

SUMMARY
Dietary potassium deficiency, common in Western diets, raises blood pressure and enhances salt sensitivity. Potassium homeostasis requires a molecular switch in the distal convoluted tubule (DCT), which fails in familial hyperkalemic hypertension (pseudohypoaldosteronism type 2), activating the thiazide-sensitive NaCl cotransporter, NCC. Here, we show that dietary potassium deficiency activates NCC, even in the setting of high salt intake, thereby causing sodium retention and a rise in blood pressure. The effect is dependent on plasma potassium, which modulates DCT cell membrane voltage and, in turn, intracellular chloride. Low intracellular chloride stimulates WNK kinases to activate NCC, limiting potassium losses, even at the expense of increased blood pressure. These data show that DCT cells, like adrenal cells, sense potassium via membrane voltage. In the DCT, hyperpolarization activates NCC via WNK kinases, whereas in the adrenal gland, it inhibits aldosterone secretion. These effects work in concert to maintain potassium homeostasis.
INTRODUCTION
Compared to diets consumed by our evolutionary ancestors, the majority of people in the world today consume a diet relatively high in salt (NaCl) and low in potassium (K+). A high dietary sodium (Na+) to K+ ratio is associated with hypertension, cardiovascular disease, and all-cause mortality. Although the DASH diet, which lowers blood pressure regardless of NaCl intake, does not specify K+ intake, it is replete with K+-rich foods, and most investigators assume that a substantial portion of its beneficial effects is mediated by K+ (Sacks et al., 2001). Recently, in two studies of individuals from around the world, low K+ (LK) intake was strongly associated with both higher blood pressure and cardiovascular death (Mente et al., 2014; O’Donnell et al., 2014). Yet, the mechanisms linking K+ intake and blood pressure remain obscure.
Although Na+ reabsorption along all nephron segments contributes to NaCl homeostasis, transport along the aldosterone-sensitive distal nephron (ASDN) plays an especially important role in K+ homeostasis. The ASDN includes a portion of the distal convoluted tubule (DCT) and the connecting tubule (CNT) and collecting duct (CD). The DCT is heterogeneous, comprising a proximal portion, the DCT1, which primarily reabsorbs NaCl, and a distal portion, the DCT2, where electroneurtral NaCl transport coexists with electrogenic Na+ and K+ transport (Subramanya and Ellison, 2014). The DCT1 does not secrete or reabsorb substantial amounts of K+, so it has been surprising that genetic diseases affecting the DCT are manifested primarily by disordered K+ metabolism. Hypokalemia is common in Gitelman and EAST/SeSAME syndromes, whereas hyperkalemia is a universal feature of familial hyperkalemic hypertension (FHHt, also called pseudohypoaldosteronism type 2, or Gordon syndrome) (Subramanya and Ellison, 2014).
The thiazide-sensitive Na-Cl cotransporter (NCC: SLC12A3) is the predominant apical Na+ entry pathway in DCT1 cells. Because DCT cells also control the delivery of NaCl into the CNT, where the epithelial sodium channel (ENaC) mediates electrogenic Na+ reabsorption and where K+ is secreted, they appear to have a substantial, albeit indirect, role in K+ secretion. The importance of the DCT in K+ secretion became apparent when the molecular solution of FHHt led to the discovery of a molecular switch comprising the with no lysine (WNK) kinases, Ste20p-related proline alanine-rich kinase (SPAK), and oxidative stress response 1 kinase (OxSR1). FHHt-causing disease mutations appear to lock this switch in the on position, driving unrelenting NCC activity, hyperkalemia, and hypertension. Although this pathogenesis is clear, the nature of the physiological switch activator has remained elusive.
Dietary NaCl restriction activates NCC via angiotensin II and aldosterone to preserve extracellular fluid (ECF) volume and arterial pressure (Subramanya and Ellison, 2014). Yet, NCC also responds to changes in dietary K+ intake. High KCl (HK) diet suppresses (Frindt and Palmer, 2010; Rengarajan et al., 2014; Sorensen et al., 2013; Vallon et al., 2009; van der Lubbe et al., 2013), and low KCl intake increases, NCC activity (Castañeda-Bueno et al., 2014; Vallon et al., 2009; Vitzthum et al., 2014). These effects contribute to systemic K+ homeostasis by altering Na+ delivery to K+ secretory segments. While the WNKs are essential to maintain systemic K+ balance, in cell systems and in vitro, they are chloride (Cr)-sensitive kinases (Piala et al., 2014; Ponce-Coria et al., 2008), which activate NCC when intracellular [Cl−] is low (Pacheco-Alvarez et al., 2006). Recently, a WNK1 crystal structure showed that WNK1 binds Cl− near the catalytic lysine in subdomain 1, inhibiting its autophosphoryla-tion and, in turn, activity (Piala et al., 2014).
Here, we sought to identify the physiological switch modulating DCT function and NCC activity. The results show that dietary K+ intake, acting via plasma [K+], is a key NCC regulator. Plasma [K+] signals NCC through changes in intracellular [Cl]−, thereby modulating WNK kinases. Our experimental data and in silico analysis suggest that plasma [K+] can enhance Cl− efflux from DCT cells through both electrogenic and electroneutral mechanisms. The results have implications for human disease and public health.
RESULTS
Low Potassium Intake Activates NCC and Raises Arterial Pressure
We first sought to determine whether ECF volume or K+ dominates NCC activity in the context of combined interventions. We confirmed that dietary NaCl loading suppresses NCC abundance in mice (Figure S1A, available online). We then showed that NCC and phosphorylated NCC (pNCC, as an index of activation) were more abundant in kidneys of mice consuming a high-salt/low-K+ (HS/LK) diet than a high-salt/normal-K+ (HS/NK) diet (Figure 1A and Table S1), suggesting that the K+ signal overrides the ECF signal. Plasma aldosterone was not different between the two groups, whereas plasma angiotensin II was lower in the HS/LK group (Figure 1B). As the effect of LK diets on pNCC was preserved in angiotensin II receptor type 1a null mice (AT1a−/−) (Figures 1C and S1B), AT1A receptors are not required for these effects. The effect is also not general, as HS/LK diet did not affect Na+-K+-2 Cl− cotransporter (NKCC2) abundance (Figure 1A and Table S1). To test whether the effect of dietary K+ restriction on pNCC is enhanced by high NaCl intake; Figure 1D confirms that, during dietary K+ deprivation, pNCC abundance is greater when mice consume a high-NaCl rather than a low-NaCl diet.
Figure 1. Effects of Dietary Potassium Intake on NCC Abundance.
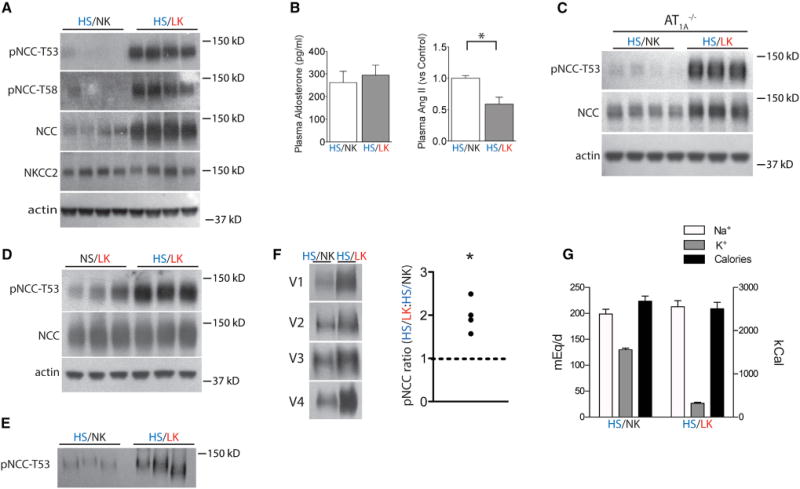
(A) Western blot of kidney from mice consuming HS/NK and HS/LK diets. The HS/LK diet increased the abundance of total NCC, pNCC-T53, and pNCC-T58 significantly (p < 0.01 for each by unpaired t test). NKCC2 was unaffected. Actin is a loading control.
(B) Plasma aldosterone and angiotensin II concentrations from the two groups. *p < 0.05 by unpaired t test.
(C) Western blot of kidney from AT1a−/− mice treated as in (A). p < 0.01 for pNCC-T53 by unpaired t test.
(D) Western blot of kidney from mice consuming NS/LK and HS/LK diets. Dietary salt loading increased the abundance of pNCC-T53 on a LK diet. p < 0.05 by unpaired t test.
(E) Western blot for pNCC-T53 of urinary exosomes from mice consuming either HS/LK or HS/NK diets. The HS/LK diet increased pNCC-T53. p < 0.05 by unpaired t test.
(F) Western blot for pNCC-T53 of urinary exosomes from volunteers consuming HS/LK and HS/NK diets. *p < 0.05 by paired t test.
(G) Dietary Na+, K+, and calorie content of volunteers consuming HS/LK and HS/NK diets. Bars represent mean ± SEM. See Table S1 for densitometry.
