

Abstract
The regenerative capacity of muscle dramatically decreases with age because old muscle stem cells fail to proliferate in response to tissue damage. Here we uncover key age-specific differences underlying this proliferative decline: namely, the genetic loci of CDK inhibitors (CDKI) p21 and p16 are more epigenetically silenced in young muscle stem cells, as compared to old, both in quiescent cells and those responding to tissue injury. Interestingly, phosphorylated ERK (pERK) induced in these cells by ectopic FGF-2 is found in association with p21 and p16 promoters, and moreover, only in the old cells. Importantly, in the old satellite cells FGF-2/pERK silences p21 epigenetically and transcriptionally, which leads to reduced p21 protein levels and enhanced cell proliferation. In agreement with the epigenetic silencing of the loci, young muscle stem cells do not depend as much as old on ectopic FGF/pERK for their myogenic proliferation. In addition, other CDKIs, such asp15INK4B and p27KIP1, become elevated in satellite cells with age, confirming and explaining the profound regenerative defect of old muscle. This work enhances our understanding of tissue aging, promoting strategies for combating age-imposed tissue degeneration.
Keywords: muscle stem cells, aging, tissue regeneration, signal transduction, MAPK, pERK, CDK inhibitor, epigenetic, chromatin
Introduction
Muscle stem cells are responsible for postnatal muscle maintenance and regeneration. During aging the regenerative capacity of skeletal muscle declines, largely due to the deteriorated myogenic capacity and the reduced proliferation of resident satellite cells [1–4]. During the growth and regeneration of skeletal muscle, myogenic proliferating cells express Pax7, Myf5 and MyoD. Upon differentiation into myotubes, the expression of Pax7 and Myf5 decline, while that of myogenin increases; terminally differentiated de-novo myotubes express eMyHC and have centrally located nuclei, the adult MHC and migration of myonuclei to the myotube periphery occurs later on [5, 6]. The local and systemic environment (known as the niche) regulates the regenerative responses of satellite cells. Some of the signaling molecules that are able to “age” young satellite cells or “rejuvenate” old satellite cells have been uncovered. For example, activation of Notch (which is lacking in the old niche), ectopic oxytocin [7] and inhibition of TGF-beta1 (which is excessive in the old niche) have been shown to enhance the myogenic capacity of satellite cells in old mice [5].
One critical early event during muscle regeneration is cell cycle re-entry in response to muscle injury. Satellite cells exhibit reduced mitotic activity with age in both breakage of G0 quiescence and subsequent cell-cycle progression [1]. FGF2 has been implicated to activate cell-cycle progression by regulating cyclin D1 expression [8, 9]. Although FGF2 has been identified to be elevated in the homeostatic aged muscle niche [10], no gross over-proliferation of aged satellite cells can be detected, and both young and old quiescent muscle stem cells are only weakly responsive to ectopic FGF2 [11]. This result is further confirmed by earlier work demonstrating an inability of myogenic cells to respond to endogenous FGF2 [12]. Moreover, the robust observation of a lack of old satellite cell proliferation is inconsistent with the known mitogenic properties of FGF2 and its role as a key mitogen in the establishment and expansion of myogenic cell lines [13]. With respect to the signal pathway downstream of FGF2, the MAPK/ERK cascade has been implicated in the regulation of satellite cell proliferation [8, 9]. The ERK1/2 signaling pathway is typically initiated by binding of growth factors to their receptors, which leads to a phosphorylation cascade culminating in phosphorylated ERK1/2. ERK1/2 activated by phosphorylation (pERK), can translocate to the nucleus and phosphorylate transcriptional factors, thereby regulating the expression of numerous downstream target genes [14].
The transition through the G1 phase or entry into S-phase of the cell cycle requires activation of the cyclin/cyclin-dependent kinase (CDK) complex, whose kinase activity is inhibited by CDKIs, including p16 and p21 [15–17]. CDKIs share some common features, such as blocking cell-cycle progression by inactivating the cyclin/CDK complexes in the G1 to S and G2 to M transitions, which leads to cell cycle arrest in both normal and malignant cells [18]. However, p16 and p21 have different CDK targets and activities. p16 binds to and inhibits CDK4/6, but p21 broadly binds to both cyclin and CDK subunits. Moreover, p16 and p21 also have distinct functions in promoting senescence versus quiescence, respectively [19, 20]. A spontaneous susceptibility to tumor formation develops at an early age in p16 deficient p53−/− mice [21, 22], but not in the p21 knockout mice. Instead, the p21 deficient mice display a defect in G1 arrest in response to DNA damage and nucleotide pool perturbation [23, 24]. CDKIs, including p21 and p16, are also involved in the regulation of cell cycle progression of stem cells. The quiescence of stem cells requires control over the G1 checkpoint, through which stem cells can stay and/or return to G0. For example, in the absence of p21, hematopoietic stem cells proliferate without self-renewal, leading to their exhaustion [19]. On the other hand, the decline in stem cell function with aging has been associated with a p16-induced blockade of cell-cycle progression, and a transition from quiescence to senescence, from which adult stem cells cannot initiate a regenerative response [25–29].
Epigenetic modification patterns are a determinant of gene expression, giving each cell its distinct characteristics and functions [30]. Several post-translational modifications of histones, including methylation, acetylation, phosphorylation and ubiquitination, lead to transcriptional regulation of gene expression [31]. For example, active promoters are generally marked by trimethylation of histone 3 lysine 4 (H3K4me3), while trimethylation of histone 3 lysine 27 (H3K27me3) is associated with transcriptional repression [32]. Furthermore, the epigenetic code can be maintained from one cell generation to the next as a memory of cell fate and identity, which is required for stem cells to maintain their potential for pluripotency and self-renewal [33, 34]. The epigenetic silencing of CDKIs is one of the mechanisms by which young stem cells and oncogenic cells acquire high rates of proliferation [29, 35].
Recent work comparing the three cohorts of (1) young quiescent cells, (2) old quiescent satellite cells, and (3) young injury-activated satellite cells, has discovered some age-imposed changes in the genome-wide epigenetic profile [36, 37]. However, the epigenomes of injury-activated young versus aged muscle stem cells have not been compared prior to our work either in general or in response to a particular biochemical signaling.
Considering the highly important topic of age-specific changes in the proliferative capacity of tissue stem cells, we studied the molecular mechanism underlying the FGF2/pERK mediated regulation of cell cycle progression in young and aged satellite cells. Our work focused on muscle stem cells that respond to tissue injury. The age-induced decline in cell proliferation is observed in injury-activated satellite cells, but those cells residing in resting muscle are equally quiescent in young and old tissue. Our data reveal that despite the elevation of FGF2 in old damaged muscle, no differences in the levels of pERK exist between young and old satellite cells. Aged cells display a profound proliferation failure as compared to young, in agreement with consistently higher expression of several CDKIs. Interestingly, ectopic FGF2 enhances the proliferation of aged satellite cells by down-regulating p21 expression, but there is only a slight trend toward diminishing p16 levels; and aged satellite cells and myofibers express more p16 protein and mRNA than young. Interestingly, experimentally activated pERK associates with the chromatin at the p16 and p21 promoters, and furthermore does so only in old muscle stem cells, but not in young. Such age-specific difference in the permissiveness of pERK is explained by an age-imposed shift toward open chromatin in the p16 and p21 promoters of the injury activated old satellite cells. Moreover, our mining of a published database [36] has identified that there is also a shift toward epigenetically open p16 and p21 chromosomal loci in old quiescent satellite cells, as compared to young.
Materials and Methods
Animals
Young (2–3 months old) and old (22–24 months old) C57BL/6J mice were purchased from the Jackson Laboratory and National Institute on Aging, respectively. All animals were housed at the University of California, Berkeley, and the animal experimental procedures were performed in accordance with the Guide for Care and Use of Laboratory Animals of the National Institutes of Health, and approved by the University of California, Berkeley, Office of Laboratory Animal Care.
