

Abstract
Objective
Acyl-CoA oxidase (ACOX1) deficiency is a rare disorder of peroxisomal very-long chain fatty acid oxidation. No reports detailing attempted treatment, longitudinal imaging, or neuropathology exist. We describe the natural history of clinical symptoms and brain imaging in two siblings with ACOX1 deficiency, including the younger sibling's response to allogeneic unrelated donor hematopoietic stem cell transplantation (HSCT).
Methods
We conducted retrospective chart review to obtain clinical history, neuro-imaging, and neuropathology data. ACOX1 genotyping were performed to confirm the disease. In vitro fibroblast and neural stem cell fatty acid oxidation assays were also performed.
Results
Both patients experienced a fatal neurodegenerative course, with late-stage cerebellar and cerebral gray matter atrophy. Serial brain magnetic resonance imaging in the younger sibling indicated demyelination began in the medulla and progressed rostrally to include the white matter of the cerebellum, pons, midbrain, and eventually subcortical white matter. The successfully engrafted younger sibling had less brain inflammation, cortical atrophy, and neuronal loss on neuroimaging and neuropathology compared to the untreated older sister. Fibroblasts and stem cells demonstrated deficient very long chain fatty acid oxidation.
Interpretation
Although HSCT did not halt the course of ACOX1 deficiency, it reduced the extent of white matter inflammation in the brain. Demyelination continued because of ongoing neuronal loss, which may be due to inability of transplant to prevent progression of gray matter disease, adverse effects of chronic corticosteroid use to control graft-versus-host disease, or intervention occurring beyond a critical point for therapeutic efficacy.
Introduction
Peroxisomes are single-membrane organelles that conduct multiple biochemical reactions including reduction of hydrogen peroxide, synthesis of plasmalogens and bile acids, and oxidation of very long chain fatty acids (VLCFA). Inherited deficiencies of peroxisomal assembly genes prohibit the formation of peroxisomes and impair function of all peroxisomal reactions. Clinical entities representing defects of peroxisomal biogenesis include Zellweger syndrome, neonatal adrenoleukodystrophy (N-ALD), and infantile Refsum syndrome. Inherited deficiencies of peroxisomal straight chain VLCFA β-oxidation include only straight-chain acyl-CoA oxidase (ACOX1) deficiency (also known as “pseudo-neonatal ALD”) and X-linked adrenoleukodystrophy (X-ALD). These two disorders share symptoms and biochemical characteristics, with varying age of onset and velocity of disease progression. The “cerebral” form of X-ALD, which manifests as neurological deterioration with inflammatory central nervous system (CNS) demyelination, is treatable using hematopoietic stem cell transplantation (HSCT), provided that the procedure is performed early in the course of the disease when demyelination is limited (Miller et al 2011; Peters et al 2004; Shapiro et al 2000. Until this report, no attempt at treatment had been made for ACOX1 deficiency. The straight-chain acyl-CoA oxidase enzyme [ACOX1, E.C. 1.3.3.6] catalyzes the first step of VLCFA β-oxidation. ACOX1 deficiency [OMIM 264470] was first recognized in a pair of siblings with severe neonatal hypotonia, seizures, and neurodegenerative course (Poll-The et al 1988). Originally diagnosed with N-ALD, their studies demonstrated intact peroxisomes and plasmalogen synthesis, but severely decreased VLCFA β-oxidation that was traced to ACOX1 deficiency.
In this report, we describe the natural history and biochemical findings in two siblings with ACOX1 deficiency. In addition, representing the first time for this disorder, we report results of hematopoietic stem cell transplantation in the younger sibling and findings from serial brain imaging, biochemical analysis of VLCFA β-oxidation from cultured neural stem cells, and CNS histopathology.
Subjects and methods
Clinical history
The CHOC Children's institutional review board (IRB) granted “exempt” status to conduct retrospective chart review of the two siblings. Hematopoietic stem cell transplantation was performed following parental consent under CHOC Children's IRB #s IH02-022 and IH02-023.
Neural stem cell culture
Parental consent was provided under CHOC Children's IRB #IH01-001 for the harvesting of neural progenitor cells. Methods of postmortem brain dissection, cell isolation, culture, and passage were previously described (Schwartz et al 2003).
Cell culture β -oxidation assay
Parental consent (John's Hopkins University IRB #83-03-01-01) was obtained to perform cell culture assays. Production of radiolabeled fatty acid degradation products was measured in cell suspensions as previously described (Jia et al 2007). Protein was measured by the method of Lowry (Lowry et al 1951).
Histopathology and immunohistochemistry
Serial sections of the brainstem and cerebellum were stained with Bielschowsky silver (axonal) and Luxol fast blue (myelin) histochemical stains, as well as neurofilament (axonal) and myelin basic protein (myelin) immunohistochemical stains.
