

Abstract
Pancreatic ductal adenocarcinoma (PDA) is the most aggressive tumor, showing incidence and mortality values almost identical. Despite remarkable advances in PDA molecular characterization, this disease is still refractory to current treatments. Desmoplastic stroma, a constant hallmark of PDA, has recently emerged as the major responsible for PDA therapeutic resistance, therefore representing a promising target. Galectin-1 (Gal1), a glycan-binding protein, is highly expressed in PDA stroma but its role remains unknown. Here, we aim to understand in vivo Gal1 functions and the molecular pathways responsible for its oncogenic properties. Genetic ablation of Gal1 in Ela-myc mice dampens tumor progression through inhibition of proliferation, angiogenesis, desmoplasia and stimulation of tumor-associated immune response, resulting in a 20% increase on the animal life span. In vitro and in vivo studies unveil that these effects are mediated by modulation of the tumor microenvironment in a non-cell autonomous manner. Importantly, acinar-to-ductal metaplasia, a crucial step for PDA initiation, is also regulated by Gal1. Finally, high-throughput gene expression studies and molecular analysis aimed at identifying the underlying mechanism revealed that Gal1 promotes Hedgehog pathway both in PDA cells and stromal fibroblasts. In summary, our studies define a novel role of Gal1 in PDA tumor epithelium-stroma crosstalk and suggest this lectin as potential molecular target for therapy of neoplasms overexpressing Gal1.
Keywords: Galectin-1, Hedgehog, pancreatic cancer, tumor microenvironment, acinar-to-ductal metaplasia
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDA), the most common type of pancreatic cancer (95%), is one of the most aggressive tumors and the fourth leading cause of cancer-related mortality across the world (1). Despite notable efforts to develop novel therapeutic targets, PDA is still highly resistant to therapy, with a median survival of 4-6 months and a 5-year survival rate lower than 5% (2). Recent genome-wide analysis has identified a complex pattern of molecular alterations in PDA including the previously well-known KRAS, CDKN2A, TRP53 and SMAD4, but also the activation of other pathways as c-Myc or Hedgehog (Hh) (3). At the histopathological level, PDA is characterized by an abundant stromal desmoplasia, a fibroinflammatory reaction composed of a dense extracellular matrix (ECM), fibroblasts/ pancreatic stellate cells, immune cells and endothelial cells. Recent data have demonstrated that PDA-associated desmoplasia plays a crucial role in promoting tumor growth and progression, and also contributes to chemotherapy resistance, emerging as a promising target for pancreatic cancer treatment (4).
Galectins are a family of lectins defined by a highly conserved carbohydrate recognition domain which can localize intracellularly or extracellularly. Galectin-1 (Gal1), one of the best-characterized members of this family, is a homodimer of 14 KDa subunits that can interact with carbohydrates from glycoconjugates located at the cell surface or in the ECM, regulating cell-cell and cell-ECM adhesion. By these interactions, it participates in different biological functions as cell cycle control, migration, invasion, angiogenesis and immune system response, both in physiological and pathological situations (5, 6). Gal1 is overexpressed in many tumors (7), including pancreatic cancer (8-14), where a positive correlation with tumor stage has been found (15). Of note, Gal1 overexpression in PDA was identified mostly in stromal cells. However, the biological relevance of these findings remains elusive.
In this study, using Ela-myc mice, a well characterized model of pancreatic cancer (16-18) and Gal1 knockout mice, we define a novel Gal1-driven mechanism controlling desmoplasia in these tumors. Our data show that partial or complete depletion of Gal1 reduces in vivo tumorigenicity, leading to a significant increase in Ela-myc mice survival. Abolishment of Gal1 expression not only prevents tumor growth but also modulates the tumor microenvironment, hampering stromal activation and angiogenesis and favoring immune surveillance. Moreover, acinar-to-ductal metaplasia (ADM), a transdifferentiation process that likely triggers PDA initiation, was severely impaired after Gal1 loss in Ela-myc mice, and further analysis suggest that EGFR and Pdx1 are the molecular pathways underlying Gal1-mediated ADM. In addition, in vivo and in vitro strategies indicate that stromal Gal1 is the major responsible for its tumoral properties. Finally, high-throughput expression analysis and in vitro molecular assays identify Hh as a key signaling pathway involved in Gal1-regulated functions in pancreatic tumor epithelial and stromal cells. Together, these data shed light on the role and molecular mechanisms of Gal1 during pancreatic cancer initiation and progression through tumor microenvironment remodeling, suggesting that targeting Gal1 represents a promising therapeutic strategy for this deadly disease.
MATERIALS AND METHODS
For animals, cell lines, histopathology and in vitro functional experiments see Supplementary Material.
Gal1 knockdown by shRNA or siRNA
PANC-1 cells were transduced with shGal1 or shCtl by lentiviral infection as described in Supplementary Material. F88.2 and HPSE cells were transfected with 50nM of Gal-1 siRNA or an irrelevant siRNA (SMARTpool® Reagents, Dharmacon). To control knockdown efficiency, cells were directly lysed 72 h after transfection and protein levels measured by Western blot (WB) using Rabbit α-Gal-1 (Sigma) or mouse α-Tubulin (Sigma), peroxidase-conjugated secondary antibodies and ECL (GE Healthcare).
Microarray Analysis
Microarray expression profiles were obtained using the Affymetrix Human Exon ST 1.0 arrays (Affymetrix) in IMIM’s Microarray facility. Detailed description and validation by RT-qPCR is provided in Supplementary Material.
In vitro Luciferase Measure
RWP-1 cells transfected with an empty pcDNA3 or with pcDNA3-Gal1, were transfected with Lipofectamine and Plus reagent with 25, 50, 100 or 150 ng of the vector pδ51LucII containing 8 Gli binding sites (19). After 48 h, cells were lysed and Luciferase and Renilla activity measured (Promega).
