

Abstract
Background
Myocarditis is an inflammatory disease of the cardiac muscle and is mainly caused by viral infections. Viral myocarditis has been proposed to be divided into 3 phases: the acute viral phase, the subacute immune phase, and the chronic cardiac remodeling phase. Although individualized therapy should be applied depending on the phase, no clinical or experimental studies have found biomarkers that distinguish between the 3 phases. Theiler’s murine encephalomyelitis virus belongs to the genus Cardiovirus and can cause myocarditis in susceptible mouse strains.
Methods and Results
Using this novel model for viral myocarditis induced with Theiler’s murine encephalomyelitis virus, we conducted multivariate analysis including echocardiography, serum troponin and viral RNA titration, and microarray to identify the biomarker candidates that can discriminate the 3 phases. Using C3H mice infected with Theiler’s murine encephalomyelitis virus on 4, 7, and 60 days post infection, we conducted bioinformatics analyses, including principal component analysis and k-means clustering of microarray data, because our traditional cardiac and serum assays, including 2-way comparison of microarray data, did not lead to the identification of a single biomarker. Principal component analysis separated heart samples clearly between the groups of 4, 7, and 60 days post infection. Representative genes contributing to the separation were as follows: 4 and 7 days post infection, innate immunity–related genes, such as Irf7 and Cxcl9; 7 and 60 days post infection, acquired immunity–related genes, such as Cd3g and H2-Aa; and cardiac remodeling–related genes, such as Mmp12 and Gpnmb.
Conclusions
Sets of molecules, not single molecules, identified by unsupervised principal component analysis, were found to be useful as phase-specific biomarkers.
Keywords: computational biology, immunology, interferons, Picornaviridae infections, systems biology, T lymphocytes, transcriptome
Myocarditis, an inflammatory disease of the cardiac muscle, affects 2 million Americans and is a major cause of sudden death (8%–20% of sudden deaths in adults).1–3 In addition, 21% of patients with myocarditis develop dilated cardiomyopathy, which can lead to cardiac failure, and sometimes requires cardiac transplantation.1 Myocarditis most commonly results from virus infection4,5; in patients with histologically proven myocarditis or dilated cardiomyopathy, 56% of patients had an endomyocardial biopsy specimen test positive for viral genome.4,6
Clinical Perspective on p 454
Although the precise pathomechanism is unclear, viral myocarditis has been suggested to be divided into 3 phases.7 The proposed pathomechanism in each phase is as follows: in phase I, the cardiotropic virus infects and replicates in the heart, leading to cardiac damage (viral pathology). Innate immune responses against the virus contribute to either viral clearance or recruitment of immune cells, the latter of which can lead to phase II pathology. In phase II, acquired antiviral immune responses can damage not only virus-infected but also uninfected cardiomyocytes (immunopathology) in a bystander fashion or through molecular mimicry between the virus and a cardiac antigen; release of cardiac antigens can induce autoimmune responses against cardiac antigens by determinant (or epitope) spreading. In phase III, the tissue damage from phases I and II results in cardiac remodeling and fibrosis, leading to dilated cardiomyopathy, in which immune cell infiltrates or virus persistence can be low or undetectable in the heart. Ideally, personalized medicine should be applied according to the phase of viral myocarditis: antiviral therapy in phase I, immune suppressive or modulating therapy in phase II, and ventricular unloading and prevention of mechanical and hormonal stresses in phase III.8 However, because there is no single biomarker that distinguishes all 3 phases, there are potential risks in viral myocarditis treatment. For example, although immunosuppressive therapy can be effective to block immunopathology in phase II, the same therapy in phase I may suppress antiviral immunity, leading to enhancement of viral replication.
Although there are several approaches to identify each phase of viral myocarditis, there are no standard methods to distinguish between all 3 phases. Cardiac troponin and creatine kinase in serum have been used as biomarkers for cardiac damage, including myocarditis.9 However, both troponin and creatine kinase lack specificity for myocarditis, because they can be released into serum, when cardiomyocytes are damaged, regardless of the cause.10 Troponin and creatine kinase are detectable in both phases I and II, but often undetectable in phase III, because dilated cardiomyopathy can develop without damage of cardiomyocytes themselves.11 Echocardiography has also been used to monitor myocarditis despite low sensitivity.1 Left ventricle dysfunction, abnormal segmental wall motion, and increased wall thickness can be observed in the early phase of myocarditis, although the progression to dilated cardiomyopathy can be observed in phase III by echocardiography.1
Endomyocardial biopsy has been used as a standard in the diagnosis of myocarditis. Dallas criteria define myocarditis by inflammatory infiltrates and associated myocyte necrosis or damage not characteristic of an ischemic event in the biopsy samples.12 However, in theory, phase I may be undetectable by Dallas criteria, if there is no change in cardiac muscle (by definition, the viral phase I precedes infiltration of acquired immune cells in the immune phase II). Indeed, clinically, a large number of articles demonstrated that virus was present in the myocardium in the hearts that had no evidence of myocarditis by Dallas criteria.13 In addition, sampling error and variations in the interpretation of histological samples often occur. Several reports suggested the failure of diagnosis by Dallas criteria in approximately one third of subjects.14 There has been dissociation between Dallas criteria myocarditis and response to immunomodulation therapy.13 Thus, identifying the phase-specific biomarkers with better specificity and sensitivity is crucial for individualized medicine for viral myocarditis.
Gene expression analyses have been conducted to identify the biomarkers of myocarditis and several reports suggested that tenascin C would be a useful marker for coxsackievirus B–induced myocarditis in mice.15 However, tenascin C cannot be used to distinguish the 3 phases of viral myocarditis from each other because tenascin C was upregulated in the hearts of infected mice when compared with that in uninfected control mice throughout the disease course.16 Microarray analyses of myocarditis were also reported in several articles.17 Although Taylor et al18 reported the gene expression profiles in the hearts of coxsackievirus B–infected mice on 3, 9, and 30 days post infection (dpi) and showed the different expression patterns among the time points, they did not identify phase-specific biomarkers. Szalay et al19 reported the different gene expression profiles among the hearts of coxsackievirus B susceptible and resistant mice at several time points and the upregulation of genes associated with immune responses. In summary, these transcriptome analyses using traditional supervised 2-way comparison of gene expressions in current animal models for myocarditis did not lead to the discovery of phase-specific biomarkers that distinguished between all 3 phases.
Theiler’s murine encephalomyelitis virus (TMEV) is a nonenveloped, single-stranded RNA virus that belongs to the genus Cardiovirus.20,21 Although intraperitoneal TMEV infection causes myocarditis in adult mice and can be useful for a viral model for myocarditis, there have been only 2 studies on TMEV-induced myocarditis.22,23 Gómez et al22 reported that intraperitoneal infection of the DA strain of TMEV caused myocarditis in ABY/SnJ and SWR/J mouse strains. Rames23 demonstrated that intracerebral infection of the GDVII and Tex (DA strain-derived) strains of TMEV into CBA mice resulted in myocardial inflammation and interstitial fibrosis in the heart tissue.
