

Abstract
Mitochondria are responsible for maintaining a variety of cellular functions. One such function is the interaction and subsequent import of proteins into these organelles via the translocase of outer membrane (TOM) complex. Antibodies have been used to analyze the presence and function of proteins comprising this complex, but have not been used to investigate variations in the abundance of TOM complex in mitochondria. Here, we report on the feasibility of using capillary cytometry with laser-induced fluorescence to detect mitochondria labeled with antibodies targeting the TOM complex and to estimate the number of antibodies that bind to these organelles. Mitochondria were fluorescently labeled with DsRed2, while antibodies targeting the TOM22 protein, one of nine proteins comprising the TOM complex, were conjugated to the Atto-488 fluorophore. At typical labeling conditions, 94 % of DsRed2 mitochondria were also immunofluorescently labeled with Atto-488 Anti-TOM22 antibodies. The calculated median number of Atto-488 Anti-TOM22 antibodies bound to the surface of mitochondria was ∼2,000 per mitochondrion. The combination of fluorescent immunolabeling and capillary cytometry could be further developed to include multicolor labeling experiments, which enable monitoring several molecular targets at the same time in the same or different organelle types.
Keywords: Mitochondria, TOM22, Fluorescence, Cytometry, Immunolabeling
Introduction
Mitochondria are heterogeneous organelles that perform a variety of cellular functions including ATP synthesis [1], the formation of reactive oxygen species [2], and the biotransformation of xenobiotics [3]. These functions are possible due to the translocation of nuclear-encoded proteins into mitochondria, through translocases. The TOM22 protein is a core component of the translocase of the mitochondrial outer membrane (TOM) protein complex [4, 5]. For the majority of nuclear-encoded proteins, the TOM complex is responsible for translocation of the protein into the intermitochondrial space, often resulting in subsequent interaction with the translocase of the mitochondrial inner membrane (TIM) and ultimate incorporation of the protein in the inner mitochondrial membrane or the mitochondrial matrix [4]. Although much is known regarding the role TOM22 has in protein import, the quantitation of this protein on individual mitochondria is critical in understanding how variations in protein import contribute to mitochondrial heterogeneity.
Previous reports regarding the heterogeneous nature of mitochondria have described variations among individual mitochondria in terms of isoelectric point, cardiolipin content, abundances of select proteins, and other mitochondrial properties being implicated in different states of mitochondrial function and viability [6–9]. However, there are no studies reporting heterogeneity in proteins associated with TOM or TIM complexes.
Most of the traditional bulk analyses of mitochondrial preparations, such as Western blots, enzyme assays, membrane potential measurements, monitoring ATP generation, and proteomics [10–12], are inadequate to describe the heterogeneous nature of mitochondria because they fail to provide information at the single-mitochondrion level [13]. Accordingly, the presence of TOM22 in mitochondria has been established using Western blots [14]. To our knowledge, there are no reports providing the abundance of TOM22 proteins on single mitochondria.
Recent technological advances including highly sensitive flow cytometry (HSFCM), capillary electrophoresis with laser-induced fluorescence, and capillary cytometry allow for high-throughput analysis of individual mitochondria that use mitochondrion-specific fluorescent labels [15]. Labels, such as 10-nonyl acridine orange, which probes for cardiolipin content, tetramethylrhodamine, a membrane potential probe, and DsRed2 used as a fusion protein targeting mitochondrial proteins, are popular for use in individual mitochondria analysis because they selectively accumulate in mitochondria at detectable levels.
On the other hand, immunofluorescent labeling, common for whole-cell flow cytometry applications [16], has not been widely used. The explanation is straightforward: Conventional flow cytometry is adequate for analysis of fluorophores in single cells because the sensitivity of such instruments is adequate for detection of sufficiently abundant fluorophores tagging single cells, but insufficient to detect fluorophores at the single organelle level. Recently, Zhang et al. reported using HSFCM for high-throughput analysis of individual mitochondria detected after immunofluorescently labeling of cytochrome c and porin [17]. Briefly, smaller probe volume lowered their background noise and extended transit time (from microseconds to milliseconds) through the laser beam increased the sensitivity characteristic of HSFCM.
In this paper, we use a custom-built system combining a capillary with laser-induced fluorescence detection, previously termed capillary cytometry [18], which is simpler than the previously reported HSFCM [17], to detect and quantify TOM22 on the surface of individual mitochondria. Labeling is based on a mitochondrion-selective Anti-TOM22 antibody with high specificity for mitochondria as recently demonstrated in affinity purification studies [19]. Fluorescent immunolabeling is a convenient approach to monitor specific proteins in biological systems [20]. As such, the Anti-TOM22 antibody has been conjugated with the Atto-488 label (excitation at 488 nm; maximum fluorescent emission at 523 nm). For simplicity, this antibody is termed Atto-488 Anti-TOM22 antibody in this report. In this paper, we characterize the ability of Atto-488 Anti-TOM22 antibodies to label mitochondria and to estimate the abundance of TOM22 in individual mitochondria, which is relevant to future studies addressing the issue of heterogeneous protein import into mitochondria. This general analytical strategy could be easily extended to characterize the abundance of other proteins in individual organelles of various types allowing for a more comprehensive characterization of subcellular heterogeneity.
