

Abstract
Translation initiation factor eIF4E mediates normal cell proliferation, yet induces tumorigenesis when overexpressed. The mechanisms by which eIF4E directs such distinct biological outputs remains unknown. We found that mouse mammary morphogenesis during pregnancy and lactation is accompanied by increased cap-binding capability of eIF4E and activation of the eIF4E-dependent translational apparatus, but only subtle oscillations in eIF4E abundance. Using a transgenic mouse model engineered so that lactogenic hormones stimulate a sustained increase in eIF4E abundance in stem/progenitor cells of lactogenic mammary epithelium during successive pregnancy/lactation cycles, eIF4E overexpression increased cell self-renewal, triggered DNA replication stress, and induced formation of pre-malignant and malignant lesions. Using complementary in vivo and ex vivo approaches, we found that increasing eIF4E levels rescued cells harboring oncogenic c-Myc or H-RasV12 from DNA replication stress and oncogene-induced replication catastrophe. Our findings indicate that distinct threshold levels of eIF4E govern its biological output in lactating mammary glands, and that eIF4E overexpression in the context of stem/progenitor cell population expansion can initiate malignant transformation by enabling cells to evade DNA damage checkpoints activated by oncogenic stimuli. Maintaining eIF4E levels below its pro-neoplastic threshold is an important anticancer defense in normal cells, with important implications for understanding pregnancy-associated breast cancer.
Keywords: eIF4E, translation, cancer incidence, DNA repair
Introduction
Dysregulation of translation initiation is frequent in malignancy (1–3). The cap-binding protein eIF4E is a rate-limiting component of the translation initiation complex eIF4F (4). When constitutively activated, eIF4E confers cells with cancer hallmark capabilities (1, 2). However, important aspects of eIF4E-mediated translational control of cancer remain undefined. Particularly vexing is the role of eIF4E dysfunction in cancer incidence. Although mice with increased amounts of eIF4E available for translation initiation due to lack of the eIF4E antagonists 4E-BP1 and 4E-BP2 are hypersensitive to oncogenic stimuli, they do not develop spontaneous tumors (5, 6). These findings point to up-regulated eIF4E as an essential, but passive participant in oncogenesis with eIF4E functioning as an integrator and downstream effector of oncogenic mutations. However, compelling data from several model systems show that enforced overexpression of deregulated eIF4E confers mammalian cells with a neoplastic phenotype in vitro (7, 8) and induces tumorigenesis in vivo (9, 10) – findings consistent with the view that aberrant eIF4E can be a cancer driver.
As a means to define the role of eIF4E overexpression versus eIF4E dysregulation in cancer incidence, it is reasonable to hypothesize that sustained activation of the eIF4E-mediated translational machinery in expanding cell populations, such as the mammary epithelium during gestation, may create a high-risk state in which relatively small increases in eIF4E expression above the physiological maximum might set the stage for oncogenesis. Pregnancy exerts a bidirectional, age-dependent effect on mammary carcinogenesis: in women older than 25, breast cancer incidence increases immediately after parturition, remains increased for 10 years and then gradually falls below the level of nulliparous women (11). Breast cancers diagnosed during or soon after pregnancy, designated pregnancy-associated breast cancer (PABC), tend to be highly aggressive (12). Explanations for PABC include aberrations in the post-partum/weaning involution process (11) and the stimulatory effect of pregnancy-related hormones on latent pro-neoplastic lesions (13). Here, we propose to model this naturally occurring high-risk state to test whether physiologically patterned eIF4E overexpression (i.e., elevated eIF4E levels controlled by lactogenic hormones) in the parity-induced mammary epithelial cell population is sufficient to cause breast tumorigenesis.
Carcinogenesis requires cells to breach the multi-layered intrinsic cancer defense program (14, 15). One such defense is triggered when oncogenes increase DNA replication stress. Stalled replication forks that collapse into double strand breaks (DSBs) activate the DNA damage response (DDR). However, persistent lesions often lead to apoptotic death or premature senescence (16). Examples include the induction of premature senescence by oncogenic Ras (17) and the activation of apoptosis by oncogenic Myc (18). The apparent exception is overexpressed eIF4E, which drives cell proliferation without triggering cell death, counteracts Myc-induced apoptosis (10, 19), and rescues mammary epithelial cells from premature senescence (20). Thus it is plausible that fluctuations of eIF4E levels just above the usual physiological maximum could drive oncogenesis by promoting excess proliferation while disabling DNA damage checkpoints.
