Ellinor PT, Sather WA, Zhang JF, Tsien RW. Molecular determinants of Ca selectivity and ion permeation in L type Ca channels. Nature. 1993;366:158–161. doi: 10.1038/366158a0. [DOI] [PubMed] [Google Scholar]
- 612.Yang J, McBride S, Mak DO, Vardi N, Palczewski K, Haeseleer F, Foskett JK. Identification of a family of calcium sensors as protein ligands of inositol trisphosphate receptor Ca2+ release channels. Proc Natl Acad Sci USA. 2002;99:7711–7716. doi: 10.1073/pnas.102006299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 613.Yang L, Liu G, Zakharov SI, Morrow JP, Rybin VO, Steinberg SF, Marx SO. Ser1928 is a common site for Cav1.2 phosphorylation by protein kinase C isoforms. J Biol Chem. 2005;280:207–214. doi: 10.1074/jbc.M410509200. [DOI] [PubMed] [Google Scholar]
- 614.Yano M, Ono K, Ohkusa T, Suetsugu M, Kohno M, Hisaoka T, Kobayahi S, Hisamatsu Y, Yamamoto T, Kohno M, Noguchi N, Takasawa S, Okamoto H, Matsuzaki M. Altered stoichiometry of FKBP12.6 versus ryanodine receptor as a cause of abnormal Ca2+ leak through ryanodine receptor in heart failure. Circulation. 2000;102:2131–2136. doi: 10.1161/01.cir.102.17.2131. [DOI] [PubMed] [Google Scholar]
- 615.Yano M, Ikeda Y, Matsuzaki M. Altered intracellular Ca2+ handling in heart failure. J Clin Invest. 2005;115:556–564. doi: 10.1172/JCI24159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 616.Yano M, Okuda S, Oda T, Tokuhisa T, Tateishi H, Mochizuki M, Noma T, Doi M, Kobayashi S, Yamamoto T, Ikeda Y, Ohkusa T, Ikemoto N, Matsuzaki M. Correction of defective inter-domain interaction within ryanodine receptor by antioxidant is a new therapeutic strategy against heart failure. Circulation. 2005;112:3633–3643. doi: 10.1161/CIRCULATIONAHA.105.555623. [DOI] [PubMed] [Google Scholar]
- 617.Yano M, Yamamoto T, Ikemoto N, Matsuzaki M. Abnormal ryanodine receptor function in heart failure. Pharmacol Ther. 2005;107:377–391. doi: 10.1016/j.pharmthera.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 618.Yeager M. Structure of cardiac gap junction membrane channels. In: Spooner PM, Joyner RW, Jalife J, editors. Discontinuous Conduction in the Heart. Armonk, NY: Futura; 1997. pp. 161–184. [Google Scholar]
- 619.Yin CC, Han H, Wei R, Lai FA. Two-dimensional crystallization of the ryanodine receptor Ca2+ release channel on lipid membranes. J Struct Biol. 2005;149:219–224. doi: 10.1016/j.jsb.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 620.Yuan W, Bers DM. Ca-dependent facilitation of cardiac Ca2+ current is due to a Ca-calmodulin dependent protein kinase. Am J Physiol Heart Circ Physiol. 1994;267:H982–H993. doi: 10.1152/ajpheart.1994.267.3.H982. [DOI] [PubMed] [Google Scholar]
- 621.Yue DT, Herzig S, Marban E. Beta adrenergic stimulation of calcium channels occurs by potentiation of high activity gating modes. Proc Natl Acad Sci USA. 1990;87:753–757. doi: 10.1073/pnas.87.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 622.Yus-Najera E, Santana-Castro I, Villarroel A. The identification and characterization of a noncontinuous calmodulin-binding site in noninactivating voltage-dependent KCNQ potassium channels. J Biol Chem. 2002;277:28545–28553. doi: 10.1074/jbc.M204130200. [DOI] [PubMed] [Google Scholar]
- 623.Zahradnikova A, Zahradnik I, Gyorke I, Gyorke S. Rapid activation of the cardiac ryanodine receptor by submillisecond calcium stimuli. J Gen Physiol. 1999;114:787–798. doi: 10.1085/jgp.114.6.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 624.Zamponi GW, Soong TW, Bourinet E, Snutch TP. Beta subunit coexpression and the alpha1 subunit domain I-II linker affect piperidine block of neuronal calcium channels. J Neurosci. 1996;16:2430–2443. doi: 10.1523/JNEUROSCI.16-08-02430.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 625.Zaza A, Rocchetti M, Brioschi A, Cantadori A, Ferroni A. Dynamic Ca2+-induced inward rectification of K+ current during the ventricular action potential. Circ Res. 1998;82:947–956. doi: 10.1161/01.res.82.9.947. [DOI] [PubMed] [Google Scholar]
- 626.Zhang YH, Youm JB, Sung HK, Lee SH, Ryu SY, Ho WK, Earm YE. Stretch-activated and background non-selective cation channels in rat atrial myocytes. J Physiol. 2000;523:607–619. doi: 10.1111/j.1469-7793.2000.00607.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 627.Zhang BX, Zhao H, Muallem S. Ca2+-dependent kinase and phosphatase control inositol 1,4,5-trisphosphate-mediated Ca2+ release. Modification by agonist stimulation. J Biol Chem. 1993;268:10997–11001. [PubMed] [Google Scholar]
- 628.Zhang JF, Ellinor PT, Aldrich RW, Tsien RW. Molecular determinants of voltage dependent inactivation in calcium channels. Nature. 1994;372:97–100. doi: 10.1038/372097a0. [DOI] [PubMed] [Google Scholar]