We aimed to identify the biomarker candidates that distinguish between the 3 phases of viral myocarditis using multivariate analyses with bioinformatics methods. We have established a novel model system using TMEV and unsupervised bioinformatics analyses, such as principal component analysis (PCA), that have not been applied to identify the phase-specific biomarkers for myocarditis. We found that PCA distinguished between all 3 phases with a set of molecules, but not a single molecule, contributing to the separation. Therefore, our model system is a powerful tool to identify phase-specific biomarker candidates of viral myocarditis.
Materials and Methods
Detailed methods are provided in the Data Supplement.
Animal Experiments
All experimental procedures involving the use of animals were reviewed and approved by the Institutional Animal Care and Use Committee of LSU and performed according to the criteria outlined by the National Institutes of Health. Samples were collected from 5 infected animals and 5 age-matched controls at each time point in all experiments, unless otherwise noted. Male C3H/HeNTac mice (5-week-old) were infected with TMEV intraperitoneally. On 0, 4, 7, 14, 30, and 60 dpi, we conducted echocardiography using the Vevo 770 High-Resolution In vivo Micro-Imaging System (Visual Sonics). The levels of cardiac troponin I in the sera were measured by ELISA using the Ultra Sensitive Mouse Cardiac Troponin-I ELISA Kit (Life Diagnostics).
Microarray Analysis and Real-Time Polymerase Chain Reaction
Total RNA was processed and hybridized to the GeneChip Mouse 1.0ST Array (Affymetrix).24 The data were normalized by robust multi-array average. The data have been deposited into the Gene Expression Omnibus repository in National Center for Biotechnology Information (accession number: GSE53607). Real-time polymerase chain reaction (PCR) was conducted by the MyiQ2 Real-Time PCR Detection System (Bio-Rad). The results were normalized using a housekeeping gene Gapd.
Bioinformatics and Statistics Analyses
We drew a volcano plot, using the OriginPro 8.1 (OriginLab Corporation) to assess significance together with fold change of transcriptome data.25 K-means clustering was conducted to clarify the variances of gene expression patterns during the disease course, using the R version 2.15.1.26 PCA can reduce the dimensionality of a data set consisting of a large number of interrelated variables, whereas retaining as much as possible of the variation present in the data set.27 PCA was also conducted using the R package prcomp. The data were showed as mean+SEM. Statistical comparisons were conducted using Student t test or ANOVA in the OriginPro 8.1. P<0.05 was considered as a significant difference.
Results
Clinical Signs and Echocardiograms of Viral Myocarditis
We induced viral myocarditis in C3H mice by intraperitoneal TMEV injection and monitored body weight changes for 2 months (Figure 1). Infected mice gained weight slower than uninfected control mice significantly from 13 to 29 dpi (P<0.05). Infected mice developed no clinical signs that suggest heart failure, such as low activity and ankle edema.28 On 4, 7, and 60 dpi, we killed 5 infected and control mice and weighed their hearts. In infected mice, the heart weight was significantly lighter on 4 dpi (P<0.05), heavier on 7 dpi (P<0.05), and same on 60 dpi compared with control mice. The heart/body weight ratio, which is an indicator of cardiac hypertrophy,29 showed a similar trend to the heart weight.
Figure 1.
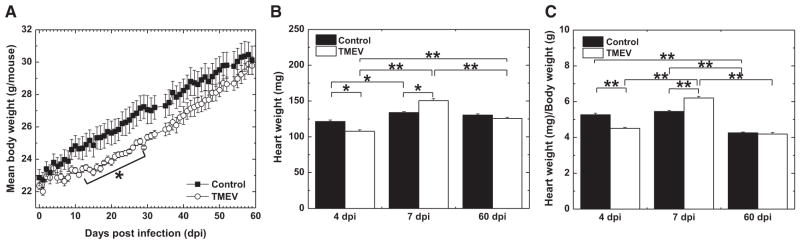
Clinical courses of Theiler’s murine encephalomyelitis virus (TMEV)–induced myocarditis. A, Time courses of body weight of age-matched control and TMEV-infected mice. B, Heart weight. C, Heart weight/body weight. n=5 per group per day post infection (dpi). *P<0.05 and **P<0.01.
We monitored the development of myocarditis for 2 months by echocardiography starting 4 dpi. On 4 dpi, we observed no morphological abnormalities in 90% of mice, using B-mode of echocardiography; only 10% (1 of 10 mice) of infected mice showed small high-intensity lesions in the left ventricle wall of the heart (Figure 2). On 7 dpi, several high-intensity lesions were observed inside the left ventricle wall of the hearts in all infected mice. On 60 dpi, multiple high-intensity lesions were observed in all hearts of infected mice. Although we also monitored cardiac functions using M-mode, no mice showed significant abnormality that suggests heart failure (data not shown).
Figure 2.

Echocardiograms of the hearts of Theiler’s murine encephalomyelitis virus (TMEV)–infected mice. A, C, and E, Echo-cardiograms (B-mode) of TMEV-infected mice on 4 (A), 7 (C), and 60 days post infection (dpi; E). B, Schematic diagram of long axis echocardiogram. There were several high-intensity lesions (arrows) in the heart on 7 and 60 dpi. White lesions (arrowheads) on the heart were observed macroscopically on 7 (D) and 60 dpi (F).
Macroscopically, we found white lesions on the surface of the hearts, which corresponded to high-intensity lesions observed in echocardiography on 7 and 60 dpi. Histologically, high-intensity lesions corresponded to inflammation, eosinophilic degeneration, calcification, or fibrosis (data not shown).
Correlation Between the Levels of Cardiac Troponin and Viral RNA
We determined the levels of cardiac troponin I in the serum as an indicator for cardiomyocyte damage (Figure 3).10 Serum troponin I was detectable in the TMEV-infected group on 4 dpi, reached a peak at 7 dpi, and became undetectable by 14 dpi. No serum troponin I was detected in control mice. We also quantified the levels of viral RNA in the hearts by real-time PCR. Interestingly, in contrast to serum troponin I levels, we detected higher levels of viral RNA on 4 dpi than on 7 dpi (10-fold; P<0.05). No viral RNA was detected on 60 dpi. To clarify whether virus replication was associated with cardio-myocyte damage on 4 and 7 dpi, we examined a correlation between the levels of serum troponin I and viral RNA. Levels of serum troponin I and viral RNA were correlated on 4 dpi (r=0.79; P<0.05), but not on 7 dpi (r=0.53; P=0.12). This suggested that cardiomyocyte damage was associated with viral replication on 4 dpi, but not on 7 dpi.
Figure 3.
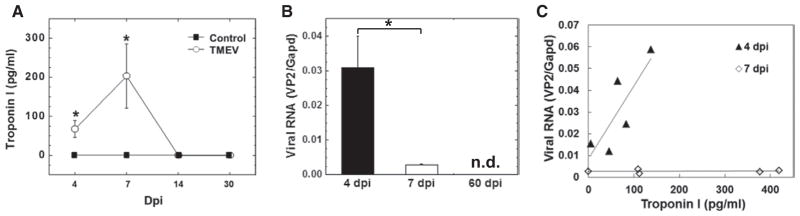
Cardiac troponin in the serum and viral RNA in the heart in Theiler’s murine encephalomyelitis virus (TMEV) infection. A, Serum troponin I levels in control and TMEV-infected mice. B, Viral RNA levels. C, Correlation between the levels of troponin I and viral RNA. n=5 per day post infection (dpi). *P<0.05. n.d. indicates not detectable.