Results
Patient 1 manifested hypotonia and poor head control at 3 months of age following an unremarkable pregnancy, delivery, and neonatal course. Parents were of Mexican descent and denied consanguinity. Brain magnetic resonance imaging (MRI) was normal at 8 months but began demonstrating white matter abnormalities at 12 months. She sat at 12 months of age, stood upright at 18 months of age, and walked without support at 3 years of age. She was responsive and babbled. At 24 months, brain MRI demonstrated extensive corticospinal tract and bi-hemispheric T2 hyperintensity with cortical atrophy. Developmental regression began at 3.5 years with rapid loss of all vision, hearing, gross motor, and verbal skills. Profound bilateral sensorineural hearing loss was found on brainstem auditory evoked response (BAER) at 3.8 years. She was referred for evaluation to one of the authors (JA) at 4.3 years. By then, T2 hyperintense demyelinating brain lesions on MRI were widespread and characteristic of ACOX1 deficiency (Fig. 1a-c). At 4.5 years, partial seizures manifested that became refractory to anti-epileptic medications. She became non-interactive and had multifocal epileptiform discharges on electroencephalography (EEG). Physical examination demonstrated horizontal nystagmus, severe truncal hypotonia, appendicular hypertonia, hyperreflexia, and spontaneous clonus. She died at age 6 years.
Fig. 1.
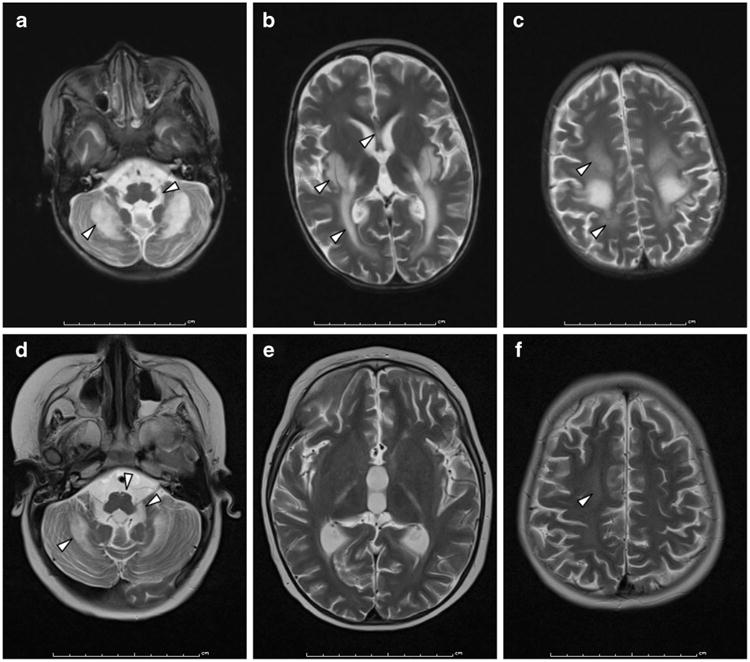
Axial T2-weighted brain MR imaging demonstrating differential demyelination (arrowheads) in patients 1 (a-c) and 2 (d-f) at similar ages (4.3 and 4 years, respectively). Patient 1 was untreated and patient 2 was 1 year post-HSCT. a, d Demyelination of the cerebellar deep white matter, cerebellar peduncles, and brainstem corticospinal tracts was more severe in patient 1 than patient 2. Cerebellar atrophy was also present in both siblings. Contrast enhancement was observed only in patient 1. b, e Demyelination of the corpus callosum, posterior limb of the internal capsule, and basal ganglia were present in patient 1, while patient 2 demonstrated no demyelination of internal capsule or basal ganglia. c, f Periventricular and subcortical demyelination, and cerebral atrophy, were again more severe in patient 1 than patient 2
Her electrolytes, transaminases, and bilirubin levels were normal. Biochemical assays indicated an isolated peroxisomal β-oxidation defect: plasma and fibroblast VLCFA (C26:0, C26:1) concentrations were elevated, fibroblast peroxisomal VLCFA β-oxidation was deficient, while other markers of peroxisomal function were intact (Table 1). VLCFA concentrations were also elevated in the medium of cultured patient fibroblasts when compared to control fibroblast medium (data not shown). Peroxisomal VLCFA β-oxidation studies performed on her neural stem cells demonstrated deficient C24:0 oxidation activity compared to unaffected control stem cells. C16:0 β-oxidation in both patient and control stem cell lines was intact (Table 1).
Table 1. Biochemical and enzymology analyses.