Statistical Analysis
Unless otherwise stated, results are expressed as mean ± SEM of triplicates. Statistical analyses were performed with SPSS version 12.0. Statistical significance cut-off has been always considered when P < .05. Kaplan-Meier analyses were used for establishing survival curves and comparisons were performed using the log-rank test. Student’s t-, Mann Whitney or Chi-squared tests were applied, as indicated.
RESULTS
Gal1 deficiency increases Ela-myc mice survival and impairs tumor proliferation
c-myc oncogene plays a key role in the initiation and progression of PDA (20),(21) and it is frequently overexpressed in human tumors (3, 22). In mouse, expression of c-myc using pancreas-specific elastase promoter (Ela-myc model) leads to the generation of acinar tumors and ductal tumors (16) as well as ADM (18) (Fig. 1A, a-c). We analyzed Gal1 expression by immunohistochemistry (IHC) in acinar and ductal tumors, as well as in metaplastic lesions (Fig. 1A, d-f). Interestingly, Gal1 was mainly expressed in the stromal compartment, as previously described in human PDA (8, 11, 13). Accordingly, high levels of Gal1 were found in ductal tumors with abundant stromal desmoplasia (Fig. 1A, f).
Figure 1. Gal1 deficiency increases pancreatic cancer survival and decreases cell proliferation in Ela-myc model.

A. H&E (a-c) and Gal1 staining (d-f) of acinar, metaplasic or ductal areas of Ela-myc transgenic mice. Bars, 50 μm. B. Kaplan-Meier survival curves (left) from Ela-myc:Gal1+/+, Ela-myc:Gal1+/− and Ela-myc:Gal1−/−. P values (log-rank test) are relative to Ela-myc:Gal1+/+. Pie charts (right) specifying the percentage of animals of each genotype that died before four months (< 4 months), between the 4th and 5th month (4-5 months), between the 5th and 6th month (5-6 months) or those animals that survived more than 6 months (> 6 months). C. Immunostaining for Ki67 in acinar and ductal parts of tumors of each genotype. Bars, 50 μm (a-c), 100 μm (d-f). Right panel: quantification of the proliferation rates in ductal areas shown by the percentage of Ki67 positive cells per field. P values (Mann-Whitney test) are relative to Ela-myc:Gal1+/+. ***P < .001.
To define the role of Gal1 in pancreatic cancer development and progression, Ela-myc transgenic mice were crossed with Gal1 knockouts to obtain Ela-myc:Gal1+/+ (n = 80), Ela-myc:Gal1+/− (n = 64) and Ela-my:Gal1−/− (n = 54). Remarkably, a significant increase in animal survival was observed after loss of either one or both Gal1 alleles (Fig. 1B, left; P < .001). These differences were even more evident when considering long-time survivors; Ela-myc:Gal1+/+ mice rarely survived more than 5 (19%) or 6 (4%) months, whereas long-term survival raised to around 50% (survival ≥ 5 months) and 20% (survival ≥ 6 months) in Ela-myc:Gal1+/− or Ela-my:Gal1−/− (Fig. 1B, right). The increased survival obtained in Ela-myc:Gal1+/− compared to wild type (WT) mice, indicates that Gal1 haploinsufficiency impairs pancreatic tumor development. Gal1 expression in Ela-myc:Gal1+/− tumors was confirmed by IHC to rule out the inactivation of the WT allele (Supplementary Fig. S1). Taken together, these data demonstrate that total or partial abolishment of Gal1 expression in vivo results in increased survival in Ela-myc mice, suggesting a role for this protein during cancer initiation and progression.
To determine whether the increase in Ela-myc mice survival after Gal1 abolishment was a result of changes in tumor growth, we analyzed the proliferation rate. Acinar tumors were highly proliferative and showed 100% of positivity for Ki67 immunostaining in all genotypes (Fig. 1C, a-c). Remarkably, ductal tumors from Ela-myc:Gal1+/− or Ela-my:Gal1−/− displayed a significant reduced Ki67 immunostaining compared to Ela-myc:Gal1+/+ counterparts (44% for Ela-myc:Gal1+/−; 39% for Ela-my:Gal1−/− compared to 61% for Ela-myc:Gal1+/+; P < .001) (Fig. 1C, d-f and right panel).
We also analyzed whether Gal1 expression can affect tumor infiltration and metastasis and no significant differences were observed (Supplementary Material and Supplementary Table 1).
Altogether, these data indicate that reduced expression of Gal1 affects proliferation of PDA tumors in Ela-myc mice, resulting in a slower tumor growth and increased survival.
Gal1 deficiency impairs ADM in Ela-myc mice
PDA is likely to originate from transdifferentiation of acinar cells into ductal cells, a process known as ADM (23). This mechanism has been also described in the Ela-myc model, where formation of ductal tumors is preceded by transdifferentiation of acinar cells (18). To assess whether Gal1 is involved in ADM in pancreatic cancer, we performed a detailed histopathological analysis and classification of acinar and ductal tumors from Ela-myc:Gal1+/+, Ela-myc:Gal1+/− and Ela-my:Gal1−/− mice (Fig. 2A, a-f). Remarkably, we found that Gal1 deficiency, either one or two alleles, results in a dramatic reduction of Ela-myc ductal tumors (Fig. 2B). Thus, while Ela-myc:Gal1+/+ mice showed 40% of tumors with ductal characteristics, this percentage decreased to the 23% or 10% in Ela-myc:Gal1+/− and Ela-my:Gal1−/−mice, respectively. Of note, the percentage of tumors with high ductal component was reduced in Ela-my:Gal1−/− mice compared to Ela-myc:Gal1+/− and Ela-myc:Gal1+/+ mice (Fig. 2C). These results strongly suggest a role for Gal1 in ADM during c-myc driven pancreatic carcinogenesis.