Antibodies
Antibodies to phosphor-ERK1/2 and total ERK1/2 were purchased from Cell Signaling Technologies. Anti-actin antibody was from Sigma. FGF2, p16, p21, myogenin, MyoD and HRP-conjugated secondary antibodies were from Santa Cruz Biotechnology. Pax7 and myosin heavy chain (MHC) antibody were from the Developmental Studies Hybridoma Bank (University of Iowa). GAPDH, Ki67, H3K4me3, H3K27me3 antibodies were from Abcam. Fluorophore-conjugated secondary antibodies were purchased from Invitrogen/Life Sciences.
Muscle fibers and satellite cell isolation
Isoflurane was used to anesthetize the animal during the muscle injury. For satellite cell activation, tibialis anterior (TA) and gastrocnemius muscle were injected with cardiotoxin 1 (Sigma-Aldrich, 200 μg/ml in PBS) at 10 μg per leg. Muscles were harvested from 3-day-post-injury (3DPI) or uninjured mice, and then myofibers and satellite cells were isolated as described [38]. Briefly, injured or uninjured TA and gastrocnemius muscles were dissected from young and old mice, with fat pad and tendon removed, and then incubated at 37°C in digestion medium (250U/ml Collagenase type II in DMEM medium, buffered with 30 mM HEPES) for 1 hour. Digested muscle was gently triturated and myofibers were collected. Myofibers were further digested with 1 U/ml Dispase and 40 U/ml Collagenase type II to liberate satellite cells. Satellite cells were pre-plated for 30 minutes, and then cultured on diluted Matrigel (BD Biosciences) coated plates in DMEM with serum from the same mouse.
Immunofluorescence
Cells were fixed with 4% PFA for 10 minutes before permeabilization with 0.1% Triton-X 100 for 30 minutes. Then cells were immunostained with primary antibody. Cells were detected with fluorophore-conjugated secondary antibody, and nuclei were visualized by Hoeschst staining. All images were captured with Zeiss AxioImager fluorescence microscope and analyzed with AxioVision software.
Western blotting
Satellite cells (2×105 cells) were treated (or not) with FGF2 and/or MEKi for 16 hours before being lysed in RIPA buffer containing 1× protease and phosphatase inhibitors (Roche) and analyzed by Western blotting. Cell or fiber lysates were mixed with an equal volume of 2× Laemmli buffer (Bio-Rad), boiled for 5 minutes and separated on precast TGX gels (Bio-Rad). The proteins were then transferred to PVDF membrane (Millipore) and blotted with the desired antibodies. Blots were subsequently developed using Amersham ECL Plus kit (GE Healthcare), and analyzed with a Bio-Rad Imaging System.
Chromatin Immunoprecipitation
Chromatin Immunoprecipitation (CHIP) was performed with the EZ CHIP Kit (Upstate) according to the manufacturer’s instructions. Briefly, 5×105 satellite cells were cultured in growth medium without FGF2 for 16 hours and then treated (or not) with FGF2 or MEKi for 1 hour. Cells were crosslinked with 1% paraformaldehyde for 10 minutes, washed with cold PBS, and then lysed in SDS lysis buffer with a protein inhibitor cocktail. DNA was sheared with a sonicator and pre-cleared with Protein G Agarose beads before immunoprecipitation with the desired antibodies. Immunoprecipitation fractions were harvested, washed, eluted and reverse-crosslinked. Then DNA was purified and used for PCR against promoter regions of each gene locus. The primers used in the PCR assay are listed in Table 1.
Table 1.
Primer sequences used for PCR amplification of CDKi genes; the primer match locations on the NCBI reference gene target, chromosome number and literature references are indicated.
| Gene | Assay | Forward Primer, 5′-3′ and match to target | Reverse Primer, 5′-3′ and match to target | NCBI gene target and References |
|---|---|---|---|---|
| p16 | QPCR | GAGTCCGCTGCAGACAGACT 85 to 104 |
CCAGGCATCGCGCACATCCA 387 to 368 |
NM_001040654.1 |
| p21 | QPCR | GCCCGAGAACGGTGGAACTT 29098537 to 29098556 |
GACAAGGCCACGTGGTCCTC 29098742 to 29098723 |
NC_000083.6, Chr. 17 |
| p15 | QPCR | AGATCCCAACGCCCTGAAC 325 to 343 |
CCCATCATCATGACCTGGATT 381 to 361 |
NM_007670.4 |
| p27 | QPCR | AGGAGAGCCAGGATGTCAGC 830 to 849 |
CAGAGTTTGCCTGAGACCCAA 895 to 875 |
NM_009875.4 |
| GAPDH | QPCR | CCACTTGAAGGGTGGAGCCA 416 to 435 |
TCATGGATGACCTTGGCCAG 583 to 564 |
NM_001289726.1 |
| p16 | CHIP | GACCCACTGGTCACACGACT 89282197 to 89282178 |
TACCCGACTGCAGATGGGAC 89281953 to 89281972 |
NC_000070.6, chr. 4, [56] |
| p21 | CHIP | CACAGTTGGTCAGGGACAGA 29093475 to 29093494 |
CAGGACCAACCCACTCCTT 29093713 to 29093695 |
NC_000083.6, chr. 17, [57] |
| p15 | CHIP | CTGCTTGGTCTAATGCTAACTGTG 89311421 to 89311398 |
GGTCTTTATTTAGCTCAGGCCTGC 89311229 to 89311252 |
NC_000070.6, chr. 4, [58] |
| p27 | CHIP | CTGGCTCTGCTCCATTTGAC 134920757 to 134920776 |
GGCTCCCGTTAGACACTCTC 134920950 to 134920931 |
NC_000072.6, chr. 6, [59] |
RNA isolation and real-time q-RT-PCR
RNA isolation was performed using RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. Reverse transcription (RT) was processed using the SuperScript III First-Strand Synthesis System (Invitrogen). Real-time PCR was performed on a Bio-Rad iQ5 real-time PCR machine. The primers used in the real-time PCR assay are listed in Table 1.
P21 promoter assay
The p21-eGFP construct (GeneCopoeia) contains the GFP reporter expressed under the promoter region of the mouse p21 gene. p21-eGFP plasmid was transiently transfected into primary mouse myoblasts with using Lipofectamine 2000 reagent (Invitrogen). 24 hours after transection, cells were treated with FGF2 for 16 hours, or left untreated, and analyzed on a Guava EasyCyte (Millipore) for the intensity of eGFP fluorescence.
Quantification and Statistical Analysis
Every experiment was repeated on 3 individual animals of each age. Data were analyzed by ANOVA followed by t-test using Statistical Analysis System (SAS Institute Inc). Results were presented as the mean±SEM. A p-value<0.05 was considered to be significant.
Results
FGF2 levels but not pERK increase in old muscle stem cells
In skeletal muscle, the microniche plays a crucial role in both maintaining the quiescence of satellite cells, and activating satellite cells when tissue is damaged. In our previous report, we found that myofibers from resting muscle have elevated levels of FGF2 [11]. To assay the expression of endogenous FGF2 in the injured/regenerating muscle, myofibers were prepared from 3-day-post-injury (3DPI) tibialis anterior muscle (TA), as previously described [38]. The levels of FGF2 and its downstream signaling molecules, pERK1 and pERK2, were analyzed by Western Blotting. As shown in Figure 1A and quantified in 1B, the protein expression of FGF2 in injured myofibers increased with age, which is in agreement with previously published work in resting myofibers [10, 11]. Similarly, levels of endogenous FGF2 are up regulated with age in both quiescent and activated satellite cells (Figure 1C, 1D and [11]). Interestingly, MAPK/ERK signaling is robustly activated by injury in both young and old cells without age-specificity, even though the levels of FGF2 are higher in the old.
Figure 1. Age-dependent comparison of FGF2 and pERK levels in muscle fibers and in satellite cells isolated from injured and resting muscle.