To test this formulation, we developed a transgenic mouse model in which naturally occurring pregnancy and lactogenic hormones controlled ectopic eIF4E expression in mammary luminal progenitor cells and their progeny. Here we show that increased eIF4E abundance during successive cycles of pregnancy and lactation is sufficient to promote pathological self-renewal of mammary luminal progenitor cells and induce neoplastic breast lesions. In companion in vitro mechanistic studies, we show that eIF4E-mediated hyperproliferation of human mammary epithelial cells is accompanied by increased DNA replication stress and an enhanced DNA damage response (DDR) that rescues cells from otherwise lethal oncogene-induced DNA damage.
Material and Methods
Transgenic Mice
FVB/N mice were obtained from the Jackson Laboratory (Bar Harbor, Maine, USA). All animal experiments were carried out under an IACUC approved protocol. The WAP vector was constructed by ligation of wild type human eIF4E sequences in frame with three hemagglutinin (HA) epitopes at the C-terminus into the pWkpbAll plasmid encoding the murine WAP promoter (a kind gift of Dr. Jeff Rosen, Baylor College of Medicine) (Figure S1A). Transgenic mice were generated by the University of Minnesota Mouse Genetics Laboratory by microinjection of this construct into FVB/N embryos. Transgenic mice were identified by Southern blotting of tail-snip genomic DNA and confirmed by PCR using the following primers: sense sequence 5′-AAGGACGGCATTGAGCCTAT-3′; anti-sense sequence 3′-GGAAGATCAACGGTCGGTAG-5′.
Cap-affinity binding assay and immunoblotting
m7GTP-Sepharose chromatography were performed as previously described (19). Primary antibodies are listed in Supplementary Material and Methods.
Polysome profiling
Colony-forming assay
We followed our previously published procedure (21). Cultures were continued for 12 days (37°C, 5% CO2) and photographed.
Cell culture and reagents
HMECs constitutively expressing human telomerase reverse transcriptase (hTERT) were provided by Robert Weinberg (Whitehead Institute, Cambridge, MA) and cultured as described (20). (See Supplementary Material and Methods for details).
Statistical analysis
ANOVA, Wilcoxon rank sum or the student’s t-test with Dunnett’s multiple comparison test (S-PLUS Guide to Statistical and Mathematical Analysis, Version 4.0, Seattle, WA) was used with 2-tails and unequal variance expressed as mean ± SE unless otherwise stated.
Results
eIF4F is activated during pregnancy and lactation
Prior studies indicate that the cap-dependent translation initiation complex eIF4F is activated in proliferating cells (22, 23). Immunoblot of mammary tissue lysates obtained during pregnancy-induced mammary morphogenesis revealed a large increase in the scaffold protein eIF4GI and a moderate increase in the mRNA cap-binding protein eIF4E (representative blot shown in Fig. 1A). To assess eIF4F activity, mammary tissue lysates were incubated with agarose-immobilized m7GTP cap analog to pull down eIF4E and its binding partners; and the amounts of cap-captured eIF4E, eIF4GI and 4E-BP1 were evaluated by immunoblot (Fig. 1B). The striking increase in cap-captured eIF4GI compared to 4E-BP1 during late pregnancy and lactation indicated marked activation of eIF4F. These data indicate that during the physiological mammary morphogenesis that occurs in pregnancy, large fluctuations in eIF4GI abundance and eIF4E activity occur; whereas oscillations in eIF4E abundance are minimized.
Figure 1.
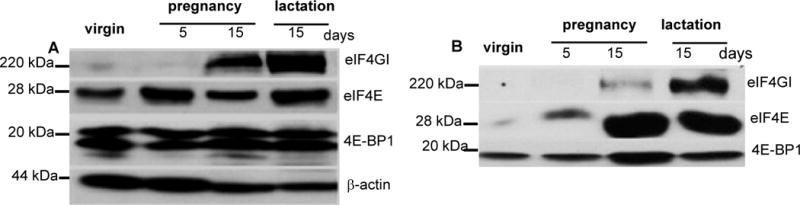
eIF4F is activated during pregnancy and lactation. A, Immunoblot showing steady state expression levels of eIF4GI, eIF4E and 4E-BP1 in mammary tissue of virgin or parous (gestation 2) wild-type FVB/N mice. In parous animals, tissue samples were obtained at pregnancy day 5 (n=2) and day 15 (n=3) and at lactation day 15 (n=3). B, cap-affinity analysis of eIF4F integrity. Cap-bound proteins were subjected to SDS-PAGE, transferred to nitrocellulose and probed for eIF4GI, eIF4E and 4E-BP1.