Figure 2. Gal1 loss impairs ADM.

A. H&E of acinar (a-c) and ductal regions (d-f) of Ela-myc:Gal1+/+, Ela-myc:Gal1+/−, and Ela-myc:Gal1−/− transgenic mice. Bars, 50 μm. B. Pie charts showing the percentage of ductal (white) versus acinar (grey) areas in tumors of each genotype. C. Quantification of the percentage of entirely ductal tumors (100%), tumors with a ductal component over the 70% (> 70%) or over the 50% (> 50%) in the different genotypes. D. Analysis of EGFR, Pdx1 and MMP7 by RT-qPCR in RNA extracts from tumors of Ela-myc:Gal1+/+, Ela-myc:Gal1+/−, and Ela-myc:Gal1−/− mice. All data are mean ± SEM of three independent experiments. P values (B: Mann Whitney; C: Chi-squared; D: Student’s t-test) are relative to Ela-myc:Gal1+/+. *P < .05; **P < .01, ***P < .001.
To get insights into the molecular signaling pathways impairing ADM after Gal1 downregulation in Ela-myc tumors, we selected several genes previously reported to be involved in this process (24) and compared their expression by RT-qPCR in tumors from Ela-myc:Gal1+/+, Ela-myc:Gal1+/− and Ela-my:Gal1−/− mice. Interestingly, EGFR and Pdx1 RNA levels were downregulated in tumors with low levels of Gal1 and MMP7 was upregulated (Fig. 2D), whereas the other genes analyzed were not affected (Supplementary Fig. S2).
Loss of Gal1 modulates pancreatic tumor microenvironment
The stroma is the major component of the tumor mass in PDA and it represents a promising target for therapy (4, 25). Considering high Gal1 levels of expression in pancreatic stroma (Fig. 1A, d-f) and our previous in vitro data supporting a role for this lectin in the tumor-stroma crosstalk (12), we evaluated the overall contribution of Gal1 to tumor microenvironment during in vivo pancreatic carcinogenesis. First, tumor vascularization was analyzed using von Willebrand factor (vWF) staining. A dramatic reduction of angiogenesis was observed in tumors from Ela-myc:Gal1−/− and Ela-myc:Gal1+/− mice compared to those from Ela-myc:Gal1+/+ (79% and 37% reduction, respectively, P < .005) (Fig. 3A, a-f and right panel). Accordingly, we observed that mice with reduced Gal1 levels displayed significantly less intraperitoneal hemorraghes (Supplementary Material and Supplementary Table 1) and increased tumoral necrosis (Supplementary Fig. S3), likely as a consequence of their reduced tumor vascular network. Second, we analyzed activated fibroblast and stellate cells in these tumors using α-SMA immunostaining (Fig. 3B, a-f). Activated stromal cells were significantly reduced after Gal1 depletion (5% and 8% in tumors from Ela-my:Gal1−/− and Ela-myc:Gal1+/− mice respectively, compared to 10% in Ela-myc:Gal1+/+ mice) (Fig. 3B, right panel), indicating that Gal1 supports PDA desmoplasia. Third, we analyzed the effects of Gal1 depletion in infiltrating tumor immune cells. Quantification of intratumoral T-lymphocytes in ductal lesions, detected by IHC against CD3, showed a significant and dose-dependent increase in this population in the absence of Gal1 (3 and 1.75 fold increase in the Ela-my:Gal1−/− and Ela-myc:Gal1+/− mice respectively, compared to Ela-myc:Gal1+/+, P < .001) (Fig. 3C a-c, right panel). Acinar lesions showed reduced immune cell infiltration, although the same pattern was observed (data not shown). Similarly, neutrophil quantification by MPO staining revealed a significant increase of this cell population in ductal tumors after Gal1 loss (2.3 fold increase in Ela-myc:Gal1−/− versus Ela-myc:Gal1+/+, P = .04) (Fig. 3C, d-f and right panel). No differences were observed in intratumoral macrophages or B cells between different genotypes (data not shown). These results demonstrate the involvement of Gal1 in maintaining pancreatic tumor immune privilege by hampering T-cell and neutrophil-mediated immune response during in vivo cancer progression.
Figure 3. Gal1 modulates tumor microenvironment.

A. Immunostaining for von Willebrand factor (vWF) in acinar and ductal parts of tumors of Ela-myc:Gal1+/+, Ela-myc:Gal1+/− and Ela-myc:Gal1−/− mice. Bars, 200 μm. B. α-SMA IHC in Ela-myc:Gal1 acinar and ductal tumors. Bars, 50 μm (a-c), 100 μm (d-f). C. IHC to detect T lymphocytes with CD3 (a-c) or neutrophils with MPO (d-f) antibodies in ductal tumors of Ela-myc:Gal1+/+, Ela-myc:Gal1+/− and Ela-myc:Gal1−/− mice. Bars, 20 μm. Bar graphs on the right (A-C), show quantifications, which are represented as the mean positively stained area ± SEM. P values (Mann Whitney test) are relative to Ela-myc:Gal1+/+. *P < .05; **P < .01, ***P < .001.
Altogether, these data indicate that modulation of Gal1 expression in pancreatic cancer has a crucial impact on remodeling in vivo the tumor microenvironment, through regulation of angiogenesis, fibroblasts activation and immune response.