(A) Total protein was isolated from fresh-derived 3-day-post-injury (3DPI) myofibers from TA muscle of young and old mice, and the level of FGF2, phospho-ERK1/2, total ERK1/2, and cytoplasmic beta-actin were analyzed by Western blotting. (B) Relative protein expression was quantified from 3 young and 3 old mice by normalization of FGF2 to beta-actin and normalization of pERK to total ERK. Significantly, higher levels of FGF2, but not pERK were detected in the old injured myofibers as compared to young (n=3, * P<0.05). (C) Quiescent and activated muscle stem cells were isolated from uninjured or 3DPI muscles of young and old mice as described. Total protein was analyzed for the expression levels of FGF2, phospho-ERK1/2, total ERK1/2 and beta-actin by Western blotting. Representative images are shown. (D) Relative protein expression of FGF2 and pERK were quantified from 3 animals of each group (n=3, * P<0.05). Significantly higher levels of FGF2 were detected in old activated muscle stem cells comparing to either young activated or old quiescent cells. Phospho-ERK levels were significantly higher in activated muscle stem cells, compared to quiescent cells from either young or old muscle, but there was no difference between young and old muscle cells without tissue injury. (E) Activated satellite cells were isolated from 3-day-post-injury (3DPI) young and old muscle were starved of growth factors for 1 hour and treated or not with FGF2 (10 ng/ml) for 30 minutes before lysis, and analyzed for the level of FGF2, phospho-ERK1/2, total ERK1/2 and beta-actin by Western blotting. Relative protein expression of pERK (F) was quantified from 3 young and 3 old mice (n=3, * P<0.05). The levels of FGF2 and pERK were equally undetectable in young and old satellite cells without FGF2 treatment. With ectopic FGF2 treatment, significantly higher levels of pERK were detected in both young and old satellite cells, and no age-specific difference was present.
To confirm the presence of equally functional FGF2/ERK signaling in young and old satellite cells, we analyzed the levels of pERK1/2 in young and old satellite cells that were starved of growth factors for 1 hour and then treated with FGF2 for 30 minutes. As shown in Figure 1E and 1F, recombinant FGF2 induced robust phosphorylation of ERK1/2 in satellite cells of both ages. Moreover, exogenous FGF2 was clearly bound to these satellite cells, and no FGF2 was detectable in untreated cells. These results suggest that both endogenous and ectopic FGF2 are able to induce pERK in satellite cells activated by muscle injury. Since the levels of pERK are not elevated with age, either the availability of active endogenous FGF2 is higher or there are other potent inducers of MAPK/ERK in the young muscle, as compared to old. In summary, the FGF2 over-expressed by old myofibers does not result in elevated MAPK/ERK signaling in old satellite cells, as compared to young, either in the injured or in uninjured muscle.
FGF2 regulates the proliferation of satellite cells in an age-specific manner
A wealth of previous reports demonstrated that the proliferative capacity of quiescent satellite cells associated with resting myofibers decreases with age [37, 39]. FGF2 failed to induce a high level of proliferation in quiescent satellite cells [11], suggesting that other factors are required for breakage of quiescence. However, it is possible that FGF2 could promote the proliferation of those aged satellite cells, which enter into the cell cycle in response to injury but struggle in their cell-cycle progression. Hence, we examined the function of FGF2 in enhancing the proliferative capacity of the satellite cells that were induced to break quiescence by muscle injury in vivo. Activated satellite cells from young and aged muscle were isolated and cultured in the presence of syngeneic mouse serum with or without FGF2 for 24 hours, then co-immunostained for the proliferation marker Ki67 and the myogenic marker, Pax7 (Figure 2, A, B). The purity of the activated satellite cells was no less than 90% and was equal between the young and old cells, as demonstrated by the immuno-detection of myogenic markers (Supplemental Figure 1). As expected, young satellite cells proliferated much better than old. Very interestingly, ectopic FGF2 enhanced the proliferation of aged satellite cells (percent of Ki67+/Pax7+ cells), while that of young satellite cells was not significantly influenced (Figure 2A and 2B), even though exogenous FGF2 robustly induced pERK both in the young and old satellite cells (Figure 1E). Furthermore, the proliferation of aged satellite cells, which was promoted by FGF-2, was pERK1/2-dependent and abolished in the presence of a MEK inhibitor (MEKi) that also reduced cell proliferation when added alone (Figure 2A and 2B). These results confirm the age-specific decline in muscle stem cell proliferation [5], and demonstrate an age-imposed difference in the functional effects of FGF2 on the properties of activated satellite cells. Finally, as we previously described [40], MAPK/ERK signaling was confirmed to be needed for the proliferation of both young and old satellite cells.
Figure 2. FGF2 regulates proliferation of satellite cells in an age-dependent manner.

(A) Muscle stem cells from injured muscle (3DPI) of young and old mice were plated in OPTI-MEM with 5% of their own serum supplemented or not with FGF2 (10 ng/ml) for 24 hours before immunostaining for Pax7 and Ki67. Scale bars span 100μm. (B) Percentage of Ki67+/Pax7+ proliferating cells was quantified. Significantly less proliferating old satellite cells were detected, as compared to young. When the old cells were treated with FGF2, there was an increase in percentage of Ki67+/Pax7+ cells, and MEKi blocked such increase. On the other hand, exogenous FGF2 did not increase Ki67+/Pax7+ cell percentage in the young satellite cell cultures. N=3, * P<0.05.
FGF2 does not change myogenic cell fate
After establishing that FGF2 preferentially enhances the proliferation of old muscle stem cells, as compared to young, we decided to determine whether the cell fate of young and old satellite cells remains unchanged upon exposure to exogenous FGF2. The expression of the myogenic marker Pax7 was assayed in young and old cells, using Western blotting (Figure 3A, 3B) and using real-time q-RT-PCR (Supplementary Figure 2A). When studied at equal cell numbers, there was no significant difference in Pax7 mRNA and protein levels between young and old satellite cells, and the expression of Pax7 was not influenced by the FGF2 treatment (Figures 3 and Supplementary 2A). Therefore, old satellite cells have reduced proliferation, but the cells, which are present are as myogenic as young, and ectopic FGF-2 does not alter the myogenicity of satellite cells, based on the expression of Pax7.
Figure 3. FGF2/pERK signaling does not change the mygenicity of the young or old satellite cells.

Activated by injury young and old satellite cells were cultured in OPTI-MEM with 5% of their own serum, supplemented (or not) with recombinant FGF-2 and/or MEKi for 24 hours, after which cells were collected for Western Blotting immediately or following an additional culture for 48 hours in DMEM,2%horse serum that is needed for differentiation into myogenin and eMyHC expressing myocites and myotubes. (A). Representative Western Blot for Pax 7 (actin – loading control), is quantified in (B). (C). Representative Western Blot for myogenin and eMyHC (actin – loading control), is quantified in (D). Treatment of satellite cells with FGF-2 and/or MEKi did not significantly change the expression of myogenic markers: Pax7, myogenin and eMyHC.
To compare these cells further along the myogenic pathway, we analyzed the expression of the differentiation markers myogenin and embryonic myosin heavy chain (eMyHC). When plated at equal numbers, young and old satellite cells display an equal capacity for myogenic differentiation [1] and these published findings are in agreement with the data shown below. Young and old satellite cells were plated in OPTI-MEM containing 5% their own sera and treated (or not) with FGF2 and/or MEKi for 24 hours, after which cells were cultured for 48 hours in conventional differentiation medium (DMEM, 2% horse serum). After normalizing for the protein loaded or cell numbers, FGF-2, MEKi and cell age did not significantly change the expression of myogenin and eMyHC (Figure 3C and D), or the percent of the multinucleated eMyHC+ myotubes (Supplementary Figure 2B).
These results determine that FGF2 can specifically enhance proliferation of old satellite cells without altering their myogenicity.