Experimental
Chemicals and reagents
Ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA), trypsin solution, phosphate-buffered saline (PBS), potassium hydroxide (KOH), hydrochloric acid, formaldehyde, poly-l-lysine, sucrose, HEPES, d(+)-mannitol, Triton X-100, Atto 488 (41051-1MG-F Lot 1480709V), and monoclonal Anti-TOM22-Atto 488 antibody (mouse IgG1 isotype, T4327, Lot 058K4766) were purchased from Sigma-Aldrich (St. Louis, MO). Mouse IgG1 isotype antibody control (Atto 488 labeled, 804-870TD-C050, Lot L28462) was purchased from Enzo Life Sciences (Farmingdale, NY). Dulbecco's modified Eagle medium (DMEM), Geneticin, fetal bovine serum, fluorescein *NIST-traceable standard* (F36915, Lot 1308448), FluoSpheres® Size Kit #2 (F8888 Lot 1306514), and ProLong Gold antifade reagent were purchased from Invitrogen Molecular Probes (Eugene, OR). Bovine serum albumin (BSA) Fraction V, heat shock, fatty acid free was purchased from Roche Diagnostics (Indianapolis, IN).
Buffers
Mitochondria isolation buffer (MIB) contained 220 mM mannitol, 70 mM sucrose, 0.5 mM EGTA, and 2 mM HEPES; the final pH was adjusted with KOH to 7.4. All buffers were prepared with Milli-Q water and filtered through a 0.22-μm membrane filter unless otherwise noted.
Cell culture
Adherent 143B human osteosarcoma cells, expressing DsRed2 targeting mitochondria (Mito-DsRed2), were prepared previously for an earlier report [21]. Briefly, these cells express Mito-DsRed2 because they were transfected with a pDsRed2-mito plasmid encoding genes that confer neomycin/ kanamycin resistance and express a fusion protein of the mitochondria targeting sequence from subunit VII of cytochrome c oxidase and the red fluorescent protein DsRed2. When Mito-DsRed2 is translocated to mitochondria, its targeting sequence is cleaved and DsRed is released to the mitochondrial matrix. These cells were cultured in DMEM medium supplemented with 10 % (v/v) fetal bovine serum, 50 μg/mL gentamicin, and 5 % CO2 in 75-cm2 vented flasks at 37 °C. Cells were split every 3–4 days when they reached confluence. Cells were rinsed with PBS and lifted with 1.00 g/L trypsin in PBS for 5 min at 37 °C and diluted with fresh medium in a 1:40 ratio for subsequent passages or diluted 1:1 in DMEM prior to use in mitochondria preparations. The typical number of cells used in an experiment was ∼1 × 107 cells.
Mitochondria preparation
Differential centrifugation was used to isolate mitochondria from 143B human osteosarcoma cells. Lifted cells were washed and resuspended in MIB before homogenization. Repeated strokes (70–100) of a 7-mL tight-fitting Dounce Homogenizer (0.0008–0.0022 in. clearance) (Wheaton, Millville, NJ) were used to mechanically disrupt cells. Disrupted cells were centrifuged at 1,000×g for 10 min to pellet out intact cells, cell debris, and the nuclear fraction. The supernatant was centrifuged at 10,000×g for 10 min to obtain a mitochondrial-enriched pellet which was resuspended in MIB. The protein content of this suspension was determined using a bicinchoninic acid protein assay according to vendor instructions (Pierce Scientific, Rockford, IL) with the following modifications. A 50:1 solution of reagent A (sodium carbonate, bicinchoninic acid)/reagent B (copper(II) sulfate, pentahydrate) was incubated with each BSA protein standard, ranging from 25 to 2,000 μg/mL and the mitochondria suspension in MIB. After a 30-min incubation at 37 °C, mixtures were loaded on a Corning 96 well plate (Corning Inc., Acton, MA). The absorbance values at 562 nm were then recorded on a Biotek Synergy 2 plate reader (BioTek Instruments Inc., Winooski, VT).
Aliquots corresponding to 5 μg of total mitochondrial protein in the sample were centrifuged at 10,000×g for 10 min, pelleting mitochondria. After discarding the supernatant, the 5-μg mitochondria pellet was resuspended with gentle pipetting in a ∼2 % BSA in MIB solution, resulting in a final concentration of 50 μg mitochondria pellet per milliliter MIB. The estimated number of mitochondria given the protein content of the mitochondrial pellet is ∼6×106. The suspension was aliquoted out and used for (a) unlabeled controls, (b) samples incubated with Atto-488 Anti-TOM22 antibodies, or (c) samples incubated with Atto-488 mouse IgG1 isotype controls. For samples subjected to antibody treatment, concentrations ranging from 1 to 32 μg/mL (or 6.7×10−9 to 2.1× 10−7 M) of the respective antibodies were incubated with 50 μg/mL mitochondria protein in a total volume of 100 μL MIB for 1 h at 4 °C in the dark; conditions typical for Anti-TOM22 antibody binding to mitochondria isolated from cell culture [19]. After incubation, mitochondria were centrifuged at 10,000×g for 10 min at 4 °C, discarding the supernatant.