Gal1 cell autonomous and non-cell autonomous effects during pancreatic tumorigenesis
Gal1 is a secreted protein, therefore Gal1 found in tumoral stroma can originate from epithelial, stromal cells or both. In fact, we have previously reported that Gal1 is highly expressed in human pancreatic cell lines (12) and primary cultures from Ela-myc (unpublished data). To analyze the cell autonomous and non-cell autonomous contribution of Gal1 in epithelial pancreatic cancer cell oncogenesis, we knocked down its expression in the human cell line PANC-1 and tested its tumorigenic properties in vitro and in vivo. Cells were transfected with an shRNA control (shCtl) or with two different Gal1 shRNA sequences (shGal1#1 and shGal1#2), which efficiently reduced Gal1 protein expression (> 90%) (Fig. 4A). In vitro characterization of these cells showed that while downregulation of Gal1 expression did not affect cell proliferation (Fig. 4B), it significantly reduced soft agar colony formation (Fig. 4C) and cell motility (Fig. 4D and Supplementary video1 and video2). These data indicate that cell autonomous expression of Gal1 in pancreatic tumoral cells may promote tumor progression by favoring migration and anchorage independent growth. On the other hand, the different effects of Gal1 downregulation in proliferation found in vivo (Fig. 1C, d-f) and in vitro (Fig. 4B) suggest that Gal1 expression promotes in vivo cell growth in a non-cell autonomous manner. Indeed, no tumorigenic differences were observed when injecting PANC-1 cells with normal or downregulated Gal1 levels into nude mice (Supplementary Material and Supplementary Fig. S4).
Figure 4. Effects of Gal1 downregulation in PDA cells in vitro.

A. Analysis of Gal1 protein levels by WB in PANC-1 cells (wt), and in cells infected with lentivirus carrying a control shRNA (shCtl) or two different Gal1 targeting sequences (shGal1#1 and shGal1#2). Tubulin is shown as the loading control. Quantification is shown on the right. B. Cell proliferation rate of shCtl or shGal1 PANC-1 cells was quantified by BrdU incorporation and immunofluorescence. C. Anchorage independent growth of PANC-1 cells expressing shCtl or shGal1 was assessed by soft agar colony formation. D. Migration of PANC-1 cells expressing shCtl or shGal1 was quantified after wound-healing experiments or time-lapse video. See also Supplementary Fig.4Dvideo1 and Fig.4Dvideo2 in online Supplementary Material. All data are mean ± SEM of three independent experiments. P values (Student’s t-test) are relative to shCtl. **P < .01, ***P < .001.
Characterization of the molecular pathways triggered by Gal1 in pancreatic carcinogenesis
To characterize the molecular pathways affected by Gal1 in PDA cells, we compared the global gene expression profile by microarray analysis of PANC-1 cells with endogenous Gal1 levels (shCtl) and after Gal1 knockdown (shGal1) (Fig. 5). We found 547 (175 upregulated and 372 downregulated) genes differentially expressed (P < .0005) in Gal1 downregulated cells (Fig. 5A and Supplementary Table 2). Gene ontology analysis of Gal1 target genes identified a significant (P < .0001) enrichment in genes involved in regulation of cell adhesion, migration and cell signaling pathways (Fig. 5B). Interestingly, we found many significantly altered pathways involved in cellular response to cytokines and inflammation/immune processes, one of the best characterized functions of Gal1 (26). Validation by RT-qPCR of several of these genes demonstrated that Gal1 levels correlates with genes involved in cell migration, adhesion and malignant transformation (Supplementary Text and Supplementary Fig. S5), and, remarkably, with genes of the Hh-Gli axis, a key pathway in the initiation and progression of pancreatic cancer (27). In particular, Disp1, which is involved in Hh ligand secretion and signaling (28), and Cyclin-D2, a downstream Gli-target gene (29), showed reduced levels in cells knocked down for Gal1 (Fig. 5C), suggesting Gal1 may regulate Hh pathway in pancreatic tumoral cells.
Figure 5. Molecular pathways triggered by Gal1 in pancreatic tumoral cells.

A. Heatmap showing gene expression of those genes differentially expressed in untransfected PANC-1 cells (wt) and cells transfected with a shCtl or with shGal1 with a P < .0005. B. Gene ontology analysis of differentially expressed genes. Barplot showing the –log10(P) of the Biological Process GO Terms obtained with differentially expressed genes with a P < .0005. C. Disp1 and Cyclin-D2 RNA levels assessed by RT-qPCR in control cells (shCtl) or PANC-1 with downregulated Gal1 levels (shGal1). P values (determined by Student’s t-test) are relative to shCtl. *P < .05.
Gal1 activates Hh signaling pathway in epithelial and fibroblastic cells
Taking into account the reported autocrine and paracrine effects reported for Hh signaling pathway, we aimed to evaluate more in detail whether Gal1 expression regulates Hh pathway in PDA epithelial cells and in fibroblastic stromal cells. First, RWP-1 control cells (RWP-1_pcDNA3) or overexpressing Gal1 (RWP-1_Gal1) were transfected with a Gli-luciferase reporter cassette in order to analyze the effects of Gal1 expression in Gli1 transcriptional activity (Fig. 6A, left). Cells overexpressing Gal1 showed a significant increase in Gli driven luciferase activity (Fig. 6A, right). These data, together with those of microarray validation, indicate that Gal1 triggers Gli expression and activity in pancreatic epithelial tumoral cells. Second, we analyzed whether Gal1 can modulate Hh-Gli pathway in tumor-associated fibroblasts. For this purpose, Gal1 expression was knocked down in F88.2 –tumor-derived fibroblasts– and HPSE –human pancreatic stellate cells– (Fig. 6B), and effects on Hh signaling were analyzed. Importantly, we found that levels of Gli and the Hh receptor Patched (PTCH) were dramatically reduced after Gal1 downregulation (Fig. 6C), indicating that Gal1 also activates Hh pathway in tumor-associated fibroblasts. Altogether these results strongly suggest a role for Gal1-mediated activation of the Hh signaling pathway in epithelium and stroma during in vivo pancreatic carcinogenesis.
Figure 6. Galectin-1 activates Hedgehog signaling pathway in pancreatic tumoral cells and fibroblasts.