FGF2 promotes the proliferation of aged muscle stem cells by negatively regulating p21 expression
To determine the mechanism of the FGF2-boosted proliferation of aged satellite cells, we examined the expression of the CDKIs p16 and p21 using real-time q-RT-PCR and Western Blotting. As shown in Figure 4A-E, aged satellite cells display elevated levels of p16 and p21 mRNA and protein as compared to young, which is consistent with the observed lack of proliferation. Notably in the old cells, ectopic FGF2 down-regulated p21 expression to young levels, while in the young cells the already low level of p21 was not further influenced by FGF2 (Figure 4A and 4D). The FGF2-induced down-regulation of p21 in the aged satellite cells was dependent on MAPK/ERK, since it was abolished by MEKi (Figure 4A and 4D). In contrast to p21, the age-induced increase in the senescence marker and effector p16 showed a tendency to decrease in response to FGF2 (Figure 4B and 4E), however, this data was not statistically significant.
Figure 4. FGF2 inhibits p21 but not p16 expression in aged muscle stem cells.

Muscle stem cells from injured muscle (3DPI) of young and old mice were isolated as described in Methods. The cells were plated in OPTI-MEM with 5% of their own serum supplemented or not with FGF2 (10 ng/ml) and/or MEKi PD98059 for 24 hours. Total RNA was isolated and mRNA levels of CDKI genes p21 (A) and p16 (B) were analyzed using real-time RT PCR with gene-specific primers. Total protein from these muscle stem cells was isolated for the analysis of p21 and p16 protein levels, using Western Blotting (C). Relative protein expression of p21 (D) and p16 (E) were quantified from 3 young and 3 old mice, using GAPDH for normalization. Expression of p21 and p16 (both mRNA and protein levels) were significantly higher in old satellite cells compared to young. Expression of p21, but not p16 in old satellite cells was significantly down-regulated by ectopic FGF2, and this down-regulation was blocked by MEKi. N=3, * P<0.05.
Furthermore, as compared to young, old muscle fibers expressed prominent levels of p16 protein, which was almost undetectable in young myofibers. p21 protein levels were also elevated in myofibers with age (Supplementary Figure 3), suggesting that similar age-imposed changes in these CDKIs occur both in muscle stem cells and in their micro-niche.
When the MEKi was added to the cells alone (without ectopic activation of pERK by FGF2), p21 and p16 protein levels did not significantly increase in young or old satellite cells. However, a tendency to increase p21 mRNA expression in young satellite cells was noticed, which is consistent with the necessity of basal MAPK/ERK signaling for key cellular functions [41].
These results reveal that aging of muscle stem cells is associated with both a trend toward cellular senescence and p21-induced inhibition of cell cycle progression in response to injury. Either of the two mechanisms could cause the observed proliferation deficiency of aged satellite cells. In old satellite cells the inhibition of proliferation was reversed by FGF2-induced activation of MAPK/ERK, through down-regulation of p21, but the expression of p16 was resistant to attenuation by ectopic FGF2.
Age-specific differences in the occupancy of ERK to p21 and p16 promoters
Upon mitogen stimulation, activated pERK rapidly translocates and accumulates in the nucleus, where pERK activates downstream transcription factors and regulates gene expression [42]. To analyze the regulation of p21 and p16 by FGF2/ERK with higher precision, we examined whether pERK interacts with the p21 and p16 promoters. An ERK-specific chromatin immunoprecipitation assay (CHIP) was performed on activated satellite cells, and DNA co-precipitated with ERK was analyzed by real-time q-PCR, using primers specific to the p21 and p16 promoter regions. Muscle stem cells were isolated from young and old injured/regenerating muscle at 3DPI and cultured with synchronic serum for 24 hours in the presence of FGF2, FGF2+MEKi, MEKi alone or no treatment. As shown in Figure 5, in young satellite cells pERK did not interact with the promoters of p21 and p16 in any of the conditions studied, including treatment with FGF2. However, in old satellite cells pERK was enriched on the p21 and p16 promoters after ectopic FGF2 treatment. The specificity of pERK for the p21 and p16 promoters upon the FGF2 treatment was confirmed by blocking the observed CHIP association with the MEKi (Figure 5A and 5B).
Figure 5. Age-specific differences in the binding of ERK to p21 and p16 promoters.
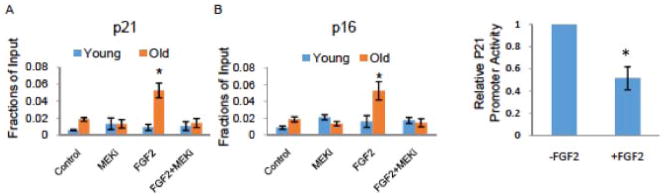
Young and old satellite cells were isolated from injured muscle (3DPI) as described in Methods. The cells were cultured in OPTI-MEM supplemented with 5% isochronic serum from the same mice for 24 hours, and then treated or not with FGF2 and/orMEKi for 1 hour. Chromatin immunoprecipitation (CHIP) with ERK1/2 antibody was then performed. CHIP and input DNA were measured using real-time PCR with specific primers targeting promoter regions of p21 (A) and p16 (B) genes. There was a significant increase in enrichment of ERK on the promoter regions of p21 and p16 genes in the old, but not young satellite cells exposed to ectopic FGF2, and MEKi blocked this enrichment. N=3, * P<0.05. (C). To confirm the repression of the p21 promoter by FGF-2/pERK, the p21-GFP reporter construct was transiently expressed in young mouse satellite cells and percent of GFP+ cells was assayed by FACS in cells treated for 24 hour with 10ng/ml of FGF-2, as compared to untreated control cultures. FGF-2 significantly reduced the percent of GFP+ satellite cells. N=3, * P<0.05.
To confirm that FGF2/pERK directly inhibits the transcription of p21, we performed promoter reporter studies. Namely, a plasmid encoding a mouse p21 promoter driven GFP reporter was transfected into satellite cells and transcriptional activation of p21 was assayed using flow cytometry for GFP fluorescence. As shown in Figure 5C, FGF2 treatment significantly decreased the percent of satellite cells expressing GFP, thus reporting a transcriptional inhibition of p21 by FGF2.
To test the regulation of other CDKIs by the FGF2/ERK pathway, we analyzed the expression levels of p15 and p27 in injury-activated satellite cells using real-time q-RT-PCR (Supplemental Figure 4A and4B). Aged satellite cells displayed elevated levels of p15 and p27 mRNA as compared to young, further explaining the age-dependent proliferative decline. Ectopic FGF2 down-regulated the expression of both p15 and p27 in the aged satellite cells, but interestingly, this down-modulation was independent of MAPK signaling, since MEKi did not attenuate it (Supplemental Figure 4A, B). Moreover, an ERK-specific CHIP assay demonstrated that treatment of satellite cells with FGF2 did not enrich pERK on the p15 and p27 promoters (Supplemental Figure 4C).
These data suggest that in old muscle stem cells ectopic FGF2 induces nuclear translocation and chromatin binding of active ERK at the p16 and p21 loci. Notably, these results reveal age-specific differences in the permissiveness of the MAPK/ERK pathway in regulating the expression of p16 and p21 genes.
The epigenetic state of p16 and p21 promoters is different between young and aged muscle stem cells
To understand the mechanism responsible for the age-specific differences in the occupancy of MAPK/ERK on the p21 and p16 promoters, we examined the epigenetic chromatin state of these genes in young versus old satellite cells responding to tissue injury. Chromatin immunoprecipitation was performed with antibodies against trimethylated histone H3 Lysine-4 (H3K4me3) and trimethylated histone H3 Lysine-27 (H3K27me3), followed by real-time q-PCR with primers specific for the p21 and p16 promoter regions. Muscle stem cells were isolated from injured muscle of young and old mice and studied immediately after isolation.
