

Abstract
Nanocarriers play an important role in targeted cancer chemotherapy. The optimal nanocarrier delivery system should provide efficient and highly specific recognition of the target cells and rapid internalization of the therapeutic cargo to reduce systemic toxicity as well as to increase the cytotoxicity to cancer cells. To this end, we developed a two-step, two-component targeted delivery system based on antibody and drug-loaded nanocarrier that uses bioorthogonal click reactions for specific internalization of nanotherapeutics. The pretargeting component, anti-HER2 humanized monoclonal antibody, trastuzumab, functionalized with azide groups labels cancer cells that overexpress HER2 surface receptors. The drug carrier component, dibenzylcyclooctyne substituted albumin conjugated with paclitaxel, reacts specifically with the pretargeting component. These two components form cross-linked clusters on the cell surface, which facilitates the internalization of the complex. This strategy demonstrated substantial cellular internalization of clusters consisted of HER2 receptors, modified trastuzumab and paclitaxel-loaded albumin nanocarriers, and subsequent significant cytotoxicity in HER2-positive BT-474 breast cancer cells. Our results show high efficacy of this strategy for targeted nanotherapeutics. We foresee to broaden the applications of this strategy using agents such as radionuclides, toxins, and interfering RNA.
Keywords: Targeted therapy, Drug delivery, HER2(+) breast cancer, Nanocarriers, Bioorthogonal click chemistry
1. Introduction
Targeted delivery of nanocarriers offers great promise to improve the efficacy of chemotherapy while reducing its systemic toxicity [1–5]. Important features of targeted nanocarriers include their biocompatibility, high drug loading capacity, possibility of loading both hydrophobic and hydrophilic compounds, and favorable pharmacokinetics [6,7]. Antibody–drug conjugates (ADCs), a new class of nanoscale therapeutic agents, where drug molecules are chemically linked to the target-specific antibody have recently been approved for targeted therapy of cancers that overexpress a specific cell surface receptor [8]. Approximately 20–30% of breast cancers overexpress HER2/neu due to the gene amplification that results in high aggressiveness and generally poor prognosis [9,10]. The HER2 receptor regulates multiple physiological pathways, including cell proliferation and differentiation [11]. The humanized anti-HER2 monoclonal antibody (mAb), trastuzumab (Herceptin®), is used as a first-line treatment for HER2/neu-positive breast cancers [12]. The cytotoxic mechanism of trastuzumab includes the inhibition of the P13K/Akt and Ras/MAPK signaling pathways, leading to cell cycle arrest [13]. Unfortunately, approximately 50% of patients with HER2-positive disease do not benefit from trastuzumab or the disease becomes refractory to the drug [14], even though the HER2 level remains high [15]. The HER2 receptor is also characterized by poor internalization capability [16], even after antibody binding and subsequent heterodimerization, perhaps due to localization of the receptor in a membrane protrusion and/or in lipid raft areas where the receptor has poor contact with the lipid bilayer. An ADC, trastuzumab emtansine conjugate (T-DM1) [17], was recently reported to have significantly improved efficacy compared to standard monotherapeutics for trastuzumab-refractory disease [18,19]. However, the direct conjugation of chemotherapeutics to mAb may reduce its therapeutic index [20,21] due to the decreased binding affinity [22]. In addition, antibody conjugation does not enhance the ADC internalization. Long circulation half-life of ADCs can also result in systemic toxicity. In fact, thrombocytopenia has been observed in a small number of patients treated with ADCs [23,24].
Here, we report a new strategy for a two-step, target-specific drug delivery that utilizes enhanced internalization of therapeutic conjugates by in situ complexation, driven by bioorthogonal click chemistry. Use of click chemistry for the synthesis of targeted nanotherapeutics was recently reported [25]. However, our two-step, two-component system for the intracellular delivery of therapeutics is based on prelabeling of target receptors with azide functionalized mAbs, followed by the delivery of dibenzylcyclooctyne (DBCO) functionalized nanocarriers. The in situ click reactions between components induce the formation of cross-linked clusters on the cell surface, leading to their rapid internalization (Fig. 1). Both components can be modified with appropriate imaging agents for tracking their delivery, internalization, and accumulation in target cells. Optimal synthetic strategy and substitution ratios for pretargeting and delivery components are of key importance for drug solubility, efficiency of delivery, and binding affinity with the targets. For this study, we conjugated paclitaxel with albumin by covalent bonding. Paclitaxel is an antineoplastic taxane drug widely used for treating advanced breast cancer and metastasis [26]. Since paclitaxel is a highly hydrophobic compound, it is typically administered as micelles made in Cremophor EL (CrEL) and dehydrated ethanol, or as encapsulated vehicles made by nanoparticle albumin-bound (nab) technology. Excipients, such as Cremophor EL, are relatively toxic and can induce severe hypersensitive reactions [27] including dyspnea, flushing, rash, chest pain, and tachycardia [28]. The nab-technology involves high-pressure homogenization of albumins in the presence of drugs [29,30], and the particle size of clinically used nab-paclitaxels, Abraxane® is in the range of 140–160 nm. This drug uses physical entrapment of paclitaxel and starts to release drug molecules before reaching the target [31]. Hence, in this study we substituted paclitaxel molecules with albumin by covalent bonds via a succinyl spacer. Unlike physical entrapment of drug molecules [32] this approach enables to control the degree of substitutions and prevent the dissociation and release of drug en route to targets. Albumin was chosen as a model platform for the delivery component because albumin is a highly soluble, chemically and thermally stable, and biodegradable plasma protein, which make it a suitable carrier for drug delivery [33].
Fig. 1.