A. Reporter plasmid pδ51/LucII showing 8 Gli binding sites, the δ-crystallin basal promoter and the luciferase gene (left). Luciferase activity driven by the Gli reporter cassette after transfection of pδ51/LucII in RWP-1 cells transfected with empty pcDNA3 or pcDNA3-Gal1 (Gal1) (right). B. WB analysis of Gal1 downregulation after siRNA Gal1 transfection in F88.2 and HPSE cells. Tubulin levels are shown as the loading control. Quantification is shown in the lower panel. C. Gli1 or PTCH RNA levels assessed by RT-qPCR in control cells (shCtl) or fibroblasts with downregulated Gal1 levels (shGal1). Data are mean ± SEM of three independent experiments. P values (determined by Student’s t-test) are relative to shCtl. *P < .05; **P < .01, ***P < .001.
DISCUSSION
Despite major efforts to unveil the molecular mechanisms underlying initiation and progression of pancreatic cancer, very little progress has been made in PDA treatment, still remaining an incurable disease. Given the persistent desmoplastic response that characterizes PDA and its role as a physical barrier for drug delivery (4, 30), tumoral stroma has emerged as a novel promising target for PDA therapy (25, 31), Here, we identify Gal1 as a novel player in tumor-stroma crosstalk in pancreatic cancer, supporting tumor progression (Fig. 7). Moreover, our data hold the in vivo therapeutic benefits of Gal1 downregulation, as its deficiency results in stroma remodeling, tumor size reduction and increased survival.
Figure 7.
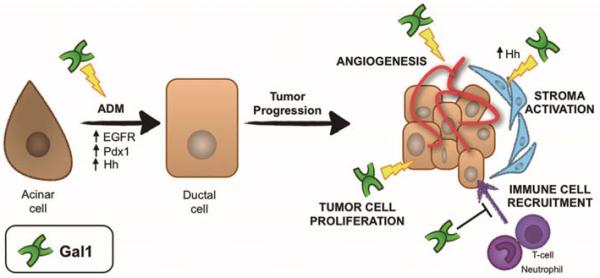
Role of Gal1 in ADM and PDA –involvement of molecular pathways. Gal1 favours ADM, a key step for PDA initiation, likely through activation of EGFR, Pdx1 and Hh pathways. In PDA, Gal1 expression favours tumor progression by promoting tumoral cell proliferation, angiogenesis and Hh-mediated stroma activation, and blocking immune surveillance.
The Ela-myc model overexpresses c-myc oncogene in acinar cells and it is the only single-transgene model that develops PDA with short latency and high penetrance. Moreover, this model displays ADM (18) and it has been one of the first models to demonstrate the acinar origin of PDA. Importantly, c-myc overexpression has been frequently found in PDA and pancreatic cell lines (3, 22) and recent data have reported that it is essential for initiation, maintenance and recurrence of PDA (20) as well as for K-Ras induced carcinogenesis (21, 32, 33). In this study, we show that loss of one or two Gal1 alleles ameliorates c-myc driven pancreatic carcinogenesis by decreasing tumor cell growth, angiogenesis and stroma activation, and increasing immune surveillance. Importantly, most of Gal1-mediated pro-tumoral functions are non-cell autonomous, as in vitro downregulation of Gal1 levels in human PANC-1 cells has no effect on cell proliferation and these cells develop tumors with similar hallmarks and progression rate than those from WT cells when injected into nude mice. These results highlight the crucial contribution of the tumor microenvironment to Gal1 effects in pancreatic carcinogenesis. The lack of effect of Gal1 knockdown in xenograft models could be due to Gal1 expressed by host stromal fibroblastic cells (Supplementary Fig. S4) or due to the absence of T-lymphocytes in nude mice, which could hide Gal1-mediated regulation of the immune system during cancer progression (6). In this regard, we have found that in Ela-myc mice, which are immune competent, pancreatic tumors with reduced Gal1 show a significant increased number of intratumoral T-lymphocytes and neutrophils in comparison to WT tumors. A role for Gal1 in tumor growth through its immunomodulatory function has been also recently proposed in other tumors (34, 35)’(36).
An important issue that comes out from our data is what type of cells produces Gal1 found in the stroma of pancreatic cancer in patients (8, 11, 13) and mice (Fig. 1, and N. Martinez-Bosch, C. Guerra and P. Navarro, unpublished data using K-Ras driven tumors). Gal1 can be secreted by a non-classical secretory pathway and can bind to the ECM through protein and keratin sulfate recognition. Activated tumor fibroblasts or stellate cells have been proposed as a source of stromal Gal1 in pancreatic cancer (37). Notwithstanding, tumoral cells may also express high Gal1 levels and secrete it to the extracellular milieu. In accordance, we have previously reported high secretion of Gal1 in conditioned medium of human tumoral pancreatic cell lines as well as Gal1 expression in ductal epithelial cells from Ela-myc tumors (12). Similarly, a paracrine mechanism involving uptake of Gal1 secreted by tumoral cells has been described in endothelial cell activation and tumor angiogenesis (38). Remarkably, recent data showing that Gal1 induces chemokine production, pancreatic stellate cell proliferation (39) and stimulation of collagen and fibronectin synthesis (40), indicates that Gal1 generates a positive feed-back loop exacerbating tumor desmoplastic reaction. In agreement with this scenario, we report here that abolishment of Gal1 expression in Ela-myc pancreatic tumors results in decreased desmoplasia.