To estimate NCC activity noninvasively, we measured the abundance of pNCC in urinary exosomes. This paralleled changes in kidney pNCC, as it was greater in mice on a HS/LK diet than those on a HS/NK diet (Figure 1E). To determine whether humans exhibit a similar response to dietary K+ intake, we analyzed pNCC in urinary exosomes (Hoorn et al., 2005; Pisitkun et al., 2004) from volunteers who consumed a HS/LK diet for 4 days followed by a HS/NK diet for 4 days. The abundance of pNCC was significantly greater after the HS/LK diet than after the HS/NK diet (Figure 1F). The diets had similar Na+ and calorie content, whereas dietary K+ differed (Figure 1G).
As these results suggested that dietary K+ deprivation activates NCC, we measured urine Na+ excretion in mice. Although food consumption was similar, body weight increased slightly (Figures S2A and S2B), and urine Na+ excretion was lower on HS/LK than on HS/NK (Figure 2A). To confirm that the LK diet also increased arterial pressure, as reported by others (Vitzthum et al., 2014), we showed that mean arterial pressure, measured telemetrically, rose when mice were switched from HS/NK to HS/LK diet. The rise in arterial pressure on the LK diet was primarily during the active period (Figure 2B), also consistent with prior reports (Vitzthum et al., 2014). To determine whether activation of NCC during LK diet contributed to the rise in blood pressure, we compared the effects of diet in Slc12a3−/− mice and wild-type (WT) controls. In Slc12a3−/− mice, blood pressure did not rise during LK diet, whereas it did in WT mice (Figures 2C and S2C). The impact of genotype on response to LK diet was significant.
Figure 2. Effects of Dietary Potassium Intake on Urine Sodium and Blood Pressure.
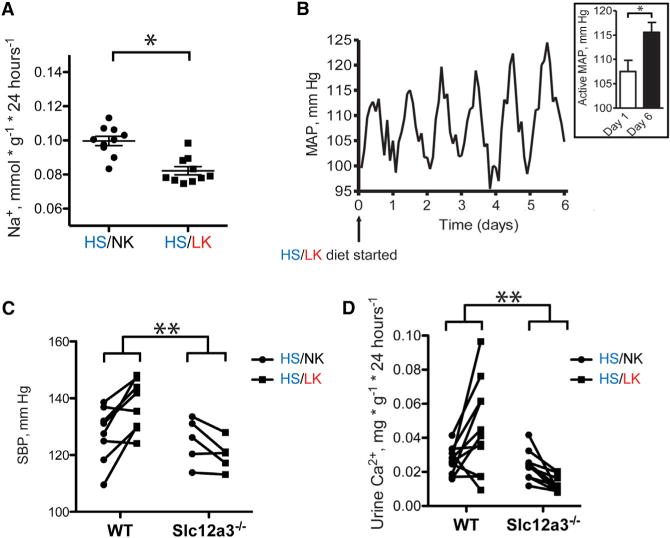
(A) Urine sodium excretion (24 hr) from mice on HS/NK and HS/LK diets. *p < 0.05 by paired t test.
(B) Mean arterial pressure (MAP) recording (24 hr) from mice on a HS/LK diet. Trace represents 2 hr averages from three animals. Mice were maintained on a HS/NK diet for 1 week until time 0, when they were switched to HS/LK. There was a statistically significant increase in MAP during the active period between day 0 and day 6 (Inset shows mean ± SEM). *p < 0.05 by paired t test.
(C) Systolic blood pressure measurements for wild-type and Slc12a3−/− mice on HS/NK and HS/LK diets. **p < 0.05 by unpaired t test for the difference between the group differences.
(D) Urine calcium excretion (24 hr) from wild-type and Slc12a3−/− mice on HS/NK and HS/LK diets. **p < 0.05 by unpaired t test for the difference between the group differences.
As modification of dietary K+ also affects urinary calcium excretion in humans (Lemann et al., 1995), we sought to determine whether NCC was involved. We confirmed that mice excreted more calcium when switched from a HS/NK to a HS/LK diet (Figure 2D). This effect was not just absent in Slc12a3−/− mice, it was reversed, so that HS/LK diet paradoxically reduced urine calcium excretion (Figure 2D).
Plasma Potassium Signals the DCT via WNK and SPAK/OxSR1
Plasma potassium was significantly lower in mice consuming a LK, compared with a NK, diet (2.37 ± 0.13 versus 3.38 ± 0.04 mM). Yet, some have suggested that K+ must be ingested orally to alter NCC (Sorensen et al., 2013). To test the role of plasma [K+] without changing K+ intake, we manipulated plasma [K+] pharmacologically with the K+-sparing diuretic amiloride. Amiloride directly inhibits ENaC, leading to K+ retention, but it does not affect NCC directly. Amiloride treatment of mice caused substantial hyperkalemia and, as indicated by a higher hematocrit (Figures 3A and 3B), ECF volume depletion. Despite the volume depletion, which would be expected to stimulate NCC, amiloride reduced both NCC and pNCC, suggesting again that the K+ signal overrides the ECF signal (Figure 3C and Table S2). Opposite effects were observed for NKCC2, confirming that these phenomena are specific to the DCT. As amiloride might have effects unrelated to the changes in plasma [K+], we treated mice with a LK diet along with amiloride to keep [K+] in the normal range. This prevented the alterations in NCC and pNCC. Interestingly, this also prevented the rise in hematocrit, suggesting that the effects of amiloride on ECF volume result from inhibiting Na+ transport along sequential nephron segments (Figures 3A–3C).
Figure 3. Plasma Potassium Signals via WNK/SPAK Independent of Diet.
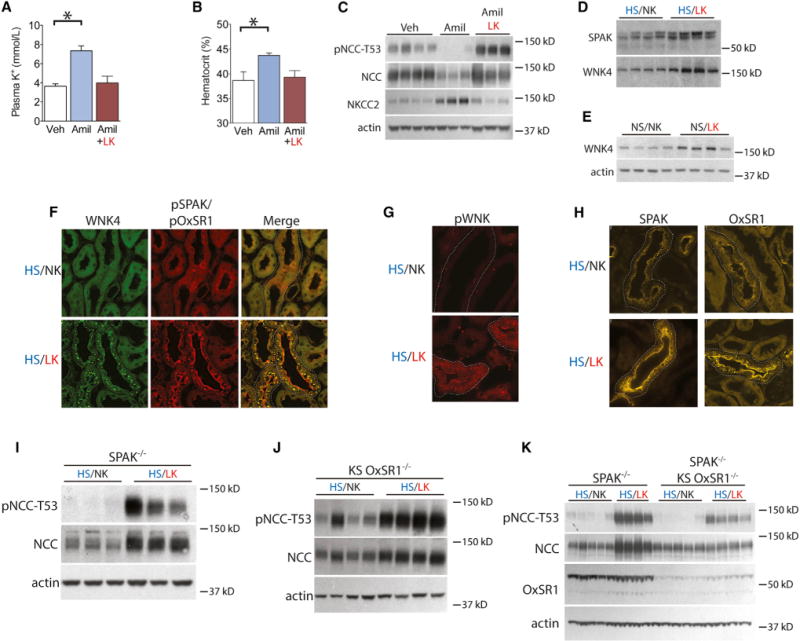
(A) Plasma potassium levels from mice treated with vehicle (Veh), amiloride (Amil), and amiloride combined with LKdiet. *p<0.01 by unpaired t test corrected for multiple comparisons.
(B) Hematocrit of mice treated with vehicle, amiloride, and amiloride combined with LK diet. *p < 0.01 by unpaired t test corrected for multiple comparisons.
(C) Western blot of kidney from mice treated with vehicle, amiloride, and amiloride combined with LK diet. p < 0.01 for each by unpaired t test (Veh versus Amil) corrected for multiple comparisons.
(D) Western blot of kidney from mice consuming HS/NK and HS/LK diets. p < 0.02 by unpaired t test for SPAK and WNK4. Actin loading control is same as Figure 1A.
(E) Western blot of kidney from mice fed NS/NK and NS/LK diets. WNK4 abundance increased on NS/LK diet. p < 0.05 by unpaired t test.
(F) WNK4 and pSPAK/pOxSR1 immunofluorescence of DCT sections (dotted lines) from mice fed HS/NK and HS/LK diets.
(G) pWNK immunofluorescence of DCT sections (dotted lines) from mice fed HS/NK and HS/LK diets.
(H) SPAK and OxSR1 immunofluorescence of DCT (dotted lines) from mice fed HS/NK and HS/LK diets.
(I) Western blot of pNCC-T53 abundance from SPAK−/− mice fed HS/NK and HS/LK diets. p < 0.01 by unpaired t test.
(J) Western blot of pNCC-T53 abundance from KS OxSR1−/− mice fed HS/NK and HS/LK diets. p < 0.01 by unpaired t test.
