

Abstract
Background
Lipopeptides (LP) are structurally diverse compounds with potent surfactant and broad-spectrum antibiotic activities. In Pseudomonas and other bacterial genera, LP biosynthesis is governed by large multimodular nonribosomal peptide synthetases (NRPS). To date, relatively little is known about the regulatory genetic network of LP biosynthesis.
Results
This study provides evidence that the chaperone ClpA, together with the serine protease ClpP, regulates the biosynthesis of the LP massetolide in Pseudomonas fluorescens SS101. Whole-genome transcriptome analyses of clpA and clpP mutants showed their involvement in the transcription of the NRPS genes massABC and the transcriptional regulator massAR. In addition, transcription of genes associated with cell wall and membrane biogenesis, energy production and conversion, amino acid transport and metabolism, and pilus assembly were altered by mutations in clpA and clpP. Proteome analysis allowed the identification of additional cellular changes associated to clpA and clpP mutations. The expression of proteins of the citrate cycle and the heat shock proteins DnaK and DnaJ were particularly affected. Combined with previous findings, these results suggest that the ClpAP complex regulates massetolide biosynthesis via the pathway-specific, LuxR-type regulator MassAR, the heat shock proteins DnaK and DnaJ, and proteins of the TCA cycle.
Conclusions
Combining transcriptome and proteome analyses provided new insights into the regulation of LP biosynthesis in P. fluorescens and led to the identification of specific missing links in the regulatory pathways.
Electronic supplementary material
The online version of this article (doi:10.1186/s12866-015-0367-y) contains supplementary material, which is available to authorized users.
Background
Lipopeptides (LPs) are biosurfactants produced by a variety of bacterial genera, including Pseudomonas and Bacillus [1,2]. LPs are composed of an (cyclic) oligopeptide moiety linked to a fatty acid tail [1]. In beneficial Pseudomonas strains, LPs play a role in colonization of seeds [3] and roots [4], in defense against competing microorganisms and predatory protozoa [5], and in swarming motility and biofilm formation [6]. LP biosynthesis is governed by large multi-modular nonribosomal peptide synthetases (NRPS) via a thiotemplate process [1,7]. Compared to our understanding of LP biosynthesis, relatively little is known about the genetic networks involved in the perception of external signals and the signal transduction pathways that drive transcription of the LP biosynthesis genes. Here we focus on the regulation of LP biosynthesis in the plant growth-promoting rhizobacterium Pseudomonas fluorescens SS101. Strain SS101 produces the LP massetolide A, a 9-amino-acid cyclic peptide linked to 3-hydroxydecanoic acid [8,9]. Massetolide A is produced in the early exponential growth phase and is essential for swarming motility and biofilm formation of strain SS101 [8]. Its biosynthesis is governed by three NRPS genes, designated massA, massB, and massC [8].
To identify the genetic networks underlying regulation of massetolide biosynthesis, P. fluorescens strain SS101 was subjected to random mutagenesis. Screening of a library of approximately 7,500 random plasposon mutants resulted in the identification of four new regulatory genes, namely phgdh, dnaK, prtR and clpA [10]. In this recent study, we focused our functional analyses on phgdh, dnaK and prtR, but not on clpA. Independently from this work, clpP had been previously identified as a regulator of massetolide biosynthesis in P. fluorescens SS101 [11]. Hence, the aims of the present study were to i) study the role of ClpA in regulation of massetolide biosynthesis, and ii) analyse the ClpA regulon at the transcriptional and proteome level in order to narrow down the role of ClpP in regulating massetolide biosynthesis.
The ATP-dependent serine protease ClpP is highly conserved in eubacteria [12] and has diverse functions, including intracellular proteolysis. ClpP associates with different ATPases that either recognize protein substrates directly or, alternatively, interact with substrates via so-called adaptor proteins [13]. Substrates are then unfolded and translocated to the proteolytic chamber of the ClpP protease [14]. ClpP consists of two heptameric rings that form a barrel-shaped proteolytic core with the active sites hidden in an interior chamber [15]. The ATPases of ClpP that have been studied in detail in various bacterial genera include ClpX, ClpB, HslU and ClpA [16,17]. In strain SS101, site-directed mutagenesis of clpX did not affect massetolide biosynthesis [11], suggesting that ClpX does not act as the chaperone of ClpP in the regulation of massetolide biosynthesis. Therefore, the focus of our present study is on the role of the ClpAP complex in the regulation of massetolide biosynthesis. ClpA is formed as a hexameric chaperone ring complex and selects the target proteins for ClpP to degrade based on the N-end rule [18]. Either misfolded or specifically tagged proteins are targeted by ClpA [19]. To unravel the cellular substrates of the ClpAP complex in E.coli, a proteomics approach [20] was adopted which revealed that several proteins involved in metabolism and energy production, cell motility and transport are potential cellular targets. In our study, we combined transcriptomic and proteomic analyses for both clpA and clpP mutants to identify putative substrates of the ClpAP complex with the ultimate goal to further elucidate the genetic regulation of massetolide biosynthesis in P. fluorescens.
Results and discussion
Role of clpA in lipopeptide biosynthesis in P. fluorescens SS101
In P. fluorescens SS101, the clpA gene is 2271 bp with 89 to 98% identity to homologs in other Pseudomonas genomes (Figure 1). Based on the drop collapse assay, a mutation in the clpA gene abolishes massetolide production (Figure 2A). RP-HPLC analysis confirmed that the clpA mutant indeed did not produce detectable levels of massetolide A or its derivatives (Figure 2B). Complementation of the clpA mutant with the stable vector pME6031-clpA restored massetolide production to wild-type level, whereas the empty-vector control did not (Figure 2B). Massetolide biosynthesis is known to be essential for swarming motility of strain SS101 [8]. The clpA mutant was not able to swarm on soft agar (0.6% w/v; Figure 2C) and this phenotype was restored by complementation with pME6031-clpA (Figure 2C). In contrast to a mutation in clpP, no effects on growth were observed for the clpA mutant (Figure 2D). Collectively, these results indicated that clpA is required for massetolide biosynthesis in P. fluorescens SS101.
Figure 1.