Two-Way Supervised Analyses and k-Means Clustering of Gene Expression Profiles in TMEV Infection
To determine which gene expressions could be altered in the hearts of TMEV-infected mice compared with age-matched control mice during disease, we conducted supervised analysis using 2-way comparison of microarray data between the infected and control groups on 4, 7, and 60 dpi. We visualized the numbers of up- or downregulated genes of the hearts from infected mice, compared with controls, using a volcano plot (Figure 4). At all time points, interestingly, the numbers of genes that were upregulated >2-fold far exceeded the numbers of genes that were downregulated <0.5-fold. Gene upregulations were most prominent on 7 dpi, where 570 genes were upregulated, whereas 169 and 38 genes were upregulated on 4 and 60 dpi, respectively. The numbers of downregulated genes were relatively small at all 3 time points; 4 dpi, 15 genes; 7 dpi, 13 genes; and 60 dpi, 7 genes. Among the up- or down-regulated genes, more than half of the genes were unidentified by the databases of Mouse Genome Informatics and NetAffx.
Figure 4.
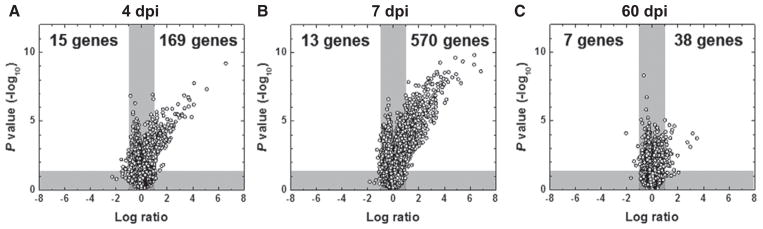
Volcano plots of microarray analyses of the hearts from Theiler’s murine encephalomyelitis virus–infected mice, compared with control mice on 4 (A), 7 (B), and 60 days post infection (dpi; C).
To compare the gene expressions between the 3 time points, we drew the heat maps for highly up- or downregulated genes (Figure 5A; Online Figure 1 in the Data Supplement). However, the heat maps did not distinguish gene expression patterns clearly between the time points. To identify genes that changed over time with similar patterns, we conducted k-means clustering (Figure 5B; Online Figure 3 in the Data Supplement). Among 20 clusters, 6 clusters showed differentially expressed patterns (Figure 5B). Radar chart visualized the different expression patterns of cluster centers in each cluster (Figure 5C; Online Figure 4 in the Data Supplement). Genes in cluster 2 were upregulated only on 60 dpi and included cardiac remodeling–related genes, such as Mmp12 and Gpnmb, and immunoglobulin-related genes, such as Igkv10-96, Igj, and Igkv6-15 (Online Table 2 in the Data Supplement). Genes in cluster 9 were highly upregulated on 4 and 7 dpi, but not on 60 dpi, and included innate immunity–related genes, such as Irf7, Ifit1, and Ifit3, and chemokines, such as Cxcl9, Cxcl10, and Ccl5 (Online Table 4 in the Data Supplement). Genes in cluster 19 were slightly upregulated only on 7 dpi and included acquired immunity–related genes, such as Cd3g and major histocompatibility complex (MHC) class II–related molecules (H2-Eaps, H2-Ab1, and H2-Aa; Online Table 6 in the Data Supplement). Clusters 8 and 20 included substantial downregulated genes only on 60 dpi and cluster 15 included genes downregulated on 4 dpi, whereas these clusters 8, 15, and 20 were mainly composed of non-immune response–related genes (Online Tables 3, 5, and 7 in the Data Supplement). Although these results showed the different expression patterns of genes in the hearts of TMEV-infected mice, we could not specify the phase-specific biomarkers to distinguish between the 3 phases of viral myocarditis.
Figure 5.

Heat map and k-means clustering of gene expression profiles in the hearts of Theiler’s murine encephalomyelitis virus (TMEV)–infected mice. A, Heat map of 20 upregulated and 16 downregulated genes based on 4 days post infection (dpi) data. Red, blue, and white indicate upregulation, downregulation, and no change, compared with control mice, respectively. Each column represents the data from 1 mouse (5 mice/dpi). A list of abbreviations of genes is shown in Table I in the Data Supplement. B, Clusters of gene with different expression patterns by k-means clustering. Shown are 6 clusters whose log ratios of cluster centers were >1 or <−1 among the total 20 clusters identified by k-means clustering (all 20 clusters are shown in Figure III in the Data Supplement). The genes in the 6 clusters are listed in Tables II to VII in the Data Supplement. Clusters 2, 9, and 19 included cardiac remodeling–related, innate immunity–related, and acquired immunity–related genes, respectively. C, Radar chart based on the values of cluster centers. The number at each vertex is the cluster number (2, 8, 9, 15, 19, and 20), whereas the numbers along the axis (−2 to 4) are log ratios, compared with age-matched controls.
Principal Component Analyses to Identify Phase-Specific Biomarkers
To find the phase-specific biomarkers of viral myocarditis, we conducted unsupervised analysis using PCA by entering microarray data from the heart samples without labeling of grouping. In PCA, each principal component (PC) is determined automatically, and PC values for each sample data can be plotted, for example, PC1 as an x axis and PC2 as a y axis. When the data of all samples from 4, 7, and 60 dpi were entered, we found that the samples were separated into 3 distinct populations (Figure 6A). The 3 populations separated by PC1 values corresponded to the 3 groups from different time points completely. The samples from 7 dpi had the highest PC1 values, whereas the samples from 60 dpi had the lowest PC1 values. PC1 seemed to associate with the changes in serum troponin I levels; we confirmed that the PC1 values correlated strongly with serum troponin I levels (r=0.81; P<0.001; Online Figure 5 in the Data Supplement). Thus, PC1 could reflect cardiomyocyte damage or viral replication. The proportions of variance of PCs showed that PC1 explained 28% of variance among 15 samples (Figure 6B). By identifying positive and negative top 10 genes of the factor loading for PC1, we determined which molecules contributed to PC1 values positively and negatively (Figure 6C). Gzmb, Irf7, and Cxcl9 contributed positively, whereas bone morphogeneic protein 10 (Bmp10), Mmp12, and other molecules contributed negatively.
Figure 6.

Principal component (PC) analyses of microarray data of the hearts from Theiler’s murine encephalomyelitis virus–infected mice. A to C, Analysis of the data from 4, 7, and 60 days post infection (dpi) without grouping. D to F, Analysis of the data from 4 and 7 dpi. G to I, Analysis of the data from 7 and 60 dpi. A, D, and G, PC1 and PC2 values of each sample, which are shown as a symbol; ▲, samples on 4 dpi; ◇, 7 dpi; and ▽, 60 dpi. B, E, and H, Proportion of variance. C, F, and I, Factor loading for the PC1.