(K) Western blot of pNCC-T53 abundance from SPAK−/− and SPAK−/−/KS OxSR1−/− mice fed HS/NK and HS/LK diets. pNCC-T53 abundance increased on HS/LK in both genotypes (p < 0.01 by unpaired t test corrected for multiple comparisons) but increased less in SPAK−/−/KS OxSR1−/− mice. Two-way ANOVA indicated p < 0.01 for diet, genotype, and interaction. Total NCC abundance increased in SPAK−/− mice (p < 0.01 by unpaired t test corrected for multiple comparisons), but not in SPAK−/−/KS OxSR1−/− mice. Two-way ANOVA indicated p < 0.01 for diet and interaction. Bars represent mean ± SEM. See Table S2 for densitometry.
In mice consuming a HS/LK diet, total renal SPAK and WNK4 abundance was significantly higher than that in mice consuming a HS/NK diet (Figure 3D). Similar effects were not observed for OxSR1, WNK1, or WNK3 (Figure S3A), although our WNK1 antibody cannot the detect KS-WNK1, which is the predominant WNK1 isoform in DCT. The same effect of dietary K+ on WNK4 abundance was observed when the dietary NaCl intake was normal, indicating that the observed effects resulted from changes in K+ intake alone (Figure 3E). Dietary K+ intake also had substantial effects on kinase activation and localization in the DCT. When mice consumed a HS/NK diet, WNK4 was diffusely expressed, at low levels, along the distal nephron. On a HS/LK diet, however, WNK4 localized to puncta, as confirmed using two different, well-validated WNK4 antibodies (Figures 3F and S3B). To confirm that this redistribution reflected WNK activation, we used an anti-phospho WNK antibody generated by Alessi and colleagues (Thastrup et al., 2012) directed against a motif containing an essential phosphorylated T-loop serine shared by WNK1 and WNK4. As shown in Figure 3G, DCT profiles from mice on a HS/LK diet exhibited a substantial increase in pWNK fluorescence intensity and punctate staining in comparison with DCT profiles from mice on a HS/NK diet.
To determine whether the WNK activation was associated with SPAK and OxSR1 activation, we first demonstrated that SPAK exhibited both an apical and a punctate cytoplasmic pattern on a HS/NK diet. On a HS/LK diet, the localization appeared similar but had greater intensity (Figure 3H). In contrast to this, OxSR1, which exhibited a diffuse DCT staining pattern in mice on a HS/NK diet, localized to both the apical membrane and puncta on HS/LK, making it similar to SPAK (Figure 3H). To confirm that this redistribution did reflect kinase activation, we used a phosphospecific antibody recognizing residues phosphorylated within the catalytic domain shared by SPAK and OxSR1 (pSPAK-T233/pOxSR1-T185) (Thastrup et al., 2012; Vitari et al., 2005). Consistent with data presented thus far, the pSPAK/pOxSR1 signal was of low intensity in mice consuming the HS/NK diet. In contrast, mice consuming the HS/LK diet exhibited an intense pSPAK/pOxSR1 signal, localized to the apical membrane and cytoplasmic puncta (Figure 3F). This indicates that on the HS/NK diet there is little activated WNK or SPAK/OxSR1 in the DCT but HS/LK diet activates both WNK and SPAK/OxSR1, causing them to appear primarily in cytoplasmic puncta; pSPAK/OxSR1 is also seen at the apical membrane, suggesting a potential sequence of activation. When costained for WNK4 and pSPAK/pOxSR1, mice on high HS/LK exhibited substantial colocalization within the puncta, but not at the apical membrane, where only pSPAK/pOxSR1 was observed (Figure 3F).
As SPAK and OxSR1 are the major activators of NCC in vivo, we used SPAK−/− and kidney-specific (KS) OxSR1−/− mice to determine if either of these kinases is essential for the observed changes in NCC. Both SPAK−/− and KS OxSR1−/− animals exhibited increased pNCC abundance on a HS/LK diet, with percentage increases similar to what was observed in wild-type mice (Figures 3I and 3J). Although mice lacking both SPAK and kidney OxSR1 still exhibited increased pNCC abundance on HS/LK compared with HS/NK (Figure 3K), the increase was substantially blunted; analysis of variance indicated a significant interaction between genotype and diet.
Extracellular Potassium Modulates NCC Directly
As plasma [K+] appeared to be the major determinant of NCC activation, we tested whether NCC is modulated directly by extracellular [K+], using cultured Flp-In NCC cells (Hoorn et al., 2011). Cells cultured in LK medium had greater pNCC abundance than cells cultured in NK (Figure 4A and Table S3). This effect appeared to be mediated by SPAK/OxSR1, as pSPAK/pOxSR1 was also more abundant in the cells cultured in LK (Figure 4B). The effects of LK were shown to be dependent on endogenous WNK kinases because WNK1 or WNK3 siRNA knockdown (these are the major WNK kinases in HEK cells) significantly attenuated the effects on pNCC and pSPAK/pOxSR1 (Figures 4C and 4D).
Figure 4. Effects of Extracellular Potassium on NCC in HEK Cells.
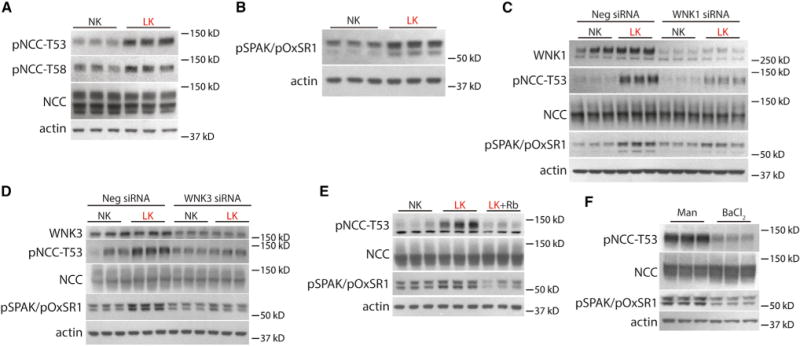
(A) Western blot of HEK cells cultured in NK or LK medium. LK increased pNCC-T53 and pNCC-T58 abundance (p < 0.01 for both by unpaired t test).
(B) Western blot of HEK cells cultured in NK or LK medium. LK increased pSPAK/pOxSR1 abundance (p < 0.05 by unpaired t test).
(C) Western blot of HEK cells cultured in NK or LK medium following WNK1 siRNA knockdown or treatment with a negative control (scramble) siRNA. WNK1 abundance was significantly decreased following knockdown (p <0.05 by unpaired t test). Cells inwhich WNK1 was knocked down exhibited a reduced increase in pNCC-T53 and pSPAK/pOxSR1 following culture in LK medium compared with cells that were transfected with a scramble siRNA. p <0.05 for interaction of K+ treatment and WNK1 abundance by two-way ANOVA.
(D) Western blot of HEK cells cultured in NK or LK medium following WNK3 siRNA knockdown or treatment with a negative control (scramble) siRNA. WNK3 abundance was significantly decreased (p < 0.05 by unpaired t test). Cells in which WNK1 was knocked down exhibited a reduced increase in pNCC-T53 and pSPAK/pOxSR1 following culture in LK medium compared with cells that were transfected with a scramble siRNA. p < 0.05 for interaction of K+ treatment and WNK3 abundance by two-way ANOVA.
(E) Western blot of HEK cells cultured in NK, LK, or LK medium with Rb+ added back. LK increased pNCC-T53 and pSPAK/pOxSR1 as before (p < 0.01 by unpaired t test corrected for multiple comparisons), whereas adding back Rb+ prevented these effects.
(F) Western blot of HEK cells cultured in mannitol (30 mM) or BaCl2 (10 mM). Treatment with BaCl2 decreased pNCC-T53 and pSPAK/pOxSR1 (p < 0.01 by unpaired t test). See Table S3 for densitometry. Representative images are shown.
As LK medium would be expected to hyperpolarize cells (Weinstein, 2005), we tested whether changes in membrane potential are required for effects on NCC. When we replaced K+ with Rb+, another cation that can traverse K+ channels and should prevent any voltage change, pNCC and pSPAK/pOxSR1 did not change; in contrast, choline chloride supplementation did not prevent effects on NCC (Figures 4E and S4A). Conversely, treating the cells with the K+ channel inhibitor Ba2+, which depolarizes cells, decreased pNCC and pSPAK/pOxSR1 (Figure 4F).
In the DCT, the inwardly rectifying K+ channel, comprising Kir4.1, likely with Kir5.1, appears to be the predominant conductive pathway for K+ exit along the basolateral membrane (Lourdel et al., 2002; Zhang et al., 2014) (Figure S4B). When Kir4.1 mutations, which lose conductive activity, are present in the DCT, a Gitelman-like tubulopathy, EAST/SeSAME syndrome, appears (Bandulik et al., 2011; Reichold et al., 2010). When EAST syndrome mutant channels were expressed in our system, the reversal potentials were less negative (Figures 5A and S4C), and the pNCC abundance was lower (Figure 5B and Table S4) than when wild-type channels were expressed.
Figure 5. Effects of Kir4.1 Mutant Channels on Whole-Cell Reversal Potential and NCC.
