

Abstract
Several advances in 2013 have improved our understanding of how epigenetic mechanisms affect autoimmune disorders. Many new insights were made into the regulation of gene expression by DNA methylation in systemic lupus erythematosus. For rheumatoid arthritis, complex interrelationships between DNA methylation and microRNAs in regulating gene expression were described.
Epigenetics is defined as heritable changes in gene expression that are not due to alteration of the DNA sequence. Epigenetic mechanisms regulate multiple aspects of chromatin structure and function, including the regulation of transcriptionally repressive and permissive configurations for gene expression. These mechanisms include DNA methylation, histone modifications including methylation and acetylation, and regulation by microRNAs (miRNA). In this 2013 Year in Review, we describe advances (Figure 1) in our understanding of DNA methylation and miRNAs in the pathogenesis of systemic lupus erythematosus (SLE)1, 2 and rheumatoid arthritis (RA).3
Figure 1. Recent advances in our understanding of epigenetics and gene expression.
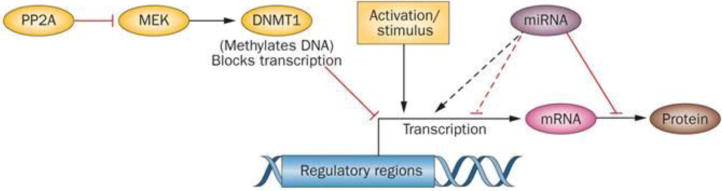
Coit et al.1 showed certain genes are ‘poised’ for transcription, based on methylation of their regulatory regions, but transcription does not occur until the cells are activated. Sunahori et al.2 demonstrated that PP2A blocks signalling pathways that maintain DNA methylation patterns in dividing immune cells, causing them to be overexpressed. De la Rica et al.3 found that DNA methylation and microRNAs can work together, or in opposition, in regulating gene expression. Abbreviations: DNMT1, DNA (cytosine-5)-methyltransferase 1; MEK, dual specificity mitogen-activated protein kinase kinase; PP2A, protein phosphatase 2A.
Genome-wide methylation studies have provided new insights into the role of DNA methylation in the pathogenesis of SLE. Coit et al.1 demonstrated that regulatory regions for interferon-responsive genes in naive CD4+ T cells from patients with SLE are hypomethylated and ‘poised’ for expression; transcription does not occur until T-cell activation. Using two independent SLE cohorts, >485,000 genomic methylation sites were analysed by array. The researchers identified 47 genes as differentially methylated, either hypomethylated or hypermethylated, in a comparison of naive CD4+ T cells from healthy individuals and patients with SLE. Methylation did not increase or decrease with disease activity, as measured by the SLE disease activity index. Bisulphite sequencing confirmed that 35 genes were hypomethylated, and most of these genes are involved in type I interferon signalling. Many of the hypomethylated genes were known to be overexpressed in total CD4+ T cells from patients with SLE, but methylation state and expression had not previously been studied in the same naive CD4+ T cell. Coit et al.1 found that none of the hypomethylated genes were overexpressed in naive CD4+ T cells. This discordance between gene accessibility and expression could be explained by the relative paucity and inactive state of transcription factors in naive CD4+ T cells. The changes in DNA methylation before activation and differentiation indicate that naive cells are ‘poised’ to express those genes even in the absence of other immune signals. This model helps explain the increased expression of IFN-α and type I interferon-inducible genes in patients with SLE. In related work, the same type of genome-wide methylation array was used to analyse B cells, monocytes and CD4+ T cells.4 Prior to this study, B cells and monocytes from patients with SLE had not been examined in this manner. Absher et al.,4 however, did not study gene expression or confirm changes in DNA methylation by bisulphite sequencing. Similar to the findings by Coit et al.,1 most of the hypomethylated genes were related to the type I interferon signalling pathway. The methylation changes in total CD4+ T cells also did not differ significantly from cell subsets, including naive CD4+ T cells, and they did not vary with disease activity.