Using sample data from 4 and 7 dpi, not from 60 dpi, we tested whether PCA could separate the samples into 2 distinct populations. Again without grouping, we entered the microarray data from 4 and 7 dpi for PCA (Figure 6D). PC1 separated the samples into 2 populations, where a population with higher PC1 values corresponded to 7 dpi samples and one with lower PC1 values was composed of 4 dpi samples. PC1 explained 27% of variance of 10 samples (Figure 6E). The factor loading for PC1 showed acquired cellular and humoral immunity–related genes, including CD3 γ-subunit (Cd3g, a T-cell marker), H2-Aa, and Igkv6-32 in the positive top 10 (Figure 6F). These results suggested that the samples between 4 and 7 dpi were distinguishable based on the expressions of a set of acquired immunity–related genes. Importantly, none of these acquired immunity–related genes were ranked among top 20 in supervised 2-way comparison analysis (Online Figure 1B in the Data Supplement); only Nkg7 and Gzmb, which can play a role in both innate and acquired immunities, were listed in top 10 molecules in Figure 6F and Online Figure 1B in the Data Supplement.
Next, we conducted PCA by entering the microarray data from samples on 7 and 60 dpi (Figure 6G). Again, PC1 separated the samples into 2 populations, where populations with higher and lower PC1 values corresponded to 7 and 60 dpi samples, respectively. PC1 explained 40% of variance among 10 samples (Figure 6H). The factor loading for PC1 showed remodeling-related genes, such as Mmp12, Bmp10, and Gpnmb, in the positive top 10 genes (Figure 6I), whereas immune-associated genes contributed to PC1 negatively. Thus, this set of remodeling genes could distinguish between the samples on 7 and 60 dpi.
Validation of the Microarray Results by Real-Time PCR
To validate the results of microarray analyses and PCAs, we conducted real-time PCR for the representative genes listed in the factor loadings (Figure 7): 2 genes each from innate immune responses (Cxcl9 and Tlr3), acquired immune responses (Gzmb and Cd3g), and cardiac remodeling (Mmp12 and Gpnmb). The expression patterns of Cxcl9, Tlr3, and Gzmb in both microarray analysis and real-time PCR were similar; their expressions were the highest on 7 dpi and the lowest on 60 dpi (Figures 5A and 7). Cd3g was upregulated only on 7 dpi. Mmp12 and Gpnmb were upregulated on 60 dpi. All of the gene expression levels were significantly different between the 3 time points.
Figure 7.

Differential gene expressions related to innate immunity (A and B), cellular immunity (C and D), and remodeling (E and F) during the disease course. Fold changes were calculated using the age-matched uninfected control mice. n=5 per day post infection (dpi). *P<0.05 and **P<0.01.
To clarify whether each gene expression was induced in response to heart damage, a correlation between serum troponin I levels and each gene expression ratios was determined (Online Figure 6 in the Data Supplement). The expressions of Cxcl9 and Tlr3 were correlated with troponin I levels on 4 dpi. The expression of Gzmb was correlated significantly with troponin I levels on 7 dpi, whereas the correlation between Cd3g expression and troponin I levels did not reach statistical significance.
Discussion
To date, although several markers are helpful to diagnose viral myocarditis, no markers can be used to discriminate all 3 phases by itself. In the present study, we conducted multivariate analysis using echocardiography, serum troponin I ELISA, viral RNA real-time PCR, and transcriptome assays on 4 (phase I), 7 (phase II), and 60 (phase III) dpi (Table). Echocardiography did not visualize cardiomyocyte damage on 4 dpi. Serum troponin I and viral RNA were not detectable on 60 dpi. We conducted the gene expression pattern analyses on 4, 7, and 60 dpi, using 2-way comparison of microarray data and Ingenuite Pathway Analysis (IPA; in the Data Supplement) as well as k-means clustering, and then attempted to identify the phase-specific biomarkers to discriminate the 3 phases of viral myocarditis using PCA. We found that a PCA of microarray data clearly separated samples from all 3 phases and identified a set of biomarker candidate genes that could contribute to distinguishing between the 3 phases. However, the traditional 2-way comparison of microarray data (heat map) and k-means clustering showed that innate immunity–related genes were upregulated on both 4 and 7 dpi, acquired immunity–related genes were upregulated on both 7 and 60 dpi, and cardiac remodeling–related genes were upregulated on 60 dpi. Here, the traditional transcriptome analysis was useful to characterize gene expression profiles in each phase, but not useful to distinguish between the 3 phases.
Table.
Phase-Specific Biomarkers in Theiler’s Murine Encephalomyelitis Virus–Induced Myocarditis
| 4 dpi (Phase I) | 7 dpi (Phase II) | 60 dpi (Phase III) | |
|---|---|---|---|
| Echocardiography | − | + | + |
| Serum troponin I | + | ++ | − |
| Viral replication | ++ | + | − |
| Microarray | |||
| Innate immunity | ++ | +++ | − |
| Acquired immunity | − | ++ | + |
| Cardiac remodeling | − | + | ++ |
− indicates undetectable; +, above the sensitivity limit; ++, moderately positive; +++, highly positive; and dpi indicates days post infection.
Similarly, Seok et al30 raised several issues on traditional transcriptome analysis based on the extent of fold changes of samples of interest compared with controls. Using peripheral blood transcriptome data from humans and mice with 3 diseases: trauma, burn, and endotoxemia, Seok et al showed that gene expression changes between humans and mouse models did not correlate. Interestingly, they also demonstrated that the transcriptome patterns, such as using heat map, among the samples from humans with the 3 diseases were similar; it seemed that traditional transcriptome analyses were not useful to discriminate the different disease samples. In our current experiment, we also found that transcriptome analyses based on the extent of fold change were not useful to discriminate the samples from 3 phases of myocarditis; heat maps between 4 and 7 dpi samples or between 7 and 60 dpi samples showed similar patterns. Seok et al and other research groups suggested that improvements of current animal model systems (rather than use of a single model) as well as several factors need to be considered in evaluating interspecies data, such as variations in sensitivity among different probe sets for genes on the microar-rays31 and identification of orthologues.32 Our current article has addressed several issues above. First, we used transcriptome data from affected organs, not from peripheral blood cells; high degree of correlation between the transcriptome data in affected organs between humans and mouse models has been reported.33 Second, our unsupervised PCA is not based on the order of levels of up- or downregulation of genes; our PCA led to identification of biomarker candidates, unlike our 2-way comparison based on the levels of up- or downregulation of genes. Third, our new TMEV-induced myocarditis model provides a clinically more relevant myocarditis model to existing model systems that have been sometimes shown to be inconsistent with findings in human cases. For example, interferon (IFN)-γ seems to play an effector role in human cases34,35 and the TMEV model (Online Table 9 in the Data Supplement), whereas IFN-γ is protective in a coxsackievirus model.36,37 Reovirus-induced cardiac damage in neonatal mice is caused by a direct cytopathic effect on cardiomyocytes, and not immune-mediated; the reovirus model represents phase I, but not phase II or III.38
On 4 dpi, the low levels of serum troponin I and high levels of viral RNA were detected in the hearts of infected mice, whereas microarray analyses showed upregulation of innate immunity–related genes. In phase I of viral myocarditis, viral entry/replication as well as innate immune responses for viral clearance have been proposed to contribute to cardiomyocyte damage, resulting in cardiac troponin release in serum.7 A significant decrease in heart weight in infected mice could be because of cardiomyocyte loss caused by direct virus lytic infection without recruitment of acquired immune cells or edema in the heart.