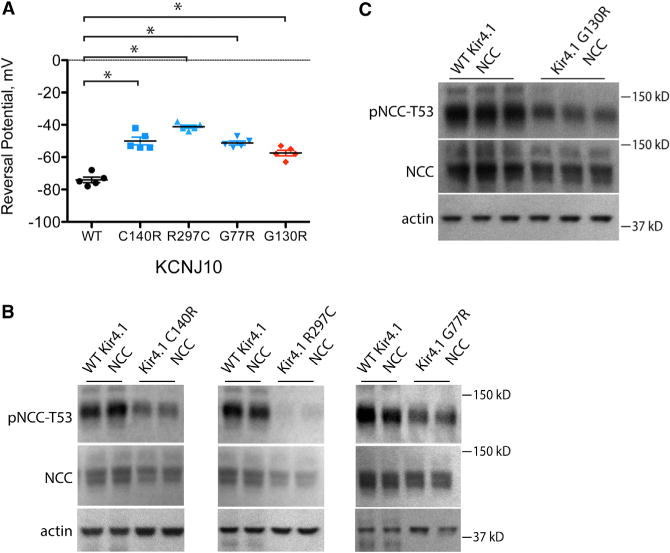
(A) Reversal potential of HEK cells expressing WT Kir4.1 or mutant Kir4.1 channels. All mutants depolarized cells compared with WT Kir4.1. *p < 0.01 by unpaired t test corrected for multiple comparisons.
(B) Western blot of HEK cells expressing WT or EAST syndrome mutant Kir4.1 channels. EAST syndrome mutants decreased pNCC-T53 compared with WT Kir4.1. p < 0.01 by unpaired t test corrected for multiple comparisons. The original image used for the first two panels is shown in Figure S4D.
(C) Western blot of HEK cells expressing WT or selectivity filter mutant Kir4.1 G130R. Expression of the mutant decreased pNCC-T53 compared with WT Kir4.1. p < 0.05 by unpaired t test. Bars represent SEM. See Table S4 for densitometry. Representative images are shown.
Mutations in the selectivity filter of a related potassium channel in adrenal cells were shown to cause hyperaldosteronism by increasing the channel Na+/K+ permeability ratio (Choi et al., 2011). As another test of the role of membrane voltage on pNCC, we mutated the homologous amino acid residue in Kir4.1 (G130). This mutated channel also depolarized the cells and decreased pNCC (Figures 5A and 5C).
Extracellular Potassium Modulates NCC via Chloride and WNK Kinases
In contemplating potential mechanisms for peritubular membrane voltage to affect NCC activity, we turned to a model of DCT cell function (Weinstein, 2005). According to the model, a decrease in extracellular [K+] reduces [Cl−] in the cell. As Cl− depletion activates NCC (Pacheco-Alvarez et al., 2006), and as WNK kinases are activated at low ambient [Cl−] (Piala et al., 2014; Ponce-Coria et al., 2008), we tested the effects of LK incubation on intracellular [Cl−], using the Cl−-sensing dye MQAE. Baseline intracellular [Cl−] was 77.6 mM, a value similar to that reported previously for human embryonic kidney 293 (HEK293) cells (Lamprecht et al., 2006). Culture in LK medium reduced intracellular [CP] significantly (Figure 6A). Conversely, intracellular [Cl−] increased with expression of each of the Kir4.1 mutants compared with WT Kir4.1 (Figure 6B). To confirm this result in a more relevant cell line, we tested the effects of LK medium on mDCT cells (Ko et al., 2012). Incubation of these cells in a LK medium also reduced intracellular [Cl−] significantly (Figure S5).
Figure 6. Role of Intracellular Chloride in Mediating K+ Effects on NCC.
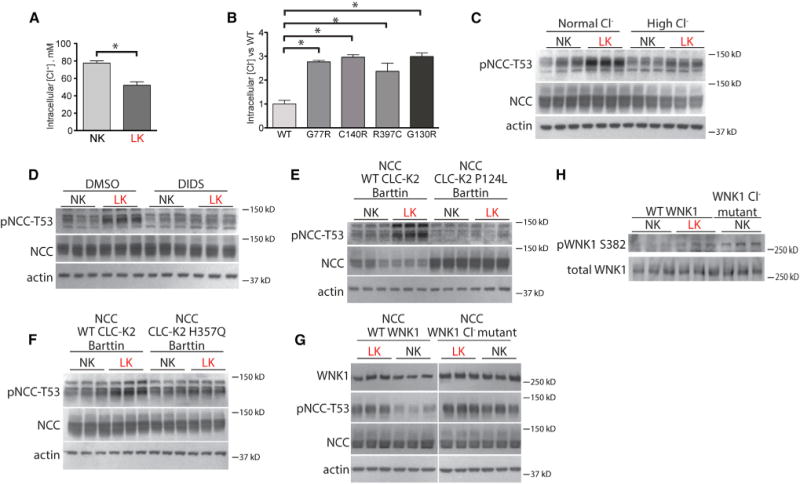
(A) Intracellular [Cl−] in HEK cells in NK or LK medium. *p < 0.05 by unpaired t test.
(B) Relative intracellular [Cr] in HEK cells expressing WT Kir4.1 or Kir4.1 mutants. *p < 0.01 by unpaired t test corrected for multiple comparisons.
(C) Western blot of HEK cells cultured in normal [Cl−] with mannitol or high [Cl−] and also treated with NK or LK medium. p< 0.05 for the interaction between K+ treatment and Cl− treatment by two-way ANOVA.
(D) Western blot of HEK cells treated with vehicle (DMSO) or DIDS and also treated with NK or LK medium. p < 0.05 for the interaction between K+ treatment and drug treatment by two-way ANOVA.
(E) Western blot of HEK cells transfected with WTCLC-K2 or Bartter syndrome type III mutant CLC-K2 P124L channels and then cultured in NK or LK medium. All cells were also transfected with NCC and barttin. p < 0.05 for the interaction between K+ treatment and CLC-K2 genotype by two-way ANOVA.
(F) Western blot of HEK cells transfected with WT CLC-K2 or Bartter syndrome type III mutant CLC-K2 H357Q channels and then cultured in NK or LK medium. All cells were also transfected with NCC and barttin. p < 0.05 for the interaction between K+ treatment and CLC-K2 genotype by two-way ANOVA.
(G) Western blot of HEK cells transfected with WT or WNK1 L369FL371F(WNK1 Cl− mutant) and then cultured in LK or NK medium. Cells expressing WTWNK1, but not WNK1 Cl− mutant, exhibited substantially lower pNCC-T53 abundance when cultured in NK compared with LK medium. p < 0.05 for the interaction between K+ treatment and WNK1 genotype by two-way ANOVA. WNK1 was detected with anti-myc antibody. See Figure S6C for the original image.
(H) Western blot of HEK cells transfected with WTWNK1 cultured in NK or LK medium and WNK1 L369F L371F in NK medium. pWNK1 abundance increased in both the WT WNK1 LK group and the WNK1 L369F L371F group. p < 0.05 for unpaired t test corrected for multiple comparisons. Total WNK1 abundance obtained before adding non-phospho-WNK1 peptide to block antibodies recognizing unphosphorylated WNK1. pWNK1 S382 abundance was obtained after addition of the non-phospho-WNK1 peptide. Bars represent mean ± SEM. See Table S5 for densitometry.
To test whether intracellular [Cl−] must decline for LK to stimulate NCC, we took three approaches. First, we showed that cells incubated in high [Cl−] medium exhibited an attenuated pNCC response to LK exposure (Figure 6C and Table S5). We next showed that the Cl− channel/antiporter inhibitor, DIDS, also attenuated the response to LK (Figure 6D). Finally, we compared the effects of transfecting wild-type and mutant ClC-K2, a Cl− channel highly expressed along the DCT. Mutations in the human ortholog, CLCNKB, cause Bartter syndrome type III, which can include the Gitelman-like features hypocalciuria and hypomagnesemia. Compared with cells expressing WT ClC-K2 channels, the LK-induced increase in pNCC was attenuated when cells were transfected with Bartter syndrome type III ClC-K2 mutants (Figures 6E, 6F, S6A, and S6B).
Recently, Piala et al. (2014) reported that Cl− inhibits WNK1 by binding to key residues in its catalytic domain. We therefore expressed WNK1 in which two of these residues, L369 and L371, were mutated to phenylalanine (WNK1 L369F L371F). In LK medium, pNCC was similar in cells expressing WT WNK1 and mutant WNK1, indicating activated WNK1. In contrast, when incubated in NK medium, the abundance of pNCC was lower in cells expressing WT, but not in cells expressing mutant WNK1 (Figure 6G). To confirm that LK medium activates WNK1, we compared the abundance of activated (phosphorylated) WNK1 in cells transfected with wild-type WNK1 in NK and LK medium and transfected with WNK1 L369F L371F in NK medium (Figure 6H). The results confirm that LK medium activates WNK1 in cells, just as mutation of the Cl− binding sites does.