Confocal microscopy
Cells were cultured overnight in DMEM in an eight-well Lab-Tek no. 1.5 chambered slide (Nunc, Rochester, NY) coated with poly-l-lysine. Cells were fixed with 4.0 % (v/v) formaldehyde in PBS for 15 min, washed with PBS, and permeabilized with 0.1 % (v/v) Triton X-100 in water.
Permeabilized cells were washed with 2 % BSA in PBS for 1 h at room temperature then washed with PBS. Working dilutions of Atto-488 antibodies, 1 μg/mL (6.7 nM) Atto-488 Anti-TOM22 and 1 μg/mL Atto-488 mouse IgG1 isotype controls (Supplementary Material, Section A, Fig. S1), were then incubated with cells at 4 °C overnight in the dark. Cells were then washed three times with PBS, set using ProLong Anti-Fade reagent, and imaged.
Confocal microscopy images were captured as previously described with minor variations [22]. Images were acquired using an Olympus IX-81 inverted microscope with a 120-W mercury lamp, a DS-IX100 disk spinning unit, and a C9100-01 EM CCD camera (Hamamatsu, Bridgewater, NJ). Images were captured using a ×60 oil immersion lens unless noted otherwise. Filter cube configurations to capture green fluorescence (460–500 nm excitation, 505 nm dichroic, 510–560 nm emission) and red fluorescence (510–560 nm excitation, 565 nm dichroic, 572–648 nm emission) were used to image subcellular regions labeled with Atto-488 Anti-TOM22 antibodies and the DsRed2 mitochondria fusion protein, respectively.
Images were subject to analysis to determine the Pearson correlation coefficient, intensity correlation quotient (ICQ), and the Manders' colocalization coefficients (M1 and M2). Images obtained with either the green or red optical filter settings were corrected for the respective background fluorescence by selecting five regions of interest of extracellular and nuclear space and subtracting the background average fluorescence intensity from these regions. The Manders' coefficients assess colocalization of both fluorophores in the same pixel, while the Pearson correlation coefficient and the ICQ correlate the fluorophore intensities of the two fluorophores in the same pixel. The Colocalization Plugin from Image J-W version 1.43s (National Institutes of Health), was used to analyze 143B human osteosarcoma cells expressing DsRed2 and labeled with Atto-488 Anti-TOM22 antibodies and to calculate the Manders' colocalization coefficients, M1 and M2. The Pearson correlation coefficient was calculated as described by Adler and Parmryd [23].
Capillary cytometry
Experiments were performed on a custom-built setup with post-capillary sheath flow dual LIF detection, which has been previously described [18, 22]. Samples were excited with an argon ion laser (10 mW, Melles Griot, Irvine, CA). Fluorescence from the DsRed2 and Atto-488 was spectrally resolved with a 560 long-pass dichroic long pass filter (DCLP560, Omega Filters, Brattleboro, VT), selected with 517.5–552.5 nm (535DF35, Omega Optical, Brattleboro, VT) and 607.5–662.5 nm (635DF55, Omega Optical) bandpass filters, respectively, and detected simultaneously by two separate photomultiplier tubes (PMT) (R1477, Hamamatsu). The output of each PMT was digitized at 200 Hz with a NiDaq I/O board (PCI-MIO-16XE-50, National Instruments, Austin, TX) run with LabVIEW (National Instruments) and stored as binary files.
Samples were passed through a 50-cm-long, 30-μm I.D., 150-μm O.D. uncoated fused silica capillary (Polymicro Technologies, Phoenix, AZ) at a rate of 20 nL/min (3 cm/min) maintained by the application of external pressure (10± 1 kPa) provided via a nitrogen pressurized filtering flask (Electronic Supplementary Material, Section E, Fig. S5 and Table S2). The optics were adjusted until the fluorescence intensities of individual alignment beads (AlignFlow™, 2.5-μm, 488-nm excitation, Invitrogen Molecular Probes, Eugene, OR) reached a maximum (S/N 1,800±200) while maintaining a relative standard deviation (∼15±3 %) within the manufacturer's specifications (∼20 % RSD).
Prior to running each sample, alignment beads were passed through the capillary and the average median intensity was calculated. This value was used to normalize the fluorescence intensities of the events detected in the subsequent run. Raw data for each run consisted of a flowgram describing the appearance of fluorescent events as a function of time. Depending on the PMT at which events were detected, they were classified as green (517.5–552.5 nm), red (607.5–662.5 nm), or dual-labeled (517.5–552.5 nm; 607.5–662.5 nm) peaks. Dual-labeled peaks had green and red peaks with maxima occurring within 20 ms of one another. Based on the labeling scheme, we anticipated that green, red, and dual-labeled peaks corresponded to mitochondria labeled with Atto-488 Anti-TOM22 antibodies, Mito-DsRed2, or both of these, respectively.
Data analysis
Raw data were processed using a combination of IgorPro and MATLAB routines (available upon request). A region of the flowgram not having any peaks (e.g., 500–1,500 ms) was used to determine a baseline standard deviation (σ). A threshold value of 5σ was used to detect peaks. Previous reports utilizing the custom-built LIF detector used in this work resulted in a false-positive rate of less than 5 % relative to the total number of the total number of mitochondrial events when using a threshold value of 5σ [6]. Typical peak widths were 53± 17 ms (N=129).