As shown in Figure 6A and 6C, the p21 promoter locus in young satellite cells activated by injury displayed a monovalent closed chromatin pattern with a low ratio of H3K4me3 to H3K27me3. In contrast, trimethylation of H3K4 was up regulated in the old muscle stem cells activated by injury, and the p21 locus adopted a more active bivalent state (Figure 6A, C). These results are consistent with the increased expression of p21 mRNA as shown in Figure 4A, and the availability of the p21 promoter for binding by a transcriptional repressor complex involving active ERK as shown in Figure 5A. When exogenous FGF2 was added to aged satellite cells, the p21 locus reverted from the bivalent to a repressive “youthful” epigenetic state (Figure 6A and 6C), in agreement with the role of active ERK1/2 in the down-regulation of p21 expression.
Figure 6. Epigenetic chromatin pattern of p21 and p16 promoters displays age-specific differences.

Young and old satellite cells were isolated from injured muscle (3DPI) and cultured in OPTI-MEM supplemented with 5% isochronic serum from the same mice for 24 hours. The cells were then treated or not with FGF2 (10 ng/ml) for 1 hour before chromatin immunoprecipitation (CHIP) analysis using H3K4me3 and H4K27me3 antibodies. CHIP and input DNA was measured using real-time PCR with specific primer targeting promoter regions of p21 (A) and p16 (B) genes. Relative ratio of enrichment of H3K4me3 to H3K27me3 at p21 and p16 promoter was quantified in 3 independent experiments. p21 and p16 promoter fragments precipitated with H3K4me3 antibody were enriched in the old satellite cells compared with young. In old satellite cells, the epigenetic pattern of H3K4me3/H3K27me3 binding was reversed to a young cell status for the p21 promoter and partially revered for the p16 promoter by FGF2 treatment. N=3, * P<0.05.
With respect to p16, in young muscle stem cells there were slightly higher levels of trimethylated H3K27 than trimethylated H3K4 (Figure 6A). With age, the levels of trimethylated H3K4 and the ratio of H3K4 to H3K27 at the p16 locus in satellite cells dramatically increased (Figure 6A and 6C), consistent with the up-regulation of transcription (shown in Figure 4B), and in agreement with the ability of pERK to occupy the p16 promoter in old muscle stem cells (as shown in Figure 5B). In contrast with the reversal of chromatin conformation of the p21 promoter, FGF2/ERK signaling only slightly silenced the p16 locus, as seen by down-regulating H3K4me3 enrichment, but the ratio of H3K4me3 to H3K27me3 did not decline to that of young satellite cells (Figure 6A and 6C). These results are consistent with an inability of FGF2 to significantly diminish p16 levels in aged satellite cells, as shown above in Figure 4B.
While no age-specific epigenome-wide data exist on the injury-activated muscle stem cells, a database comparing epigenomes of young and old quiescent satellite cells has been recently published [36]. Considering our results on the age-imposed epigenetic differences in p16 and p21 promoter regions, we analyzed this database [36] for the broader chromosomal loci of these genes. As shown in Supplemental Figure 5A and B, shifts toward repressive H3K27me3 chromatin state are evident in young quiescent muscle stem cells, as compared to old, for the entire chromosomal locus of p16 and p21 genes. While both p21 and p16 loci become more open with age, only p21 mRNA, but not p16, is elevated in old quiescent satellite cells residing in non-injured muscle (Supplementary Figure 5C and D). This is in contrast to the cells activated by muscle injury where both p21 and p16 become up-regulated in the old satellite cells, as compared to young (Figure 4).
These findings demonstrate age-specific epigenetic differences for the p21 and p16 gene regulation in muscle stem cells responding to tissue injury, as well as basal state, age-specific changes in the epigenome of these chromosomal areas that predispose aged cells for diminished proliferative responses. These results also suggest a mechanism for the differential age-specific permissiveness of FGF2-induced, active ERK presence on the p21 and p16 promoters, and clarify the molecular mechanisms by which the expression of these cell-cycle regulators is controlled in young versus old muscle stem cells.
Discussion
This study is the first to establish age-specific changes in the epigenetic status of the senescence marker and effector p16 and the quiescence regulator p21, in muscle stem cells responding to tissue injury. Notably, this work identifies a clear impact of aging on the epigenome of muscle stem cells and on their responsiveness to and dependence on FGF2/ERK signaling. The positive effects of ectopic FGF-2 on adult myogenesis in vivo and on the myogenic cultures have been known for a number of years [13], but until our work have not been well understood in cellular and molecular terms.
FGF-2 is one of the known mitogens that specifically enhance myogenic cell proliferation, and in agreement with previous work we found that the levels of FGF-2 increase with age in both uninjured and injured muscle [10, 11]. A previous study reported an age-imposed FGF-implicated proliferation of satellite cells in homeostatic uninjured muscle with a link to the loss of myogenicity, perhaps by slowly losing self-renewing stem cells [10]. However, the percent of affected satellite cells at any given time appeared to be low, although it is noted that the effect of such a low percent over time might accumulate to be large. If persistent FGF signaling leads to stem cell loss, and FGF-2 has known oncogenic properties [8, 9], one should be cautious in considering promoting FGF2 signaling for translational therapeutic purposes.
Another publication demonstrated that in uninjured muscle, the bulk of both young and old satellite cells remain quiescent, and moreover are poorly responsive to ectopic FGF-2 [11]. Selection by flow cytometry for a subset of cell surface markers [10] can potentially produce results that are different from studies of the entire population of fiber-associated muscle stem cells [38], particularly, because satellite cells are notoriously heterogeneous. As shown in our work and as we typically find [5], [7], the bulk-isolated satellite cells which we study are at least 90% myogenic. Therefore, the differences in the isolation and sub-selection of satellite cells could account for the different data on the age-specific myogenicity in the starting cell populations. How with age individual satellite cells change their myogenicity and respond or not to FGF signaling remains to be studied further.
With respect to the muscle regenerating after injury, results of this work and a number of previous publications [5], [7], suggest that old satellite cells have a marked proliferative decline, as compared to young. Such a decline is consistent with the age-specific up-regulation of all the examined CDK inhibitors (p15, p16, p21 and p27). Very interestingly, our mining of the published data-base [36] has identified that even in quiescence, large areas of the genome at the p16 and p21 loci are more open in old satellite cells as compared to young. Therefore, when numerous molecular signals modulate the expression of cell-cycle inhibitors upon tissue damage, the “open” basal state in the old muscle stem cells may facilitate a shift away from cell proliferation.
In further support for our conclusions, the levels of pERK1/2, a key signaling molecule downstream of FGF2, remain very low in resting myofibers despite high endogenous FGF2 [11], but become elevated in both young and aged myofibers upon tissue injury. Since FGF2 does not have a signal peptide and is not secreted via the conventional Golgi pathway [43], one possibility is that intracellular FGF2 is stored in intact myofibers and is released upon myofiber damage, hence becoming available for binding to its receptor and activating MAPK/ERK signaling in neighboring satellite cells. An enigma remains as to why the endogenous pERK, which increases in old satellite cells after muscle injury, fails to promote the proliferation of aged satellite cells, while the pERK that is induced by ectopic FGF2 is able to enhance such cell proliferation. It appears that endogenous FGF2 does not act in the same manner as ectopic FGF in old satellite cells and in primary myogenic cells [12].
It has been demonstrated that FGF2 promotes the proliferation of many cell types, primarily through down-regulation of cyclin D1 expression [8, 9]. The expression of cyclin D1 is tightly regulated at several levels, including transcription, translation, protein stability, complex formation, and the abundance of CDKIs [44]. p21 is the major negative regulator of cyclins and CDKs, which inhibits cell cycle progression by inhibiting cyclin/CDK activity [24]. Here, we studied whether FGF2 promotes the proliferation of aged satellite cell by regulating p21. Several growth factors promote cell proliferation by down-modulating p21, either by inhibiting its transcription or by phosphorylating p21 and interrupting its interaction with CDKs [45–47]. Our data suggest that FGF2/pERK enhances the proliferation of aged satellite cells by changing the epigenetic status of p21, but post-transcriptional regulation of p21 may also be involved in this process. These findings also raise the question of how active ERK binds to chromatin at p21 and p16 loci and changes their epigenetic states. It has been demonstrated that activation of MAPK/ERK recruits downstream substrates, including MSK1/2 and Rsk-2, which phosphorylate histone H3[48, 49]. Our data suggest that in aged satellite cells, FGF2-induced active ERK forms chromatin-bound protein complexes at the p16 and p21 promoters, which might change the epigenetic permissiveness of these loci, causing attenuation of p21 expression, but not sufficing for a significant down-modulation of p16. The identification of such protein complexes that regulate gene expression in an age-specific way would be very interesting in the future. Some possible candidates include the Polycomb group proteins, known for their role in repressive chromatin modifications and maintenance [35].