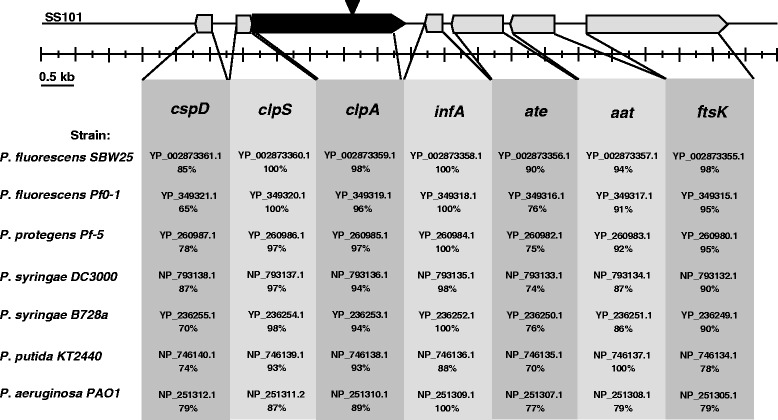
Genomic organization of clpA and flanking genes in P. fluorescens SS101. The clpA gene (PflSS101_ 3193) and flanking genes in P. fluorescens SS101 and the percentages of amino acid identity with their corresponding homologues in other Pseudomonas species and strains are indicated. The triangle indicates the position of the plasposon insertion in the clpA gene. Abbreviations: cspD: cold shock domain protein; clpS: ATP-dependent Clp protease adaptor protein; clpA: ATP-dependent Clp protease ATP-binding subunit; infA: translation initiation factor IF-1; ate: putative arginyl-tRNA-protein transferase; aat: leucyl/phenylalanyl-tRNA-protein transferase; ftsK: DNA translocase.
Figure 2.
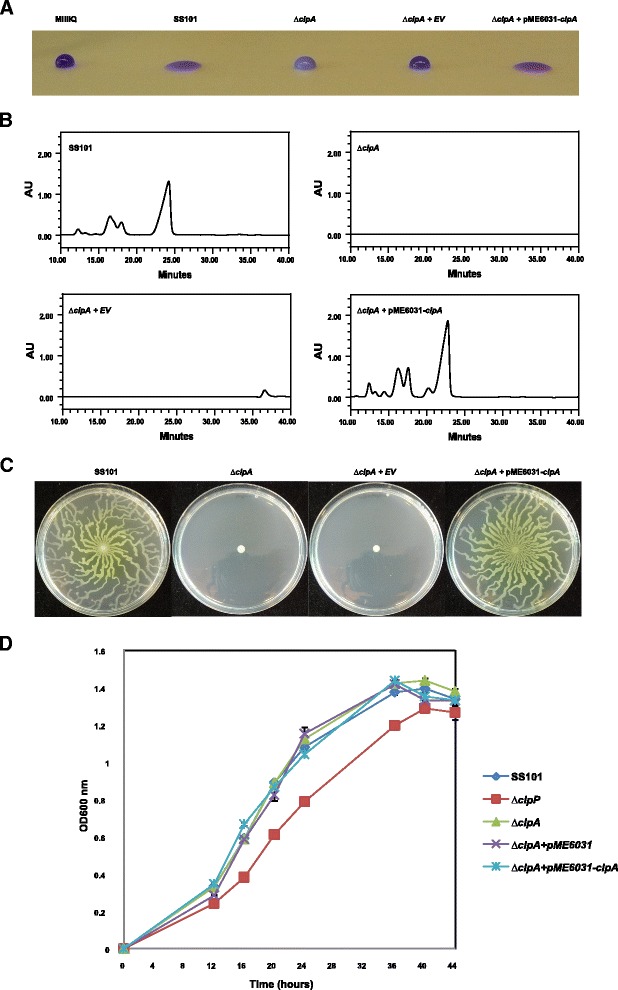
Phenotypic and chemical analyses of P. fluorescens strain SS101, and its clpA mutant. (A) Drop collapse assay with cell suspensions of wild-type strain SS101, clpA plasposon mutant, clpA mutant + pME6031 (empty vector control) and clpA mutant + pME6031-clpA. Bacterial cultures grown for 2 days at 25°C on KB agar plates were suspended in sterile water to a final density of 1x1010 cells/ml. 10-μl droplets were spotted on parafilm and crystal violet was added to the droplets to facilitate visual assessment. A flat droplet is a highly reliable proxy for the production of the surface-active lipopeptide massetolide A. (B) RP-HPLC chromatograms of cell-free culture extracts of the wild-type strain SS101, clpA plasposon mutant, clpA + pME6031 (empty vector control) and clpA + pME6031-clpA as described in panel A. The wild-type strain SS101 produces massetolide A (retention time of approximately 23–25 min) and various other derivatives of massetolide A (minor peaks with retention times ranging from 12 to 18 min) which differ from massetolide A in the amino acid composition of the peptide moiety. (C) Swarming motility of the wild-type strain SS101, clpA plasposon mutant, clpA mutant + pME6031 (empty vector control) and clpA mutant + pME6031-clpA on soft (0.6% wt/vol) agar plates. Five microliter (1×1010 cells/ml) of washed cells from overnight cultures was spot-inoculated in the center of a soft agar plate and incubated for 48–72 h at 25°C. (D) Growth of the wild-type SS101 strain, clpA plasposon mutant, clpA mutant + pME6031 (empty vector control), clpA mutant + pME6031-clpA and clpP site-directed mutagenesis mutant in liquid medium at 25°C. The optical density of the cell cultures was measured spectrophotometrically (600 nm) at different time points. Mean values of four biological replicates are given; the error bars represent the standard error of the mean.
Transcriptome analysis
To further investigate the genetic basis for ClpAP-mediated regulation of massetolide biosynthesis, whole-genome transcriptome analyses were performed for the clpA (Additional file 1: Figure S1A) and clpP (Additional file 1: Figure S1B) mutants. Given the differences in growth kinetics between the mutants and wild-type SS101 (Figure 2D), cells were harvested in the exponential growth phase (OD600nm = 0.6). In the clpA mutant, transcription of 14 and 37 genes increased and decreased, respectively, by at least 2-fold (PFDR < 0.05) (Additional file 2: Table S1). Apart from the massetolide biosynthesis genes, several of the differentially regulated genes were associated with energy production and conversion, amino acid transport and metabolism, cell wall and membrane biogenesis and pilus assembly. Several of the other differentially regulated genes could not be assigned to clusters of orthologous groups (COGs). Two pili gene clusters were significantly down-regulated in the clpA mutant. The first was the csu gene cluster (PflSS101_3282-3285) which is known to affect biofilm formation in Acinetobacter baumannii [21]. The second was the type IVb pili gene cluster PflSS101_0648-0655 and the regulator pprB (Additional file 2: Table S1). In Pseudomonas aeruginosa, type IVb pili are required for adhesion to abiotic surfaces and to eukaryotic cells [22]. Further experiments will be needed to explore the functions of both pili gene clusters in P. fluorescens SS101.