Due to the cross-talk between PMTs used to detect green and red events, controls with only one fluorophore (DsRed2 and Atto488) were used to determine a correction factor. This factor was determined empirically as a third-order polynomial fit (Electronic Supplementary Material, Section B, Fig. S2). These controls were mitochondria isolated from 143B cells (a) containing DsRed2 and (b) not containing DsRed2 but labeled with Atto-488 Anti-TOM22 antibodies. Green peak intensities were corrected for cross-talk from DsRed2 fluorescence as follows:
| (1) |
where GT is the corrected green intensity, Gm is the observed green intensity, Rm is the observed coincident red peak intensity, and C(Rm) is the correction factor given by the polynomial fit at a given Rm. Likewise, red peak intensities were corrected for cross-talk from Atto-488 fluorescence as follows:
| (2) |
where RT is the corrected red intensity, Rm is the observed red intensity, Gm is the observed coincident green peak intensity and C(Gm) is the correction factor given by the polynomial fit at a given Gm. The corrected red and green intensities were again compared to the 5σ threshold value. If RT or GT<5σ, threshold value the peak value was excluded from the analysis.
Atto-488 Anti-TOM22 antibodies per mitochondrion
To estimate the number of Atto-488 Anti-TOM22 Antibodies per mitochondrion, the following steps were taken: (a) assigning molecular equivalent of soluble fluorophore (MESF) values to a fluorescein microbead standard as previously described by Poe et al. [24], (b) determining the relative fluorescence response between fluorescein and Atto-488, (c) comparing the fluorescent response of fluorescein microbeads and Alignflow 488 flow cytometry alignment beads, and (d) calculating the number of Atto-488 Anti-TOM22 antibodies labeling each mitochondrion (Electronic Supplementary Material, Section C). These calculations allow us to estimate the number of Atto-488 Anti-TOM22 antibodies per mitochondrion.
Statistical analysis
To ensure the events analyzed represented single organelles rather than multiple organelles simultaneously traveling through the detection window, flowgrams were analyzed using statistical overlap theory as previously described [25]. Briefly, flowgrams were split into N bins, where the determination of N was based on the number of peaks detected in the flowgram, ptot.
| (3) |
The threshold m value (maximum number of observed peaks that do not represent an overlap; Bt=0.90) for each bin was calculated and compared to the respective number of observed peaks, p, in the same bin. If the number of peaks in any bin was greater than the m value calculated, then p is a poor estimate of m and any peaks detected in the bin were eliminated from further analysis. This data treatment ensures that peaks in the retained bins have a 90 % probability of lying within the 95 % confidence interval of representing a single event.
Results and discussion
Labeling of mitochondria with Atto-488 Anti-TOM22 antibodies
Initial assessment of the binding of Atto-488 Anti-TOM22 antibodies to mitochondria was based on confocal fluorescence microscopy of cells expressing DsRed2, a fusion protein that localizes in the matrix of mitochondria to release DsRed2 upon cleavage of the targeting sequence. An example of colocalization of Atto-488 Anti-TOM22 antibodies and DsRed2 is shown in Fig. 1. Not all the cells observed expressed DsRed2 because transient transfections used here result in highly variable numbers of cells that are positive for the expression of DsRed2. Furthermore, it is common to observe heterogeneous distributions of mitochondrial proteins among the various subcellular regions [26]. Thus, it is not surprising that the distribution of DsRed2 is heterogeneous among mitochondria. These two factors explain the labeling patterns observed in Fig. 1.
Fig. 1.

Colocalization of mito-DsRed and Atto-488 anti-TOM22 antibody in transiently transfected 143B cells. Imaging by confocal fluorescence microscopy detection of (i) Atto-488 Anti-TOM22 antibodies and (ii) DsRed2. The overlay (iii) illustrates the overlap of (i) and (ii). Note that DsRed2 localization is not equally distributed in all mitochondria. Scale bar, 10 μm
To assess the colocalization of Atto-488 fluorescence with that of mito-DsRed2, a subset of cells that appear to express DsRed2 were analyzed (N=3 cells), and a Pearson correlation coefficient of 0.78 and an ICQ value of 0.28 were indicative of a positive colocalization of the labels. Based on the Manders' M1 coefficient, 92 % of pixels positive for DsRed2 were also Atto-488 positive. The remaining 8 % of pixels positive for DsRed2 but not for Atto-488 may be attributed to low levels of TOM22 in mitochondria at different subcellular regions [26]. Similarly, the Manders' M2 coefficient indicated 72 % of pixels positive for Atto-488 were also DsRed2 positive. The remaining 28 % of pixels positive for Atto-488 were not DsRed2 positive within a cell expressing DsRed2 mitochondria, which is expected from nonspecific binding of this antibody to other subcellular regions or the lack of homogeneous mito-DsRed2 expression within the cell, which may result in some mitochondria having undetectable DsRed2 levels. Indeed, these results would be highly dependent on the transient expression of DsRed2 in the subset of cells chosen. On the other hand, it illustrates that both Atto-488 and DsRed2 are distinct fluorophores which are adequate for capillary cytometric analysis of TOM22 in individual mitochondria that is described below.