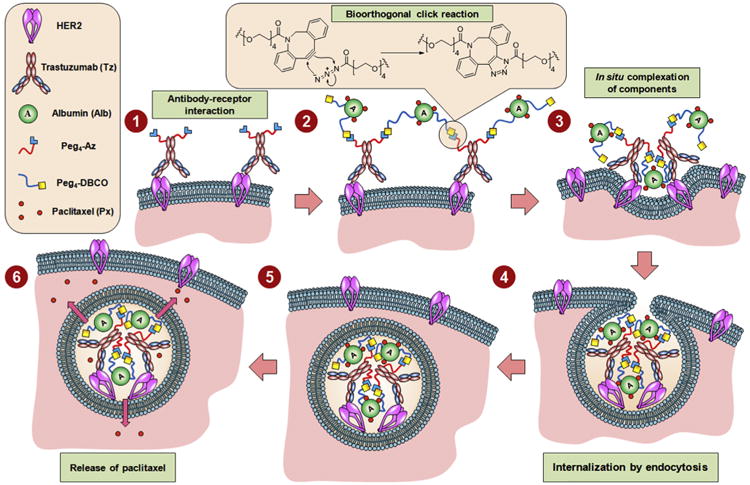
Schematics of the therapeutic strategy based on in situ bioorthogonal click chemistry. The strategy proceeds via interaction between functionalized trastuzumab and HER2 receptors on the cell surface, and bioorthogonal multiple click reactions between azide functional groups in trastuzumab and DBCO groups in albumin, followed by cluster formation, internalization, and release of drug molecules.
In our study, in situ complexation was achieved by multiple bioorthogonal click reactions between azido-trastuzumab and a model nanocarrier, DBCO-functionalized albumin–paclitaxel conjugate. Copper-free, strain-promoted click chemistry [34–36], or alternative bioorthogonal strategies [37,38], have received considerable attention for imaging applications [33–37] but, to the best of our knowledge, this is the first study to employ bioorthogonal click chemistry for chemotherapy. This strategy was evaluated in HER2-positive and HER2-negative breast cancer cell lines. We have demonstrated that this delivery system provides high efficacy and highly efficient intracellular drug accumulation in HER2-overexpressing cells.
2. Materials and methods
2.1. Cell lines
BT-474 and MDA-MB-231 cells were purchased from the American Type Culture Collection (ATCC) and cultured according to the manufacturer's direction using ATCC® 46-X and DMEM (Cellgro) media, respectively. Both media were supplemented with 1% Penicillin–Streptomycin, and 10% FBS. Cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2 unless otherwise mentioned. Third or fourth passages of cells with 70–80% confluency were used for imaging experiments.
2.2. Therapeutics and chemicals
Paclitaxel and Taxol® were purchased from LKT Laboratories, Inc. and Sagent Pharmaceuticals, Inc., respectively. Trastuzumab (Herceptin®) was purchased from Genentech Inc., or kindly provided by Dr. Robert Ivkov (The Johns Hopkins University School of Medicine) and used after purification. Appropriate amounts of Taxol® were diluted in DPBS+ (Dulbecco's phosphate buffered saline with 1% FBS) to prepare Px2.2 (paclitaxel 1.2 μM) and Px3.3 (paclitaxel 1.8 μM) for control experiments. The azide precursors, NHS-Peg4-Azide and NHS-Rhodamine, were purchased from Thermo Fisher Scientific. The DBCO precursor, NHS-Peg4-DBCO, was purchased from Click Chemistry Tools. The dyes, Click-iT® TAMRA and NHS-Alexa Fluor® 488, were purchased from Invitrogen, Inc. Dry or HPLC grade solvents were purchased from Sigma–Aldrich and used without further purification. The WST-8 cell viability kit was purchased from Dojindo Molecular Technologies, Inc.
2.3. Ultrafiltration and HPLC purification
Amicon Ultra centrifugal units (0.5 mL/3 kDa and 15 mL, 30 kDa) were used for the initial purification, buffer exchange, desalting or to concentrate the samples. A Waters binary pump/dual absorbance HPLC system was used with a YMC-Pack Diol-300 (300 × 8.0 mm I.D.; Partial size, 5 μm; Pore size, 30 nm) size exclusion column for the purification of modified trastuzumab and albumin using 0.1 m PBS with 0.2 m NaCl (pH 7.2) as the mobile phase.
2.4. Formulation of components
2.4.1. Modification of trastuzumab
For the optimization of pegylated azide substitution, five samples (500 μL of 2.5 mg/mL, ∼8.5 × 10−9 mol each) of pure trastuzumab were treated with different equivalents of NHS-Peg4-Azide (20, 50, 100, 200, and 500 equiv. dissolved in 10 μL of DMSO) in 2% DMSO/PBS. The mixture was vortexed and allowed to conjugate for 2 h at room temperature (Fig. 2A). Excess reactants were removed by ultrafiltration and the product was further purified by HPLC size exclusion chromatography. Pure fractions were concentrated by ultrafiltration. The degree of functionalization (DOF) was determined based on the change in molecular weights measured by MALDI-TOF/MS (Fig. 3A). The availability of azide functional groups was verified by Click-iT® TAMRA labeling. Briefly, approximately 100 μL of 0.2 mg/mL modified trastuzumab was reacted with excess (300–500 equiv.) Click-iT® TAMRA DIBO alkyne, and incubated for 2 h at room temperature in 2% DMSO/PBS. Excess reagents were removed by ultrafiltration and the products were further purified by HPLC size exclusion chromatography. Pure fractions were concentrated by ultrafiltration. The number of azides substituted per antibody was determined by manufacturer's protocol recommended to calculate the degree of fluorescence labeling. The changes of DOF measured by MALDI-TOF/MS and Click-iT® TAMRA methods with respect to the equiv. of NHS-Peg4-Azide are shown in the Fig. 3B with a linear correlation (Fig. 3C).
Fig. 2.