The molecular mechanisms by which DNA methylation is regulated in patients with SLE is an area of intense investigation. DNA (cytosine-5)-methyltransferase 1 (DNMT1) is an ERK-regulated and JNK-regulated enzyme required for maintaining methylation patterns in dividing cells. In CD4+ T cells from patients with SLE, the expression of DNMT1 is decreased and the ERK and JNK pathways are disrupted.5 Disrupting ERK or JNK signalling in vitro decreases Dnmt1 expression and consequently increases expression of CD70 and other proinflammatory methylation-sensitive genes.6 Furthermore, disrupting either ERK or JNK signalling in transgenic mice results in an SLE-like disorder characterized by anti-double stranded DNA antibodies and glomerulonephritis.6, 7 Despite these findings, the mechanisms by which ERK and JNK signalling are disrupted are only partially understood. Work by Sunahori et al.2 demonstrated that protein phosphatase 2A (PP2A) contributes to disrupted signalling of these pathways in SLE.
Evidence indicates that PP2A is overexpressed in T cells from patients with SLE, compared with T cells from healthy individuals.8 Protein phosphatases can regulate ERK and JNK signalling, but the specific role of PP2A, if any, in SLE pathogenesis was not previously known. Sunahori et al.2 developed a model linking the overexpression of PP2A in T cells from patients with SLE with changes in methylation-sensitive proinflammatory gene expression. Blocking PP2A expression in vitro increases ERK and MEK activation and, consequently, DNMT1 expression and activity. CD70 and CD11a expression, which are regulated by DNA methylation, were also decreased when PP2A was silenced. These findings suggest that PP2A contributes to SLE pathogenesis by blocking ERK and JNK signalling and DNMT1 activity to enable aberrant overexpression of methylation-sensitive proinflammatory genes. This hypothesis would be amenable to testing in an animal model, similar to previous studies in which the ERK and JNK pathways were individually disrupted in transgenic mouse models.6, 7
DNA methylation and histone modifications regulate gene expression at the level of mRNA transcription, and miRNAs regulate gene expression post-transcriptionally.9 The role of miRNAs in autoimmune diseases is increasingly recognized, and a major advance was reported in 2013. De la Rica et al.3 linked DNA methylation, miRNA expression, and mRNA expression in synovial fibroblasts by using a genome-wide methylation array to identify >1,200 genes that are differentially methylated in six patients with osteoarthritis (OA) compared with six patients with RA. An analogous study by Nakano et al.10 reported similar changes in DNA methylation patterns. De la Rica et al.3 integrated the methylation data with two different data sets. First, methylation data were compared with mRNA expression data from synovial fibroblasts from patients with RA and OA. With some exceptions, most of the hypomethylated genes had overexpressed mRNAs and many of the hypermethylated genes had underexpressed mRNAs, as would be predicted. Next, miRNA expression in synovial fibroblasts from patients with OA or RA was then compared with the methylation data, in particular the location of hypermethylated and hypomethylated CpG sites in the genome. 11 miRNAs that are downregulated in patients with RA, compared to patients with OA, were located near hypermethylated CpG sites, and four miRNAs that are upregulated were located near hypomethylated CpG sites. Finally, all three data sets (genomic methylation, mRNAs and miRNAs) were compared simultaneously.
De la Rica et al.3 split the results of their complex analysis into groups that suggest the roles of DNA methylation and miRNAs in regulating gene expression. One group consisted of downregulated mRNAs with hypermethylated DNA and increased expression of targeting miRNAs. In this group, DNA methylation and miRNAs work together to suppress gene expression. In a second group of downregulated mRNAs, corresponding DNA elements were hypomethylated, but the number of miRNAs that specifically target that mRNA were increased. In this second group, miRNAs seemed to have a dominant effect on gene expression. In a third group of downregulated mRNAs, corresponding DNA elements were hypermethylated and targeting miRNA levels were decreased. In this group, the dominant effector of gene expression was DNA methylation. Analogous groups of upregulated mRNAs were also found. This is the first report to study the integration of effects of DNA methylation and miRNAs on the pathogenesis of an autoimmune disorder. Despite the small sample sizes involved, many of the results from the methylation screen and mRNA expression profiles overlap with previously reported work. This research highlights the importance of multiple layers of control in regulating gene expression, and it identifies many new potential therapeutic targets. It also supports the role of other, as yet unidentified, molecules in the pathogenesis of RA, possibly explaining the discordant effects that DNA methylation and miRNAs can have on gene expression.