| Plasma VLCFA | Patient 1 | Patient 2 | Unaffected reference range | X-ALD hemizygote reference range | Zellweger syndrome reference range |
|---|---|---|---|---|---|
| C26:0 (μg/ml) | 2.22* | 1.04* | 0.24±0.14 | 1.30±0.45 | 3.93±1.50 |
| C26:1 (μg/ml) | 1.13* | 1.69* | 0.11 ±0.04 | 0.34±0.16 | 4.08±2.30 |
| C26/C22 ratio | 0.11* | 0.12* | 0.01±0.003 | 0.07±0.03 | 0.50±0.16 |
| C24/C22 ratio | 1.582* | 1.284* | 0.78±0.10 | 1.71±0.23 | 2.07±0.28 |
| Phytanic acid (μg/ml) | 0.47 | 0.04 | 0.54±0.29 | 0.57±0.46 | 0.40±0.28 |
| Pristanic acid (μg/ml) | 0.04 | 0.05 | 0.05±0.04 | 0.06±0.06 | 0.09±0.16 |
| Fibroblast studies | |||||
| Phytanic acid oxidation (pmol/48 h/mg protein) | 832.3 | 942.6±111.4 | |||
| C26:1 (μg/mg protein) | 0.537* | 0.09±0.07 | |||
| C26:0 (μg/mg protein) | 0.448* | 0.07±0.04 | |||
| C22:0 (μg/mg protein) | 0.33* | 0.90±0.40 | |||
| C26:0/C22:0 | 1.355* | 0.08±0.03 | |||
| C16 oxidation capacity (nmol/hr/mg prot) | 1.67 | 1.68 | |||
| C24 oxidation capacity (nmol/hr/mg prot) | 0.11* | 0.29 | |||
| C24:C16 oxidation ratio | 0.068* | 0.17 | |||
| Neural stem cell studies | |||||
| C16 oxidation capacity (pmol/hr/mg protein) | 703 | 1336 | |||
| C24 oxidation capacity (pmol/hr/mg protein) | 255* | 981 | |||
| C24:C16 oxidation ratio | 0.362* | 0.739 |
Plasma VLCFA levels and ratios were elevated in both patients compared to normal, above the ranges of X-ALD hemizygotes and below the ranges seen in Zellweger syndrome. Phytanic and pristanic acid levels were normal. β-Oxidation of [1-14 C]C16:0 or [1-14 C]C24:0 in skin fibroblasts or neural stem cells demonstrated intact mitochondrial long chain fatty acid oxidation but defective peroxisomal oxidation of C24:0 VLCFA in both fibroblasts and neural stem cells from patient 1. Abnormal values are highlighted with asterisks
Major gross postmortem findings were prominent atrophy and inflammatory demyelination of the cerebrum and cerebellum (Fig. 2). The right hemibrain weighed only 323 g (expected 600–650 g). The cerebral (Fig. 2a), cerebellar (Fig. 2c), and brainstem (Fig. 2e) white matter were severely myelin-deficient. In the brainstem, severe myelin pallor with volume loss was grossly evident in the cerebral peduncle and medullary pyramid; the basis pontis was also small. Within the cerebrum, myelin deficiency was more pronounced occipitally than frontally, with relative sparing of intragyral white matter and arcuate fibers. Microscopic observations (Fig. 3 and Supplemental Fig. 1) in the white matter were consistent with the gross findings and were virtually indistinguishable from those seen in X-ALD. There was myelin pallor (Fig. 3a), prominent frontal perivascular and intraparenchymal infiltrates composed of lymphocytes (Fig. 3c and e), activated macrophages (Fig. 3g), and microglia. Gray matter throughout the CNS was marked by profound neuronal loss and gliosis (Supplemental Fig. 1A and 1C).
Fig. 2.
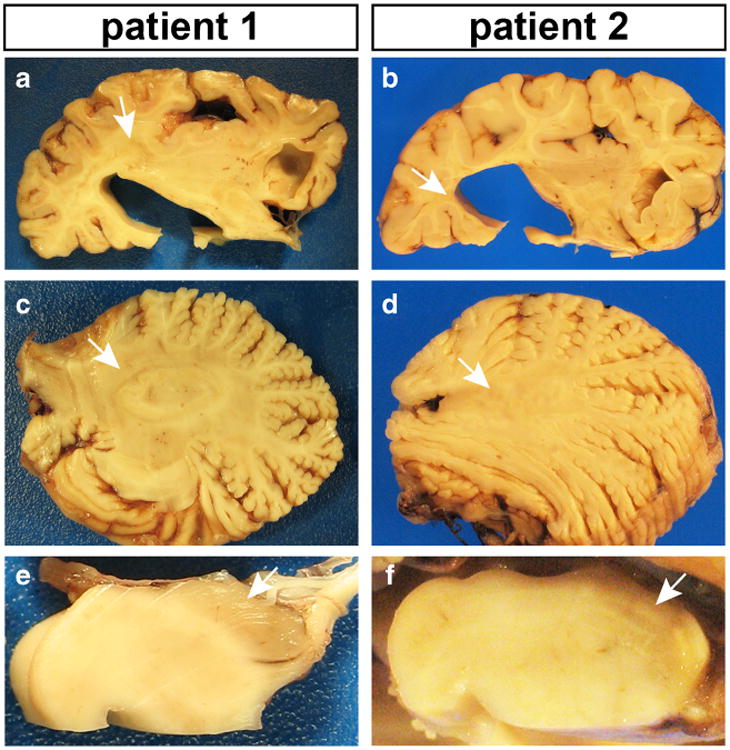
Gross neuropathology of the right hemispheres. Overall, patient 2 appeared to have less demyelination compared to patient 1. In each panel, white arrows point to areas of myelin pallor. a, b Coronal sections revealed pallor and loss of cerebral white matter volume in both patients, as well as striking cortical atrophy in patient 1. Subcortical U-fibers were spared. c, d Myelin pallor was observed in cerebellar white matter, with significant atrophy of the vermis. e, f There was also profound myelin pallor in the cerebral peduncle
Fig. 3.