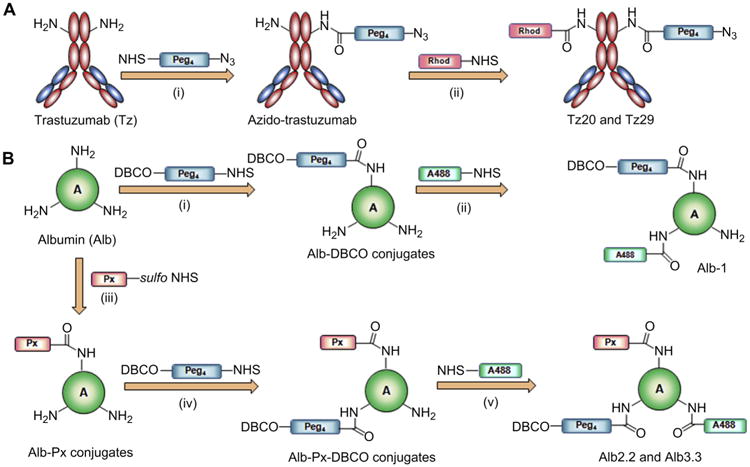
Syntheses and formulation of pretargeting and drug loaded nanocarriers. (A) Modification of trastuzumab for pretargeting. Antibody was first substituted with pegylated azide using NHS-Peg4-Azide reagent (i), and selected azido-trastuzumab conjugates were labeled with rhodamine to obtain Tz20 and Tz29 (ii). Tz-1 was synthesized by directly labeling pure Tz with rhodamine. (B) Modification of albumin for imaging and therapeutic studies. For imaging studies, albumin was functionalized with pegylated DBCO using NHS-Peg4-DBCO (i) followed by fluorescent labeling with Alexa Fluor® 488 to obtain Alb-1 (ii). For therapeutic studies, paclitaxel was derivatized to sulfo-NHS-paclitaxel analogue (Scheme S1). Then albumin was first reacted with sulfo-NHS-paclitaxel to obtain albumin–paclitaxel conjugates (iii). Purified soluble albumin–paclitaxel conjugates were functionalized with pegylated DBCO using NHS-Peg4-DBCO (iv) followed by fluorescent labeling with Alexa Fluor® 488 to obtain Alb2.2 and Alb3.3 (v).
Fig. 3.
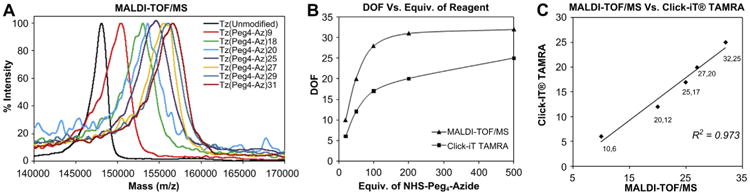
Characterization of trastuzumab conjugates. (A) MALDI-TOF/MS spectra of Tz(Peg4-Az)n conjugates (n = DOF). (B) Changes of DOFs determined by MALDI-TOF/MS and Click-iT® TAMRA methods. (C) Linear correlation of DOF determined by MALDI-TOF/MS and Click-iT® TAMRA methods.
2.4.2. Formulation of albumin nanocarriers
For the optimization of pegylated DBCO substitution, five samples (500 μL of 10 mg/mL) of albumin were treated with NHS-Peg4-DBCO (50, 100, 200, 500, and 1000 equiv. in 10 μL of DMSO) in 2% DMSO/PBS, vortexed and allowed to conjugate at room temperature for 2 h (Fig. 2B). Excess reactants were removed by ultrafiltration and the product was further purified by HPLC size exclusion chromatography. The pure fractions were concentrated by ultrafiltration. The molecular weights of the products were determined by MALDI-TOF/MS (Fig. 4A and B). The DOF was calculated based on the change in molecular weight. Albumin modified with DBCO (DOF of 15) was labeled with Alexa Fluor® 488 and used for in vitro imaging experiments.
Fig. 4.
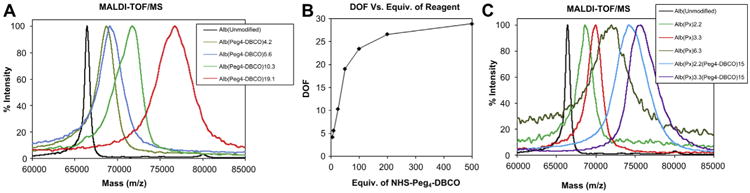
Characterization of albumin conjugates. (A) MALDI-TOF/MS spectra of Alb(Peg4-DBCO)n (n = DOF) conjugates. (B) Change of DOF measured by MALDI-TOF/MS with respect to the molar equiv. of NHS-Peg4-DBCO. (C) MALDI-TOF/MS spectra of Alb–paclitaxel conjugates and Alb–Paclitaxel–DBCO conjugates.
For the therapeutic experiments, the model drug delivery nanocarrier, albumin, was conjugated with paclitaxel. First paclitaxel was derivatized to amine reactive sulfo-NHS-paclitaxel following a previously reported procedure (Scheme S1) [39]. Three samples of albumin (4 mg, 0.06 μmol each in PBS) were treated with sulfo-NHS-paclitaxel (0.6, 1.2, and 2.4 μmol i.e. 10, 20, and 40 equiv.) and stirred for 1 h at room temperature. The reaction mixtures treated with 0.6 and 1.2 μmol of activated drug were purified by PD-10 desalting column. The 40 equiv. reaction (2.4 μmol of activated drug) mixture resulted in precipitation and was purified by centrifugation. The albumin–paclitaxel conjugates were analyzed by MALDI-TOF/MS and determined the degree of substitution (DOS) of paclitaxel (Fig. 4C). The conjugates with 2.2 and 3.3 DOS of paclitaxel were substituted with DBCO and labeled with Alexa Fluor® 488 to obtain Alb2.2 and Alb3.3 as drug delivery nanocarriers (Fig. 2B). Efforts taken for the preassembling of two components to use as a control were failed due to the formation of cross-linked clusters, which resulted in precipitation.
2.5. MALDI analysis
For the MALDI analysis, samples were desalted by ultrafiltration. Samples (1 μL of 5-10 mg/mL) were loaded on a Voyager teflon-coated plate, using a “dried drop” method with sinapinic acid (10 mg/mL in 50% acetonitrile/0.3% trifluoroacetic acid) as the matrix. Samples were laser-irradiated with 500 shots, and the corresponding molecular weight was determined following standard MALDI-TOF/MS method used for protein analysis. The control IgG, γ-globulin or pure albumin was used to standardize and calibrate the instrument. The DOS was calculated based on the change in molecular weight with respect to unmodified trastuzumab or albumin.
2.6. Confocal microscopic imaging
BT-474 cells (5 × 105 cells/well in 0.5 mL of 46-X medium) or MDA-MB-231 cells (2.5 × 105 cells/well in 0.5 mL of DMEM/high glucose) were placed in four-well chamber slides and grown for 24–48 h to 70–80% confluency. First, cells were incubated with modified trastuzumab (20 μg/mL, 130 nm) in DPBS+ for 20 min and washed twice with DPBS+ to remove the unbound trastuzumab. As the model delivery nanocarrier, modified albumin (40 μg/mL, 550 nm) in DPBS+ was added and incubated at 37 °C for 2 h. Cells were washed twice with DPBS+ and fixed by 4% PFA. Cells were counterstained by Hoechst 33342(1 μg/mL in PBS) to visualize nuclei, and wet-mounted for confocal microscopic imaging on a Zeiss Axiovert 200 system with an LSM 510-Meta confocal module.