- 629.Zhang J, Liu Z, Masumiya H, Wang R, Jiang D, Li F, Wagenknecht T, Chen SR. Three-dimensional localization of divergent region 3 of the ryanodine receptor to the clamp-shaped structures adjacent to the FKBP binding sites. J Biol Chem. 2003;278:14211–14218. doi: 10.1074/jbc.M213164200. [DOI] [PubMed] [Google Scholar]
- 630.Zhang XQ, Tillotson DL, Moore RL, Zelis R, Cheung JY. Na/Ca exchange currents and SR Ca2+ contents in postinfarction myocytes. Am J Physiol Cell Physiol. 1996;271:C1800–C1807. doi: 10.1152/ajpcell.1996.271.6.C1800. [DOI] [PubMed] [Google Scholar]
- 631.Zhang Y, Miura M, ter Keurs HEDJ. Triggered propagated contractions in rat cardiac trabeculae: inhibition by octanol and heptanol. Circ Res. 1996;79:1077–1085. doi: 10.1161/01.res.79.6.1077. [DOI] [PubMed] [Google Scholar]
- 632.Zhang Y, ter Keurs HEDJ. Effects of gadolinium on twitch force and triggered propagated contractions in rat cardiac trabeculae. Cardiovasc Res. 1996;32:180–188. [PubMed] [Google Scholar]
- 633.Zhang Z, Xu Y, Song H, Rodriguez J, Tuteja D, Namkung Y, Shin HS, Chiamvimonvat N. Functional roles of Cav1.3 (alpha1D) calcium channel in sinoatrial nodes. Insight gained using gene-targeted null mutant mice. Circ Res. 2002;90:981–987. doi: 10.1161/01.res.0000018003.14304.e2. [DOI] [PubMed] [Google Scholar]
- 634.Zhang Z, He Y, Tuteja D, Xu D, Timofeyev V, Zhang Q, Glatter KA, Xu Y, Shin HS, Low R, Chiamvimonvat N. Functional roles of Cav1.3(α1D) calcium channels in atria: insights gained from gene-targeted null mutant mice. Circulation. 2005;112:1936–1944. doi: 10.1161/CIRCULATIONAHA.105.540070. [DOI] [PubMed] [Google Scholar]
- 635.Zhou H, Kim SA, Kirk EA, Tippens AL, Sun H, Haeseleer F, Lee A. Ca2+-binding protein-1 facilitates and forms a postsynaptic complex with Cav1.2 (L-type) Ca2+ channels. J Neurosci. 2004;24:4698–4708. doi: 10.1523/JNEUROSCI.5523-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 636.Zhou H, Yu K, McCoy KL, Lee A. Molecular mechanism for divergent regulation of Cav1.2 Ca2+ channels by calmodulin and Ca2+-binding protein-1. J Biol Chem. 2005;280:29612–29619. doi: 10.1074/jbc.M504167200. [DOI] [PubMed] [Google Scholar]
- 637.Zhou J, Olcese R, Qin N, Noceti F, Birnbaumer L, Stefani E. Feedback inhibition of Ca2+ channels by Ca2+ depends on a short sequence of the C terminus that does not include the Ca2+ binding function of a motif with similarity to Ca2+ binding domains. Proc Natl Acad Sci USA. 1997;94:2301–2305. doi: 10.1073/pnas.94.6.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 638.Zhou Z, Lipsius SL. NaCa exchange current in latent pacemaker cells isolated from cat right atrium. J Physiol. 1993;466:263–285. [PMC free article] [PubMed] [Google Scholar]
- 639.Zhou Z, Lipsius SL. T-type calcium currents in latent pacemaker cells isolated from cat right atrium. J Mol Cell Cardiol. 1994;26:1211–1219. doi: 10.1006/jmcc.1994.1139. [DOI] [PubMed] [Google Scholar]
- 640.Zhou Z, Matlib MA, Bers DM. Cytosolic and mitochondrial Ca2+ signals in patch clamped mammalian ventricular myocytes. J Physiol. 1998;507:379–403. doi: 10.1111/j.1469-7793.1998.379bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 641.Zhu Y, Nosek TM. Inositol trisphosphate enhances Ca2+ oscillations but not Ca2+-induced Ca2+ release from cardiac sarcoplasmic reticulum. Pflügers Arch. 1991;418:1–6. doi: 10.1007/BF00370444. [DOI] [PubMed] [Google Scholar]
- 642.Zima AV, Blatter LA. Inositol-1,4,5-trisphosphate-dependent Ca2+ signaling at cat atrial excitation-contraction coupling and arrhythmias. J Physiol. 2004;555:607–615. doi: 10.1113/jphysiol.2003.058529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 643.Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- 644.Zygmunt AC. Intracellular calcium activates a chloride current in canine ventricular myocytes. Am J Physiol Heart Circ Physiol. 1994;267:H1984–H1995. doi: 10.1152/ajpheart.1994.267.5.H1984. [DOI] [PubMed] [Google Scholar]
- 645.Zygmunt AC, Gibbons WR. Calcium-activated chloride current in rabbit ventricular myocytes. Circ Res. 1991;68:424–437. doi: 10.1161/01.res.68.2.424. [DOI] [PubMed] [Google Scholar]
- 646.Zygmunt AC, Gibbons WR. Properties of the calcium-activated chloride current in heart. J Gen Physiol. 1992;99:391–414. doi: 10.1085/jgp.99.3.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 647.Zygmunt AC, Goodrow RJ, Antzelevitch C. INaCa contributes to electrical heterogeneity within the canine ventricle. Am J Physiol Heart Circ Physiol. 2000;278:H1671–H1678. doi: 10.1152/ajpheart.2000.278.5.H1671. [DOI] [PubMed] [Google Scholar]