LIF analysis of individual immunolabeled mitochondria
Flowgrams were obtained with the capillary cytometry-LIF detection system, previously used for capillary electrophoretic analysis of individual organelles. Flowgrams are similar to electropherograms obtained in capillary electrophoretic analyses, but differ in that the time of appearance of an individual event in a flowgram cannot be used to calculate the electrophoretic mobility of the detected organelle (Fig. 2). Capillary electrophoresis requires an injection of a small sample volume (∼nanoliter), which contains the organelles that will be detected during a separation that typically lasts 10–20 min. In contrast, capillary cytometry uses a continuous introduction of sample into the capillary, making it possible to collect data of as many organelles as necessary from a given sample. The flowgrams obtained here proved that capillary cytometry-LIF is adequate for the analysis of mitochondria fluorescently labeled with DsRed2 (red peaks) and Atto-488 Anti-TOM22 antibodies (green peaks) (Fig. 2).
Fig. 2.

Flowgram of mitochondria isolated from DsRed2 transfected 143B cells incubated with Atto-488 Anti-TOM22 antibodies. Panel (A) shows examples of mitochondria labeled with both DsRed2 and Atto-488 anti-TOM22 antibodies. Panel (B) shows an expanded view illustrating events observed in a typical flowgram which may include the following: (a) Green fluorescent peaks represent mitochondria events from Atto-488 anti-TOM22 antibody labeling. (b) Red fluorescent peaks represent mitochondrial events from DsRed2 labeling. (c) Simultaneous detection of green and red fluorescent peaks represents mitochondria labeled with both DsRed2 and Atto-488 anti-TOM22 antibodies
Flow cytometry is also potentially useful for the analysis of individual mitochondria and provides results similar to those obtained by capillary cytometry-LIF [21]. However, use of flow cytometry is feasible only when each organelle has a relatively high number of fluorophores, which is the case when using probes such as 10-N-nonyl acridine orange [27] or MitoTracker dyes [28]. Conventional flow cytometry cannot be used to detect immunolabeled mitochondria due to the limited number of fluorophores attached to each organelle. On the other hand, the previously reported HSFCM [17] and capillary cytometry-LIF reported here have sufficient sensitivity to detect immunolabeled organelles. As such, the capillary cytometer used here was capable of detecting single mitochondria labeled with Atto-488 Anti-TOM22 antibodies (green peaks, Fig. 2).
Taking DsRed2 as a reference label, we evaluated immunofluorescence labeling of mitochondria with Atto-488 Anti-TOM22 antibodies. As seen in Fig. 2, flowgrams displayed three types of organelles: (a) exclusively labeled with DsRed2 (red peaks), (b) exclusively labeled with Atto-488 Anti-TOM22 antibodies (green peaks), or (c) dual-labeled with DsRed2 and Atto-488 Anti-TOM22 antibodies (red and green peaks). The third type of event indicates successful detection of TOM22 in mitochondria.
Relative to all Atto-488 Anti-TOM22 antibody labeled events, 81±3 % (N=643) were positive for detection of DsRed2; only 19±4 % (N=643) did not have detectable levels of DsRed2. The respective values in the fluorescence microscopy images (Fig. 1) were statistically different, with a Manders' M2 coefficient of 72 % (chi-square 25, P<0.0001). In theory, a smaller fraction of Atto-488 Anti-TOM22 antibody labeled events would be expected to be positive for DsRed2 in the capillary cytometry-LIF results relative to those from fluorescence microscopy because of some mitochondria are disrupted and lose DsRed2 from the matrix during mechanical homogenization of the cells. However, the differences between the microscopy and the capillary cytometry-LIF analysis are not surprising because different preparations were used for each experiment and because the efficiency of protein expression obtained with transient transfections in each preparation is highly variable. Furthermore, the differences may result from the absence of nonspecific binding of Atto-488 Anti-TOM22 antibodies to non-mitochondrial regions in the images observed or differences in sensitivity of the two techniques that favor improved detection of DsRed2-labeled mitochondria by capillary cytometry-LIF. Despite these differences, these results confirm that the majority of events are dual-labeled with DsRed2 and Atto-488 Anti-TOM22 antibodies, which confirms that these events are most likely mitochondria.
Most importantly, relative to 542 DsRed2 events detected at five different antibody concentrations, 94±2 % were positive for detection of TOM22; only 6±2 % of the DsRed2 organelles were not positive for TOM22. This is statistically similar to 92 % for the Manders' M1 coefficient obtained from the microscopy images (chi-square value of 2.6 and P=0.1). Together, these results suggest that only a relative small number of mitochondria have undetectable levels of TOM22 (<∼60 antibodies per mitochondrion).
Two additional observations were needed to confirm that indeed dual-labeled events correspond to mitochondria. These are negligible levels of (1) native fluorescence of mitochondria and (2) nonspecific binding of the Atto-488 Anti-TOM22 antibodies to organelles other than mitochondria.
It is unlikely that green events are caused by native fluorescence because of the high threshold used here to select peaks, which eliminate small peaks associated with native fluorescence (data not shown).