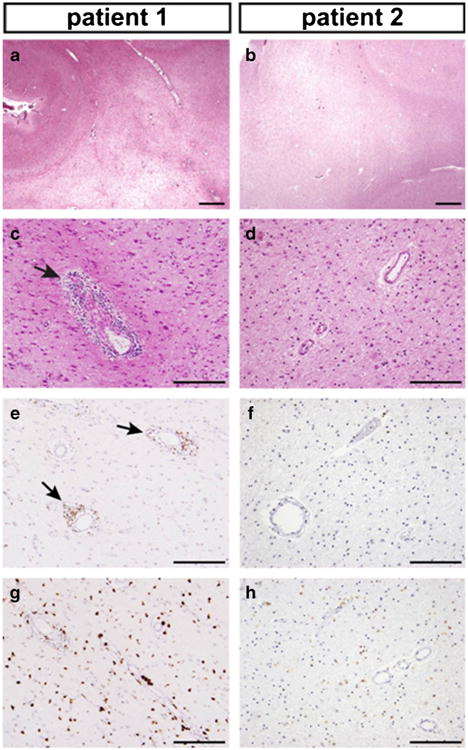
Microscopic neuropathology of the cerebral white matter, superior frontal regions (h & e stains and DAB immunohistochemistry). a, b Low power views revealed marked pallor of cerebral white matter, which was more severe in patient 1, along with sparing of subcortical U-fibers. c, d Perivascular inflammatory infiltrates were prominent in patient 1 (arrow), but sparse in patient 2. Gliosis was marked in both sections. e, f CD3 immunostains revealed perivascular T cells in patient 1 (arrows), as well as scattered infiltrating Tcells in both patients. g, h CD68 immunostains showed marked macrophage infiltration, with much stronger staining intensity in patient 1. Scale bars: 1 mm (a, b), 200 um (c-h)
Patient 2 was born after the diagnosis of ACOX1 deficiency was made in his older sister. Prenatal and postnatal testing for ACOX1 deficiency were offered, but parents declined. He was hypotonic at birth, and then experienced a seizure at 3 months prompting metabolic evaluation showing elevated plasma straight-chain VLCFA levels (Table 1). Regular surveillance with hearing, vision, brain MRI, and developmental assessments began thereafter. Visual evoked responses were normal at 3 months, but BAER at 4 months indicated bilateral delayed conduction through the auditory nerve and lower brainstem. Transaminase, bilirubin, and cortisol levels were normal. He began rolling at 6 months; babbling, sitting, and crawling at 12 months; pulled to stand at 17 months; walked with assistance at 28 months; and spoke his first word at 30 months.
Supportive care was initially recommended due to the degenerative natural history of the disorder. However, when neuropathology resembling X-ALD was appreciated after his sister's passing, HSCT was considered as a possible disease-arresting therapeutic intervention. Brain MRIs were normal until 23 months, when pontine medial lemnisci began to demonstrate nonenhancing symmetrical T2 hyperintensities ascending into the midbrain by 33 months. There was also incipient demyelination in the cerebellar white matter and brainstem corticospinal tracts (Supplemental Fig. 2A-C). His Loes score (Loes et al 1994), used to assess radiographic severity of cerebral involvement and ranging from 0 (no demyelination) to 34 (globally severe demyelination), was 2. Due to consistently successful neurologic outcomes reported in cerebral X-ALD patients transplanted with Loes scores less than 9 (Peters et al 2004), we proceeded with HSCTat 3 years. His conditioning regimen consisted of intravenous immune globulin × 1 day, fludarabine 30 mg/m2 × 3 days, pharmacokinetically targeted busulfan 0.8 mg/kg Q6hr× 4 days, and cyclophosphamide 50 mg/kg/day×4 days. He then received an allogeneic unrelated (7/10 HLA allele match) marrow transplant with 9 × 108 nucleated cells/kg (9.3 × 106 CD34+ cells/kg; 1.05 × 108 CD3+ cells/kg) followed by cyclosporine and methotrexate for graft-versus-host disease (GvHD) prophylaxis and ursodiol for prevention of hepatic veno-occlusive disease. Standard prophylaxis against Pneumocystis carinii, fungal, and viral infections were used; 200 mg fish oil twice per day was given for docosahexaenoic acid supplementation. He achieved full engraftment, with a recovery course complicated by recurrent bacterial central venous catheter infections, hypertension, and grade I cutaneous graft-versus-host disease.
Afterwards, he continued to be hypotonic with stagnation of psychomotor development. Ophthalmologic examination demonstrated early pigmentary retinopathy. At age 3.5 years (+6 months post-transplant), he experienced precipitous psychomotor regression following a respiratory infection and exacerbation of cutaneous GvHD, which was controlled only following high-dose corticosteroid therapy for 5 months. Mechanical ventilation and tracheostomy placement were required due to respiratory failure. A gastrostomy tube was placed because dysphagia and aspiration developed. Plasma VLCFA levels and ratios remained elevated (Supplemental Fig. 3). Chronic grade I cutaneous GvHD persisted, requiring ongoing corticosteroid treatment that resulted in transient cytomegalo-virus (CMV) reactivation and adrenal cortical suppression. Mineralocorticoid production and response were intact. Transaminases were normal except during the CMV reactivation. Despite consistently full donor engraftment, he remained minimally interactive, hypotonic, and became areflexic at 3.8 years. He died at age 6.75 years from pneumonia.