With 195 and 154 genes significantly up and down regulated, respectively, the clpP mutation had a much bigger impact, as expected, on the overall gene expression in strain SS101 than a mutation in clpA (Additional file 2: Table S2, Additional file 1: Figure S1B). Combining the transcriptome data of the clpA and clpP mutants revealed that seven and thirteen genes were up and down-regulated, respectively, in both mutants (Figure 3). These include the massetolide biosynthesis genes massA, massB, massC and their flanking genes consisting of the LuxR-type transcriptional regulator massAR and the efflux-associated genes PflSS101_3398, PflSS101_2189 and PflSS101_2190. Among the genes differentially regulated in both clpA and clpP mutants were also the thiF_moeB gene cluster (PflSS101_3967-3970) as well as genes encoding a FAD-binding domain protein (PflSS101_0033) and an auto-inducer-binding LuxR-type transcriptional regulator (PflSS101_4691) (Figure 3). Expression of the previously identified regulatory genes of massetolide biosynthesis, phgdh, dnaK, and prtR [10], was not affected in the clpA and clpP mutants. This suggests that, at the transcriptional level, clpAP-mediated regulation of massetolide biosynthesis operates downstream or operates independently from these other regulatory genes.
Figure 3.
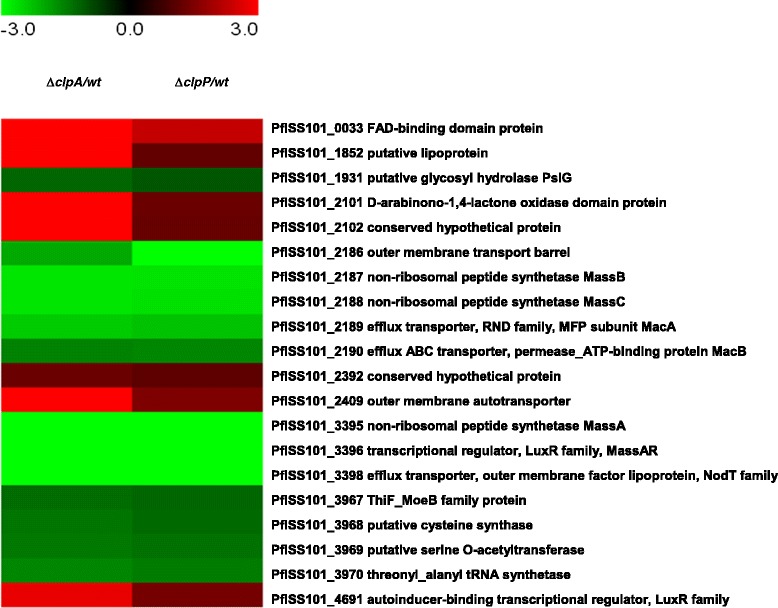
Heatmaps showing log 2 -fold changes in the expression of genes that are differently expressed in the clpA or clpP mutants of Pseudomonas fluorescens SS101. See supplementary Additional file 2: Tables S1 and S2 for the list of all genes differentially regulated in the clpA or clpP mutant versus wild-type SS101.
Proteome analysis
Total cell proteomic analyses were performed to further decipher the potential cellular substrates and target proteins of ClpAP (Additional file 1: Figure S2). The culture conditions and ‘harvest’ time of the bacterial cells (OD600 = 0.6) were identical to those used in the transcriptome analyses described above. It should be noted that the ClpAP system is a degradative protease thereby complicating the interpretation of proteomics data. While transcriptomics can validly argue that mRNAs (and hence proteins) are up- or down-regulated, the higher abundance of a particular protein in the clpA and or clpP mutants can also be due to an inherent up- or down-regulation by other modulated pathways. Hence, the proteomics results described below should be interpreted with caution.
Proteins differentially expressed in the clpA mutant or clpP mutant
iTRAQ-based proteome analyses allowed the identification of a total of 596 proteins in the clpA mutant (Additional file 2: Table S3): 68 proteins were significantly up-regulated (Fold change > 1.2) while 132 were down-regulated (Additional file 2: Table S3). Gap2 (PflSS101_4355), encoding a glyceraldehyde-3-phosphate dehydrogenase, was up-regulated in the clpA mutant, which was consistent with the earlier report [20] that reported a similar GapA protein as one of the substrates of ClpAP in E. coli. All three protein groups from the ‘intracellular trafficking and secretion’ COG category were up-regulated in the clpA mutant, including SecA, SecB, and the Tol-Pal system protein TolB (Additional file 1: Figure S2A, Additional file 2: Table S3).
In line with the findings in E. coli [20], we observed that the cell division protein FtsZ and the isocitrate lyase AceA were up-regulated in the clpP mutant (Additional file 1: Figure S2B; Additional file 2: Table S4), suggesting that these proteins might be substrates of ClpP in strain SS101. Moreover, we detected five transcriptional regulators and five chaperons that were uniquely up-regulated in the clpP mutant (Table 1). One of the up-regulated transcriptional regulators was MvaT (PflSS101_4330), which is known to regulate the biosynthesis of specific secondary metabolites in the rhizobacterium Pseudomonas protegens CHA0 [23]. Furthermore, the heat shock proteins DnaK and DnaJ, the chaperonin GroS, GroL and the chaperon HtpG were significantly up-regulated in the clpP mutant. Also CheA, a histidine kinase that mediates chemotaxis signaling events in many prokaryotes [24], was 1.49-fold up-regulated, suggesting it may be a substrate of ClpP in strain SS101.
Table 1.
Regulator and chaperon proteins differentially expressed in the clpP mutant of Pseudomonas fluorescens SS101
| Locus | Gene | Gene description | Fold changes in ∆c lpP /SS101 | |
|---|---|---|---|---|
| PflSS101_1716 | cysB | HTH-type transcriptional regulator CysB | 1.25 | up |
| PflSS101_3936 | transcriptional regulator, GntR family | 1.35 | up | |
| PflSS101_4330 | mvaT | transcriptional regulator MvaT | 1.26 | up |
| PflSS101_4600 | cbrB | two-component response regulator CbrB | 1.50 | up |
| PflSS101_5275 | rnk | regulator of nucleoside diphosphate kinase | 1.65 | up |
| PflSS101_1812 | htpG | chaperone protein HtpG | 1.2 | up |
| PflSS101_4373 | groL | chaperonin GroL | 1.22 | up |
| PflSS101_4374 | groS | chaperonin GroS | 1.32 | up |
| PflSS101_4632 | dnaJ | chaperone protein DnaJ | 1.21 | up |
| PflSS101_4633 | dnaK | chaperone protein DnaK | 1.32 | up |
Proteins differentially expressed in both clpA and clpP mutants
In both clpA and clpP mutants, 32 and 39 proteins were up- and down-regulated, respectively (Table 2, Additional file 2: Table S5). The most up-regulated was CspD (PflSS101_3195), a gene encoding one of the cold shock protein CspA family members in E. coli. CspD is known to be induced by nutritional deprivation [25]. Moreover, the response regulator CbrB and the transcriptional regulator GntR were up-regulated in both mutants. The CbrA-CbrB two-component system is known to control the utilization of different carbon and nitrogen sources in P. aeruginosa [26] and affects chemotaxis, stress tolerance and biofilm development in Pseudomonas putida [27]. GntR is a transcriptional regulator that controls antibiotic production in both Streptomyces and Serratia [28,29]. None of these proteins and their corresponding genes were found in genome-wide screening for massetolide-deficient mutants, except DnaK [10]. In our proteome analyses, the DnaK protein was found at higher concentrations in the clpP mutant and its chaperon DnaJ protein was up-regulated in both clpA and clpP mutants. Given that DnaK and DnaJ also regulate putisolvin biosynthesis in P. putida [30], our results suggest that ClpAP regulates LP biosynthesis in multiple Pseudomonas species at least in part, via DnaK and DnaJ (Figure 4).