To determine the extent to which nonspecific binding results in green peaks, an Atto-488 mouse IgG1 isotype control, which does not have any binding specificity, was incubated with DsRed2-labeled mitochondria and analyzed via capillary cytometry-LIF (Fig. 3). A wide range of identical antibody concentrations (from 4 to 32 μg/mL or 2.7×10−8 to 2.1× 10×7 M) of isotype antibody control and the Atto-488 Anti-TOM22 antibody were used to analyze nonspecific binding. Out of 202 organelles that were labeled when using five different concentrations of the isotype antibody, only 31± 17 % labeled mitochondria, while 69±17 % labeled other organelles or cellular debris. Of 708 DsRed2-labeled mitochondria detected in five samples with different isotype antibody concentrations, the percentage that was labeled with the isotype antibody control was 10±6 % (Electronic Supplementary Material, Section D, Fig. S4 and Table S1). Even at the highest antibody concentrations tested, 32 μg/mL (2.1× 10×7 M), the level of nonspecific antibody binding remained the same. Similarly, the medians of Atto-488 fluorescence intensities of individual coincident events indicated that nonspecific binding is ∼4 % (Electronic Supplementary Material, Section D). Together, these observations confirm that Atto-488 Anti-TOM22 Antibodies are adequate to selectively label and detect mitochondria by capillary cytometry-LIF.
Fig. 3.

A graphical representation of the percentage of mitochondrial events labeled either (i) exclusively with DsRed, as indicated with red bars, (ii) exclusively with Atto-488 Anti-TOM22 antibodies, as indicated with green bars, or (iii) with both DsRed2 and Atto-488 Anti-TOM22 antibodies, is presented. At concentrations of 2.7 × 10−8 to 2.1 × 10−7 M antibody, 542 DsRed2 events were detected. From this total, the percent of DsRed2 mitochondria also labeled with antibody is 94±2 % (N=5 antibody concentrations)
Number of Atto-488 Anti-TOM22 antibodies per mitochondrion
The antibody concentration relative to the amount of protein in isolated mitochondria was critical to maximize immunolabeling of the TOM22 targets on the surface of mitochondria. Antibody concentrations greater than 4 μg/mL (2.7×10−8 M) were needed for 50 μg/mL protein resuspensions of mitochondria pellets to maximize the fraction of organelles with both DsRed2 and Atto-488 Anti-TOM22 antibodies (Fig. 3).
Similarly, Atto-488 Anti-TOM22 antibody fluorescence of dual-labeled peaks suggests that antibody concentrations greater than 4 μg/mL (2.7×10×8 M) were needed to maximize the fluorescence response when labeling mitochondria in suspension with protein contents of 50 μg/mL (Fig. 4).
Fig. 4.

Distribution of individual fluorescence intensities of dual-labeled events. The box plot presents the 25th, 50th, and 75th percentile and whiskers represent the minimum and maximum values of green fluorescence (RFU) of dual-labeled events. At concentrations of 2.7 × 10−8 to 2.1 × 10−7 M antibody, there is a significant increase in RFU of green fluorescence in dual-labeled events (P<0.0001, α=0.05)
Based on the individual peak heights (c.f. Fig. 4) at optimized antibody concentrations, we estimated the number of Atto-488 Anti-TOM22 antibodies per mitochondrion (Fig. 5). The number of Atto-488 Anti-TOM22 Antibodies per mitochondrion ranged from 60 to 10,000 with a median of ∼ 2,000 (N=512).
Fig. 5. Distribution of Atto-488 anti-TOM22 antibodies per mitochondrion. Bin width is 935.
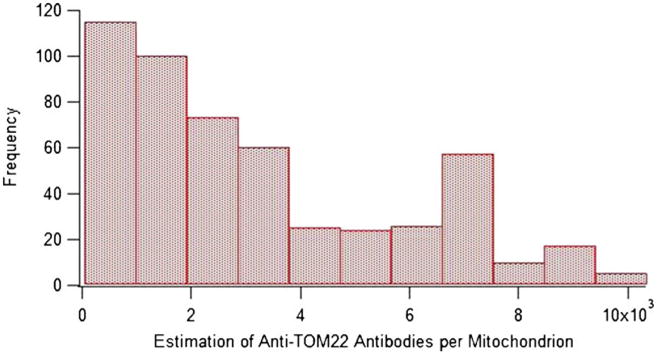
Assuming that TOM22 molecules are not within ∼100 Å [29], which is the distance between the two paratopes of an IgG, each anti-TOM22 antibody binds to only one TOM22, and it is safe to assume univalent binding between the Atto-488 Anti-TOM22 antibody and the TOM22 protein [30]. Therefore, we can assume that the estimate on the number of antibodies corresponds to the average abundance of TOM22 on the surface of single mitochondria.
Using super-resolution microscopy techniques, Wurm et al. observed densities of TOM clusters of ∼112, ∼102, and ∼90/μm2 in HeLa, Vero, and PtK2 cells, respectively [26]. From these values, the number of TOM complexes per cluster (10), the number of TOM22 molecules per TOM complex (3), and the expected surface area of an isolated mitochondrion (0.5 μm2), we estimated that there are ∼1,500 TOM22 molecules per mitochondrion. Although Wurm et al. used different cell types, the value based on their data compares well with the median of ∼2,000 anti-TOM22 antibodies that we determined for 143B cells by capillary cytometry-LIF.