Serial brain MRI examinations offered the opportunity to study the progression of the CNS pathology. There was progressive atrophy of the cortical gray matter and cerebellar dentate nuclei. T2 hyperintensity of the corticospinal tracts were first seen in the medullary pyramids and basis pontis at 36 months (Supplemental Fig. 2A), ascended into posterior limbs at 46 months (Fig. 1d), and subcortical white matter at 60 months (Supplemental Fig. 2F). Contrast enhancement was only noted in the corticospinal tracts and basis pontis at 36 months, with none following transplant. White matter abnormalities were not as severe as his sister's brain MRI performed at a similar chronologic age (Fig. 1d-f), but eventually were widespread throughout all the CNS (Supplemental Fig. 2D-F). Prior to his death, brain imaging demonstrated continued gray matter atrophy and non-enhancing demyelination.
The neuropathology of patient 2 was similar, but less severe, than what was observed in patient 1. Right hemibrain weight was 525 g (expected 600–650 g). There was perisylvian polymicrogyria that was most severe in the insula. The overall pattern of myelin pallor in the cerebrum (Fig. 2b), cerebellum (Fig. 2d), and brainstem (Fig. 2f) was similar to that in patient 1, but cerebral demyelination appeared less severe. Cerebral atrophy was relatively mild (Fig. 2b), but more pronounced in the cerebellum (Fig. 2d) and brainstem (Fig. 2f). Microscopically, there was deficient myelin (Fig. 3b), but perivascular lymphocyte (Fig. 3d and f), activated macrophage (Fig. 3h) infiltration, and neuronal loss (Supplemental Fig. 1B) were less striking compared to what was observed in patient 1.
Sequence analysis of all exons and flanking intronic sequences of the ACOX1 gene in patient 2 demonstrated a previously unreported, presumed homozygous c.1195C>T (p.His399Tyr) mutation. Parental genotypes could not be obtained. The histidine at amino acid position 399 is highly conserved across many species and immediately adjacent to an active site residue (Tyr401). The c.1195C>T mutation is not present in the SNP or control chromosome databases, and is predicted by PolyPhen-2 to be “probably damaging” and by SIFT to “affect protein function” (Adzhubei et al 2010; Kumar et al 2009).
Discussion
Straight-chain acyl-CoA oxidase deficiency is a rare disorder of peroxisomal very long chain fatty acid β-oxidation. Since the disorder was first recognized, a total of 31 patients have been reported (Carrozzo et al 2008; Ferdinandusse et al 2007, 2010; Kurian et al 2004; Rosewich et al 2006; Suzuki et al 1994, 2002; Wanders et al 1990; Watkins et al 1995). With the exception of two ACOX1 adults, the natural history of the disease follows a fairly consistent pattern of infantile-onset hypotonia, seizures, delayed acquisition of early developmental milestones, followed by rapid developmental regression between 24 and 48 months of age. Advanced patients become non-interactive, non-ambulatory, areflexic, and die in early childhood. Retinitis pigmentosa, vision abnormalities, and hearing loss are also common manifestations, while facial dysmorphism, hepatic dysfunction, and adrenal insufficiency are less frequently reported findings. There are no clear genotype-phenotype correlations as the p.Arg210His mutation found in the ACOX1 adults and the p.His399Tyr mutation found in our patients are both highly conserved mutations predicted in silico to be deleterious, and result in impaired ACOX1 catalytic activity (Ferdinandusse et al 2010).
In contrast to the well-described neuropathology of more common peroxisomal disorders, there was essentially none regarding that of ACOX1 deficiency until this report. The posterior predominance of demyelination; sparing of arcuate and intragyral myelin; presence of lesions at different stages of evolution; and inflammatory infiltrates consisting mostly of activated macrophages, microglia, and lymphocytes are also observed in X-ALD, N-ALD, and bifunctional enzyme deficiency (Powers and Moser 1998; Schaumberg et al 1975). In contrast, certain neuropathological features from our cases distinguish ACOX1 deficiency from other peroxisomal disorders. Cerebral and cerebellar cortical atrophy are prominent early findings, in contrast to X-ALD, where gray matter atrophy occurs in advanced disease. The combined gray and white matter disease in ACOX1 likely reflects its expression in oligodendrocytes, astrocytes, and CNS neurons (Fouquet et al 1997). In rat CNS neurons, ACOX1 expression is most prominent in the giant neurons of neocortical layer V, pontine nuclei, cerebellar Golgi and Purkinje neurons (Farioli-Vecchioli et al 2001), which mirror the sites of greatest gray matter damage in the patients presented. Expression of ABCD1, the transporter deficient in X-ALD, is limited to astrocytes and oligodendrocytes but not neurons (Fouquet et al 1997), consistent with X-ALD as primarily a white matter disorder. The caudal-rostral progression of myelin loss observed in our patients resembles X-ALD, while the perisylvian polymicrogyria in patient 2 appears to be characteristic of ACOX1 deficiency.