TMEV is a single-stranded RNA virus that has been shown to be recognized by melanoma differentiation–associated protein 5 (MDA5), which is also known as IFN induced with helicase C domain 1 (Ifih1), and toll-like receptors (Tlr3 and Tlr7).39–41 Our current transcriptome data showed significant upregulation of MDA5 and toll-like receptors on both 4 and 7 dpi (Ifih1, 2.7- and 4.4-fold; Tlr3, 2.2- and 2.8-fold; and Tlr7, 1.4- and 1.7-fold, compared with control mice on 4 and 7 dpi, respectively; P<0.001). Although we did not find the change in the expression of mitochondrial antiviral signaling protein (Mavs), a key molecule in the signaling from MDA5, we detected significant upregulation of Irf7, downstream gene of Mavs, on both 4 and 7 dpi (17.3-fold on 4 dpi and 24.4-fold on 7 dpi; P<0.001). Thus, the pathway of innate immune responses mediated by MDA5 seemed to be involved in in vivo TMEV infection in the heart. An interesting result of 4 dpi transcriptome data was that although there was a strong interferon-stimulated gene signature, upregulation of type I IFN genes themselves were curiously completely missing (Online Figure 7 in the Data Supplement). This gene patterns suggest that interferon regulatory factor (IRF)-dependent induction of interferon-stimulated gene that is independent of type I IFN production. Similar patterns have been reported by others.42,43 However, we also found upregulation of other pattern recognition receptors, such as retinoic acid–inducible gene-I (RIG-I) gene (DEAD [Asp-Glu-Ala-Asp] box polypeptide 58 [Ddx58]) and laboratory of genetics and physiology 2 (LGP2) gene (DEXH [Asp-Glu-X-His] box polypeptide 58 [Dhx58]; Ddx58, 2.7- and 3.7-fold; Dhx58, 3.0- and 4.4-fold, compared with control mice on 4 and 7 dpi, respectively; P<0.001). Thus, TMEV might be recognized by toll-like receptors and MDA5, stimulating expressions of other receptors.44
On 7 dpi, we detected high levels of troponin I in serum and reduced levels of viral RNA in the hearts of infected mice. Granzyme B (Gzmb) was the most highly upregulated gene in the heat map on 7 dpi, whereas Gzmb was also upregulated on 4 dpi. Granzyme B belongs to a family of neutral serine proteases and can be produced by natural killer (NK) cells, cytotoxic T lymphocytes, and natural killer T (NKT) cells.45–47 NK cells and cytotoxic T lymphocytes are innate and acquired immune cells, respectively, whereas NKT cells can bridge between innate and acquired immune responses. In viral infections, granzyme B produced by NK cells and cytotoxic T lymphocytes has been reported to contribute to killing of infected cells, leading to viral clearance.45 The expression levels of Gzmb were correlated with serum troponin I levels, suggesting that Gzmb could be an effector molecule that damages cardiomyocytes. In addition, NK cell group 7 sequence (Nkg7), which is a cytotoxic granule protein found in activated NK cells and a subpopulation of CD8+ T cells, was also upregulated (2.5-fold on 4 dpi and 15.6-fold on 7 dpi; P<0.001).48 This also supports the effector roles of NK cells and cytotoxic T lymphocytes in viral clearance on 4 and 7 dpi, respectively. Innate immunity–related genes were also ranked among highly upregulated genes on 7 dpi, although we initially expected that acquired immunity–related genes should be ranked among highly upregulated genes, because virus-specific T- and B-cell responses (acquired immunities) play pathogenic roles in viral myocarditis on 7 dpi (phase II) mainly.7 Here, high upregulation of innate immunity–related genes masked the modest upregulation of acquired immunity–related genes. Thus, using standard 2-way comparison and their ranking, acquired immune response genes were not identified as major molecules that distinguish phase II from other phases.
By analyzing samples from 4 and 7 dpi with PCA, we were able to find that upregulation of a set of acquired immunity–related genes that contributed to the separation of the 2 groups. In PCA, Cd3g, which encodes for the γ-subunit of the CD3-T-cell receptor (TCR) complex, was listed as the most contributing to the separation (ie, PC1 value). CD3 antigens (Cd3d, Cd3e, and Cd3g), which are markers for T cells and NKT cells,49 were upregulated only on 7 dpi (Cd3d, 1.5-fold; Cd3e, 2.7-fold; and Cd3g, 8.8-fold; P<0.001). The expression levels of Cd3g were not correlated with serum troponin I levels, suggesting that CD3+ T-and NKT cell–mediated cytotoxicity was not the only effector mechanism to damage cardiomyocytes. Several MHC class II–related genes, such as H2-Ea-ps and H2-Aa, and immunoglobulin variant chain (Igkv6-32) were also listed as contributing genes. Spp1, which is also known as osteopontin, can play a role in the differentiation of T-helper 1 and T-helper 17 cells, which can cause tissue damage by producing proinflammatory cytokines (immunopathology).50 These results suggested that acquired immune responses, particularly T-helper 1 and T-helper 17 responses, could be active in the hearts of infected mice, because MHC class II molecules on antigen-presenting cells contribute to the activation of MHC class II–restricted CD4+ T cells, which can promote immuno-globulin production from B cells. Thus, in this phase, both innate and acquired immune responses, particularly cellular immunity (T cells, NK cells, and NKT cells), rather than humoral immunity or viral replication, seemed to play effector roles. A significant increase in heart weight on 7 dpi could be because of infiltration of immune cells and edema in the heart.
On 60 dpi, we did not detect serum troponin I or viral RNA in the heart, suggesting neither ongoing cardiomyocyte damage nor viral replication contributes to the pathogenesis in phase III. The findings in our model are consistent with those in phase III of human myocarditis, because ongoing cardio-myocyte damage or active virus replication is not required for the progression to phase III.10 In the heat map, remodeling-related genes, such as Mmp12 and Gpnmb, were the most highly upregulated genes. Mmp12 may contribute to fibrosis, because we found fibrillar collagen in the hearts of infected mice but not age-matched control mice histologically (data not shown). However, Gpnmb may act as a feedback regulator of cardiac inflammation and fibrosis in mice with TMEV-induced myocarditis, because Gpnmb has been shown to regulate proinflammatory responses and hepatic fibrosis in vivo.51,52 In PCA, Bmp10 contributed most to PC1 values based on the factor loading for PC1. Bmp10 is a member of transforming growth factor-β and plays a critical role in regulating the development of the heart.53 Cybb, which is also known as NADPH oxidase 2 (Nox2) and is associated with cardiac remodeling and cardiac fibrosis, was listed as a cardiac fibrosis–related gene.54,55 These data suggested that cardiac remodeling was active on 60 dpi. Of note, MHC class II–related (H2-Ea-ps, H2-Aa, and H2-Ab1) and immunoglobulin genes (Igkv10-96, Ighv1-43, and Igj) were upregulated during phase III, suggesting the presence of B cells and dendritic cells in the heart. Because B cells express MHC class II molecules and contribute to immunoglobulin production, humoral immunity, but not cellular immunity mediated by T cells, may play a role in phase III. A pathogenic role of autoantibodies in myocarditis has been shown in human myocarditis and its animal models.8,56,57 Thus, in our model, there may be residual viral or cardiac antigens, driving some antibody-dependent immunopathology. Dendritic cells may play a role in tissue protection and repair.58 This will be addressed in our future experiments.