Table 2.
Up-regulated proteins in both clpA and clpP mutants of Pseudomonas fluorescens SS101
| Locustag | Gene | Gene descriptions | ∆ clpA /SS101 | ∆ clpP /SS101 |
|---|---|---|---|---|
| PflSS101_0002 | dnaN | DNA polymerase III, beta subunit | 1.3 | 1.3 |
| PflSS101_0021 | qor | NADPH_quinone reductase | 1.25 | 1.6 |
| PflSS101_0364 | secB | protein-export chaperone SecB | 1.34 | 1.42 |
| PflSS101_0509 | thiC | thiamine biosynthesis protein ThiC | 1.33 | 1.28 |
| PflSS101_0546 | rnr | ribonuclease R | 1.27 | 1.27 |
| PflSS101_0920 | hisC_1 | histidinol-phosphate transaminase | 1.3 | 1.2 |
| PflSS101_0926 | mqo_1 | malate_quinone-oxidoreductase | 1.32 | 1.21 |
| PflSS101_1161 | argG | argininosuccinate synthase | 1.3 | 1.2 |
| PflSS101_1203 | TIGR00730 family protein | 1.22 | 1.22 | |
| PflSS101_1209 | fpr_2 | ferredoxin--NADP+ reductase | 1.28 | 1.24 |
| PflSS101_1348 | fabD | acyl-carrier-protein S-malonyltransferase | 1.26 | 1.32 |
| PflSS101_1554 | LamB_YcsF family protein | 1.25 | 1.27 | |
| PflSS101_1626 | short-chain alcohol dehydrogenase family protein | 1.53 | 1.23 | |
| PflSS101_1652 | cmk | cytidylate kinase | 1.35 | 1.36 |
| PflSS101_1729 | 3-deoxy-7-phosphoheptulonate synthase | 1.28 | 2 | |
| PflSS101_2196 | AP endonuclease, family 2 | 1.65 | 2.12 | |
| PflSS101_3195 | cold shock domain protein CspD | 2.14 | 3.15 | |
| PflSS101_3348 | bkdA2 | 2-oxoisovalerate dehydrogenase E1 component, beta subunit | 1.26 | 1.23 |
| PflSS101_3776 | flagellin domain protein | 1.21 | 2.14 | |
| PflSS101_3786 | phhA | phenylalanine-4-hydroxylase | 1.24 | 1.81 |
| PflSS101_3936 | transcriptional regulator, GntR family | 1.25 | 1.35 | |
| PflSS101_4181 | conserved hypothetical protein | 1.2 | 1.2 | |
| PflSS101_4298 | tolB | Tol-Pal system beta propeller repeat protein TolB | 1.33 | 1.29 |
| PflSS101_4316 | PF04461 family protein | 1.21 | 1.55 | |
| PflSS101_4394 | thrC | threonine synthase | 1.29 | 1.43 |
| PflSS101_4600 | cbrB | two-component response regulator CbrB | 1.25 | 1.5 |
| PflSS101_4631 | dapB | dihydrodipicolinate reductase | 1.5 | 1.55 |
| PflSS101_4632 | dnaJ | chaperone protein DnaJ | 1.26 | 1.22 |
| PflSS101_4676 | conserved hypothetical protein | 1.31 | 1.21 | |
| PflSS101_4945 | rpsU | ribosomal protein S21 | 1.25 | 1.25 |
| PflSS101_5275 | rnk | regulator of nucleoside diphosphate kinase | 1.52 | 1.65 |
| PflSS101_5280 | lysA | diaminopimelate decarboxylase | 1.23 | 1.27 |
Figure 4.
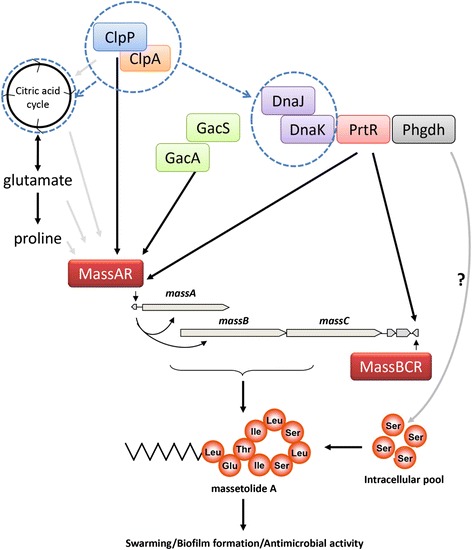
Proposed model for the genetic regulation of massetolide biosynthesis in P. fluorescens strain SS101. The darkly shaded arrows are based on experimental data obtained earlier [10] and in this study; the lightly shaded arrows are hypothetical and not based on experimental data. The blue dashed arrows and circles represent translational regulation whereas the other arrows represent transcriptional regulation.
TCA cycle proteins were expressed differently in both clpP and clpA mutants
Our proteome analyses also revealed that several proteins from the TCA cycle were differentially expressed in both the clpA and the clpP mutants (Additional file 1: Figure S3). Five proteins were down-regulated and two were up-regulated in the clpA mutant. Similar numbers of down-regulated (6) and up-regulated (2) proteins were found in the clpP mutant (Additional file 1: Figure S3). In the TCA cycle, PckA (PflSS101_0285) encodes phosphoenolpyruvate carboxykinase ATP and transfers oxaloacetate to phosphoenolpyruvate. This protein was 1.20 up- and 1.47 down-regulated in the clpA and clpP mutants, respectively. Mqo_1 (PflSS101_0926), a malate quinone oxidoreductase, was up-regulated in both mutants. Malate quinone oxidoreductase is known to be essential for growth on ethanol or acetate in Pseudomonas aeruginosa [31]. It is also required for virulence of Pseudomonas syringae pv. tomato strain DC3000 on Arabidopsis thaliana [32]. Its function in P. fluorescens SS101, however, is not yet known.