2.7. Cytotoxicity study
BT-474 cells (5000 cells/well in 200 μL of 46-X media) or MDA-MB-231 cells (5000 cells/well in 200 μL in DMEM/high glucose media) were seeded in a 96-well plate and incubated at 37 °C for 24 h. Cultured cells were treated with 100 μL of DPBS+ with or without modified or unmodified trastuzumab (20 μg/mL, 130 nm), and were incubated at 37 °C for 20 min. Cells were washed twice with DPBS+ and incubated in 100 μL of DPBS+ with or without a drug delivery nanocarrier, Alb2.2 or Alb3.3 (40 μg/mL, 550 nm), or the equiv. amount of paclitaxel as dilute Taxol® Px2.2 (1.2 μm) or Px3.3 (1.8 μm) for 2 h. Cells were washed twice with DPBS+, and refilled with fresh media (200 μL). After the incubation at 37 °C for 72 h cell viability was determined using a WST-8 assay following the manufacturer's protocol, Briefly, cells in 100 μL of media was treated with 10 μL of WST-8 reagent per well, incubated at 37 °C for 4 h, and measured the absorbance at 450 nm. For receptor blocking experiments, corresponding cells were treated with pure trastuzumab (20 μg/mL, 130 nm) and incubated for 20 min at 37 °C and washed twice with DPBS+ before treating with pretargeting modified Tz and drug delivery nanocarriers. In monotherapeutic experiments, cells were incubated with pretargeting modified trastuzumabs (20 μg/mL, 130 nm) at 37 °C for 20 min or drug loaded nanocarriers Alb2.2, Alb3.3 (40 μg/mL, 550 nm), Px2.2 (paclitaxel 1.2 μm), or Px3.3 (paclitaxel 1.8 μm), respectively, at 37 °C for 2 h and the WST-8 cell viability assay was performed as described above.
2.8. Statistical analysis
The WST-8 assay test was quadruplicated per plate and triplicate independent experiments were carried out for the statistical analysis. The one-way Analysis of Varian (ANOVA) was used for omnibus F-test, and the Scheffé's test was undertaken for post hoc analysis (StatPlus®:mac, AnalystSoft Inc., Alexandria, VA, USA). Changes in cell viability were considered significant (p-value <0.05) when the F value is greater than the critical value of the F-distribution.
3. Results and discussion
3.1. Design and formulation of components
Trastuzumab was functionalized with terminal azide functional groups using amine-reactive NHS-Peg4-Azide (Fig. 2A). Trastuzumab was modified with variable DOF up to 30 azide groups to investigate and optimize the binding affinity and efficiency of click reaction. Molecular weights of modified trastuzumabs were measured by MALDI-TOF/MS (Fig. 3A). The DOF was optimized by changing the equiv. of NHS-Peg4-Azide reagent (Fig. 3A). The number of available azide groups was quantified by Click-iT® TAMRA dye labeling and good linear correlation with DOF measured by the change of molecular weights was observed (R2 = 0.97, Fig. 3B, C). High DOF enhances the rate of the bioorthogonal click reaction but can also reduce the antibody binding affinity. No significant decrease in the binding affinity of functionalized trastuzumab was detected at a DOF of ≤20. Selected azido–trastuzumab conjugates as well as control, unmodified trastuzumab were labeled by rhodamine (two fluorophores per antibody).
The delivery platform, albumin was functionalized by DBCO using NHS-Peg4-DBCO (Fig. 2B) and the molecular weights were determined by MALDI-TOF/MS (Fig. 4A). The DOF of DBCO was optimized by changing the equiv. of NHS-Peg4-DBCO (Fig. 4B). Since DBCO is a comparatively hydrophobic moiety, the DOF of DBCO was maintained at 15. This albumin–DBCO conjugate was further labeled with Alexa Fluor® 488 fluorescent dye to track the agent and evaluate its delivery and internalization (Fig. 2B). For therapeutic study albumin was substituted with paclitaxel. First, paclitaxel was reacted with succinyl anhydride forming a terminal carboxyl group and an ester bond with the hydroxyl group of the drug molecule. The resulted 2′-succinyl paclitaxel was derivatized to amine-reactive and aqueous soluble sulfo-NHS-paclitaxel analogue (Scheme S1) [39]. The activated sulfo-NHS-paclitaxel was reacted with albumin (Figs. 2B and 4C). The DOS of paclitaxel higher than 5 drastically reduced the solubility of the conjugates in PBS. Therefore albumin conjugates with a DOS of ∼2–3 were used for further studies. The complete list of the pretargeting and delivery components used in the study is presented in Table 1. Preassembling of the pretargeting and delivery components resulted in rapid formation of cross-linked clusters followed by precipitation. Therefore preassembled components were not used in the study.
Table 1.
Components used for imaging and therapeutic studies.