Several reports have involved transdifferentiation of acinar cells into ductal cells in the onset of pancreatic cancer (18, 23, 41-44). Our data show that loss of expression of Gal1 in Ela-myc mice leads to ADM blockade and consequently a dramatic reduction in the number of ductal tumors. These results unveil Gal1 as a novel gene to be added to the increasing list of molecules triggering ADM (24). Furthermore, our molecular analysis from Ela-myc tumors suggests that Gal1-mediated ADM involves EGFR activation and subsequent alteration of ADM-key transcription factors, as Pdx1 (45). In contrast, the opposite correlation found between Gal1 and MMP7 levels suggest that MMP7 pathway should be upstream of Gal1 and may be upregulated in the absence of the lectin as a compensatory mechanism. Altogether, these data identify for the first time Gal1 and EGFR-Pdx1 axis in ADM and PDA initiation.
We further evaluated the molecular pathways underlying Gal1 pro-malignant functions during pancreatic carcinogenesis. We found that Gal1 downregulation in PANC-1 cells results in increased expression of fibronectin-1, integrin-α5, thrombospondin-1 and E-cadherin, which could be responsible for the inhibition of cell motility. Interestingly, Gal1 downregulation in glioblastoma cells also increases ECM and cell adhesion molecules (46), suggesting a common Gal1-related gene signature between different tumors.
One of our major findings is the activation of Hh-Gli pathway by Gal1. In PDA, deregulated Hh signaling has been found in precursor lesions and primary tumors, involving this pathway in the initiation and progression of the disease (27). Interestingly, Hh signaling in PDA can be activated in autocrine and paracrine ways supporting tumor epithelium-stroma crosstalk and tumor progression (47, 48). Our findings demonstrate that Gal1 has a direct role in activating Hh-Gli pathway in pancreatic tumoral epithelium and fibroblasts, as shRNA downregulation of Gal1 decreases the expression of upstream and downstream effectors of Hh-Gli axis (i.e. Disp1 and CyclinD2 in PANC-1 cells; and Gli and Patched in fibroblasts and pancreatic stellate cells) and overexpression of Gal1 in PDA cells leads to increased Gli activity in a luciferase reporter assay. Importantly, Hh activation has been previously related to stroma activation and ADM (49) making tempting to speculate that in vivo Gal1 effects on stroma remodeling and ADM reported here are triggered by Hh signaling.
In conclusion, our data identify a crucial role for Gal1 in promoting pancreatic carcinogenesis through the activation of tumor and microenvironment crosstalk, favoring key steps of cancer progression as proliferation, angiogenesis, desmoplasia, immune evasion and ADM (Fig. 7). These pleiotropic functions of Gal1 in PDA, together with increasing evidences supporting the tumoral stroma – where Gal1 is highly expressed– as a potential target to surmount PDA resistance to therapy, lead us to propose Gal1 as a promising molecular target for pancreatic cancer treatment. For instance, our results showing that heterozygous deletion of Gal1 also inhibits pancreatic tumor progression indicate that partial abolishment of this lectin may be therapeutically effective.
Moreover, lessons from Gal1 knockout mice indicate that this protein is not essential for animal survival, suggesting that Gal1 inhibitors should be a safe therapy lacking associated undesirable side-effects. Last but not least, given Gal1 overexpression in many different tumors, our data have broader implications in the use of this lectin as a novel molecular target for general cancer diagnosis and therapy.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to acknowledge E. Sandgren (University of Wisconsin-Madison, Madison, WI) for kindly providing Ela-myc mice, FX. Real (CNIO, Madrid, Spain) for critical reading of the manuscript and helpful suggestions, L. Nonell and E. Puigdecanet from the IMIM Microarrays Analysis Unit for their valuable technical assistance and J. Martín-Caballero and the staff of the PRBB Animal house for their help with animal care.
Financial Support: Supported by research grants ISCII-FEDER (PI080421 and PI11/01562) from MICINN, Fundació La MaratóTV3 (051110), AICR (11-0086) and Generalitat de Catalunya (2009SGR1409) to P.N.; Instituto de Salud Carlos III FEDER (RD09/0076/00036) and Xarxa de Bancs de tumors sponsored by Pla Director d'Oncologia de Catalunya (XBTC); Ligue contre le cancer, Comité de Paris to F.P.; and EC GlycoHIT program (no. 260600) to H-J.G. N.M-B. has been supported by a grant from Fundación Ramón Areces. ME.F-Z. was supported by Mayo Clinic Pancreatic SPORE P50 CA102701, and Mayo Clinic Center for Cell Signaling in Gastroenterology P30 DK84567.
Abbreviations
- ADM
acinar-to-ductal metaplasia
- ECM
extracellular matrix
- Gal1
Galectin-1
- Hh
Hedgehog
- IHC
Immunohistochemistry
- PDA
pancreatic ductal adenocarcinoma
- WB
Western blot
- WT
wild type
Footnotes
Disclosures: The authors declare that no conflict of interest exists
Supplementary Material is available for this article.