These three reports indicate an important role for miRNA and DNA methylation dysregulation in the pathogenesis of SLE and RA. The maintenance of DNA methylation patterns is sensitive to environmental influences7 and miRNAs might serve as biomarkers and therapeutic targets.3 These data suggest new approaches for the diagnosis and treatment of autoimmune diseases.
Key Advances.
Abnormal DNA methylation exists in CD4+ T cells from patients with SLE before activation and differentiation, although the genes are not expressed until they are activated1
Overexpression of protein phosphatase 2A in T cells increases expression of proinflammatory methylation-sensitive genes important in SLE pathogenesis2
DNA methylation and microRNAs can operate in conjunction with, or in opposition to, each other in regulating gene expression in synovial fibroblasts3
Acknowledgments
This work was supported by PHS grants AR042525 and ES017885, and a Merit Grant from the Department of Veterans Affairs.
Footnotes
Competing interests statement
Dr. Richardson is on the Scientific Advisory Board for IGNYTA. Dr. Patel declares no competing interests.
References
- 1.Coit P, et al. Genome-wide DNA methylation study suggests epigenetic accessibility and transcriptional poising of interferon-regulated genes in naive CD4+ T cells from lupus patients. J Autoimmun. 2013;43:78–84. doi: 10.1016/j.jaut.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sunahori K, et al. The catalytic subunit of protein phosphatase 2A (PP2Ac) promotes DNA hypomethylation by suppressing the phosphorylated mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) kinase (MEK)/phosphorylated ERK/DNMT1 protein pathway in T-cells from controls and systemic lupus erythematosus patients. J Biol Chem. 2013;288:21936–21944. doi: 10.1074/jbc.M113.467266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de la Rica L, et al. Identification of novel markers in rheumatoid arthritis through integrated analysis of DNA methylation and microRNA expression. J Autoimmun. 2013;41:6–16. doi: 10.1016/j.jaut.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 4.Absher DM, et al. Genome-wide DNA methylation analysis of systemic lupus erythematosus reveals persistent hypomethylation of interferon genes and compositional changes to CD4+ T-cell populations. PLoS Genet. doi: 10.1371/journal.pgen.1003678. http://dx.doi.org/10.1371/journal.pgen.1003678. [DOI] [PMC free article] [PubMed]
- 5.Deng C, et al. Decreased Ras-mitogen-activated protein kinase signaling may cause DNA hypomethylation in T lymphocytes from lupus patients. Arthritis Rheum. 2001;44:397–407. doi: 10.1002/1529-0131(200102)44:2<397::AID-ANR59>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 6.Sawalha AH, et al. Defective T-cell ERK signaling induces interferon-regulated gene expression and overexpression of methylation-sensitive genes similar to lupus patients. Genes Immun. 2008;9:368–378. doi: 10.1038/gene.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gorelik GJ, Yarlagadda S, Richardson BC. Protein kinase Cδ oxidation contributes to ERK inactivation in lupus T cells. Arthritis Rheum. 2012;64:2964–2974. doi: 10.1002/art.34503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Katsiari CG, Kyttaris VC, Juang YT, Tsokos GC. Protein phosphatase 2A is a negative regulator of IL-2 production in patients with systemic lupus erythematosus. J Clin Invest. 2005;115:3193–3204. doi: 10.1172/JCI24895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 10.Nakano K, Whitaker JW, Boyle DL, Wang W, Firestein GS. DNA methylome signature in rheumatoid arthritis. Ann Rheum Dis. 2013;72:110–117. doi: 10.1136/annrheumdis-2012-201526. [DOI] [PMC free article] [PubMed] [Google Scholar]