| Compound | Abbreviation |
|---|---|
| Trastuzumab (Tz) | Tz |
| Tz(Rhod)2 | Tz-1 |
| Tz(Peg4-Az)20(Rhod)2 | Tz20 |
| Tz(Peg4-Az)29(Rhod)2 | Tz29 |
| Alb(Peg4-DBCO)15(Alexa488)1 | Alb-1 |
| Alb(Px)2.2(Peg4-DBCO)15(Alexa488)1 | Alb2.2 |
| Alb(Px)3.3(Peg4-DBCO)15(Alexa488)1 | Alb3.3 |
| Taxol® equivalent to paclitaxel content in Alb2.2 | Px2.2 |
| Taxol® equivalent to paclitaxel content in Alb3.3 | Px3.3 |
3.2. Evaluation of internalization by fluorescence imaging
The two-step, two-component delivery strategy was evaluated in HER2-positive and HER2-negative breast cancer cell lines. The intracellular delivery strategy was first validated for the pretargeting and the delivery components without drug loading by confocal microscopy using red and green channels, respectively. The colocalization was detected by merging the images. The click treatment of HER2 overexpressing BT-474 cells, i.e., the sequential addition of azido-trastuzumab (Tz20) and DBCO conjugated delivery component (Alb-1), demonstrated efficient cellular internalization and colocalization of components at 37 °C (Fig. 5A, Treated). Neither colocalization nor cellular internalization by BT-474 cells was detected when unmodified trastuzumab Tz-1 was used as a control at 37 °C (Fig. 5A, Control). These findings were confirmed by flow cytometry analysis of two-component delivery system (Figure S1). Neither surface labeling by Tz20 nor colocalization with Alb-1 was observed in the HER2-negative cell line, MDA-MB-231 at 37 °C (Figure S2). Overall these data support our hypothesis that bioorthogonal click reaction driven delivery enhances the internalization of clusters at physiological conditions. The internalization likely occurs via clathrin-mediated endocytosis that depends on the membrane tension, temperature [40], and the size of particles [41]. Collectively, this experiment proved that (a) only cells with an overexpression of cell surface receptors provide moieties for multiple click reactions, and (b) bioorthogonal click chemistry is the key mechanism for colocalization and in situ assembling of the components. The results also suggested that the system can be used for targeted drug delivery and is expected to provide efficacious therapy [42].
Fig. 5.
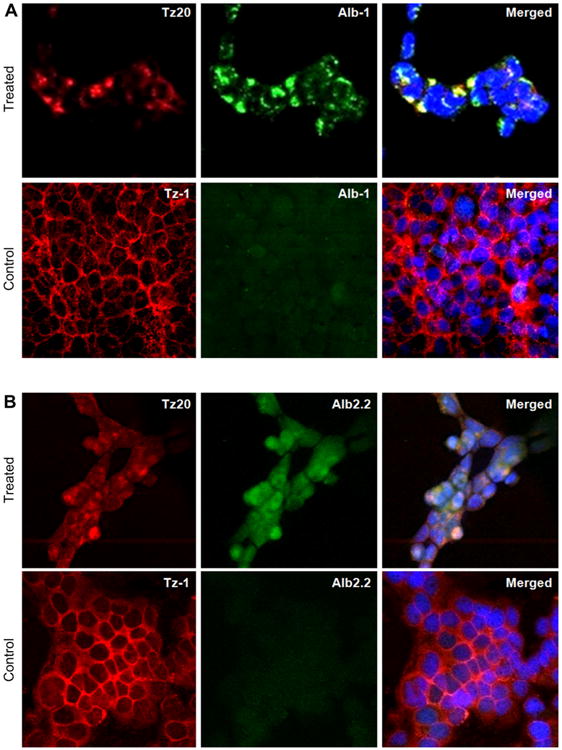
Evaluation of the internalization of two-component delivery system driven by in situ click reactions with confocal fluorescent microscopy. (A) Albumin-based carrier without paclitaxel (Alb-1). Treated: BT-474 cells were treated with Tz20 (trastuzumab functionalized with azide), followed by Alb-1 at 37 °C Control: BT-474 cells were treated with Tz-1 (trastuzumab without azide), followed by Alb-1 at 37 °C. (B) Albumin-based carrier conjugated with paclitaxel (Alb2.2). Treated: BT-474 cells treated with Tz20, followed by Alb2.2 at 37 °C Control: BT-474 cells treated with Tz-1, followed by Alb2.2 at 37 °C
To evaluate the specific internalization of the drug-loaded delivery component, HER2 overexpressing BT-474 cells were treated with azido-trastuzumab, Tz20 (Fig. 5B, Treated) or control Tz-1 without azide modification (Fig. 5B, Control) followed by the drug delivery nanocarrier Alb2.2 at 37°C. A significant colocalization and internalization of clusters were observed when Tz20 and Alb2.2 were used for the delivery. Neither significant cellular uptake nor colocalization of Alb2.2 was observed with control trastuzumab (Tz-1) as pretargeting component. These results revealed that the drug conjugation step used did not interfere with the specific antibody binding and/or bioorthogonal click reaction through possible steric hindrance.
3.3. Evaluation of the strategy in vitro
The therapeutic efficacy was assessed in BT-474 and MDA-MB-231 cells in vitro to corroborate our hypothesis that therapeutic efficacy can be improved by enhanced, target-specific internalization of drugs. First, the therapeutic effect of the single components, Tz-1, Tz20, Tz29, Px2.2, Px3.3, Alb2.2, and Alb3.3 were evaluated (Fig. 6, Table S1). A marginal change in cell viability was observed for treatment of BT-474 cells with trastuzumab (85%) and antibodies functionalized with azides, Tz20 (90%) and Tz29 (93%). No significant effects of trastuzumab and its variants were detected in HER2-negative MDA-MB-231 cells (Fig. 6B).
Fig. 6.
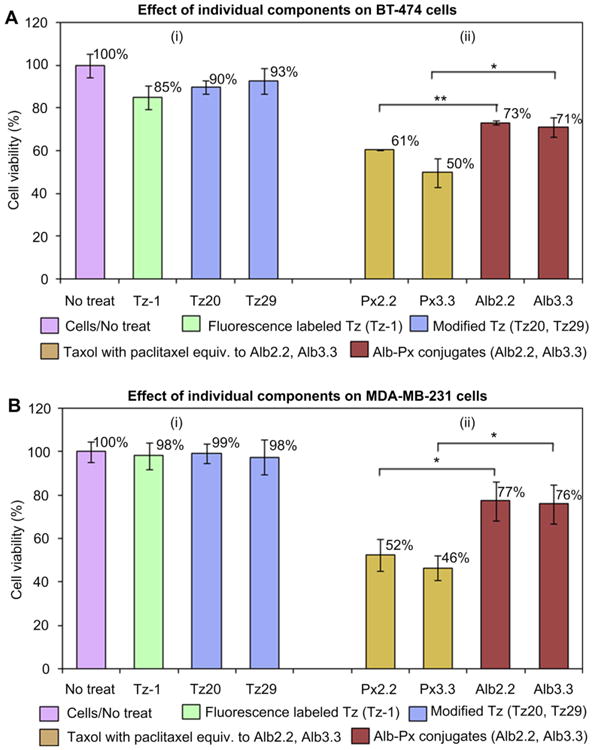
Therapeutic effect of individual components on (A) HER2-positive BT-474 cells and (B) HER2-negative MDA-MB-231 cells. Cells were treated with (i) trastuzumab-based pretargeting components (Tz-1, Tz20, or Tz29) for 20 min at 37 °C, or (ii) paclitaxel-containing therapeutic components (Px2.2, Px3.3, Alb2.2, or Alb3.3) for 2 h at 37 °C. Cell viability was measured using WST-8 cell viability kit The changes in cell viability were considered significant when p-values were lower than 0.05 (*p < 0.05, **p < 0.01).