The results indicate that plasma [K+], acting through intracellular [Cl−], likely affects NCC activity via WNK kinases; this effect should have important physiological effects on distal K+ secretion. We therefore introduced [Cl−] sensitivity into the previously developed DCT model (Figure 7A). In the calculations for Figure 7, the fractional decrease in luminal membrane NCC activity (compared with baseline) is 4-fold greater than the factional increase in cytosolic [Cl−] compared with baseline (see Experimental Procedures). As in the prior model (black lines), reducing peritubular [K+], within the physiological range, hyperpolarizes the peritubular membrane (Figure 7A) and reduces intracellular [Cl−] (Figure 7B). Now, however, fluxes of Na+ and Cl− (Figure 7B) are affected more substantially by the change in peritubular [K+]. When these effects are integrated along a modeled tubule, the effect of peritubular [K+] to modulate Na+ delivery to the CNT is greatly enhanced (Figure 7D).
Figure 7. Effects of Cl− Sensitivity on Modeled DCT Function.
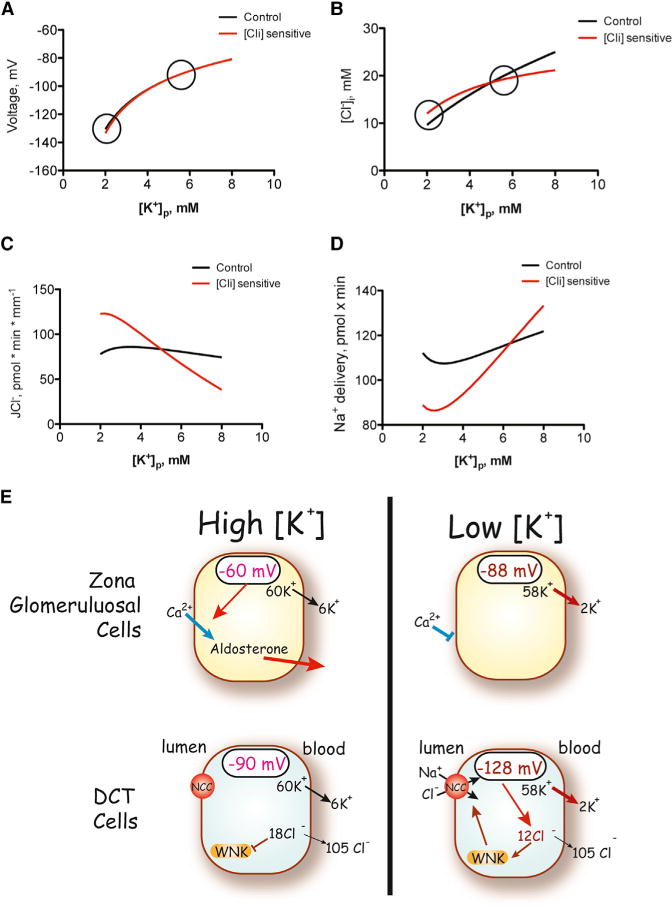
(A–C) Membrane voltage (A), intracellular chloride concentration (B), and transepithelial chloride flux (JCl) (C) as functions of peritubular [K+] concentration. Circled are values at [K+]P, as shown in the cartoon.
(D) The results of peritubular [K+] concentration on sodium delivery to the CNT. The black lines are taken from Weinstein (2005). The red lines show results modified by the inclusion of NCC sensitivity to intracellular [Cl−].
(E) Cartoon showing the effects of plasma [K+] on cells from the adrenal zona glomerulosa and renal distal convoluted tubule. Directionally similar changes in membrane voltage elicit opposite effects to activate or inhibit cell activity. Note that, for simplicity, effects of angiotensin II have been omitted. In this case, angiotensin II may have similar effects to stimulate both cell populations.
DISCUSSION
WNK kinases comprise a molecular switch in the mammalian distal nephron that modulates systemic electrolyte balance and blood pressure. This switch is most potent within DCT cells, where it acts through the intermediary kinases SPAK and OxSR1 to modulate NCC. When switched on, as in WNK4-FHHt, hypertrophy of the DCT ensues, along with increased pNCC abundance (Subramanya and Ellison, 2014). When switched on by anomalous WNK1 expression, as in WNK1-FHHt, pNCC abundance is also increased (Vidal-Petiot et al., 2013). In Gitelman syndrome, although the switch is likely on, it cannot prevent K+ losses because the effector, NCC, is defective, and K+ wasting is therefore relentless. Although these clinical syndromes indicate that WNKs, within the DCT, play a central role in systemic K+ balance, the mechanisms and activators of this pathway have been unclear. WNK kinases are Cl−, but not K+, sensitive (Piala et al., 2014; Ponce-Coria et al., 2008), and the DCT itself secretes very little K+ (Subramanya and Ellison, 2014). The current results show the dominant role that plasma [K+] plays in regulating NCC and indicate that extracellular [K+] affects NCC indirectly, by modulating intracellular [Cl−]. They indicate that plasma [K+] affects K+ secretion by altering Na+ delivery to the CNT and suggest that the health benefits of dietary K+ supplementation are dependent on NCC.
A blood pressure lowering effect of dietary K+ in humans was reported in 1928 (Addison, 1928). Yet, dietary K+ has typically received less attention than salt, perhaps because the traditional body fluid model for blood pressure control assigns primacy to Na+. It is now clear that dietary K+ intake associates inversely with blood pressure (Mente et al., 2014), often just as strongly as dietary NaCl does. Recent reviews suggest myriad and complex mechanisms for effects of K+ on arterial pressure, including effects on the sympathetic nervous system, on the vasculature, and on circulating factors, such as endogenous ouabain; K+-induced natriuresis is rarely cited as essential (Adrogue and Madias, 2007; He et al., 2010).
NCC is activated during ECF volume depletion via angiotensin II, aldosterone, and renal nerves (Terker et al., 2014). The current results confirm that dietary K+ intake also modulates pNCC abundance and show that dietary K+ intake is more powerful in this regard than ECF volume. These effects on NCC lead to blood pressure changes, which are secondary to NCC activity. They are mediated largely by changes in plasma [K+], as the effects were reproduced when plasma [K+] was altered using drugs or diet, a conclusion consistent with K+ infusion experiments (Rengarajan et al., 2014). In contrast, NKCC2, the major transporter mediating Na+ reabsorption in the thick ascending limb (TAL), was affected differently, suggesting that the DCT responds preferentially to K+, while the TAL responds to other stimuli.
The results indicate that extracellular [K+], independent of hormonal effects, directly modulates the WNK-SPAK/OxSR1-NCC axis. Our cell model provides insight into the mechanisms involved, as reducing extracellular [K+] activated WNK1, SPAK/OxSR1, and NCC in cells, and the effect required the WNK kinases. In vivo, the effect of LK on pNCC abundance was substantially attenuated in mice lacking both SPAK and OxSR1, supporting a role for these kinases. The remaining stimulation observed in these animals could have resulted from WNK- and Cab39-dependent, but SPAK-independent, activation (Ponce-Coria et al., 2014). We documented that changes in membrane voltage are involved, as maneuvers that alter membrane voltage produced changes in pNCC abundance consistent with voltage dependence. In contemplating how membrane voltage might affect WNK kinases, we reflected on their sensitivity to Cl−. Recently, crystallographic analysis revealed that Cl− binding to WNK1 inhibits its autophosphorylation and activation (Piala et al., 2014). The DCT model proposed by Weinstein predicts that reducing peritubular [K+] lowers intracellular [Cl−] (Weinstein, 2005). In that model, decreased peritubular [K+] could enhance Cl− exit both via peritubular potassium chloride cotransport and, by hyperpolarizing the cell, via Cl− channels. We therefore used several approaches to determine whether the effects of extracellular [K+] are mediated by changes in intracellular [Cl−]; all were consistent with a Cl−-dependent effect. Finally, we also show that overexpressing a WNK1 mutant with altered Cl-binding residues abrogates effects of [K+] on NCC. When we introduced Cl− sensing into the DCT model (Figure 7A), the potent effects of physiological variation in plasma [K+] on Na+ delivery to K+ secretory segments of the nephron become clear. Although increasing NaCl delivery to distal segments may have limited effects on K+ secretion during normokalemic euvolemia (Hunter et al., 2014), the effects will be large when these segments are primed by exposure to aldosterone (Weinstein, 2012), as occurs during hyperkalemia.
Our in vivo and in vitro data are largely concordant, suggesting that the HEK-NCC cell model is suitable for mechanistic studies, but they are corroborated by our mDCT studies and by reports that peritubular [K+] affects the reversal potential of intact DCT segments (Lourdel et al., 2002). Recently, Wang and colleagues showed that DCT cells from neonatal mice lacking KCNJ10 are substantially depolarized (Zhang et al., 2014), supporting the relevance of our results. Furthermore, Markadieu et al. (2012) showed that incubating primary cultures of mouse DCT cells in low Cl− hypotonic medium increases thiazide-sensitive 22Na flux across monolayers. Although Sorensen et al. (2013) reported that high extracellular [K+] did not affect the pNCC abundance of sorted mouse DCT cells in vitro, an effect may have been missed for technical reasons.