REFERENCES
- (1).Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- (2).Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–17. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- (3).Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–29. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Camby I, Le Mercier M, Lefranc F, Kiss R. Galectin-1: a small protein with major functions. Glycobiology. 2006;16:137R–57R. doi: 10.1093/glycob/cwl025. [DOI] [PubMed] [Google Scholar]
- (6).Perillo NL, Pace KE, Seilhamer JJ, Baum LG. Apoptosis of T cells mediated by galectin-1. Nature. 1995;378:736–9. doi: 10.1038/378736a0. [DOI] [PubMed] [Google Scholar]
- (7).Demydenko D, Berest I. Expression of galectin-1 in malignant tumors. Exp Oncol. 2009;31:74–9. [PubMed] [Google Scholar]
- (8).Berberat PO, Friess H, Wang L, Zhu Z, Bley T, Frigeri L, et al. Comparative analysis of galectins in primary tumors and tumor metastasis in human pancreatic cancer. J Histochem Cytochem. 2001;49:539–49. doi: 10.1177/002215540104900414. [DOI] [PubMed] [Google Scholar]
- (9).Grutzmann R, Pilarsky C, Ammerpohl O, Luttges J, Bohme A, Sipos B, et al. Gene expression profiling of microdissected pancreatic ductal carcinomas using high-density DNA microarrays. Neoplasia. 2004;6:611–22. doi: 10.1593/neo.04295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Iacobuzio-Donahue CA, Ashfaq R, Maitra A, Adsay NV, Shen-Ong GL, Berg K, et al. Highly expressed genes in pancreatic ductal adenocarcinomas: a comprehensive characterization and comparison of the transcription profiles obtained from three major technologies. Cancer Res. 2003;63:8614–22. [PubMed] [Google Scholar]
- (11).Shen J, Person MD, Zhu J, Abbruzzese JL, Li D. Protein expression profiles in pancreatic adenocarcinoma compared with normal pancreatic tissue and tissue affected by pancreatitis as detected by two-dimensional gel electrophoresis and mass spectrometry. Cancer Res. 2004;64:9018–26. doi: 10.1158/0008-5472.CAN-04-3262. [DOI] [PubMed] [Google Scholar]
- (12).Roda O, Ortiz-Zapater E, Martinez-Bosch N, Gutierrez-Gallego R, Vila-Perello M, Ampurdanes C, et al. Galectin-1 is a novel functional receptor for tissue plasminogen activator in pancreatic cancer. Gastroenterology. 2009;136:1379–5. doi: 10.1053/j.gastro.2008.12.039. [DOI] [PubMed] [Google Scholar]
- (13).Pan S, Chen R, Reimel BA, Crispin DA, Mirzaei H, Cooke K, et al. Quantitative proteomics investigation of pancreatic intraepithelial neoplasia. Electrophoresis. 2009;30:1132–44. doi: 10.1002/elps.200800752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Martinez-Bosch N, Navarro Pilar. Glycans and galectins: sweet new approaches in pancreatic cancer diagnosis and treatment. In: Srivastava S, editor. Pancreatic cancer: molecular mechanism and targets. InTech; Rijeka, Croatia: 2012. pp. 305–28. [Google Scholar]
- (15).Chung JC, Oh MJ, Choi SH, Bae CD. Proteomic analysis to identify biomarker proteins in pancreatic ductal adenocarcinoma. ANZ J Surg. 2008;78:245–51. doi: 10.1111/j.1445-2197.2008.04429.x. [DOI] [PubMed] [Google Scholar]
- (16).Sandgren EP, Quaife CJ, Paulovich AG, Palmiter RD, Brinster RL. Pancreatic tumor pathogenesis reflects the causative genetic lesion. Proc Natl Acad Sci U S A. 1991;88:93–7. doi: 10.1073/pnas.88.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Liao DJ, Wang Y, Wu J, Adsay NV, Grignon D, Khanani F, et al. Characterization of pancreatic lesions from MT-tgf alpha, Ela-myc and MT-tgf alpha/Ela-myc single and double transgenic mice. J Carcinog. 2006;5:19. doi: 10.1186/1477-3163-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Grippo PJ, Sandgren EP. Acinar-to-ductal metaplasia accompanies c-myc-induced exocrine pancreatic cancer progression in transgenic rodents. Int J Cancer. 2012;131:1243–8. doi: 10.1002/ijc.27322. [DOI] [PubMed] [Google Scholar]
- (19).Sasaki H, Hui C, Nakafuku M, Kondoh H. A binding site for Gli proteins is essential for HNF-3beta floor plate enhancer activity in transgenics and can respond to Shh in vitro. Development. 1997;124:1313–22. doi: 10.1242/dev.124.7.1313. [DOI] [PubMed] [Google Scholar]
- (20).Lin WC, Rajbhandari N, Liu C, Sakamoto K, Zhang Q, Triplett AA, et al. Dormant cancer cells contribute to residual disease in a model of reversible pancreatic cancer. Cancer Res. 2013;73:1821–30. doi: 10.1158/0008-5472.CAN-12-2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ischenko I, Zhi J, Moll UM, Nemajerova A, Petrenko O. Direct reprogramming by oncogenic Ras and Myc. Proc Natl Acad Sci U S A. 2013;110:3937–42. doi: 10.1073/pnas.1219592110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Schleger C, Verbeke C, Hildenbrand R, Zentgraf H, Bleyl U. c-MYC activation in primary and metastatic ductal adenocarcinoma of the pancreas: incidence, mechanisms, and clinical significance. Mod Pathol. 2002;15:462–9. doi: 10.1038/modpathol.3880547. [DOI] [PubMed] [Google Scholar]
- (23).Parsa I, Longnecker DS, Scarpelli DG, Pour P, Reddy JK, Lefkowitz M. Ductal metaplasia of human exocrine pancreas and its association with carcinoma. Cancer Res. 1985;45:1285–90. [PubMed] [Google Scholar]
- (24).Reichert M, Rustgi AK. Pancreatic ductal cells in development, regeneration, and neoplasia. J Clin Invest. 2011;121:4572–8. doi: 10.1172/JCI57131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18:4266–76. doi: 10.1158/1078-0432.CCR-11-3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Rabinovich GA, Toscano MA. Turning 'sweet' on immunity: galectin-glycan interactions in immune tolerance and inflammation. Nat Rev Immunol. 2009;9:338–52. doi: 10.1038/nri2536. [DOI] [PubMed] [Google Scholar]