Free paclitaxel (Px2.2 and Px3.3) and albumin–paclitaxel conjugates (Alb2.2 and Alb3.3) demonstrated significant efficacy against both cell lines (Fig. 6). Interestingly, the therapeutic effect of Alb2.2 and Alb3.3 conjugates (73% and 71% respectively) was significantly lower than that of the equivalent dose of free drug (61% and 50% for Px2.2 and Px3.3, respectively) in HER2-positive BT-474 cells. The effects of paclitaxel containing monotherapeutic agents in MDA-MB-231 cells are similar to the effects observed in BT-474 cells. However, Alb2.2 and Alb3.3 agents alone show certain cytotoxic effects in both cell lines (Fig. 6). This observation was supported by imaging that demonstrated moderate non-specific internalization of Alb2.2 (data not shown).
The therapeutic effect of the two-component system was studied in BT-474 (Fig. 7A) and MDA-MB-231 (Fig. 7B). The targeted delivery system, Tz20/Alb2.2, using azide functionalized Tz20 and Alb2.2 components, resulted in dramatic reduction of BT-474 cell viability to 41% following the treatment. Combinations of non-functionalized control trastuzumab, Tz-1, with free paclitaxel or albumin–paclitaxel conjugates produced the therapeutic effect similar to that of the combined effects of the individual components, which was significantly lower than the therapy via click reaction. The difference in cell survival between Tz20/Alb2.2 (41%) and Tz-1/Alb2.2 (64%) was highly significant with p = 0.003. Blocking HER2 receptors with trastuzumab prior to the treatment Tz/Tz20/Alb2.2 significantly reduced therapeutic efficacy with cell survival of 62%, p = 0.006. The delivery system Tz29/Alb2.2 also reduced cell viability to 53%, and the reduction was significant with respect to receptor-blocked control, Tz/Tz29/Alb2.2 (62%, p = 0.023). The effects between Tz20/Alb2.2 (41%) and Tz29/Alb2.2 (53%) systems were not significant, suggesting that the DOF of 20 or 29 of trastuzumab resulted in a similar efficacy.
Fig. 7.
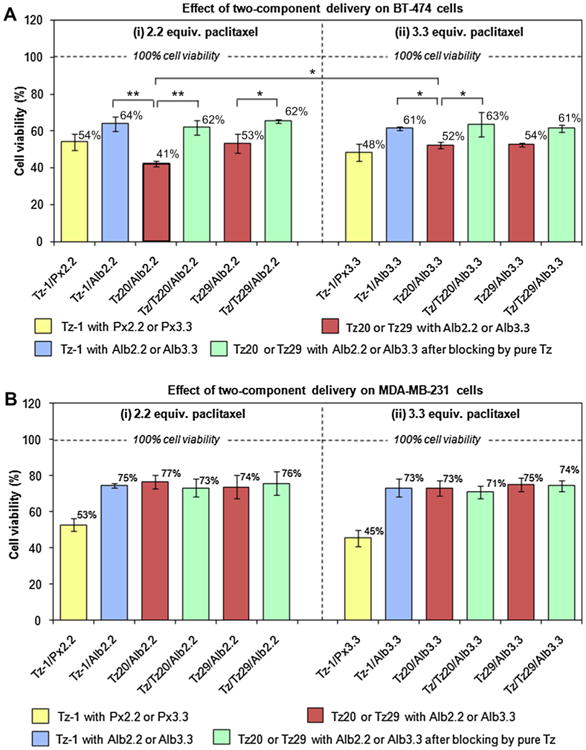
Therapeutic effect of two-component delivery strategy on (A) HER2-positive BT-474 cells and (B) HER2-negative MDA-MB-231 cells. Cells were treated with trastuzumab-based pretargeting components (Tz-1, Tz20, or Tz29), followed by paclitaxel-containing therapeutic components ((i) for Px2.2 or Alb2.2 and (ii) for Px3.3 or Alb3.3). Receptor blocking with unlabeled trastuzumab was also performed prior to the pretargeting step to verify the specific HER2 receptor-mediated targeting. Cell viability was measured using WST-8 cell viability kit. The changes in cell viability were considered significant when p-values were lower than 0.05 (*p < 0.05, **p < 0.01).
The Tz20/Alb3.3 combination reduced the cell viability to 52% compared to the control Tz-1/Alb3.3 (61%) in BT-474 cells, however this effect was significantly less efficacious than the Tz20/Alb2.2, despite the higher drug content of the former combination (p = 0.014). This observation indicates that the therapeutic efficacy of two-component delivery is not exclusively dependent on the drug content in the carrier, but rather, on the efficiency of the entire delivery complex. Albumin conjugated with 2.2 units of paclitaxel (Alb2.2) apparently undergoes a click reaction and forms internalized complexes more efficiently than Alb3.3. Similarly, the Tz29/Alb3.3 combination resulted in a significant reduction of cell viability to 54% with respect to the negative controls,Tz-1/Alb3.3 (61%, p = 0.043)and the receptor-blocking Tz/Tz29/Alb3.3 (61%, p = 0.041). Cell viability of MDA-MB-231 cells treated with the two-component delivery system was comparable to the effect of individual components (Figs. 6B and 7B). This effect on HER2-negative cells was attributable to the nonspecific cell uptake of paclitaxel-loaded albumin and no synergistic effect between trastuzumab and paclitaxel–albumin conjugates was observed. Based on our experimental results, the Tz20/Alb2.2 combination provided the highest efficacy in HER2-overexpressing BT-474 human breast cancer cells presumably due to the efficient internalization of drug carriers driven by bioorthogonal click reactions.