Two important differences between HEK293 cells and DCT cells should be emphasized, however. First, NCC activity is driven largely by endogenous full-length WNK1 and WNK3 in HEK cells, whereas WNK4 plays a central role in vivo (Takahashi et al., 2014). Given the remarkable shift in WNK4 expression pattern, the current data suggest that WNK4 is involved in sensing [K+] in vivo, but they do not exclude roles for WNK1 and WNK3. Our WNK1 antibody does not detect kidney-specific WNK1, which comprises more than 90% of WNK1 message in the DCT (Vidal-Petiot et al., 2013), and WNK3 did accumulate in the puncta, in the HS/LK mice (data not shown). Additionally, as WNK kinases regulate ROMK, and probably ENaC (McCormick and Ellison, 2011), effects on these channels might also contribute. Second, [Cl]i in HEK cells is substantially higher than in the DCT (Lamprecht et al., 2006). Both WNKs, however, contain the Cl−-sensing motif (Piala et al., 2014), suggesting that the same signals activate both. Thus, our results help to resolve the paradox that cell-based and in vitro studies have indicated that WNK kinases are primarily Cl− sensitive, whereas in vivo studies suggest regulation by plasma K+.
The current work adds to evidence that NCC is activated by LK diet and that the effects of LK diet on NCC activity are enhanced by HS intake. While this conclusion seems counterintuitive, it corroborates results from others (Vitzthum et al., 2014). Further, it likely accounts for the observation that HK diet has a greater effect to lower pressure in humans during high, compared with low, NaCl intake (Sacks et al., 2001). As the effects of HS/LK diet on arterial pressure are strikingly blunted when NCC has been deleted genetically (Figure 2C), NCC plays an essential and nonredundant role in the antihypertensive effects of dietary K+. As LK diet in humans also stimulates NCC, the results are relevant to human health.
Overall, our results indicate that dietary K+ is the dominant force dictating NCC activity. Recently, van der Lubbe et al. (2013) reported that a LS/HK diet, the inverse manipulation to our dietary model, decreased NCC levels and induced a natriuresis, even in the setting of continued low NaCl intake. This brings the total to three models where K+ overrides ECF volume and the renin-angiotensin-aldosterone system (RAAS) status to dictate NCC activity (van der Lubbe et al., 2013; Vitzthum et al., 2014). Although effects of LK are consistent between studies, the effects of HK depend on the accompanying anion (Castañeda-Bueno et al., 2014).
In EAST/SeSAME syndrome, ECF volume depletion and hypokalemia should activate NCC (Bandulik et al., 2011). Instead, the effects of K+-channel dysfunction on membrane voltage, and in turn intracellular [Cl−], suppress NCC activity, a finding consistent with decreased pNCC abundance in KCNJ10−/− mice (Zhang et al., 2014). The current results also help to explain the phenotype of individuals with Bartter syndrome type III, caused by mutations in CLCNKB. In contrast to other forms of Bartter syndrome, where NCC is strikingly activated (Wagner et al., 2008), the individuals exhibit a combined loop-DCT phenotype, with suppressed DCT activity. Here, we show that expression of disease-causing CLC-K2 mutants prevents LK from activating NCC, likely because intracellular [Cl−] remains high.
Prevention of recurrent nephrolithiasis often includes supplemental dietary K+. Potassium deficit enhances urinary calcium excretion, an effect dependent on ECF volume (Lemann et al., 1995). The current results show that NCC activation plays an essential role in the effects of dietary K+ on urinary calcium excretion; not only was K+-induced calcium retention lacking in Slc12a3−/− mice, but the effects of dietary K+ on calcium excretion were reversed. This suggests that effects of dietary K+ supplementation and thiazide diuretics on the same transport pathway, NCC, mediate their similar, and complementary, effects on hypercalciuria.
Lastly, the results establish the DCT as a renal K+ sensor, which acts in concert with the adrenal gland to preserve K+ homeostasis. In this respect, DCT cells and adrenal zona glomerulosa cells both sense the same signal, membrane voltage, but respond in opposite directions (Figure 7C). A rise in plasma [K+] depolarizes zona glomerulosa cells, as the membrane conductance is dominated by KCNJ5 and TASK-2. This opens voltage-activated calcium channels and stimulates aldosterone production and release. In the kidney, a rise in plasma [K+] also depolarizes DCT cells, as the basolateral membrane conductance is dominated by KCNJ10. In this case, however, the ensuing rise in intracellular [Cl−] binds to WNK kinases, switching them into the off position and shutting down NCC. To produce kaliuresis, events in adrenal and DCT must occur pari passu. This is assured by having the same signal, extracellular [K+], regulate both cell types. As adrenal cells respond to the ECF volume signal angiotensin II, DCT cells likely do too. In this case, however, the signals in the two organs must be consonant. Thus Cl−-sensing WNK kinases, which likely evolved to protect cell volume and osmolality (McCormick and Ellison, 2011), now play central roles in systemic K+ balance in mammals. That systemic K+ balance is defended more avidly than NaCl balance (see above and Vitzthum et al., 2014) indicates its physiological importance. As hyperkalemia is often a major factor limiting therapeutic RAAS blockade in humans, these insights provide opportunities to intervene.
EXPERIMENTAL PROCEDURES
Antibodies
The antibodies employed have all been characterized and reported before. Details are provided in the Supplemental Information.
Animals
Studies were approved by Oregon Health and Science University’s Animal Care and Usage Committee (Protocol IS918). All mice were 12–24 weeks old, 25–30 g, and had a C57Bl/6 background, except for NCC−/− mice, which were on a BALB/c background. All strains are backcrossed to the appropriate wild-type mice every ten generations to maintain genetic backgrounds. Additional details can be found in the Supplemental Information.
PCR Genotyping
Tail-snip genotyping for NCC−/− (Schultheis et al., 1998), KS OxSR1−/−, and AT1a−/− mice (Terker et al., 2014) used previously reported primers. A previously reported approach was used (McCormick et al., 2011) to genotype SPAK−/− mice. Details are provided in the Supplemental Information.
Animal Diets
All diets used in K+ studies were prepared in our lab by modifying a K+-deficient diet (Harlan Laboratories K+-deficient diet; TD.88239). High salt was obtained by adding NaCl to make a 6% NaCl diet. Low-K+ diets had no additional K+ added, and normal K+ was obtained by adding KCl to make a 1% K+ diet. Low-NaCl diet used was also purchased from Harlan Laboratories (Na+-deficient diet; TD.90228).
Blood Pressure Measurement
Noninvasive systolic blood pressures were measured with tail cuffs using volumetric pressure recording (CODA-16; Kent Scientific); details are provided in the Supplemental Information. Radiotelemetry studies were performed using carotid probes. Animals were allowed to recover from surgery for 1 week, during which the animals were fed a HS/NK diet. Following the 7-day recovery period, animals were changed to a HS/LK diet.
Blood Analysis
Whole blood was collected via cardiac puncture following indicated treatments. Plasma K+ and hematocrit values were obtained by iSTAT just after collection by loading whole blood into a chem 8 cartridge (Abbot Point of Care). The remaining blood was centrifuged for 5 min at 2000 × g in heparinized tubes. Plasma was removed and frozen at −80°C until future use. Plasma aldosterone was measured by ELISA (IBL America), and plasma angiotensin II was measured by EIA (Phoenix Pharmaceuticals).
Urinary Electrolyte Measurement
Mice were maintained on HS/NK for 7 days. For the final 3 days, mice were individually housed in metabolic cages, and urine was collected under water-saturated light mineral oil over the final 24 hr period. Animals were then switched to the HS/LK diet, and the procedure was repeated. Body weight was monitored during the metabolic cage period. Urine was frozen at −20°C until Na+ was measured by flame photometry and Ca2+ by o-Cresolphthalein Complexone method (Pointe Scientific).
Urinary Exosome Preparation in Mice
Wild-type animals were fed either a high-salt/normal-K+ or high-salt/low-K+ diet for 7 days. For the last 3 days, animals were housed in metabolic cages, and urine was collected under water-saturated light mineral oil over the final 24 hr period. Exosomes were then obtained from one-third of the total urine volume according to a previously published protocol (van der Lubbe et al., 2012). The entire exosome preparation was then loaded onto a 3%–8% Tris-acetate gel (Invitrogen), and western blot was performed.
Immunoblotting
Mice were maintained on indicated diets for 7–10 days or treated with amiloride (50 mg/l drinking water) for 5–7 days, after which kidneys were harvested and snap-frozen in liquid nitrogen. Kidneys were then homogenized on ice in chilled buffer containing protease and phosphatase inhibitors. Protein (20–80 μg) was separated on a 4%–12% Bis-Tris gel or a 3%–8% Tris-acetate gel (Invitrogen). Densitometry was performed using ImageJ (http://rsbweb.nih.gov/ij/).
Immunofluorescence
Mice were anesthetized and kidneys perfusion-fixed by retrograde abdominal aortic perfusion of 3% paraformaldehyde in PBS (pH 7.4). After cryoprotection with 800 mOsm sucrose and freezing, 5 μm sections were cut and stored at −80°C until use. Details are provided in the Supplemental Information.