The number of anti-TOM22 antibodies labeling individual mitochondria was highly heterogeneous (c.f. Fig. 5). The overall shape of the observed distribution may be influenced by the limit of detection of the technique (60 anti-TOM22 antibodies per mitochondrion), which will truncate the distribution excluding mitochondria with fewer TOM22 molecules. The appearance of the distribution is also biased by the selection of bin width: Fewer bins reduce the magnitude of the bin at ∼7,000 Anti-TOM22 antibodies per mitochondrion; if there were more events, more bins could reveal more detail on the bin at ∼7,000 Anti-TOM22 antibodies. Despite potential issues with limits of detection and the construction of the histogram representing the data, it is clear that isolated mitochondria are highly heterogeneous in regards to their bound Anti-TOM22 antibodies. It would not be surprising if the lack of normality of the data in Fig. 5 reflects variable protein translocation activity into mitochondria, which ultimately would be associated with variable mitochondrial demands in different subcellular regions.
Concluding remarks
We have demonstrated detection and analysis of immunofluorescent labeled individual mitochondria using a relatively simple custom-built capillary cytometry-LIF. At optimal labeling conditions, the estimated median number of Atto-488 Anti-TOM22 molecules per mitochondrion was ∼2,000, detecting as little as 60 antibodies per mitochondrion. This technique could be used to determine the levels of TOM22 per mitochondrion under different states of mitochondrial dysfunction or in models of protein import deficiencies based on knockdown of TOM22 via commercially available TOM22 siRNA. Given similar labeling conditions, proteins with similar abundance on mitochondria or other organelle types such as peroxisomes, lysosomes, and autophagosomes would be detectable provided that antibodies and corresponding fluorophores are available.
The analysis of immunolabeled individual mitochondria via capillary cytometry-LIF suggests potential of immunolabeling of organelles for analysis via individual organelle capillary electrophoresis analyses as well as analysis of organelle subpopulations characterized by differences in abundance of surface molecular targets.
Supplementary Material
Acknowledgments
This work was supported by NIH AG020866. THT was supported in part by the Center for Analysis of Biomolecular Signaling, University of Minnesota, NIH T32GM008700 and NIH GM063533.
Footnotes
Electronicsupplementary material The online version of this article (doi:10.1007/s00216-013-7593-7) contains supplementary material, which is available to authorized users.
References
- 1.Hatefi Y. The mitochondrial electron-transport and oxidative-phosphorylation system. Annu Rev Biochem. 1985;54:1015–1069. doi: 10.1146/annurev.biochem.54.1.1015. [DOI] [PubMed] [Google Scholar]
- 2.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol Lond. 2003;552(2):335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Omura T. Mitochondrial P450s. Chem Biol Interact. 2006;163(1–2):86–93. doi: 10.1016/j.cbi.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 4.Ryan MT, Wagner R, Pfanner N. The transport machinery for the import of preproteins across the outer mitochondrial membrane. Int J Biochem Cell Biol. 2000;32(1):13–21. doi: 10.1016/s1357-2725(99)00114-4. [DOI] [PubMed] [Google Scholar]
- 5.Neupert W. Protein import into mitochondria. Annu Rev Biochem. 1997;66:863–917. doi: 10.1146/annurev.biochem.66.1.863. [DOI] [PubMed] [Google Scholar]
- 6.Wolken GG, Kostal V, Arriaga EA. Capillary isoelectric focusing of individual mitochondria. Anal Chem. 2011;83(2):612–618. doi: 10.1021/ac102712r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen HC, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. 2005;280(28):26185–26192. doi: 10.1074/jbc.M503062200. [DOI] [PubMed] [Google Scholar]
- 8.Fuller KM, Duffy CF, Arriaga EA. Determination of the cardiolipin content of individual mitochondria by capillary electrophoresis with laser-induced fluorescence detection. Electrophoresis. 2002;23(11):1571–1576. doi: 10.1002/1522-2683(200206)23:11<1571::AID-ELPS1571>3.0.CO;2-3. doi:10.1002/1 522-2683(200206)23:11<1571∷aid-elps1571>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 9.Anand RK, Chiu DT. Analytical tools for characterizing heterogeneity in organelle content. Curr Opin Chem Biol. 2012;16(3–4):391–399. doi: 10.1016/j.cbpa.2012.05.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barrientos A. In vivo and in organello assessment of OXPHOS activities. Methods. 2002;26(4):307–316. doi: 10.1016/s1046-2023(02)00036-1. [DOI] [PubMed] [Google Scholar]
- 11.Ly JD, Grubb DR, Lawen A. The mitochondrial membrane potential (Delta psi m) in apoptosis; an update. Apoptosis. 2003;8(2):115–128. doi: 10.1023/a:1022945107762. [DOI] [PubMed] [Google Scholar]