Conclusions
ClpA is a chaperon protein that is highly conserved in bacteria and eukaryotes [33,34]. Together with the serine protease ClpP, it plays an important role in intracellular refolding and degradation of proteins, an essential process for the viability and growth of cells. In this study, we cloned and sequenced clpA from the plant growth-promoting bacterium P. fluorescens strain SS101 and showed that clpA plays an important role in the regulation of massetolide biosynthesis. The combined results of the transcriptomic and proteomic analyses suggest that the ClpAP complex regulates massetolide biosynthesis via the pathway-specific LuxR-type transcriptional regulator MassAR, the heat shock proteins DnaK and DnaJ and via proteins involved in the TCA cycle. These findings extend our previous regulatory model for LP biosynthesis in P. fluorescens SS101 (Figure 4) which, to a large extent, may also apply to the regulatory networks of LP biosynthesis in other Pseudomonas species and strains.
Methods
Bacterial strains and culture conditions
P. fluorescens strain SS101 and its clpP and clpA mutants were cultured in King’s medium B (KB) broth at 25°C. The clpA and clpP mutants were obtained in our previous studies [10,11]. Escherichia coli strain DH5α was the host for the plasmids used for genetic complementation. E. coli strains were grown on Luria-Bertani (LB) plates or in LB broth amended with the appropriate antibiotics.
Identification of the clpA cluster
clpA was identified by partial sequencing of the regions flanking the plasposon insertion as described by Song et al. [10]. The complete flanking regions of clpA were obtained from the genome sequence of P. fluorescens SS101 [35]. Open reading frames (ORFs) were identified with the Softberry FGENESB program (http://www.softberry.com/berry.phtml). The ORFs were analyzed using BlastX in the NCBI database and Pseudomonas.com (http://pseudomonas.com). For genetic complementation, the pME6031-clpA construct was generated according to methods described previously [11]. Briefly, a 2,870-bp fragment, including the promoter and terminator, was subcloned into the shuttle vector pME6031 and transformed into. E. coli DH5α. The pME6031-clpA construct was subsequently electroporated into the clpA plasposon mutant of P. fluorescens SS101. Transformed cells were plated on KB supplemented with tetracycline (25 μg/ml), and the presence of pME6031-clpA was verified by PCR analysis with primers specific for pME6031.
Lipopeptide extraction and RP-HPLC separation
Massetolide extractions and RP-HPLC analysis were performed as described earlier [8,10,11]. Briefly, Pseudomonas strains were grown on Pseudomonas isolation agar plates (Pseudomonas agar 38 g/L, glycerol 10 g/L) for 48 h at 25°C. The cells were suspended in sterile de-mineralized water (~40 ml per plate), transferred to 50 mL tubes, shaken vigorously for 2 min and then centrifuged (30 min, 5292 g, 4°C). The culture supernatant was transferred to a new tube and acidified to pH 2.0 with 9% HCl. The precipitate was recovered by centrifugation (30 min, 5292 g, 4°C) and washed three times with acidified dH2O (pH 2.0). It was then resuspended in 5 mL dH2O and the pH adjusted to 8.0 with 0.2 M NaOH until complete dissolution. The solution was centrifuged (30 min, 5292 g, 4°C) and the supernatant transferred to a new tube, subjected to lyophilisation and RP-HPLC analysis according to methods described previously [36].
Swarming motility
Swarming motility assays of the wild-type and mutants strains were performed as described earlier [10]. Swarming motility of the wild-type SS101 strain and the mutants was assessed on soft [0.6% wt/vol] standard succinate agar medium (SSM) consisting of 32.8 mM K2HPO4, 22 mM KH2PO4, 7.6 mM (NH4)2SO4, 0.8 mM MgSO4, and 34 mM succinic acid. The pH of the medium was adjusted to 7 with NaOH. Cells from overnight cultures of the wild-type and mutant strains were washed three times with 0.9% NaCl, and 5 μL of the washed cell suspensions (1 × 1010 cells/ml) was spot inoculated in the centre of the soft SSM agar plate and incubated for 48–72 h at 25°C.
Transcriptome analysis
The wild-type SS101 strain and the clpA and clpP mutants were grown in KB broth in 24-well plates, and harvested for RNA isolation at an OD600nm = 0.6. For each strain, three biological replicates were used. Total RNA was extracted with Trizol reagent (Invitrogen) and further purified with the NucleoSpin RNA kit. A tiling microarray for P. fluorescens SS101 was developed by the Dutch Genomics Service & Support Provider, University of Amsterdam (UvA, Amsterdam, the Netherlands). In total, 134,276 probes (60-mer) were designed with, in general, a gap of 32 nucleotides between adjacent probes on the same strand and an overlap of 14 nucleotides for both strands. In addition, 5,000 custom negative control probes were hybridized and used as an internal control to validate the designed probes in a CGH experiment of 4 arrays. Probes were annotated and assembled into probe sets for known genes based on location information retrieved from the Pathosystems Resource Integration Center (PATRIC, http://patricbrc.org). Probes outside of known gene sequences were labeled as InterGenic Region (IGR). cDNA labelling was conducted as described previously [37]. Briefly, cDNA was synthesized in presence of Cy3-dUTP (Cy3) for the test samples and with Cy5-dUTP (Cy5) for the common reference. The common reference consisted of an equimolar pool of the test samples (3 μg per sample). 5 μg of total RNA per reaction was used and yielded 1.5-2.5 μg cDNA for each sample with larger than 16 pmol of Cy3 or Cy5 dye per microgram. Hybridizations were performed as described elsewhere [38]. Slides were washed according to the procedures described in the Nimblegen Arrays User’s Guide - Gene Expression Arrays Version 5.0 and scanned in an ozone-free room with an Agilent DNA microarray scanner G2565CA (Agilent Technologies). Feature extraction was performed with NimbleScan v2.5 (Roche Nimblegen). Data pre-processing consisted of log2-transformation of the raw probe-intensity data, followed by a within slide Lowess normalization. Thus normalized sample (Cy3) channel intensities were summarized into probe sets values and normalized between arrays using the RMA (Robust Multi-Array Analysis) algorithm [39]. Analysis of the gene expression data was conducted using the Arraystar software. All results described were found to be significant using a false discovery rate of less than 5%.