Our group was fully cognizant that HSCT had never been performed in ACOX1 deficiency but we reasoned that transplantation offered a chance to prevent him from following the same clinical course as his older sibling. We ensured that his parents fully understood the risks of HSCTand the uncertainty of neurologic outcome before proceeding. Unfortunately, HSCT did not alter the clinical course of ACOX1 deficiency in patient 2, but comparison of neuroimaging and brain pathology in the siblings provide insights into the effects of HSCT upon the CNS. While patient 2 did experience progressive demyelination, he demonstrated less demyelination and cortical atrophy compared to brain MRIs obtained at similar ages in his untransplanted sister. Additionally, the absence of MRI contrast enhancement and marked reduction of macrophage, microglia, and lymphocytic infiltrates point to transplant-induced amelioration of CNS inflammation. Other positive effects of HSCT included preservation of choroid plexus epithelial cell count and reduction of CNS macrophages bearing membrane-bound spiculated inclusions (Supplemental Fig. 4), which are thought to be modified VLCFA-containing myelin remnants and are indistinguishable from those described in X-ALD (Ghatak et al 1981; Powers et al 1982).
There are several possibilities for the clinical deterioration and progressive demyelination in patient 2 despite HSCT. First, patients with X-ALD demonstrate continued demyelination for 12–18 months post-transplant before the process stops because the therapeutic effect of donor microglial re-constitution is not immediate (Aubourg et al 1990). In addition, gray matter atrophy may have continued as a sequela of the conditioning regimen, chronic GvHD, or subsequent corticosteroid therapy, all findings that have been reported in HSCT survivors (Padovan et al 1998). Chronic corticosteroid use is known to cause cerebral atrophy by potentiating neuronal apoptosis, suppressing dendritic mass and synaptic junction formation (Zivadinov 2005). The “critical window” for transplant efficacy in ACOX1 deficiency may be much earlier than that of X-ALD. Prior to transplant, patient 2 already had hypotonia and developmental delay; X-ALD patients with neurological involvement at HSCT demonstrate worse outcomes compared to those who are asymptomatic (Miller et al 2011). One final possibility is that HSCT, while able to address demyelination arising from CNS inflammation, cannot prevent neuronal loss caused by ACOX1 deficiency and the secondary loss of axons and myelin that follows. This possibility is supported by the observations in both siblings of bilateral, symmetric white matter tract degeneration and equal loss of both axons and myelin, which are findings highly characteristic of primary neuroaxonal loss with secondary demyelination. This is further supported by the progression of pyramidal tract degeneration from medullary to subcortical levels, and sequential loss of white matter proceeding from the dentate nuclei and superior cerebellar peduncles to involve the cerebellar white matter and remaining peduncles, and confirms the report of Suzuki et al and recent brain MRI data from N-ALD and bifunctional enzyme deficiency (Suzuki et al 2002; van der Knaap et al 2012). This type of “dying-back” axonopathy due to neuronal degeneration and loss in motor cortex has been previously demonstrated in amyotrophic lateral sclerosis (Brownell et al 1970; Terao et al 1995; Iwata et al 2011).
Recognizing the hazards of drawing conclusions from one patient, we deduce from our experience with HSCT in patient 2 and the collective experience of HSCT for X-ALD that transplant for disorders of VLCFA accumulation may only be effective for treating inflammatory white matter disease, not for mitigating VLCFA toxicity to gray matter (Berciano 1982). Since we demonstrated that ACOX1-deficient neural stem cells from patient 1 accumulate VLCFA, it is consistent with the observation that VLCFA has toxic and pro-inflammatory effects on neurons, leading to progressive gray matter atrophy and secondary demyelination (Hein et al 2008; El Hajj et al 2012). In summary, each patient reported herein provides unique insight into the nature of ACOX1 deficiency. Our report provides first-time outcome of hematopoietic stem cell transplantation, as well as longitudinal neuroimaging and detailed neuropathology of the condition. Finally, our development of an ACOX1-deficient neural stem cell line provides a powerful tool to study the pathogenesis of, and test potential treatments for, peroxisomal disorders arising from VLCFA accumulation.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
This study was supported by CHOC Children's (PHS). We would like to express our gratitude to Nuriel Abdenur for document typesetting and formatting, and to the Commission for Families and Children of Orange County for its support of our clinical work (RYW, JEA).
Footnotes
Communicated by: Piero Rinaldo
Electronic supplementary material The online version of this article (doi:10.1007/s10545-014-9698-3) contains supplementary material, which is available to authorized users.
Conflict of interest None.
Contributor Information
Raymond Y. Wang, Email: rawang@choc.org, Division of Metabolic Disorders, CHOC Children's, 1201 W. La Veta Blvd., Orange, CA 92868, USA; Department of Pediatrics, University of California-Irvine School of Medicine, Irvine, CA, USA.
Edwin S. Monuki, Department of Pathology and Laboratory Medicine, University of California-Irvine School of Medicine, Irvine, CA, USA
James Powers, Department of Pathology and Laboratory Medicine, University of Rochester School of Medicine and Dentistry, Rochester, NY, USA.