4. Conclusions
We have developed a two-step, two-component drug delivery system based on the functionalized trastuzumab pretargeting component and paclitaxel-loaded nanocarriers, and have demonstrated an enhanced cellular internalization, as well as cytotoxic effects of the system in HER2-overexpressing breast cancer cells. The strategy is based on two targeting steps: (1) recognition of the biological target by the antibody component; and (2) a chemoselective bioorthogonal click chemistry reaction between the pre-targeting component and the drug delivery nanocarrier. These steps provide highly target-specific, chemoselective, and efficient drug delivery. Multiple reactions between the pretargeting modified trastuzumab, bound to HER2 cell surface receptors, and the drug delivery nanocarriers result in the formation of cross-linked clusters on the cell. The untargeted drug carriers demonstrated low cytotoxicity in our studies. Thus, the formation, internalization, and cytoplasmic accumulation of component-clusters occur preferably in surface receptor-overexpressed cells. In addition, the method produced significantly less cytotoxicity in HER2-negative cells. Therefore, this strategy has a potential to reduce systemic toxicity while retaining efficacy against target cells. The components of the delivery system can be further independently optimized to improve the efficacy. Thus, the targeting component can be optimized for circulation time, binding affinity, and functionality, whereas the second therapeutic component can be optimized to minimize nonspecific toxicity comparing to the free drug and/or ADC formulations by adjusting the composition and circulation time of the drug delivery nanocarrier in vivo. Promising findings obtained with our bioorthogonal click chemistry-based delivery system provide the basis for further testing of the system at preclinical stage.
Supplementary Material
Acknowledgments
This work was supported by research grant KG100594 from Susan G. Komen for the Cure. The authors would like to acknowledge Dr. Farhad Vesuna for flow cytometry measurements. The authors thank Ms. Mary McAllister for her help with preparation of the manuscript.
Footnotes
Appendix A. Supplementary data: Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.biomaterials.2013.11.075
References
- 1.Gelperina S, Kisich K, Iseman MD, Heifets L. The potential advantages of nanoparticle drug delivery systems in chemotherapy of tuberculosis. Am J Respir Crit Care Med. 2005;172:1487–90. doi: 10.1164/rccm.200504-613PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuykendall DW, Zimmerman SC. Nanoparticles – a very versatile nanocapsule. Nat Nanotechnol. 2007;2:201–2. doi: 10.1038/nnano.2007.90. [DOI] [PubMed] [Google Scholar]
- 3.Sanhai WR, Sakamoto JH, Canady R, Ferrari M. Seven challenges for nanomedicine. Nat Nanotechnol. 2008;3:242–4. doi: 10.1038/nnano.2008.114. [DOI] [PubMed] [Google Scholar]
- 4.Singh S, Sharma A, Robertson GP. Realizing the clinical potential of cancer nanotechnology by minimizing toxicologic and targeted delivery concerns. Cancer Res. 2012;72:5663–8. doi: 10.1158/0008-5472.CAN-12-1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yokoyama M. Drug targeting with nano-sized carrier systems. J Artif Organs. 2005;8:77–84. doi: 10.1007/s10047-005-0285-0. [DOI] [PubMed] [Google Scholar]
- 6.De Jong WH, Borm PJA. Drug delivery and nanoparticles: applications and hazards. Int J Nanomed. 2008;3:133–49. doi: 10.2147/ijn.s596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peer D, Karp JM, Hong S, FaroKHzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751–60. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 8.Sievers EL, Senter PD. Antibody–drug conjugates in cancer therapy. Annu Rev Med. 2013;64:15–29. doi: 10.1146/annurev-med-050311-201823. [DOI] [PubMed] [Google Scholar]
- 9.Cho HS, Mason K, Ramyar KX, Stanley AM, Gabelli SB, Denney DW, et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421:756–60. doi: 10.1038/nature01392. [DOI] [PubMed] [Google Scholar]
- 10.Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, Mcguire WL. Human-breast cancer – correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–82. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 11.Lee-Hoeflich ST, Crocker L, Yao E, Pham T, Munroe X, Hoeflich KP, et al. A central role for HER3 in HER2-amplified breast cancer: implications for targeted therapy. Cancer Res. 2008;68:5878–87. doi: 10.1158/0008-5472.CAN-08-0380. [DOI] [PubMed] [Google Scholar]
- 12.Zaczek A, Brandt B, Bielawski KP. The diverse signaling network of EGFR, HER2, HER3 and HER4 tyrosine kinase receptors and the consequences for therapeutic approaches. Histol Histopathol. 2005;20:1005–15. doi: 10.14670/HH-20.1005. [DOI] [PubMed] [Google Scholar]
- 13.zum Buschenfelde CM, Hermann C, Schmidt B, Peschel C, Bernhard H. Anti-human epidermal growth factor receptor 2 (HER2) monoclonal antibody trastuzumab enhances cytolytic activity of class I-restricted HER2-specific T lymphocytes against HER2-overexpressing tumor cells. Cancer Res. 2002;62:2244–7. [PubMed] [Google Scholar]
- 14.Zhang SY, Huang WC, Li P, Guo H, Poh SB, Brady SW, et al. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat Med. 2011;17:461–70. doi: 10.1038/nm.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pohlmann PR, Mayer IA, Mernaugh R. Resistance to trastuzumab in breast cancer. Clin Cancer Res. 2009;15:7479–91. doi: 10.1158/1078-0432.CCR-09-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hommelgaard AM, Lerdrup M, van Deurs B. Association with membrane protrusions makes ErbB2 an internalization-resistant receptor. Mol Biol Cell. 2004;15:1557–67. doi: 10.1091/mbc.E03-08-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Girish S, Gupta M, Wang B, Lu D, Krop IE, Vogel CL, et al. Clinical pharmacology of trastuzumab emtansine (T-DM1): an antibody-drug conjugate in development for the treatment of HER2-positive cancer. Cancer Chemother Pharmacol. 2012;69:1229–40. doi: 10.1007/s00280-011-1817-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burris HA, Rugo HS, Vukelja SJ, Vogel CL, Borson RA, Limentani S, et al. Phase II study of the antibody drug conjugate trastuzumab-DM1 for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer after prior HER2-directed therapy. J Clin Oncol. 2011;29:398–405. doi: 10.1200/JCO.2010.29.5865. [DOI] [PubMed] [Google Scholar]