Proteome analysis
The wild-type SS101 strain and the clpA and clpP mutants were grown in KB broth in 24-well plates, and cells were harvested for protein extraction at an OD600nm = 0.6. Three biological replicates were used for each strain. The cells were harvested by centrifugation and resuspended in 15 mL ice-cold 1 x PSB buffer containing the protease Inhibitor Cocktail from Sigma-Aldrich, as instructed by the manufacturer. The following steps were performed at 4°C. The cells were disrupted twice in a French pressure cell press (SLM Instruments Inc) at 14,000 psi and centrifuged for 30 min at 47,000 g. Protein concentration was determined using the Bradford assay followed by iTRAQ labeling in a 4-plex experiment according to the manufacturer’s protocol (AB Sciex Pte. Ltd). Briefly, 100 μg of protein in 100–400 μL were successively reduced in the presence of 1 μL TCEP (tris(2-carboxyethyl)phosphine), alkylated using 2 μL 85 mM iodoacetamide, and hydrolyzed with 2.5 μg trypsin. A further addition of 2.5 μg trypsin 1 h after the initial addition of the protease was performed prior to an overnight incubation. Each of the reaction mixtures was then freeze-dried, redissolved in 100 μL 125 mM TEAB (triethylammonium bicarbonate) in 75% ethanol and transferred to one vial of iTRAQ reagent (4-plex, 114–117). After 1 h incubation, 100 μL of H20 was added followed by 15 min incubation in order to hydrolyze the excess of iTRAQ reagent. The resulting samples were pooled together and desalted using SepPak C18 cartridges (Waters Corporation). The pooled samples (800 μL) were diluted to 3.6 mL in 0.1% formic acid (FA) and loaded onto pre-wetted (95% acetonitrile (ACN) containing 0.1% FA) and equilibrated (0.1% FA) cartridges. After washing the loaded cartridges 5 times with 1 mL 0.1% FA, elution was performed in 1 mL 50% ACN/0.1% FA followed by 95% ACN/0.1% FA. Eluates were combined and evaporated to dryness.
The evaporated iTRAQ-labeled samples were resolubilized (10 μL) in the sample loading buffer (5 mM ammonium acetate containing 5% ACN) and injected (4.9 μL) using the partial loop mode on a liquid chromatograph (nanoAcquity UPLC system, Waters Corporation) plumbed for two-pump trapping and two-dimensional strong-cation exchange and reversed-phase (SCX-RP) separation. Salt plugs (10, 20, 30, 40, 50, 80, 150, 200 mM ammonium acetate in 5% ACN, followed by 200 mM in 30% ACN and 350 mM in 50% ACN) were injected using the full loop mode. Sample and salt plugs were loaded in trap mode (SCXtrap-C18trap-waste) onto the SCXtrap column (18x20mm, 5 μm particle size, P/N 186003507) using the sample and loading buffer for 10 min at 5 μL/min. Subsequently, an analytical separation was performed in analytical mode (C18trap-C18Analytical-ESI source) at 400 nL/min with the following consecutive steps and gradient: 1% B (100% ACN, 0.1% FA) (0–1 min); 1–40% B (1–50 min); 40-60% B (50–65 min); 60-85% B (65–66 min); 85% B (66–70 min); 85-1% B (70–71 min).
The gradient flow from the nanoAcquity was delivered into the Nano ESI ion source of a Xevo Q-TOF mass spectrometer from Waters Corporation (source voltage 4 kV; source temperature 80°C; cone voltage 35 V; cone gas flow 20 L/h; nano flow gas 0.8 bar). Data were acquired in data dependent mode with one full scan (350–1400 m/z) followed by maximum 5 MS/MS scans (50–1800 m/z) on doubly and triply charged peptides only. External TOF mass calibration was performed prior to the UPLC-MS analysis. This was obtained by direct infusion of a solution containing 2 g/L sodium iodide in 50% isopropanol, and data acquisition in TOF-MS mode over the m/z range 50–2000.
Proteome data analysis
Raw data files were treated using the trans-proteomic pipeline (TPP) software package for proteomic data analysis supplied by the Seattle Proteome Centre [40]. The processing of data through the TPP modules was automated by in-house java-based software. Initially, raw files were converted into uncentroided mzXML files using MSConvert. Before search all data was centroided and processed to only keep the top 100 peaks in each fragment spectra. Centroided data was then analysed using X!tandem with native scoring. Search hits from each individual replicate were assigned probabilities using Peptide Prophet [41] utilizing the semi-parametric model, at this stage each technical-replicate was assigned a unique experiment ID to allow iProphet [42] to utilize the number of replicate experiments model. Libra (TPP module) was then used to extract iTRAQ reporter ion signals from the uncentroided data, in each replicate the four different iTRAQ reporter channels were normalized to account for 25% of the total signal.
Each set of technical replicates were then combined into a single output pep.xml using iProphet [43] and final protein lists were assembled using Protein Prophet [44] and Libra was used to calculate iTRAQ protein ratios. Parameters used for analysis were as follows; X!tandem searches were ran against the P. fluorescens SS101 amino acid sequence database, concatenated to its own reversed sequences for use as decoy hits. Searches used trypsin specificity, a precursor ion tolerance of 50 ppm, a fragment monoisotopic tolerance of 0.4 Da and the following post-translational modifications were assigned; fixed carbamidomethyl cystein, fixed iTRAQ (N-term), fixed iTRAQ (K), variable oxidation (M), variable iTRAQ (Y), variable phosphorylation (S/T). Libra protein ratios were extracted using intensity weighted average, using normalization by sum of reagent profiles, minimum reporter ion intensity of 20 and a reporter ion mass tolerance of 0.05.
Acknowledgements
We are very grateful to the Graduate School of Experimental Plant Sciences (EPS) for financing this project. We thank the Dutch Genomics Service & Support Provider for conducting the microarray analysis. The authors of this manuscript have no conflicts of interest to declare. This publication is no.5771 of the Netherlands Institute of Ecology (NIOO-KNAW).
Additional files
Differential gene transcription between the wild-type P. fluorescens SS101 strain and the clpA (A) or clpP (B) mutant at exponential phase (OD600 = 0.6), assessed by microarray analyses. The transcription chart shows log2-based fold changes of transcripts of clpA or clpP mutant compared to the wild-type strain SS101. Each dot in the chart represents each of the 5374 annotated genes in the SS101 genome with the x-axis showing gene order, and the y-axis showing the log2 of relative transcripts abundance for each gene in the clpA or clpP mutant compared to the wild-type strain SS101. Gene clusters whose members are discussed in the main text are shown. Figure S2. Differential protein expression between wild-type P. fluorescens SS101 and the clpA (A) or the clpP (B) mutant at exponential phase (OD600 = 0.6), assessed using isobaric tag labeling for relative and absolute quantitation (iTRAQ) experiments. The expression chart shows fold changes of protein expression in the clpA or clpP mutant compared to the wild-type strain SS101. Each dot in the chart represents the 200 and 223 proteins that significantly accumulated in the clpA and clpP mutants, respectively. The x-axis shows gene order and the y-axis shows fold changes. Figure S3. TCA cycle pathway of P. fluorescens SS101 (adjusted from KEGG with P. fluorescens A506, the most related strain of SS101). Red boxes indicate up-regulation; green boxes indicate down-regulation; empty boxes stand for “not detected”. The left and right boxes stand for protein expression in the clpA and clpP mutants, respectively.