Phillip H. Schwartz, Research Institute, CHOC Children's, Orange, CA, USA; Centers for Neuroscience and Translational Research, CHOC Children's, Orange, CA, USA
Paul A. Watkins, Department of Neurology, Johns Hopkins School of Medicine, Baltimore, MD, USA; Kennedy Krieger Institute, Baltimore, MD, USA
Yang Shi, Department of Pathology and Laboratory Medicine, University of California-Irvine School of Medicine, Irvine, CA, USA.
Ann Moser, Kennedy Krieger Institute, Baltimore, MD, USA.
David A. Shrier, Department of Imaging Sciences, University of Rochester Medical Center, Rochester, NY, USA
Hans R. Waterham, Laboratory Genetic Metabolic Diseases, Academic Medical Center, University of Amsterdam, Amsterdam, The Netherlands
Diane J. Nugent, Division of Hematology, CHOC Children's, Orange, CA, USA
Jose E. Abdenur, Division of Metabolic Disorders, CHOC Children's, 1201 W. La Veta Blvd., Orange, CA 92868, USA; Department of Pediatrics, University of California-Irvine School of Medicine, Irvine, CA, USA
References
- Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubourg P, Blanche S, Jambaqué I, et al. Reversal of early neurologic and neuroradiologic manifestations of X-linked adrenoleuko-dystrophy by bone marrow transplantation. N Engl J Med. 1990;322:1860–1866. doi: 10.1056/NEJM199006283222607. [DOI] [PubMed] [Google Scholar]
- Berciano J. Olivopontocerebellar atrophy. A review of 117 cases. J Neurol Sci. 1982;53:253–272. doi: 10.1016/0022-510x(82)90011-9. [DOI] [PubMed] [Google Scholar]
- Brownell B, Oppenheimer D, Hughes J. The central nervous system in motor neurone disease. J Neurol Neurosurg Psychiatry. 1970;33:338–357. doi: 10.1136/jnnp.33.3.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrozzo R, Bellini C, Lucioli S, et al. Peroxisomal acyl-CoA-oxidase deficiency: two new cases. Am J Med Genet A. 2008;146A:1676–1681. doi: 10.1002/ajmg.a.32298. [DOI] [PubMed] [Google Scholar]
- El Hajj HI, Vluggens A, Andreoletti P, et al. The inflammatory response in acyl-CoA oxidase 1 deficiency (pseudoneonatal adrenoleukodystrophy) Endocrinology. 2012;153:2568–2575. doi: 10.1210/en.2012-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farioli-Vecchioli S, Moreno S, Cerù MP. Immunocytochemical localization of acyl-CoA oxidase in the rat central nervous system. J Neurocytol. 2001;30:21–33. doi: 10.1023/a:1011913223541. [DOI] [PubMed] [Google Scholar]
- Ferdinandusse S, Denis S, Hogenhout EM, et al. Clinical, biochemical, and mutational spectrum of peroxisomal acyl-coenzyme A oxidase deficiency. Hum Mutat. 2007;28:904–912. doi: 10.1002/humu.20535. [DOI] [PubMed] [Google Scholar]
- Ferdinandusse S, Barker S, Lachlan K, et al. Adult peroxisomal acyl-coenzyme A oxidase deficiency with cerebellar and brainstem atrophy. J Neurol Neurosurg Psychiatry. 2010;81:310–312. doi: 10.1136/jnnp.2009.176255. [DOI] [PubMed] [Google Scholar]
- Fouquet F, Zhou JM, Ralston E, et al. Expression of the adrenoleukodystrophy protein in the human and mouse central nervous system. Neurobiol Dis. 1997;3:271–285. doi: 10.1006/nbdi.1997.0127. [DOI] [PubMed] [Google Scholar]
- Ghatak NR, Nochlin D, Peris M, Myer EC. Morphology and distribution of cytoplasmic inclusions in adrenoleukodystrophy. J Neurol Sci. 1981;50:391–398. doi: 10.1016/0022-510x(81)90151-9. [DOI] [PubMed] [Google Scholar]
- Hein S, Schönfeld P, Kahlert S, Reiser G. Toxic effects of X-linked adrenoleukodystrophy-associated, very long chain fatty acids on glial cells and neurons from rat hippocampus in culture. Hum Mol Genet. 2008;17:1750–1761. doi: 10.1093/hmg/ddn066. [DOI] [PubMed] [Google Scholar]
- Iwata NK, Kwan JY, Danielian LE, et al. White matter alterations differ in primary lateral sclerosis and amyotrophic lateral sclerosis. Brain. 2011;134:2642–2655. doi: 10.1093/brain/awr178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Z, Moulson CL, Pei Z, et al. FATP4 is the principal very long-chain fatty acyl-CoA synthetase in skin fibroblasts. J Biol Chem. 2007;282:20573–20583. doi: 10.1074/jbc.M700568200. [DOI] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;5:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- Kurian MA, Ryan S, Besley GT, et al. Straight-chain acyl-CoA oxidase deficiency presenting with dysmorphia, neurodevelopmental autistic-type regression and a selective pattern of leukodystrophy. J Inherit Metab Dis. 2004;27:105–108. doi: 10.1023/b:boli.0000016687.88818.6d. [DOI] [PubMed] [Google Scholar]