A recent study showed that aged satellite cells switch from quiescence to pre-senescence, which is caused by elevated expression of p16 [29]. Notably, we observed that while there was a trend toward diminished expression, FGF2/pERK was not capable of significantly down-regulating p16 mRNA or silencing the p16 promoter, which suggests that additional factors are needed for the attenuation of this marker and effector of cellular senescence. Such factors would be very interesting to look for and a synergy with active ERK is expected based on the occupancy of the p16 promoter and the slight decrease in the trimethylated H3K4 to trimethylated H3K27 ratio. An additional interesting finding of this study is the bivalent chromatin state on the p21 and p16 promoters in muscle stem cells that are engaged in tissue regeneration. Bivalent epigenetic regulation is a hallmark of pluripotent embryonic stem cells [50] and is thought to be replaced by monovalent (open or closed) chromatin conformations upon cell differentiation [50, 51]. However, based on our work, a bivalent epigenome might play a role in the regulation of gene expression in adult stem cells.
The age-specific decline in the proliferative capacity of old satellite cells has been not only functionally confirmed but molecularly justified by this work, based on the significant up-regulation of all studied CDKIs: p15, p16, p21 and p27, in cells responding to muscle damage. While ectopic FGF2 down-regulated all these CDKIs in old muscle stem cells, the attenuation of p15 and p27 was MAPK-independent, since it was not blocked by MEKi. MAPK/ERK is the canonical evolutionary and developmentally conserved pathway for FGF2 signaling, however, this growth factor is also known to signal via PI3 kinase, PLCγ/PIP2 and even to be internalized reaching the cytosol and nuclear compartments [11, 52–54]. Thus, it would be very interesting to study the MAPK-independent activities of FGF2 in young versus aged satellite cells. In addition, a clarification of the signal pathway downstream of FGF2 in young and old satellite cells might help to better understand the differences between endogenous and ectopic FGF2 ligand in boosting the proliferation of old muscle stem cells, as well as the differences in FGF-2 effects on satellite cells resident to non-injured versus injured old muscle.
MAPK/ERK signaling plays multiple roles in such diverse cellular responses as senescence, activation, proliferation, and differentiation [55]. All of these processes are very important for tissue stem cells that differentiate and replenish the quiescent stem cell population through self-renewal in a life-long process that unfortunately becomes deteriorated with age. This work demonstrates that the epigenetic status of CDKIs becomes altered in aged satellite cells in response to injury, such that when the regenerative responses are most needed, the proliferation of old muscle stem cells is actively inhibited by elevated levels of p15, p16, p21 and p27. Ectopic mitogens (e.g. FGF2) are thus, required to boost the proliferation of aged satellite cells. As expected, no changes in the myogenic cell fate of the satellite cells occurred when FGF2/pERK has boosted their proliferation, in agreement with a number of other treatments that enhance the proliferation of old satellite cells [5], [7].
The age-specific functional epigenetics and differential permissiveness of active ERK for the chromatin loci of p16 and p21 CDKIs uncovered in our work provides novel understanding and therefore, theoretical ramifications and clinical strategies for enhancing and rejuvenating tissue regeneration.
Supplementary Material
Supplementary Figure 1. Young and old satellite cells are highly pure, based on the expression of myogenic markers. Muscle stem cells from injured muscle (3DPI) of young and old mice were plated in OPTI-MEM with 5% of their own serum for 24 hours before immunostaining for Pax7 and MyoD. At least 90% of isolated young and old cells were positive for the myogenic marker Pax7 and/or MyoD. Scale bars span 100μm.
Supplementary Figure 2 FGF2/pERK signaling does not change the myogenicity of young or old satellite cells. (A) Young and old satellite cells activated by injury were cultured in OPTI-MEM with 5% of their own serum, supplemented (or not) with recombinant FGF-2 and/or MEKi for 24 hours, after which cells were collected for real-time RT-PCR for Pax7. (B) Satellite cells were also cultured in DMEM with 2% horse serum for myogenic differentiation for 48 hours before immunostaining for eMyHC. No statistically significant differences were detected between satellite cells of different ages or with FGF-2 and/or MEKi. Thus, FGF2 does not change the myogenic lineage commitment of either young or old satellite cells.
Supplementary Figure 3. Age-specific difference of p21 and p16 levels in myofibers. (A) Total protein was isolated from freshly-derived uninjured, and 3 day-post-injury (3DPI), TA myofibers of young and old mice. The expression levels of p16, p21 and GAPDH were analyzed by Western Blotting. Representative images are shown. (B) Relative protein expression of p16 and p21 were quantified from 3 young and 3 old mice, normalized to GAPDH. The levels of p16 and p21 were significantly higher in old myofibers as compared to young, from both uninjured and 3DPI muscle. N=3, * P<0.05.
Supplementary Figure 4. FGF2 inhibits p15INK4B and p27KIP1 gene expression in aged muscle stem cells independently of the MAPK/pERK pathway. Muscle stem cells from injured muscle (3DPI) of young and old mice were isolated as described in Methods. The cells were plated in OPTI-MEM with 5% of their own serum, supplemented or not with FGF2 (10 ng/ml) and MEK inhibitor PD98059 for 24 hours. Total RNA was isolated and transcription of CDKI genes p15INK4B (A) and p27KIP1 (B) were analyzed by q-RT-PCR with gene-specific primers. Expression of p15 and p27 were significantly higher in old satellite cells as compared to young, and the expression of both genes was attenuated by ectopic FGF2 in a MAPK-independent manner (the FGF-2 effect was not reversed by MEK inhibition). (C) A portion of the isolated cells was cultured in OPTI-MEM, supplemented with isochronic serum from the same mice, for 24 hours, and then treated or not with FGF2 for 1 hour before CHIP assay with ERK1/2 antibody. CHIP and input DNA were measured using q-PCR with specific primers targeting promoter regions of the p15INK4B and p27WIF1 genes. In contrast to p16 and p21, there was no FGF-2-induced enrichment of pERK on the promoter regions of p15 and p27 genes in either young or old satellite cells. N=3, * P<0.05.
Supplementary Figure 5. Epigenetic status and transcriptional expression of p16 and p21 genes in quiescent satellite cells reveals age-specific differences.
The distribution of H3K4me3 and H3K27me3 at p21 (A) and p16 (B) genomic loci were analyzed, using the CHIP-seq database GSE47362, (Liu et al., 2013). Expression of p21 (C) and p16 (D) were analyzed, using the microarray database GSE47177, (Liu et al., 2013) and compared to the q-RT-PCR performed in our studies (E): namely, quiescent muscle stem cells from uninjured muscle of young and old mice were isolated as described in Methods, total RNA was isolated and transcription of p21 and p16 was analyzed by q-RT-PCR with gene-specific primers. A significant shift to repressive H3K27me3 modification in the p21 and p16 genes was found in the young quiescent satellite cells, as compared to old; and significant up-regulation of p21, but not p16 expression, were observed in the old quiescent satellite, as compared to young. These results provide evidence for an epigenetically regulated age-imposed inhibition of satellite cell proliferation that is detectable even in the state of quiescence without muscle injury. N=3, * P<0.05
Acknowledgments
This work was supported by grants from the National Institute of Health R01 AG02725201, the Keck Foundation and the California Institute of Regenerative Medicine RN1-00532 to IMC. We thank Professor Thomas Rando (Stanford) for helpful discussion of our manuscript and providing the published database CHIP-seq histograms for quiescent satellite cells. We thank Michael Conboy and Christian Elabd for helpful discussion of this study and editing the manuscript, Mary West and the CIRM/QB3 shared Stem Cell Facility at UC Berkeley for providing help with fluorescent microscopy and real-time PCR, and Jacqueline Loo, Thomas Chow, Daniel Chung and Calvin Chang for providing technical assistance with this work.