PCA is an unsupervised approach to analyze large data sets, such as microarray data to find the key molecules for the variance among data without grouping.59 Thus, PCA can be used to identify an unbiased set of molecules; this is in contrast to a supervised approach that identifies a single molecule with significant expression ratio by comparing 2 sets of data (experimental group versus control group). In this study, PCA resulted in clear separation of samples from all 3 phases of viral myocarditis. Factor loading for PC1 identified a set of phase-specific biomarker candidates to distinguish the phases. In most clinical setting in viral myocarditis, determinations of the phase or transition from one phase to another phase in individual patients are currently difficult. Here, a traditional 2-way comparison between the phases is not applicable. Using microarray and PCA, we may be able to discriminate the phases of viral myocarditis in humans, which will set the stage for phase-specific treatments for patients in the future. Determination of the myocarditis phase by PCA might have limitation depending on the phase, because the proportion of variance of PC1 in the current study was higher when we analyzed the samples from phases II and III (41%) than the samples from all 3 phases (28%) or samples from phases I and II (27%). Thus, PCA could be most powerful for discrimination between phases II and III. Once the roles of immune effector cells in our TMEV model and phase-specific immunopathomechanisms (and their biomarker candidates) are elucidated, information can be applied clinically to patients with myocarditis, as a means of personalized medicine in prediction of disease courses (eg, development of cardiomyopathy) or determination of treatment (eg, responses to therapies, which depend on the disease phase and the roles of immune effector cells).
To diagnose viral myocarditis, Dallas criteria have been used as a standard method. Dallas criteria require the inflammatory cellular infiltrate with or without associated myocyte necrosis on the heart tissue sections (at least 4–5 samples of 1–2 mm3) stained by immunohistochemistry against surface antigens, such as CD3.5,60 However, sampling error and variation in expert interpretation occur. As shown in our results, CD3+ T cells may be not present in phase I or III. We also found that myocyte necrosis or CD3+ T cells were not detectable histologically on 4 dpi (data not shown). In our microarray analysis, we required 2 mg of heart samples, which is obtainable in human biopsy. Here, innate immune responses against viral infection can be detectable from 2 mg of heart tissue, even in the area where no abnormality is detected in echocardiography or histology in phase I. Although the limitations of microarray are high cost and investment of time (≥3 days), these limitations would be resolved using custom arrays that are less expensive and more sensitive to diagnose viral myocarditis.
In summary, we successfully separated samples from all 3 phases of viral myocarditis and identified the sets of genes contributing to separation, using a bioinformatics approach. Only innate immunity–related genes were upregulated significantly in phase I, whereas both innate immunity–related and acquired immunity–related genes were upregulated in phase II. Both acquired immunity–related and cardiac remodeling–related genes were upregulated in phase III. The set of genes that was identified in our model may be useful as phase-specific biomarkers. The translational application of information about the sets of the biomarker candidates from our model system and bioinformatics approach will aid the development of phase-specific therapy for viral myocarditis.
Supplementary Material
CLINICAL PERSPECTIVE.
Myocarditis most commonly results from virus infections, affects 2 million Americans, and is a major cause of sudden death. Viral myocarditis has been proposed to be a triphasic disease: in phase I, the disease is triggered by viral infection in the heart; in phase II, uncontrolled antiviral immune and autoimmune responses damage cardiomyocytes; and in phase III, as a result of phases I and II, cardiac remodeling leads to dilated cardiomyopathy. Ideally, each patient with myocarditis should be treated depending on the phase: phase I, antiviral; phase II, immunomodulation; and phase III, standard heart failure therapy (eg, immunosuppression may be appropriate for phase II but will enhance virus replication in phase I). For appropriate treatment of myocarditis, the identification of phase-specific biomarkers is crucial to develop effective phase-specific therapy. Although most human and experimental myocarditis studies have used traditional supervised transcriptome analyses with 2-way comparison based on P values between controls versus predefined myocarditis samples, they did not lead to a discovery of the phase-specific biomarkers that clearly distinguish all 3 phases of myocarditis. Using a novel mouse model for viral myocarditis induced with cardiovirus, we conducted unsupervised bioinformatics transcriptome analyses (eg, principal component analysis) along with multivariate conventional analyses that have been used clinically (eg, echo-cardiography, viral titration, cardiac troponin, and histology). We found a set of phase-specific biomarkers in the heart. Our approach can be applied clinically to patients with myocarditis, as a means of personalized medicine in prediction of disease course (eg, development of cardiomyopathy) or determination of treatment.
Acknowledgments
We thank Drs Matthew D. Woolard, Michelle M. Arnold, Viromi Fernando, Pratap C. Reddy, D. Neil Granger, Fereidoon Shafiei, and Liam A. Morris for helpful discussions and Sadie Faith Pearson, Lesya Ekshyyan, and Paula Polk for excellent technical assistance. We also thank Dr Paola Sebastiani, Department of Biostatistics, Boston University, for helpful statistical comments.
Sources of Funding
This work was supported by the fellowships (to Drs Omura and Sato) from the Malcolm Feist Cardiovascular Research Endowment, Louisiana State University Health Sciences Center-Shreveport, and grant from the National Institute of General medical Sciences Centers of Biomedical Research Excellence (COBRE) Grant (8P20GM103433 and P30GM110703).
Footnotes
The Data Supplement is available at http://circgenetics.ahajournals.org/lookup/suppl/doi:10.1161/CIRCGENETICS.114.000505/-/DC1.
Disclosures
None.