Cell Culture
Flp-In NCC cells as reported (Hoorn et al., 2011) were used for experiments in which only NCC expression was required. NCC was cotransfected in HEK293 cells using Lipofectamine (Invitrogen) in studies that required coexpression of NCC with other proteins or in siRNA studies. Additional information about transfection and conditions is provided in the Supplemental Information.
Intracellular Cl− Measurement
Cells were plated in poly D-Lysine coated 96-well plates on day 0 (Becton Dickinson Labware). On day 1, the indicated medium was added, and measurements were performed on day 2. For Kir4.1 experiments, stably transfected cell lines were made by subcloning wild-type or mutant Kir4.1 into pIRES-puro3 (Clontech) and selecting cells with puromycin (1 ug/ml). Stable cells were plated in normal K+ Dulbecco’s modified Eagle’s medium (DMEM), and Cl− was measured. Additional details are provided in the Supplemental Information.
Human Studies
Human studies were approved by Oregon Health & Science University’s Internal Review Board (Protocol IRB9934). Volunteers consumed a high-salt/low-K+ diet and a high-salt/K+-replete diet, based on food package nutritional facts label or the USDA nutrient database (http://ndb.nal.usda.gov/), each for a 4-day period. Additional information is included in the Supplemental Information.
Measurement of K+ Reversal Potentials
HEK293T cells (American Type Culture Collection) were grown in DMEM (Invitrogen) supplemented with 10% FBS (Invitrogen) in 5% CO2 and 95% air at 37°C. Cells were grown to 50%–70% confluence for transfection, and the corresponding cDNAs were simultaneously applied to the cells using TurboFect transfection reagent (Fermentas).
For the measurement of K+ reversal potential, an Axon 200A patch-clamp amplifier was used. We carried out the perforated whole-cell patch-clamp experiments on Kcnj10/mutant transfected HEK293T cells at room temperature. Details are provided in the Supplemental Information.
Model of Distal Convoluted Tubule Cells
Model calculations were performed using the DCT model of Weinstein (2005), with modified model parameters provided in the Supplemental Information.
Statistical Analyses
All values are expressed as mean ± SEM. Between-group comparisons were performed using Student’s t test (paired or unpaired, as indicated in figure legends), corrected for multiple comparisons when appropriate. Variable interactions were determined by two-way ANOVA. Body weight was analyzed by one-way ANOVA with repeated measures.
Supplementary Material
Acknowledgments
This work was funded in part by grants from the NIH (DK51496 to D.H.E., DK54983 to W.-H.W., NIH DK-29857 to A.M.W., and DK098141 to J.A.M.) and the Department of Veterans Affairs (1I0BX002228-01A1 to D.H.E.). A.S.T. (3PRE14090030) and R.A.L. (14PRE18330021) were recipients of American Heart Association predoctoral fellowship awards. Chong Zhang was supported by the Shanghai Municipal Education Commission and a Shanghai Jiao Tong University K.C. Wong Medical Fellowship fund. This work was performed by A.S.T. in partial fulfillment for a PhD in Cell & Developmental Biology from Oregon Health and Science University. It was also supported by Shared Instrumentation Grant Number S10-RR023432 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and its contents are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH. We wish to acknowledge Aurelie Snyder and the Advanced Light Microscopy Core at The Jungers Center, OHSU, for technical assistance.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, six figures, and five tables and can be found with this article online at http://dx.doi.org/10.1016/j.cmet.2014.12.006.
AUTHOR CONTRIBUTIONS
A.S.T. conceived the study, carried out most of the experiments, analyzed the data, and wrote and edited the manuscript. Chong Zhang conducted the WNK1 cell studies. J.A.M. generated the KS OxSR1−/− and SPAK−/−/KS OxSR1−/− mice. R.A.L and D.A.S. performed animal surgeries. Chengiao Zhang and W.-H.W. performed whole-cell patch-clamp experiments. N.P.M. and H.J.P. assisted with animal studies. Y.F. and D.M.C. set up the intracellular chloride measurement assay. A.M.W. generated the DCT computer model. C.-LY. helped plan experiments and supervised the work. D.H.E. conceived the study, supervised the work, and edited the manuscript.
References
- Addison WLT. The Use of Sodium Chloride, Potassium Chloride, Sodium Bromide, and Potassium Bromide in Cases of Arterial Hypertension which are Amenable to Potassium Chloride. Can Med Assoc J. 1928;18:281–285. [PMC free article] [PubMed] [Google Scholar]
- Adrogué HJ, Madias NE. Sodium and potassium in the pathogenesis of hypertension. N Engl J Med. 2007;356:1966–1978. doi: 10.1056/NEJMra064486. [DOI] [PubMed] [Google Scholar]
- Bandulik S, Schmidt K, Bockenhauer D, Zdebik AA, Humberg E, Kleta R, Warth R, Reichold M. The salt-wasting phenotype of EAST syndrome, a disease with multifaceted symptoms linked to the KCNJ10 K+ channel. Pflugers Arch. 2011;461:423–435. doi: 10.1007/s00424-010-0915-0. [DOI] [PubMed] [Google Scholar]
- Castañeda-Bueno M, Cervantes-Perez LG, Rojas-Vega L, Arroyo-Garza I, Vázquez N, Moreno E, Gamba G. Modulation of NCC activity by low and high K(+) intake: insights into the signaling pathways involved. Am J Physiol Renal Physiol. 2014;306:F1507–F1519. doi: 10.1152/ajprenal.00255.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M, Scholl UI, Yue P, Björklund P, Zhao B, Nelson-Williams C, Ji W, Cho Y, Patel A, Men CJ, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science. 2011;331:768–772. doi: 10.1126/science.1198785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frindt G, Palmer LG. Effects of dietary K on cell-surface expression of renal ion channels and transporters. Am J Physiol Renal Physiol. 2010;299:F890–F897. doi: 10.1152/ajprenal.00323.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He FJ, Marciniak M, Carney C, Markandu ND, Anand V, Fraser WD, Dalton RN, Kaski JC, MacGregor GA. Effects of potassium chloride and potassium bicarbonate on endothelial function, cardiovascular risk factors, and bone turnover in mild hypertensives. Hypertension. 2010;55:681–688. doi: 10.1161/HYPERTENSIONAHA.109.147488. [DOI] [PubMed] [Google Scholar]
- Hoorn EJ, Pisitkun T, Zietse R, Gross P, Frokiaer J, Wang NS, Gonzales PA, Star RA, Knepper MA. Prospects for urinary proteomics: exosomes as a source of urinary biomarkers. Nephrology (Carlton) 2005;10:283–290. doi: 10.1111/j.1440-1797.2005.00387.x. [DOI] [PubMed] [Google Scholar]
- Hoorn EJ, Walsh SB, McCormick JA, Fürstenberg A, Yang CL, Roeschel T, Paliege A, Howie AJ, Conley J, Bachmann S, et al. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med. 2011;17:1304–1309. doi: 10.1038/nm.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter RW, Craigie E, Homer NZ, Mullins JJ, Bailey MA. Acute inhibition of NCC does not activate distal electrogenic Na+ reabsorption or kaliuresis. Am J Physiol Renal Physiol. 2014;306:F457–F467. doi: 10.1152/ajprenal.00339.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko B, Mistry AC, Hanson L, Mallick R, Cooke LL, Hack BK, Cunningham P, Hoover RS. A new model of the distal convoluted tubule. Am J Physiol Renal Physiol. 2012;303:F700–F710. doi: 10.1152/ajprenal.00139.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamprecht G, Schaefer J, Dietz K, Gregor M. Chloride and bicarbonate have similar affinities to the intestinal anion exchanger DRA (down regulated in adenoma) Pflugers Arch. 2006;452:307–315. doi: 10.1007/s00424-006-0049-6. [DOI] [PubMed] [Google Scholar]
- Lemann J, Jr, Pleuss JA, Hornick L, Hoffman RG. Dietary NaCl-restriction prevents the calciuria of KCl-deprivation and blunts the calciuria of KHCO3-deprivation in healthy adults. Kidney Int. 1995;47:899–906. doi: 10.1038/ki.1995.134. [DOI] [PubMed] [Google Scholar]
- Lourdel S, Paulais M, Cluzeaud F, Bens M, Tanemoto M, Kurachi Y, Vandewalle A, Teulon J. An inward rectifier K(+) channel at the basolateral membrane of the mouse distal convoluted tubule: similarities with Kir4-Kir5.1 heteromeric channels. J Physiol. 2002;538:391–404. doi: 10.1113/jphysiol.2001.012961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markadieu N, San-Cristobal P, Nair AV, Verkaart S, Lenssen E, Tudpor K, van Zeeland F, Loffing J, Bindels RJ, Hoenderop JG. A primary culture of distal convoluted tubules expressing functional thiazide-sensitive NaCl transport. Am J Physiol Renal Physiol. 2012;303:F886–F892. doi: 10.1152/ajprenal.00114.2012. [DOI] [PubMed] [Google Scholar]