Footnotes
Disclosure of potential Conflict of Interest
All authors declare that they have no conflict of interest.
Author Contributions
J. L.: design, collection and assembly of data, data analysis, manuscript writing, final approval of manuscript; S.H.: collection and assembly of data, final approval of manuscript; WC: collection and assembly of data for figure 5C and supplemental figure 2, final approval of manuscript; I.M.C.: concept and design, financial support, data interpretation, manuscript writing, final approval of manuscript.
References
- 1.Conboy IM, Conboy MJ, Smythe GM, et al. Notch-mediated restoration of regenerative potential to aged muscle. Science. 2003;302:1575–1577. doi: 10.1126/science.1087573. [DOI] [PubMed] [Google Scholar]
- 2.Schultz E, Lipton BH. Skeletal muscle satellite cells: changes in proliferation potential as a function of age. Mech Ageing Dev. 1982;20:377–383. doi: 10.1016/0047-6374(82)90105-1. [DOI] [PubMed] [Google Scholar]
- 3.Doherty TJ. Invited review: Aging and sarcopenia. J Appl Physiol (1985) 2003;95:1717–1727. doi: 10.1152/japplphysiol.00347.2003. [DOI] [PubMed] [Google Scholar]
- 4.Conboy IM, Rando TA. Aging, stem cells and tissue regeneration: lessons from muscle. Cell Cycle. 2005;4:407–410. doi: 10.4161/cc.4.3.1518. [DOI] [PubMed] [Google Scholar]
- 5.Conboy IM, Rando TA. Heterochronic parabiosis for the study of the effects of aging on stem cells and their niches. Cell Cycle. 2012;11:2260–2267. doi: 10.4161/cc.20437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yablonka-Reuveni Z. The skeletal muscle satellite cell: still young and fascinating at 50. J Histochem Cytochem. 2011;59:1041–1059. doi: 10.1369/0022155411426780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elabd C, Cousin W, Upadhyayula P, et al. Oxytocin is an age-specific circulating hormone that is necessary for muscle maintenance and regeneration. Nat Commun. 2014;5:4082. doi: 10.1038/ncomms5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weber JD, Cheng J, Raben DM, et al. Ablation of Goalpha overrides G1 restriction point control through Ras/ERK/cyclin D1-CDK activities. J Biol Chem. 1997;272:17320–17326. doi: 10.1074/jbc.272.28.17320. [DOI] [PubMed] [Google Scholar]
- 9.Lavoie JN, L’Allemain G, Brunet A, et al. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem. 1996;271:20608–20616. doi: 10.1074/jbc.271.34.20608. [DOI] [PubMed] [Google Scholar]
- 10.Chakkalakal JV, Jones KM, Basson MA, et al. The aged niche disrupts muscle stem cell quiescence. Nature. 2012;490:355–360. doi: 10.1038/nature11438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yousef H, Conboy MJ, Li J, et al. hESC-secreted proteins can be enriched for multiple regenerative therapies by heparin-binding. Aging (Albany NY) 2013;5:357–372. doi: 10.18632/aging.100559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fedorov YV, Rosenthal RS, Olwin BB. Oncogenic Ras-induced proliferation requires autocrine fibroblast growth factor 2 signaling in skeletal muscle cells. J Cell Biol. 2001;152:1301–1305. doi: 10.1083/jcb.152.6.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson SE, Allen RE. Activation of skeletal muscle satellite cells and the role of fibroblast growth factor receptors. Exp Cell Res. 1995;219:449–453. doi: 10.1006/excr.1995.1251. [DOI] [PubMed] [Google Scholar]
- 14.Keshet Y, Seger R. The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods Mol Biol. 2010;661:3–38. doi: 10.1007/978-1-60761-795-2_1. [DOI] [PubMed] [Google Scholar]
- 15.Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 16.Xiong Y, Hannon GJ, Zhang H, et al. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 17.Ezoe S, Matsumura I, Satoh Y, et al. Cell cycle regulation in hematopoietic stem/progenitor cells. Cell Cycle. 2004;3:314–318. [PubMed] [Google Scholar]
- 18.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 19.Cheng T, Rodrigues N, Shen H, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- 20.Brown JP, Wei W, Sedivy JM. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science. 1997;277:831–834. doi: 10.1126/science.277.5327.831. [DOI] [PubMed] [Google Scholar]
- 21.Serrano M, Lee H, Chin L, et al. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- 22.Krimpenfort P, Quon KC, Mooi WJ, et al. Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature. 2001;413:83–86. doi: 10.1038/35092584. [DOI] [PubMed] [Google Scholar]
- 23.Deng C, Zhang P, Harper JW, et al. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–684. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- 24.Brugarolas J, Chandrasekaran C, Gordon JI, et al. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature. 1995;377:552–557. doi: 10.1038/377552a0. [DOI] [PubMed] [Google Scholar]
- 25.Janzen V, Forkert R, Fleming HE, et al. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature. 2006;443:421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- 26.Krishnamurthy J, Ramsey MR, Ligon KL, et al. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- 27.Molofsky AV, Slutsky SG, Joseph NM, et al. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443:448–452. doi: 10.1038/nature05091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stepanova L, Sorrentino BP. A limited role for p16Ink4a and p19Arf in the loss of hematopoietic stem cells during proliferative stress. Blood. 2005;106:827–832. doi: 10.1182/blood-2004-06-2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sousa-Victor P, Gutarra S, Garcia-Prat L, et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014;506:316–321. doi: 10.1038/nature13013. [DOI] [PubMed] [Google Scholar]
- 30.Spivakov M, Fisher AG. Epigenetic signatures of stem-cell identity. Nat Rev Genet. 2007;8:263–271. doi: 10.1038/nrg2046. [DOI] [PubMed] [Google Scholar]
- 31.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 32.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 33.Kamminga LM, de Haan G. Cellular memory and hematopoietic stem cell aging. Stem Cells. 2006;24:1143–1149. doi: 10.1634/stemcells.2005-0345. [DOI] [PubMed] [Google Scholar]
- 34.Elabd C, Cousin W, Chen RY, et al. DNA methyltransferase-3-dependent nonrandom template segregation in differentiating embryonic stem cells. J Cell Biol. 2013;203:73–85. doi: 10.1083/jcb.201307110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6:107–116. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- 36.Liu L, Cheung TH, Charville GW, et al. Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell Rep. 2013;4:189–204. doi: 10.1016/j.celrep.2013.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rando TA, Chang HY. Aging, rejuvenation, and epigenetic reprogramming: resetting the aging clock. Cell. 2012;148:46–57. doi: 10.1016/j.cell.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Conboy MJ, Conboy IM. Preparation of adult muscle fiber-associated stem/precursor cells. Methods Mol Biol. 2010;621:149–163. doi: 10.1007/978-1-60761-063-2_10. [DOI] [PubMed] [Google Scholar]
- 39.Conboy IM, Conboy MJ, Wagers AJ, et al. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;433:760–764. doi: 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]
- 40.Conboy IM, Yousef H, Conboy MJ. Embryonic anti-aging niche. Aging (Albany NY) 2011;3:555–563. doi: 10.18632/aging.100333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gorospe M, Wang X, Holbrook NJ. Functional role of p21 during the cellular response to stress. Gene Expr. 1999;7:377–385. [PMC free article] [PubMed] [Google Scholar]
- 42.Pouyssegur J, Lenormand P. Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Eur J Biochem. 2003;270:3291–3299. doi: 10.1046/j.1432-1033.2003.03707.x. [DOI] [PubMed] [Google Scholar]