References
- 1.Kindermann I, Barth C, Mahfoud F, Ukena C, Lenski M, Yilmaz A, et al. Update on myocarditis. J Am Coll Cardiol. 2012;59:779–792. doi: 10.1016/j.jacc.2011.09.074. [DOI] [PubMed] [Google Scholar]
- 2.Fabre A, Sheppard MN. Sudden adult death syndrome and other non-ischaemic causes of sudden cardiac death. Heart. 2006;92:316–320. doi: 10.1136/hrt.2004.045518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu ZL, Liu ZJ, Liu JP, Kwong JS. Herbal medicines for viral myocarditis. Cochrane Database Syst Rev. 2013;8:CD003711. doi: 10.1002/14651858.CD003711.pub5. [DOI] [PubMed] [Google Scholar]
- 4.Guglin M, Nallamshetty L. Myocarditis: diagnosis and treatment. Curr Treat Options Cardiovasc Med. 2012;14:637–651. doi: 10.1007/s11936-012-0204-7. [DOI] [PubMed] [Google Scholar]
- 5.Cooper LT., Jr Myocarditis. N Engl J Med. 2009;360:1526–1538. doi: 10.1056/NEJMra0800028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Archard LC, Richardson PJ, Olsen EG, Dubowitz V, Sewry C, Bowles NE. The role of Coxsackie B viruses in the pathogenesis of myocarditis, dilated cardiomyopathy and inflammatory muscle disease. Biochem Soc Symp. 1987;53:51–62. [PubMed] [Google Scholar]
- 7.Martinez NE, Sato F, Kawai E, Omura S, Chervenak RP, Tsunoda I. Regulatory T cells and Th17 cells in viral infections: implications for multiple sclerosis and myocarditis. Future Virol. 2012;7:593–608. doi: 10.2217/fvl.12.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu PP, Mason JW. Advances in the understanding of myocarditis. Circulation. 2001;104:1076–1082. doi: 10.1161/hc3401.095198. [DOI] [PubMed] [Google Scholar]
- 9.Shauer A, Gotsman I, Keren A, Zwas DR, Hellman Y, Durst R, et al. Acute viral myocarditis: current concepts in diagnosis and treatment. Isr Med Assoc J. 2013;15:180–185. [PubMed] [Google Scholar]
- 10.Elamm C, Fairweather D, Cooper LT. Pathogenesis and diagnosis of myocarditis. Heart. 2012;98:835–840. doi: 10.1136/heartjnl-2012-301686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith SC, Ladenson JH, Mason JW, Jaffe AS. Elevations of cardiac troponin I associated with myocarditis. Experimental and clinical correlates. Circulation. 1997;95:163–168. [PubMed] [Google Scholar]
- 12.Aretz HT. Myocarditis: the Dallas criteria. Hum Pathol. 1987;18:619–624. doi: 10.1016/s0046-8177(87)80363-5. [DOI] [PubMed] [Google Scholar]
- 13.Baughman KL. Diagnosis of myocarditis: death of Dallas criteria. Circulation. 2006;113:593–595. doi: 10.1161/CIRCULATIONAHA.105.589663. [DOI] [PubMed] [Google Scholar]
- 14.Chow LH, Radio SJ, Sears TD, McManus BM. Insensitivity of right ventricular endomyocardial biopsy in the diagnosis of myocarditis. J Am Coll Cardiol. 1989;14:915–920. doi: 10.1016/0735-1097(89)90465-8. [DOI] [PubMed] [Google Scholar]
- 15.Imanaka-Yoshida K, Hiroe M, Yasutomi Y, Toyozaki T, Tsuchiya T, Noda N, et al. Tenascin-C is a useful marker for disease activity in myocarditis. J Pathol. 2002;197:388–394. doi: 10.1002/path.1131. [DOI] [PubMed] [Google Scholar]
- 16.Ruppert V, Meyer T, Pankuweit S, Jonsdottir T, Maisch B. Activation of STAT1 transcription factor precedes up-regulation of coxsackievirus-adenovirus receptor during viral myocarditis. Cardiovasc Pathol. 2008;17:81–92. doi: 10.1016/j.carpath.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 17.Ruppert V, Maisch B. Molecular signatures and the study of gene expression profiles in inflammatory heart diseases. Herz. 2012;37:619–626. doi: 10.1007/s00059-012-3662-5. [DOI] [PubMed] [Google Scholar]
- 18.Taylor LA, Carthy CM, Yang D, Saad K, Wong D, Schreiner G, et al. Host gene regulation during coxsackievirus B3 infection in mice: assessment by microarrays. Circ Res. 2000;87:328–334. doi: 10.1161/01.res.87.4.328. [DOI] [PubMed] [Google Scholar]
- 19.Szalay G, Meiners S, Voigt A, Lauber J, Spieth C, Speer N, et al. Ongoing coxsackievirus myocarditis is associated with increased formation and activity of myocardial immunoproteasomes. Am J Pathol. 2006;168:1542–1552. doi: 10.2353/ajpath.2006.050865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knowles NJ, Hovi T, Hyypiä T, King AMQ, Lindberg AM, Pallansch MA, et al. Part II - The Positive Sense Single Stranded RNA Viruses: Genus Cardiovirus. In: King AMQ, Adams MJ, Carstens EB, Lefkowiz EJ, editors. Virus Taxonomy: Classification and Nomenclature of Viruses, Ninth Report of the International Committee on Taxonomy of Viruses. Virology Division, International Union of Microbiological Societies. Waltham, MA: Elsevier Academic Press; 2012. pp. 862–863. [Google Scholar]
- 21.Sato F, Tanaka H, Hasanovic F, Tsunoda I. Theiler’s virus infection: patho-physiology of demyelination and neurodegeneration. Pathophysiology. 2011;18:31–41. doi: 10.1016/j.pathophys.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gómez RM, Rinehart JE, Wollmann R, Roos RP. Theiler’s murine encephalomyelitis virus-induced cardiac and skeletal muscle disease. J Virol. 1996;70:8926–8933. doi: 10.1128/jvi.70.12.8926-8933.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rames DS. The Etiopthogenesis of Theiler’s Murine Encephalomyelitis Virus (TMEV)-Induced Cardiomyopathy, Including Characterization of New Strain of TMEV [doctoral thesis] College Station, TX: Texas A&M University; 1995. [Google Scholar]
- 24.Omura S, Koike E, Kobayashi T. Microarray analysis of gene expression in rat alveolar epithelial cells exposed to fractionated organic extracts of diesel exhaust particles. Toxicology. 2009;262:65–72. doi: 10.1016/j.tox.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 25.Li W. Volcano plots in analyzing differential expressions with mRNA microarrays. J Bioinform Comput Biol. 2012;10:1231003. doi: 10.1142/S0219720012310038. [DOI] [PubMed] [Google Scholar]
- 26.R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; http://www.R-project.org/ [Google Scholar]
- 27.Jolliffe IT. Principal Component Analysis. Briefings in Bioinfomatics. 2. Vol. 12. New York: Springer; 2011. pp. 714–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim J, Jacobs DR, Jr, Luepker RV, Shahar E, Margolis KL, Becker MP. Prognostic value of a novel classification scheme for heart failure: the Minnesota Heart Failure Criteria. Am J Epidemiol. 2006;164:184–193. doi: 10.1093/aje/kwj168. [DOI] [PubMed] [Google Scholar]
- 29.Greer JJ, Ware DP, Lefer DJ. Myocardial infarction and heart failure in the db/ db diabetic mouse. Am J Physiol Heart Circ Physiol. 2006;290:H146–H153. doi: 10.1152/ajpheart.00583.2005. [DOI] [PubMed] [Google Scholar]