- 12.Taylor SW, Fahy E, Zhang B, Glenn GM, Warnock DE, Wiley S, Murphy AN, Gaucher SP, Capaldi RA, Gibson BW, Ghosh SS. Characterization of the human heart mitochondrial proteome. Nat Biotechnol. 2003;21(3):281–286. doi: 10.1038/nbt793. [DOI] [PubMed] [Google Scholar]
- 13.Kuznetsov AV, Margreiter R. Heterogeneity of mitochondria and mitochondrial function within cells as another level of mitochondrial complexity. Int J Mol Sci. 2009;10(4):1911–1929. doi: 10.3390/ijms10041911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dekker PJT, Ryan MT, Brix J, Muller H, Honlinger A, Pfanner N. Preprotein translocase of the outer mitochondrial membrane: molecular dissection and assembly of the general import pore complex. Mol Cell Biol. 1998;18(11):6515–6524. doi: 10.1128/mcb.18.11.6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Satori CP, Kostal V, Arriaga EA. Review on recent advances in the analysis of isolated organelles. Anal Chim Acta. 2012;753:8–18. doi: 10.1016/j.aca.2012.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Veal DA, Deere D, Ferrari B, Piper J, Attfield PV. Fluorescence staining and flow cytometry for monitoring microbial cells. J Immunol Methods. 2000;243(1–2):191–210. doi: 10.1016/s0022-1759(00)00234-9. [DOI] [PubMed] [Google Scholar]
- 17.Zhang SY, Zhu SB, Yang LL, Zheng Y, Gao M, Wang S, Zeng JZ, Yan XM. High-throughput multiparameter analysis of individual mitochondria. Anal Chem. 2012;84(15):6421–6428. doi: 10.1021/ac301464x. [DOI] [PubMed] [Google Scholar]
- 18.Poe BG, Duffy CF, Greminger MA, Nelson BJ, Arriaga EA. Detection of heteroplasmy in individual mitochondrial particles. Anal Bioanal Chem. 2010;397(8):3397–3407. doi: 10.1007/s00216-010-3751-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hornig-Do HT, Gunther G, Bust M, Lehnartz P, Bosio A, Wiesner RJ. Isolation of functional pure mitochondria by superparamagnetic microbeads. Anal Biochem. 2009;389(1):1–5. doi: 10.1016/j.ab.2009.02.040. [DOI] [PubMed] [Google Scholar]
- 20.Chattopadhyay PK, Hogerkorp CM, Roederer M. A chromatic explosion: the development and future of multiparameter flow cytometry. Immunology. 2008;125:441–449. doi: 10.1111/j.1365-2567.2008.02989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Navratil M, Poe BG, Arriaga EA. Quantitation of DNA copy number in individual mitochondrial particles by capillary electrophoresis. Anal Chem. 2007;79(20):7691–7699. doi: 10.1021/ac0709192. [DOI] [PubMed] [Google Scholar]
- 22.Kostal V, Arriaga EA. Capillary electrophoretic analysis reveals subcellular binding between individual mitochondria and cytoskeleton. Anal Chem. 2011;83(5):1822–1829. doi: 10.1021/ac200068p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adler J, Parmryd I. Quantifying colocalization by correlation: the Pearson correlation coefficient is superior to the Mander's overlap coefficient. Cytometry A. 2010;77A(8):733–742. doi: 10.1002/cyto.a.20896. [DOI] [PubMed] [Google Scholar]
- 24.Poe BG, Navratil M, Arriaga EA. Analysis of subcellular sized particles—capillary electrophoresis with post-column laser-induced fluorescence detection versus flow cytometry. J Chromatogr A. 2006;1137(2):249–255. doi: 10.1016/j.chroma.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 25.Davis JM, Arriaga EA. Evaluation of peak overlap in migration-time distributions determined by organelle capillary electrophoresis: type-II error analogy based on statistical-overlap theory. J Chromatogr A. 2009;1216(35):6335–6342. doi: 10.1016/j.chroma.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wurm CA, Neumann D, Lauterbach MA, Harke B, Egner A, Hell SW, Jakobs S. Nanoscale distribution of mitochondrial import receptor Tom20 is adjusted to cellular conditions and exhibits an inner-cellular gradient. Proc Natl Acad Sci USA. 2011;108(33):13546–13551. doi: 10.1073/pnas.1107553108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andreyev D, Arriaga EA. Simultaneous laser-induced fluorescence and scattering detection of individual particles separated by capillary electrophoresis. Anal Chem. 2007;79(14):5474–5478. doi: 10.1021/ac070770u. [DOI] [PubMed] [Google Scholar]
- 28.Strack A, Duffy CF, Malvey M, Arriaga EA. Individual mitochondrion characterization: a comparison of classical assays to capillary electrophoresis with laser-induced fluorescence detection. Anal Biochem. 2001;294(2):141–147. doi: 10.1006/abio.2001.5148. [DOI] [PubMed] [Google Scholar]
- 29.Sosnick TR, Benjamin DC, Novotny J, Seeger PA, Trewhella J. Distances between the antigen-binding sites of 3 murine antibody subclasses measured using neutron and x-ray-scattering. Biochemistry. 1992;31(6):1779–1786. doi: 10.1021/bi00121a028. [DOI] [PubMed] [Google Scholar]
- 30.Mason DW, Williams AF. The kinetics of antibody-binding to membrane-antigens in solution and at the cell-surface. Biochem J. 1980;187(1):1–20. doi: 10.1042/bj1870001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.