Whole genome transcriptome analysis of ∆clpA/wt. Table S2. Whole genome transcriptome analysis of ∆clpP/wt. Table S3. Whole genome proteome analysis of ∆clpA/wt. Table S4. Whole genome proteome analysis of ∆clpP/wt. Table S5. Down-regulated proteins in both clpA and clpP mutants of Pseudomonas fluorescens SS101.
Footnotes
Chunxu Song and Gustav Sundqvist contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CS and JMR designed the experiments and drafted the manuscript. CS carried out the experiments. GS, EM and VB participated in design and carried out the proteomic analysis and statistical analysis. JvdM generated the transposon mutant library. IdB and AK helped to screen the mutant library. All authors read and approved the final manuscript.
Contributor Information
Chunxu Song, Email: c.song@nioo.knaw.nl.
Gustav Sundqvist, Email: gustav_sundqvist@waters.com.
Erik Malm, Email: erik.malm@biotech.kth.se.
Irene de Bruijn, Email: i.deBruijn@nioo.knaw.nl.
Aundy Kumar, Email: kumar@iari.res.in.
Judith van de Mortel, Email: J.vandeMortel@has.nl.
Vincent Bulone, Email: bulone@kth.se.
Jos M Raaijmakers, Email: j.raaijmakers@nioo.knaw.nl.
References
- 1.Raaijmakers JM, de Bruijn I, de Kock MJ. Cyclic lipopeptide production by plant-associated Pseudomonas spp.: diversity, activity, biosynthesis, and regulation. Mol Plant Microbe Interact. 2006;19(7):699–710. doi: 10.1094/MPMI-19-0699. [DOI] [PubMed] [Google Scholar]
- 2.Ongena M, Jacques P. Bacillus lipopeptides: versatile weapons for plant disease biocontrol. Trends Microbiol. 2008;16(3):115–25. doi: 10.1016/j.tim.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 3.Nielsen TH, Nybroe O, Koch B, Hansen M, Sorensen J. Genes involved in cyclic lipopeptide production are important for seed and straw colonization by Pseudomonas sp. strain DSS73. Appl Environ Microbiol. 2005;71(7):4112–6. doi: 10.1128/AEM.71.7.4112-4116.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tran H, Ficke A, Asiimwe T, Hofte M, Raaijmakers JM. Role of the cyclic lipopeptide massetolide A in biological control of Phytophthora infestans and in colonization of tomato plants by Pseudomonas fluorescens. New Phytol. 2007;175(4):731–42. doi: 10.1111/j.1469-8137.2007.02138.x. [DOI] [PubMed] [Google Scholar]
- 5.Mazzola M, de Bruijn I, Cohen MF, Raaijmakers JM. Protozoan-induced regulation of cyclic lipopeptide biosynthesis is an effective predation defense mechanism for Pseudomonas fluorescens. Appl Environ Microbiol. 2009;75(21):6804–11. doi: 10.1128/AEM.01272-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raaijmakers JM, De Bruijn I, Nybroe O, Ongena M. Natural functions of lipopeptides from Bacillus and Pseudomonas: more than surfactants and antibiotics. FEMS Microbiol Rev. 2010;34(6):1037–62. doi: 10.1111/j.1574-6976.2010.00221.x. [DOI] [PubMed] [Google Scholar]
- 7.Finking R, Marahiel MA. Biosynthesis of nonribosomal peptides1. Annu Rev Microbiol. 2004;58:453–88. doi: 10.1146/annurev.micro.58.030603.123615. [DOI] [PubMed] [Google Scholar]
- 8.de Bruijn I, de Kock MJ, de Waard P, van Beek TA, Raaijmakers JM. Massetolide A biosynthesis in Pseudomonas fluorescens. J Bacteriol. 2008;190(8):2777–89. doi: 10.1128/JB.01563-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Souza JT, de Boer M, de Waard P, van Beek TA, Raaijmakers JM. Biochemical, genetic, and zoosporicidal properties of cyclic lipopeptide surfactants produced by Pseudomonas fluorescens. Appl Environ Microbiol. 2003;69(12):7161–72. doi: 10.1128/AEM.69.12.7161-7172.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song C, Aundy K, van de Mortel J, Raaijmakers JM. Discovery of new regulatory genes of lipopeptide biosynthesis in Pseudomonas fluorescens. FEMS Microbiol Lett. 2014;356(2):166–75. doi: 10.1111/1574-6968.12404. [DOI] [PubMed] [Google Scholar]
- 11.de Bruijn I, Raaijmakers JM. Regulation of cyclic lipopeptide biosynthesis in Pseudomonas fluorescens by the ClpP protease. J Bacteriol. 2009;191(6):1910–23. doi: 10.1128/JB.01558-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maurizi MR, Clark WP, Kim SH, Gottesman S. Clp P represents a unique family of serine proteases. J Biol Chem. 1990;265(21):12546–52. [PubMed] [Google Scholar]
- 13.Kirstein J, Moliere N, Dougan DA, Turgay K. Adapting the machine: adaptor proteins for Hsp100/Clp and AAA+ proteases. Nat Rev Microbiol. 2009;7(8):589–99. doi: 10.1038/nrmicro2185. [DOI] [PubMed] [Google Scholar]
- 14.Gottesman S. Proteolysis in bacterial regulatory circuits. Annu Rev Cell Dev Biol. 2003;19:565–87. doi: 10.1146/annurev.cellbio.19.110701.153228. [DOI] [PubMed] [Google Scholar]
- 15.Reid BG, Fenton WA, Homwich AL, Weber-Ban EU. ClpA mediates directional translocation of substrate proteins into the ClpP protease. Proc Natl Acad Sci U S A. 2001;98(7):3768–72. doi: 10.1073/pnas.071043698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoskins JR, Pak M, Maurizi MR, Wickner S. The role of the ClpA chaperone in proteolysis by ClpAP. Proc Natl Acad Sci U S A. 1998;95(21):12135–40. doi: 10.1073/pnas.95.21.12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gottesman S. Proteases and their targets in Escherichia coli. Annu Rev Genet. 1996;30:465–506. doi: 10.1146/annurev.genet.30.1.465. [DOI] [PubMed] [Google Scholar]
- 18.Mogk A, Schmidt R, Bukau B. The N-end rule pathway for regulated proteolysis: prokaryotic and eukaryotic strategies. Trends Cell Biol. 2007;17(4):165–72. doi: 10.1016/j.tcb.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 19.Moore SD, Sauer RT. The tmRNA system for translational surveillance and ribosome rescue. Annu Rev Biochem. 2007;76:101–24. doi: 10.1146/annurev.biochem.75.103004.142733. [DOI] [PubMed] [Google Scholar]
- 20.Flynn JM, Neher SB, Kim YI, Sauer RT, Baker TA. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol Cell. 2003;11(3):671–83. doi: 10.1016/S1097-2765(03)00060-1. [DOI] [PubMed] [Google Scholar]