- 19.Muller V, Witzel I, Stickeler E. Immunological approaches in the treatment of metastasized breast cancer. Breast Care. 2009;4:358–66. doi: 10.1159/000262454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamblett KJ, Senter PD, Chace DF, Sun MMC, Lenox J, Cerveny CG, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res. 2004;10:7063–70. doi: 10.1158/1078-0432.CCR-04-0789. [DOI] [PubMed] [Google Scholar]
- 21.Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol. 2008;26:925–32. doi: 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- 22.Janthur WD, Cantoni N, Mamot C. Drug conjugates such as antibody drug conjugates (ADCs), immunotoxins and immunoliposomes challenge daily clinical practice. Int J Mol Sci. 2012;13:16020–45. doi: 10.3390/ijms131216020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krop IE, Beeram M, Modi S, Jones SF, Holden SN, Yu W, et al. Phase I study of trastuzumab-DM1, an HER2 antibody–drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J Clin Oncol. 2010;28:2698–704. doi: 10.1200/JCO.2009.26.2071. [DOI] [PubMed] [Google Scholar]
- 24.Lyass O, Uziely B, Ben-Yosef R, Tzemach D, Heshing NI, Lotem M, et al. Correlation of toxicity with pharmacokinetics of pegylated liposomal doxorubicin (Doxil) in metastatic breast carcinoma. Cancer. 2000;89:1037–47. doi: 10.1002/1097-0142(20000901)89:5<1037::aid-cncr13>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 25.Chan DPY, Deleavey GF, Owen SC, Damha MJ, Shoichet MS. Click conjugated polymeric immuno-nanoparticles for targeted siRNA and antisense oligonu-cleotide delivery. Biomaterials. 2013;34:8408–15. doi: 10.1016/j.biomaterials.2013.07.019. [DOI] [PubMed] [Google Scholar]
- 26.Wiseman LR, Spencer CM. Paclitaxel – an update of its use in the treatment of metastatic breast cancer and ovarian and other gynaecological cancers. Drugs Aging. 1998;12:305–34. doi: 10.2165/00002512-199812040-00005. [DOI] [PubMed] [Google Scholar]
- 27.Boddy AV, Plummer ER, Todd R, Sludden J, Griffin M, Robson L, et al. A phase I and pharmacokinetic study of paclitaxel poliglumex (XYOTAX), investigating both 3-weekly and 2-weekly schedules. Clin Cancer Res. 2005;11:7834–40. doi: 10.1158/1078-0432.CCR-05-0803. [DOI] [PubMed] [Google Scholar]
- 28.Gelderblom H, Verweij J, Nooter K, Sparreboom A, Cremophor EL. The drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer. 2001;37:1590–8. doi: 10.1016/s0959-8049(01)00171-x. [DOI] [PubMed] [Google Scholar]
- 29.Fu Q, Sun J, Zhang WP, Sui XF, Yan ZT, He ZG. Nanoparticle albumin-bound (NAB) technology is a promising method for anti-cancer drug delivery. Recent Pat Anti-Canc. 2009;4:262–72. doi: 10.2174/157489209789206869. [DOI] [PubMed] [Google Scholar]
- 30.Stinchcombe TE. Nanoparticle albumin-bound paclitaxel: a novel Cremphor-EL-free formulation of paclitaxel. Nanomedicine-UK. 2007;2:415–23. doi: 10.2217/17435889.2.4.415. [DOI] [PubMed] [Google Scholar]
- 31.Yamada K. Clinical development of abraxane, albumin-bound paclitaxel. Drug Deliv Syst. 2009;24:38–44. [Google Scholar]
- 32.Gradishar WJ. Albumin-bound paclitaxel: a next-generation taxane. Expert Opin Pharmacother. 2006;7:1041–53. doi: 10.1517/14656566.7.8.1041. [DOI] [PubMed] [Google Scholar]
- 33.Kratz F. Albumin as a drug carrier: design of prodrugs, drug conjugates and nanoparticles. J Control Release. 2008;132:171–83. doi: 10.1016/j.jconrel.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 34.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, et al. Copper-free click chemistry for dynamic in vivo imaging. Proc Natl Acad Sci U S A. 2007;104:16793–7. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang PV, Prescher JA, Sletten EM, Baskin JM, Miller IA, Agard NJ, et al. Copper-free click chemistry in living animals. Proc Natl Acad Sci U S A. 2010;107:1821–6. doi: 10.1073/pnas.0911116107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koo H, Lee S, Na JH, Kim SH, Hahn SK, Choi K, et al. Bioorthogonal copper-free click chemistry in vivo for tumor-targeted delivery of nanoparticles. Angew Chem Int Ed Engl. 2012;51:11836–40. doi: 10.1002/anie.201206703. [DOI] [PubMed] [Google Scholar]
- 37.Haun JB, Devaraj NK, Hilderbrand SA, Lee H, Weissleder R. Bioorthogonal chemistry amplifies nanoparticle binding and enhances the sensitivity of cell detection. Nat Nanotechnol. 2010;5:660–5. doi: 10.1038/nnano.2010.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peterson VM, Castro CM, Lee H, Weissleder R. Orthogonal amplification of nanoparticles for improved diagnostic sensing. ACS Nano. 2012;6:3506–13. doi: 10.1021/nn300536y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dosio F, Brusa P, Crosasso P, Arpicco S, Cattel L. Preparation, characterization and properties in vitro and in vivo of a paclitaxel-albumin conjugate. J Control Release. 1997;47:293–304. [Google Scholar]
- 40.Hong GS, Wu JZ, Robinson JT, Wang HL, Zhang B, Dai HJ. Three-dimensional imaging of single nanotube molecule endocytosis on plasmonic substrates. Nat Commun. 2012;3 doi: 10.1038/ncomms1698. [DOI] [PubMed] [Google Scholar]
- 41.Zhang SL, Li J, Lykotrafitis G, Bao G, Suresh S. Size-dependent endocytosis of nanoparticles. Adv Mater. 2009;21:419–24. doi: 10.1002/adma.200801393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hillaireau H, Couvreur P. Nanocarriers' entry into the cell: relevance to drug delivery. Cell Mol Life Sci. 2009;66:2873–96. doi: 10.1007/s00018-009-0053-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.