- Loes DJ, Hite S, Moser H, et al. Adrenoleukodystrophy: a scoring method for brain MR observations. Am J Neuroradiol. 1994;15:1761–1766. [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Miller WP, Rothman SM, Nascene D, et al. Outcomes after allo-geneic hematopoietic cell transplantation for childhood cerebral adrenoleukodystrophy: the largest single-institution cohort report. Blood. 2011;118:1971–1978. doi: 10.1182/blood-2011-01-329235. [DOI] [PubMed] [Google Scholar]
- Padovan CS, Yousry TA, Schleuning M, et al. Neurological and neuroradiological findings in long-term survivors of allogeneic bone marrow transplantation. Ann Neurol. 1998;43:627–633. doi: 10.1002/ana.410430511. [DOI] [PubMed] [Google Scholar]
- Peters C, Charnas LR, Tan Y, et al. Cerebral X-linked adrenoleukodystrophy: the international hematopoietic cell transplantation experience from 1982 to 1999. Blood. 2004;104:881–888. doi: 10.1182/blood-2003-10-3402. [DOI] [PubMed] [Google Scholar]
- Poll-The BT, Roels F, Ogier H, et al. A new peroxisomal disorder with enlarged peroxisomes and a specific deficiency of acyl-CoA oxidase (pseudo-neonatal adrenoleukodystrophy) Am J Hum Genet. 1988;42:422–434. [PMC free article] [PubMed] [Google Scholar]
- Powers JM, Moser HW. Peroxisomal disorders: genotype, phenotype, major neuropathologic lesions, and pathogenesis. Brain Pathol. 1998;8:101–120. doi: 10.1111/j.1750-3639.1998.tb00139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers JM, Moser HW, Moser AB, Schaumburg HH. Fetal adrenoleukodystrophy: the significance of pathologic lesions in adrenal gland and testis. Hum Pathol. 1982;13:1013–1019. doi: 10.1016/s0046-8177(82)80093-2. [DOI] [PubMed] [Google Scholar]
- Rosewich H, Waterham HR, Wanders RJ, et al. Pitfall in metabolic screening in a patient with fatal peroxisomal beta-oxidation defect. Neuropediatrics. 2006;37:95–98. doi: 10.1055/s-2006-923943. [DOI] [PubMed] [Google Scholar]
- Schaumberg HH, Powers JM, Raine CS, et al. Adrenoleukodystrophy. A clinical and pathological study of 17 cases. Arch Neurol. 1975;32:577–591. doi: 10.1001/archneur.1975.00490510033001. [DOI] [PubMed] [Google Scholar]
- Schwartz PH, Bryant PJ, Fuja TJ, et al. Isolation and characterization of neural progenitor cells from post-mortem human cortex. J Neurosci Res. 2003;74:838–851. doi: 10.1002/jnr.10854. [DOI] [PubMed] [Google Scholar]
- Shapiro E, Krivit W, Lockman L, et al. Long-term effect of bone-marrow transplantation for childhood-onset cerebral X-linked adrenoleukodystrophy. Lancet. 2000;356:713–718. doi: 10.1016/S0140-6736(00)02629-5. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Shimozawa N, Yajima S, et al. Novel subtype of peroxisomal acyl-CoA oxidase deficiency and bifunctional enzyme deficiency with detectable enzyme protein: identification by means of complementation analysis. Am J Hum Genet. 1994;54:36–43. [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Iai M, Kamei A, et al. Peroxisomal acyl CoA oxidase deficiency. J Pediatr. 2002;140:128–130. doi: 10.1067/mpd.2002.120511. [DOI] [PubMed] [Google Scholar]
- Terao S, Sobue G, Yasuda T, et al. Magnetic resonance imaging of the corticospinal tracts in amyotrophic lateral sclerosis. J Neurol Sci. 1995;133:66–72. doi: 10.1016/0022-510x(95)00143-p. [DOI] [PubMed] [Google Scholar]
- van der Knaap MS, Wassmer E, Wolf NI, et al. MRI as diagnostic tool in early-onset peroxisomal disorders. Neurology. 2012;78:1304–1308. doi: 10.1212/WNL.0b013e31825182dc. [DOI] [PubMed] [Google Scholar]
- Wanders RJ, Schelen A, Feller N, et al. First prenatal diagnosis of acyl-CoA oxidase deficiency. J Inherit Metab Dis. 1990;13:371–374. doi: 10.1007/BF01799398. [DOI] [PubMed] [Google Scholar]
- Watkins PA, McGuinness MC, Raymond GV, et al. Distinction between peroxisomal bifunctional enzyme and acyl-CoA oxidase deficiencies. Ann Neurol. 1995;38:472–477. doi: 10.1002/ana.410380322. [DOI] [PubMed] [Google Scholar]
- Zivadinov R. Steroids and brain atrophy in multiple sclerosis. J Neurol Sci. 2005;233:73–81. doi: 10.1016/j.jns.2005.03.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.