- 43.Grounds MD. Age-associated changes in the response of skeletal muscle cells to exercise and regeneration. Ann N Y Acad Sci. 1998;854:78–91. doi: 10.1111/j.1749-6632.1998.tb09894.x. [DOI] [PubMed] [Google Scholar]
- 44.Coqueret O. Linking cyclins to transcriptional control. Gene. 2002;299:35–55. doi: 10.1016/s0378-1119(02)01055-7. [DOI] [PubMed] [Google Scholar]
- 45.Gartel AL, Tyner AL. Transcriptional regulation of the p21((WAF1/CIP1)) gene. Exp Cell Res. 1999;246:280–289. doi: 10.1006/excr.1998.4319. [DOI] [PubMed] [Google Scholar]
- 46.Coleman ML, Marshall CJ, Olson MF. Ras promotes p21(Waf1/Cip1) protein stability via a cyclin D1-imposed block in proteasome-mediated degradation. EMBO J. 2003;22:2036–2046. doi: 10.1093/emboj/cdg189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ostrovsky O, Bengal E. The mitogen-activated protein kinase cascade promotes myoblast cell survival by stabilizing the cyclin-dependent kinase inhibitor, p21WAF1 protein. J Biol Chem. 2003;278:21221–21231. doi: 10.1074/jbc.M211357200. [DOI] [PubMed] [Google Scholar]
- 48.Vicent GP, Ballare C, Nacht AS, et al. Induction of progesterone target genes requires activation of Erk and Msk kinases and phosphorylation of histone H3. Mol Cell. 2006;24:367–381. doi: 10.1016/j.molcel.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 49.Sassone-Corsi P, Mizzen CA, Cheung P, et al. Requirement of Rsk-2 for epidermal growth factor-activated phosphorylation of histone H3. Science. 1999;285:886–891. doi: 10.1126/science.285.5429.886. [DOI] [PubMed] [Google Scholar]
- 50.Bernstein BE, Mikkelsen TS, Xie X, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 51.Cui K, Zang C, Roh TY, et al. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell. 2009;4:80–93. doi: 10.1016/j.stem.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goldfarb M. Signaling by fibroblast growth factors: the inside story. Sci STKE. 2001:pe37. doi: 10.1126/stke.2001.106.pe37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matsumoto T, Turesson I, Book M, et al. p38 MAP kinase negatively regulates endothelial cell survival, proliferation, and differentiation in FGF-2-stimulated angiogenesis. J Cell Biol. 2002;156:149–160. doi: 10.1083/jcb.200103096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wiedlocha A, Sorensen V. Signaling, internalization, and intracellular activity of fibroblast growth factor. Curr Top Microbiol Immunol. 2004;286:45–79. doi: 10.1007/978-3-540-69494-6_3. [DOI] [PubMed] [Google Scholar]
- 55.Boucher MJ, Jean D, Vezina A, et al. Dual role of MEK/ERK signaling in senescence and transformation of intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2004;286:G736–746. doi: 10.1152/ajpgi.00453.2003. [DOI] [PubMed] [Google Scholar]
- 56.Nakade K, Pan J, Yamasaki T, et al. JDP2 (Jun Dimerization Protein 2)-deficient mouse embryonic fibroblasts are resistant to replicative senescence. J Biol Chem. 2009;284:10808–10817. doi: 10.1074/jbc.M808333200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schreiber M, Kolbus A, Piu F, et al. Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev. 1999;13:607–619. doi: 10.1101/gad.13.5.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iordanskaia T, Nawshad A. Mechanisms of transforming growth factor beta induced cell cycle arrest in palate development. J Cell Physiol. 2011;226:1415–1424. doi: 10.1002/jcp.22477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carlson ME, Hsu M, Conboy IM. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature. 2008;454:528–532. doi: 10.1038/nature07034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Young and old satellite cells are highly pure, based on the expression of myogenic markers. Muscle stem cells from injured muscle (3DPI) of young and old mice were plated in OPTI-MEM with 5% of their own serum for 24 hours before immunostaining for Pax7 and MyoD. At least 90% of isolated young and old cells were positive for the myogenic marker Pax7 and/or MyoD. Scale bars span 100μm.
Supplementary Figure 2 FGF2/pERK signaling does not change the myogenicity of young or old satellite cells. (A) Young and old satellite cells activated by injury were cultured in OPTI-MEM with 5% of their own serum, supplemented (or not) with recombinant FGF-2 and/or MEKi for 24 hours, after which cells were collected for real-time RT-PCR for Pax7. (B) Satellite cells were also cultured in DMEM with 2% horse serum for myogenic differentiation for 48 hours before immunostaining for eMyHC. No statistically significant differences were detected between satellite cells of different ages or with FGF-2 and/or MEKi. Thus, FGF2 does not change the myogenic lineage commitment of either young or old satellite cells.
Supplementary Figure 3. Age-specific difference of p21 and p16 levels in myofibers. (A) Total protein was isolated from freshly-derived uninjured, and 3 day-post-injury (3DPI), TA myofibers of young and old mice. The expression levels of p16, p21 and GAPDH were analyzed by Western Blotting. Representative images are shown. (B) Relative protein expression of p16 and p21 were quantified from 3 young and 3 old mice, normalized to GAPDH. The levels of p16 and p21 were significantly higher in old myofibers as compared to young, from both uninjured and 3DPI muscle. N=3, * P<0.05.
Supplementary Figure 4. FGF2 inhibits p15INK4B and p27KIP1 gene expression in aged muscle stem cells independently of the MAPK/pERK pathway. Muscle stem cells from injured muscle (3DPI) of young and old mice were isolated as described in Methods. The cells were plated in OPTI-MEM with 5% of their own serum, supplemented or not with FGF2 (10 ng/ml) and MEK inhibitor PD98059 for 24 hours. Total RNA was isolated and transcription of CDKI genes p15INK4B (A) and p27KIP1 (B) were analyzed by q-RT-PCR with gene-specific primers. Expression of p15 and p27 were significantly higher in old satellite cells as compared to young, and the expression of both genes was attenuated by ectopic FGF2 in a MAPK-independent manner (the FGF-2 effect was not reversed by MEK inhibition). (C) A portion of the isolated cells was cultured in OPTI-MEM, supplemented with isochronic serum from the same mice, for 24 hours, and then treated or not with FGF2 for 1 hour before CHIP assay with ERK1/2 antibody. CHIP and input DNA were measured using q-PCR with specific primers targeting promoter regions of the p15INK4B and p27WIF1 genes. In contrast to p16 and p21, there was no FGF-2-induced enrichment of pERK on the promoter regions of p15 and p27 genes in either young or old satellite cells. N=3, * P<0.05.
Supplementary Figure 5. Epigenetic status and transcriptional expression of p16 and p21 genes in quiescent satellite cells reveals age-specific differences.
The distribution of H3K4me3 and H3K27me3 at p21 (A) and p16 (B) genomic loci were analyzed, using the CHIP-seq database GSE47362, (Liu et al., 2013). Expression of p21 (C) and p16 (D) were analyzed, using the microarray database GSE47177, (Liu et al., 2013) and compared to the q-RT-PCR performed in our studies (E): namely, quiescent muscle stem cells from uninjured muscle of young and old mice were isolated as described in Methods, total RNA was isolated and transcription of p21 and p16 was analyzed by q-RT-PCR with gene-specific primers. A significant shift to repressive H3K27me3 modification in the p21 and p16 genes was found in the young quiescent satellite cells, as compared to old; and significant up-regulation of p21, but not p16 expression, were observed in the old quiescent satellite, as compared to young. These results provide evidence for an epigenetically regulated age-imposed inhibition of satellite cell proliferation that is detectable even in the state of quiescence without muscle injury. N=3, * P<0.05