- 21.de Breij A, Gaddy J, van der Meer J, Koning R, Koster A, van den Broek P, et al. CsuA/BABCDE-dependent pili are not involved in the adherence of Acinetobacter baumannii ATCC19606(T) to human airway epithelial cells and their inflammatory response. Res Microbiol. 2009;160(3):213–8. doi: 10.1016/j.resmic.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 22.Bernard CS, Bordi C, Termine E, Filloux A, de Bentzmann S. Organization and PprB-dependent control of the Pseudomonas aeruginosa tad locus, involved in Flp pilus biology. J Bacteriol. 2009;191(6):1961–73. doi: 10.1128/JB.01330-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baehler E, de Werra P, Wick LY, Pechy-Tarr M, Mathys S, Maurhofer M, et al. Two novel MvaT-like global regulators control exoproduct formation and biocontrol activity in root-associated Pseudomonas fluorescens CHA0. Mol Plant Microbe Interact. 2006;19(3):313–29. doi: 10.1094/MPMI-19-0313. [DOI] [PubMed] [Google Scholar]
- 24.Stewart RC. Protein histidine kinases: assembly of active sites and their regulation in signaling pathways. Curr Opin Microbiol. 2010;13(2):133–41. doi: 10.1016/j.mib.2009.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamanaka K, Inouye M. Growth-phase-dependent expression of cspD, encoding a member of the CspA family in Escherichia coli. J Bacteriol. 1997;179(16):5126–30. doi: 10.1128/jb.179.16.5126-5130.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nishijyo T, Haas D, Itoh Y. The CbrA-CbrB two-component regulatory system controls the utilization of multiple carbon and nitrogen sources in Pseudomonas aeruginosa. Mol Microbiol. 2001;40(4):917–31. doi: 10.1046/j.1365-2958.2001.02435.x. [DOI] [PubMed] [Google Scholar]
- 27.Amador CI, Canosa I, Govantes F, Santero E. Lack of CbrB in Pseudomonas putida affects not only amino acids metabolism but also different stress responses and biofilm development. Environ Microbiol. 2010;12(6):1748–61. doi: 10.1111/j.1462-2920.2010.02254.x. [DOI] [PubMed] [Google Scholar]
- 28.Hillerich B, Westpheling J. A new GntR family transcriptional regulator in Streptomyces coelicolor is required for morphogenesis and antibiotic production and controls transcription of an ABC transporter in response to carbon source. J Bacteriol. 2006;188(21):7477–87. doi: 10.1128/JB.00898-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fineran PC, Everson L, Slater H, Salmond GPC. A GntR family transcriptional regulator (PigT) controls gluconate-mediated repression and defines a new, independent pathway for regulation of the tripyrrole antibiotic, prodigiosin, in Serratia. Microbiol-Sgm. 2005;151:3833–45. doi: 10.1099/mic.0.28251-0. [DOI] [PubMed] [Google Scholar]
- 30.Dubern JF, Lagendijk EL, Lugtenberg BJ, Bloemberg GV. The heat shock genes dnaK, dnaJ, and grpE are involved in regulation of putisolvin biosynthesis in Pseudomonas putida PCL1445. J Bacteriol. 2005;187(17):5967–76. doi: 10.1128/JB.187.17.5967-5976.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kretzschmar U, Ruckert A, Jeoung JH, Gorisch H. Malate : quinone oxidoreductase is essential for growth on ethanol or acetate in Pseudomonas aeruginosa. Microbiol-Sgm. 2002;148:3839–47. doi: 10.1099/00221287-148-12-3839. [DOI] [PubMed] [Google Scholar]
- 32.Mellgren EM, Kloek AP, Kunkel BN. Mqo, a tricarboxylic acid cycle enzyme, is required for virulence of Pseudomonas syringae pv. tomato strain DC3000 on Arabidopsis thaliana. J Bacteriol. 2009;191(9):3132–41. doi: 10.1128/JB.01570-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wong P, Houry WA. Chaperone networks in bacteria: analysis of protein homeostasis in minimal cells. J Struct Biol. 2004;146(1–2):79–89. doi: 10.1016/j.jsb.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 34.Yu AYH, Houry WA. ClpP: A distinctive family of cylindrical energy-dependent serine proteases. Febs Lett. 2007;581(19):3749–57. doi: 10.1016/j.febslet.2007.04.076. [DOI] [PubMed] [Google Scholar]
- 35.Loper JE, Hassan KA, Mavrodi DV, Davis EW, Lim CK, Shaffer BT, et al. Comparative genomics of plant-associated Pseudomonas spp.: insights into diversity and inheritance of traits involved in multitrophic interactions. PloS Genet. 2012;8(7):e1002784. doi: 10.1371/journal.pgen.1002784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song C, Van der Voort M, Van de Mortel J, Hassan KA, Elbourne LDH, Paulsen IT, Loper JE, Raaijmakers JM. The Rsm regulon of plant growth-promoting Pseudomonas fluorescens SS101: role of small RNAs in regulation of lipopeptide biosynthesis. Microbial biotechnology. 2014. [DOI] [PMC free article] [PubMed]
- 37.de Knegt GJ, Bruning O, ten Kate MT, de Jong M, van Belkum A, Endtz HP, et al. Rifampicin-induced transcriptome response in rifampicin-resistant Mycobacterium tuberculosis. Tuberculosis. 2013;93(1):96–101. doi: 10.1016/j.tube.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 38.Pennings JLA, Rodenburg W, Imholz S, Koster MPH, van Oostrom CTM, Breit TM, et al. Gene expression profiling in a mouse model identifies fetal liver- and placenta-derived potential biomarkers for down syndrome screening. PloS One. 2011;6(4):e18866. doi: 10.1371/journal.pone.0018866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 40.Keller A, Eng J, Zhang N, Li XJ, Aebersold R. A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol Syst Biol. 2005;1:2005 0017. doi: 10.1038/msb4100024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kessner D, Chambers M, Burke R, Agusand D, Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24(21):2534–6. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20(9):1466–7. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 43.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74(20):5383–92. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 44.Shteynberg D, Deutsch EW, Lam H, Eng JK, Sun Z, Tasman N, et al. iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol Cell Proteomics. 2011;10(12):M111 007690. doi: 10.1074/mcp.M111.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]