- 30.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, et al. Inflammation and Host Response to Injury, Large Scale Collaborative Research Program. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci USA. 2013;110:3507–3512. doi: 10.1073/pnas.1222878110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Osterburg AR, Hexley P, Supp DM, Robinson CT, Noel G, Ogle C, et al. Concerns over interspecies transcriptional comparisons in mice and humans after trauma. Proc Natl Acad Sci USA. 2013;110:E3370. doi: 10.1073/pnas.1306033110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perlman H, Budinger GRS, Ward PA. Humanizing the mouse: in defense of murine models of critical illness. Am J Respir Crit Care Med. 2013;187:898–900. doi: 10.1164/rccm.201303-0489ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu M, Eckart MR, Morgan AA, Mukai K, Butte AJ, Tsai M, et al. Identification of an IFN-γ/mast cell axis in a mouse model of chronic asthma. J Clin Invest. 2011;121:3133–3143. doi: 10.1172/JCI43598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Noutsias M, Rohde M, Göldner K, Block A, Blunert K, Hemaidan L, et al. Expression of functional T-cell markers and T-cell receptor Vbeta repertoire in endomyocardial biopsies from patients presenting with acute myocarditis and dilated cardiomyopathy. Eur J Heart Fail. 2011;13:611–618. doi: 10.1093/eurjhf/hfr014. [DOI] [PubMed] [Google Scholar]
- 35.Fuse K, Kodama M, Aizawa Y, Yamaura M, Tanabe Y, Takahashi K, et al. Th1/Th2 balance alteration in the clinical course of a patient with acute viral myocarditis. Jpn Circ J. 2001;65:1082–1084. doi: 10.1253/jcj.65.1082. [DOI] [PubMed] [Google Scholar]
- 36.Fairweather D, Stafford KA, Sung YK. Update on coxsackievirus B3 myocarditis. Curr Opin Rheumatol. 2012;24:401–407. doi: 10.1097/BOR.0b013e328353372d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yuan J, Yu M, Lin QW, Cao AL, Yu X, Dong JH, et al. Th17 cells contribute to viral replication in coxsackievirus B3-induced acute viral myocarditis. J Immunol. 2010;185:4004–4010. doi: 10.4049/jimmunol.1001718. [DOI] [PubMed] [Google Scholar]
- 38.Dermody TS, Parker JSL, Sherry B. Orthoreoviruses: heart, chap 44. In: Knipe DM, Howley PM, editors. Fields Virology. 6. II. Philadelphia, PA: Wolters Kluwer/Lippincott Williams & Wilkins; 2013. pp. 1335–1336. [Google Scholar]
- 39.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 40.Schulz O, Pichlmair A, Rehwinkel J, Rogers NC, Scheuner D, Kato H, et al. Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe. 2010;7:354–361. doi: 10.1016/j.chom.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jin YH, Kim SJ, So EY, Meng L, Colonna M, Kim BS. Melanoma differentiation-associated gene 5 is critical for protection against Theiler’s virus-induced demyelinating disease. J Virol. 2012;86:1531–1543. doi: 10.1128/JVI.06457-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dixit E, Boulant S, Zhang Y, Lee AS, Odendall C, Shum B, et al. Peroxisomes are signaling platforms for antiviral innate immunity. Cell. 2010;141:668–681. doi: 10.1016/j.cell.2010.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hasan M, Koch J, Rakheja D, Pattnaik AK, Brugarolas J, Dozmorov I, et al. Trex1 regulates lysosomal biogenesis and interferon-independent activation of antiviral genes. Nat Immunol. 2013;14:61–71. doi: 10.1038/ni.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bowie AG, Unterholzner L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat Rev Immunol. 2008;8:911–922. doi: 10.1038/nri2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Edwards KM, Davis JE, Browne KA, Sutton VR, Trapani JA. Anti-viral strategies of cytotoxic T lymphocytes are manifested through a variety of granule-bound pathways of apoptosis induction. Immunol Cell Biol. 1999;77:76–89. doi: 10.1046/j.1440-1711.1999.00799.x. [DOI] [PubMed] [Google Scholar]
- 46.Pham CTN, Ley TJ. The role of granzyme B cluster proteases in cell-mediated cytotoxicity. Semin Immunol. 1997;9:127–133. doi: 10.1006/smim.1997.0060. [DOI] [PubMed] [Google Scholar]
- 47.Metelitsa LS, Weinberg KI, Emanuel PD, Seeger RC. Expression of CD1d by myelomonocytic leukemias provides a target for cytotoxic NKT cells. Leukemia. 2003;17:1068–1077. doi: 10.1038/sj.leu.2402943. [DOI] [PubMed] [Google Scholar]
- 48.Zhou L, Li X, Church RL. The mouse lens fiber-cell intrinsic membrane protein MP19 gene (Lim2) and granule membrane protein GMP-17 gene (Nkg7): Isolation and sequence analysis of two neighboring genes. Mol Vis. 2001;7:79–88. [PubMed] [Google Scholar]
- 49.Tsunoda I, Libbey JE, Fujinami RS. TGF-β1 suppresses T cell infiltration and VP2 puff B mutation enhances apoptosis in acute polioencephalitis induced by Theiler’s virus. J Neuroimmunol. 2007;190:80–89. doi: 10.1016/j.jneuroim.2007.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Santamaría MH, Corral RS. Osteopontin-dependent regulation of Th1 and Th17 cytokine responses in Trypanosoma cruzi-infected C57BL/6 mice. Cytokine. 2013;61:491–498. doi: 10.1016/j.cyto.2012.10.027. [DOI] [PubMed] [Google Scholar]
- 51.Ripoll VM, Irvine KM, Ravasi T, Sweet MJ, Hume DA. Gpnmb is induced in macrophages by IFN-γ and lipopolysaccharide and acts as a feedback regulator of proinflammatory responses. J Immunol. 2007;178:6557–6566. doi: 10.4049/jimmunol.178.10.6557. [DOI] [PubMed] [Google Scholar]
- 52.Abe H, Uto H, Takami Y, Takahama Y, Hasuike S, Kodama M, et al. Transgenic expression of osteoactivin in the liver attenuates hepatic fibrosis in rats. Biochem Biophys Res Commun. 2007;356:610–615. doi: 10.1016/j.bbrc.2007.03.035. [DOI] [PubMed] [Google Scholar]
- 53.Huang J, Elicker J, Bowens N, Liu X, Cheng L, Cappola TP, et al. Myocardin regulates BMP10 expression and is required for heart development. J Clin Invest. 2012;122:3678–3691. doi: 10.1172/JCI63635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Johar S, Cave AC, Narayanapanicker A, Grieve DJ, Shah AM. Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB J. 2006;20:1546–1548. doi: 10.1096/fj.05-4642fje. [DOI] [PubMed] [Google Scholar]
- 55.Zhao Y, McLaughlin D, Robinson E, Harvey AP, Hookham MB, Shah AM, et al. Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with Doxorubicin chemotherapy. Cancer Res. 2010;70:9287–9297. doi: 10.1158/0008-5472.CAN-10-2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu M, Wen S, Wang M, Liang W, Li HH, Long Q, et al. TNF-α-secreting B cells contribute to myocardial fibrosis in dilated cardiomyopathy. J Clin Immunol. 2013;33:1002–1008. doi: 10.1007/s10875-013-9889-y. [DOI] [PubMed] [Google Scholar]
- 57.Matsumori A. Lessons learned from experimental myocarditis. Herz. 2012;37:817–821. doi: 10.1007/s00059-012-3692-z. [DOI] [PubMed] [Google Scholar]
- 58.Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327:291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Loke P, Favre D, Hunt PW, Leung JM, Kanwar B, Martin JN, et al. Correlating cellular and molecular signatures of mucosal immunity that distinguish HIV controllers from noncontrollers. Blood. 2010;115:e20–e32. doi: 10.1182/blood-2009-12-257451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Andréoletti L, Lévêque N, Boulagnon C, Brasselet C, Fornes P. Viral causes of human myocarditis. Arch Cardiovasc Dis. 2009;102:559–568. doi: 10.1016/j.acvd.2009.04.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.