- McCormick JA, Ellison DH. The WNKs: atypical protein kinases with pleiotropic actions. Physiol Rev. 2011;91:177–219. doi: 10.1152/physrev.00017.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick JA, Mutig K, Nelson JH, Saritas T, Hoorn EJ, Yang CL, Rogers S, Curry J, Delpire E, Bachmann S, Ellison DH. A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab. 2011;14:352–364. doi: 10.1016/j.cmet.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mente A, O’Donnell MJ, Rangarajan S, McQueen MJ, Poirier P, Wielgosz A, Morrison H, Li W, Wang X, Di C, et al. Association of urinary sodium and potassium excretion with blood pressure. N Engl J Med. 2014;371:601–611. doi: 10.1056/NEJMoa1311989. [DOI] [PubMed] [Google Scholar]
- O’Donnell M, Mente A, Rangarajan S, McQueen MJ, Wang X, Liu L, Yan H, Lee SF, Mony P, Devanath A, et al. Urinary sodium and potassium excretion, mortality, and cardiovascular events. N Engl J Med. 2014;371:612–623. doi: 10.1056/NEJMoa1311889. [DOI] [PubMed] [Google Scholar]
- Pacheco-Alvarez D, Cristóbal PS, Meade P, Moreno E, Vazquez N, Muñoz E, Díaz A, Juárez ME, Giménez I, Gamba G. The Na+:Cl− cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J Biol Chem. 2006;281:28755–28763. doi: 10.1074/jbc.M603773200. [DOI] [PubMed] [Google Scholar]
- Piala AT, Moon TM, Akella R, He H, Cobb MH, Goldsmith EJ. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci Signal. 2014;7:ra41. doi: 10.1126/scisignal.2005050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisitkun T, Shen RF, Knepper MA. Identification and proteomic profiling of exosomes in human urine. Proc Natl Acad Sci USA. 2004;101:13368–13373. doi: 10.1073/pnas.0403453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponce-Coria J, San-Cristobal P, Kahle KT, Vazquez N, Pacheco-Alvarez D, de Los Heros P, Juárez P, Muñoz E, Michel G, Bobadilla NA, et al. Regulation of NKCC2 by a chloride-sensing mechanism involving the WNK3 and SPAK kinases. Proc Natl Acad Sci USA. 2008;105:8458–8463. doi: 10.1073/pnas.0802966105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponce-Coria J, Markadieu N, Austin TM, Flammang L, Rios K, Welling PA, Delpire E. A novel Ste20-related proline/alanine-rich kinase (SPAK)-independent pathway involving calcium-binding protein 39 (Cab39) and serine threonine kinase with no lysine member 4 (WNK4) in the activation of Na-K-Cl cotransporters. J Biol Chem. 2014;289:17680–17688. doi: 10.1074/jbc.M113.540518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichold M, Zdebik AA, Lieberer E, Rapedius M, Schmidt K, Bandulik S, Sterner C, Tegtmeier I, Penton D, Baukrowitz T, et al. KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc Natl Acad Sci USA. 2010;107:14490–14495. doi: 10.1073/pnas.1003072107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengarajan S, Lee DH, Oh YT, Delpire E, Youn JH, McDonough AA. Increasing plasma [K+] by intravenous potassium infusion reduces NCC phosphorylation and drives kaliuresis and natriuresis. Am J Physiol Renal Physiol. 2014;306:F1059–F1068. doi: 10.1152/ajprenal.00015.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacks FM, Svetkey LP, Vollmer WM, Appel LJ, Bray GA, Harsha D, Obarzanek E, Conlin PR, Miller ER, 3rd, Simons-Morton DG, et al. DASH-Sodium Collaborative Research Group Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. N Engl J Med. 2001;344:3–10. doi: 10.1056/NEJM200101043440101. [DOI] [PubMed] [Google Scholar]
- Schultheis PJ, Lorenz JN, Meneton P, Nieman ML, Riddle TM, Flagella M, Duffy JJ, Doetschman T, Miller ML, Shull GE. Phenotype resembling Gitelman’s syndrome in mice lacking the apical Na+-Cl− cotransporter of the distal convoluted tubule. J Biol Chem. 1998;273:29150–29155. doi: 10.1074/jbc.273.44.29150. [DOI] [PubMed] [Google Scholar]
- Sorensen MV, Grossmann S, Roesinger M, Gresko N, Todkar AP, Barmettler G, Ziegler U, Odermatt A, Loffing-Cueni D, Loffing J. Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int. 2013;83:811–824. doi: 10.1038/ki.2013.14. [DOI] [PubMed] [Google Scholar]
- Subramanya AR, Ellison DH. Distal convoluted tubule. Clin J Am Soc Nephrol. 2014;9:2147–2163. doi: 10.2215/CJN.05920613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi D, Mori T, Nomura N, Khan MZ, Araki Y, Zeniya M, Sohara E, Rai T, Sasaki S, Uchida S. WNK4 is the major WNK positively regulating NCC in the mouse kidney. Biosci Rep. 2014;34:e00107. doi: 10.1042/BSR20140047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terker AS, Yang CL, McCormick JA, Meermeier NP, Rogers SL, Grossmann S, Trompf K, Delpire E, Loffing J, Ellison DH. Sympathetic stimulation of thiazide-sensitive sodium chloride cotransport in the generation of salt-sensitive hypertension. Hypertension. 2014;64:178–184. doi: 10.1161/HYPERTENSIONAHA.114.03335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thastrup JO, Rafiqi FH, Vitari AC, Pozo-Guisado E, Deak M, Mehellou Y, Alessi DR. SPAK/OSR1 regulate NKCC1 and WNK activity: analysis of WNK isoform interactions and activation by T-loop trans-autophosphorylation. Biochem J. 2012;441:325–337. doi: 10.1042/BJ20111879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallon V, Schroth J, Lang F, Kuhl D, Uchida S. Expression and phosphorylation of the Na+-Cl− cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am J Physiol Renal Physiol. 2009;297:F704–F712. doi: 10.1152/ajprenal.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lubbe N, Jansen PM, Salih M, Fenton RA, van den Meiracker AH, Danser AH, Zietse R, Hoorn EJ. The phosphorylated sodium chloride cotransporter in urinary exosomes is superior to prostasin as a marker for aldosteronism. Hypertension. 2012;60:741–748. doi: 10.1161/HYPERTENSIONAHA.112.198135. [DOI] [PubMed] [Google Scholar]
- van der Lubbe N, Moes AD, Rosenbaek LL, Schoep S, Meima ME, Danser AH, Fenton RA, Zietse R, Hoorn EJ. K+-induced natriuresis is preserved during Na+ depletion and accompanied by inhibition of the Na+-Cl− cotransporter. Am J Physiol Renal Physiol. 2013;305:F1177–F1188. doi: 10.1152/ajprenal.00201.2013. [DOI] [PubMed] [Google Scholar]
- Vidal-Petiot E, Elvira-Matelot E, Mutig K, Soukaseum C, Baudrie V, Wu S, Cheval L, Huc E, Cambillau M, Bachmann S, et al. WNK1-related Familial Hyperkalemic Hypertension results from an increased expression of L-WNK1 specifically in the distal nephron. Proc Natl Acad Sci USA. 2013;110:14366–14371. doi: 10.1073/pnas.1304230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitari AC, Deak M, Morrice NA, Alessi DR. The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J. 2005;391:17–24. doi: 10.1042/BJ20051180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitzthum H, Seniuk A, Schulte LH, Müller ML, Hetz H, Ehmke H. Functional coupling of renal K+ and Na+ handling causes high blood pressure in Na+ replete mice. J Physiol. 2014;592:1139–1157. doi: 10.1113/jphysiol.2013.266924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner CA, Loffing-Cueni D, Yan Q, Schulz N, Fakitsas P, Carrel M, Wang T, Verrey F, Geibel JP, Giebisch G, et al. Mouse model of type II Bartter’s syndrome. II. Altered expression of renal sodium- and water-transporting proteins. Am J Physiol Renal Physiol. 2008;294:F1373–F1380. doi: 10.1152/ajprenal.00613.2007. [DOI] [PubMed] [Google Scholar]
- Weinstein AM. A mathematical model of rat distal convoluted tubule. I. Cotransporter function in early DCT. Am J Physiol Renal Physiol. 2005;289:F699–F720. doi: 10.1152/ajprenal.00043.2005. [DOI] [PubMed] [Google Scholar]
- Weinstein AM. Potassium excretion during antinatriuresis: perspective from a distal nephron model. Am J Physiol Renal Physiol. 2012;302:F658–F673. doi: 10.1152/ajprenal.00528.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Wang L, Zhang J, Su XT, Lin DH, Scholl UI, Giebisch G, Lifton RP, Wang WH. KCNJ10 determines the expression of the apical Na-Cl cotransporter (NCC) in the early distal convoluted tubule (DCT1) Proc Natl Acad Sci USA. 2014;111:11864–11869. doi: 10.1073/pnas.1411705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.