- (27).Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851–6. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Burke R, Nellen D, Bellotto M, Hafen E, Senti KA, Dickson BJ, et al. Dispatched, a novel sterol-sensing domain protein dedicated to the release of cholesterol-modified hedgehog from signaling cells. Cell. 1999;99:803–15. doi: 10.1016/s0092-8674(00)81677-3. [DOI] [PubMed] [Google Scholar]
- (29).Yoon JW, Kita Y, Frank DJ, Majewski RR, Konicek BA, Nobrega MA, et al. Gene expression profiling leads to identification of GLI1-binding elements in target genes and a role for multiple downstream pathways in GLI1-induced cell transformation. J Biol Chem. 2002;277:5548–55. doi: 10.1074/jbc.M105708200. [DOI] [PubMed] [Google Scholar]
- (30).Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–61. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Erkan M, Hausmann S, Michalski CW, Fingerle AA, Dobritz M, Kleeff J, et al. The role of stroma in pancreatic cancer: diagnostic and therapeutic implications. Nat Rev Gastroenterol Hepatol. 2012;9:454–67. doi: 10.1038/nrgastro.2012.115. [DOI] [PubMed] [Google Scholar]
- (32).Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–70. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, et al. Modelling Myc inhibition as a cancer therapy. Nature. 2008;455:679–83. doi: 10.1038/nature07260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Banh A, Zhang J, Cao H, Bouley DM, Kwok S, Kong C, et al. Tumor galectin-1 mediates tumor growth and metastasis through regulation of T-cell apoptosis. Cancer Res. 2011;71:4423–31. doi: 10.1158/0008-5472.CAN-10-4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Dalotto-Moreno T, Croci DO, Cerliani JP, Martinez-Allo VC, Dergan-Dylon S, Mendez-Huergo SP, et al. Targeting galectin-1 overcomes breast cancer-associated immunosuppression and prevents metastatic disease. Cancer Res. 2013;73:1107–17. doi: 10.1158/0008-5472.CAN-12-2418. [DOI] [PubMed] [Google Scholar]
- (36).Compagno D, Laderach DJ, Gentilini L, Jaworski FM, Rabinovich GA. Delineating the "galectin signature" of the tumor microenvironment. Oncoimmunology. 2013;2:e23565. doi: 10.4161/onci.23565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Xue X, Lu Z, Tang D, Yao J, An Y, Wu J, et al. Galectin-1 secreted by activated stellate cells in pancreatic ductal adenocarcinoma stroma promotes proliferation and invasion of pancreatic cancer cells: an in vitro study on the microenvironment of pancreatic ductal adenocarcinoma. Pancreas. 2011;40:832–9. doi: 10.1097/MPA.0b013e318217945e. [DOI] [PubMed] [Google Scholar]
- (38).Thijssen VL, Barkan B, Shoji H, Aries IM, Mathieu V, Deltour L, et al. Tumor cells secrete galectin-1 to enhance endothelial cell activity. Cancer Res. 2010;70:6216–24. doi: 10.1158/0008-5472.CAN-09-4150. [DOI] [PubMed] [Google Scholar]
- (39).Masamune A, Satoh M, Hirabayashi J, Kasai K, Satoh K, Shimosegawa T. Galectin-1 induces chemokine production and proliferation in pancreatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2006;290:G729–G736. doi: 10.1152/ajpgi.00511.2005. [DOI] [PubMed] [Google Scholar]
- (40).Wu MH, Hong HC, Hong TM, Chiang WF, Jin YT, Chen YL. Targeting Galectin-1 in Carcinoma-Associated Fibroblasts Inhibits Oral Squamous Cell Carcinoma Metastasis by Downregulating MCP-1/CCL2 Expression. Clin Cancer Res. 2011;17:1306–16. doi: 10.1158/1078-0432.CCR-10-1824. [DOI] [PubMed] [Google Scholar]
- (41).Perez-Mancera PA, Guerra C, Barbacid M, Tuveson DA. What we have learned about pancreatic cancer from mouse models. Gastroenterology. 2012;142:1079–92. doi: 10.1053/j.gastro.2012.03.002. [DOI] [PubMed] [Google Scholar]
- (42).De La OJ, Emerson LL, Goodman JL, Froebe SC, Illum BE, Curtis AB, et al. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc Natl Acad Sci U S A. 2008;105:18907–12. doi: 10.1073/pnas.0810111105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Wagner M, Greten FR, Weber CK, Koschnick S, Mattfeldt T, Deppert W, et al. A murine tumor progression model for pancreatic cancer recapitulating the genetic alterations of the human disease. Genes Dev. 2001;15:286–93. doi: 10.1101/gad.184701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Crawford HC, Scoggins CR, Washington MK, Matrisian LM, Leach SD. Matrix metalloproteinase-7 is expressed by pancreatic cancer precursors and regulates acinar-to-ductal metaplasia in exocrine pancreas. J Clin Invest. 2002;109:1437–44. doi: 10.1172/JCI15051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Miyatsuka T, Kaneto H, Shiraiwa T, Matsuoka TA, Yamamoto K, Kato K, et al. Persistent expression of PDX-1 in the pancreas causes acinar-to-ductal metaplasia through Stat3 activation. Genes Dev. 2006;20:1435–40. doi: 10.1101/gad.1412806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Camby I, Decaestecker C, Lefranc F, Kaltner H, Gabius HJ, Kiss R. Galectin-1 knocking down in human U87 glioblastoma cells alters their gene expression pattern. Biochem Biophys Res Commun. 2005;335:27–35. doi: 10.1016/j.bbrc.2005.07.037. [DOI] [PubMed] [Google Scholar]
- (47).Bailey JM, Swanson BJ, Hamada T, Eggers JP, Singh PK, Caffery T, et al. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin Cancer Res. 2008;14:5995–6004. doi: 10.1158/1078-0432.CCR-08-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Bailey JM, Mohr AM, Hollingsworth MA. Sonic hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene. 2009;28:3513–25. doi: 10.1038/onc.2009.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Pasca dM, Sekine S, Ermilov A, Ferris J, Dlugosz AA, Hebrok M. Hedgehog/Ras interactions regulate early stages of pancreatic cancer. Genes Dev. 2006;20:3161–73. doi: 10.1101/gad.1470806. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.