Generation of transgenic mice expressing increased levels of eIF4E during pregnancy and lactation
To test whether larger hormone-driven fluctuations of eIF4E abundance might induce neoplastic changes, we developed a transgenic mouse model in which expression of exogenous eIF4E (tagged with hemagglutinin to distinguish it from the endogenous protein, designated HA-eIF4E) is controlled by the mammary-specific whey acidic protein (WAP) promoter (Supplementary Fig. S1A). In this system, transgene expression is activated by lactogenic hormones and directs expression of HA-eIF4E to a subpopulation of hormone-responsive WAP-expressing mammary cells (24). These cells survive post-weaning involution of mammary tissue, self-renew and form lactating ductal/alveolar units during subsequent gestations (25). We established three lines of hemizygous transgenic mice (designated WAP-4E) expressing different levels of HA-tagged eIF4E (Supplementary Fig. S1B). Line 3 demonstrated the most stable pattern of gene expression in mammary glands, and was used in all experiments. The presence of exogenous HA-eIF4E can be probed with HA antibody (Fig. 2A) or with antibody against eIF4E (Fig. 2D and in Supplementary Fig. S1C). In virgin females (Fig. 2A) and in wild-type (WT) animals (Fig. 2D left panel) expression of HA-eIF4E was below the limit of detection, and its expression in transgenic females followed the same temporal pattern as the fluctuations in levels of endogenous eIF4GI and eIF4F integrity; turned on during lactation and turned off upon weaning (Figs. 2A and 2D left panel, and Supplementary Fig. S1C).
Figure 2.
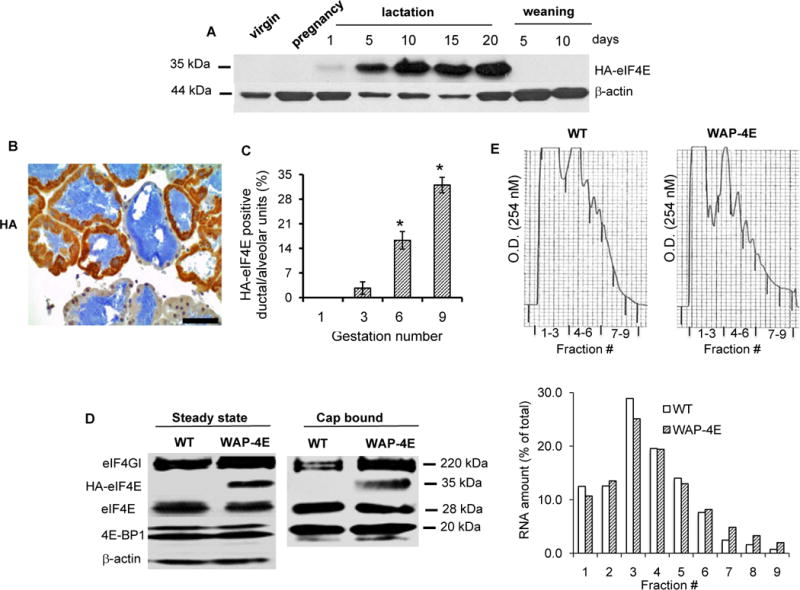
Expression of WAP-driven HA-eIF4E in lactating mammary glands promotes hyperactivation of eIF4F and stimulates recruitment of ribosomes to mRNA. A, expression of WAP-driven HA-eIF4E in mammary glands of virgin, pregnant (day 17), lactating and post-weaning females. Crude mammary tissue lysates were probed with HA antibody. B, formalin-fixed and paraffin-embedded sections of mammary glands (gestation 6, 3 animals) were immunostained for HA expression (HA-eIF4E positive cells are brown). C, Shown are the number of HA-eIF4E positive mammary acinar/ductal structural units during consecutive gestations (mean ± SE, n=2 for each gestation), *p ≤ 0.001. D, representative immunoblot of major eIF4F constituents at steady state and cap-bound (gestation 6, 3 animals). Endogenous eIF4E and exogenous HA-eIF4E were detected using anti-eIF4E antibody. E, ribosome bound RNA from the mammary glands of WT and WAP-4E females (gestation 6, 2 animals) was resolved into discrete fractions by sucrose gradient ultracentrifugation. The top panel shows optical density (O.D.) as a function of sedimentation velocity with fractions 4 to 9 corresponding to RNA associated with a progressively increasing number of ribosomes. The bottom panel shows the amounts of RNA relative to total RNA in each fraction.
To determine whether physiologically patterned overexpression of eIF4E is sufficient to induce breast oncogenesis, transgenic hemizygous females were continuously housed with wild-type males beginning after puberty resulting in almost incessant pregnancy/lactation cycles. WAP promoter-driven transgenes typically show a mosaic expression pattern in hemizygous mice (26). Accordingly, we found that transgene expression in multiparous WAP-4E females was organized into clusters of HA-eIF4E positive and HA-eIF4E negative ductal/alveolar units (representative image shown in Fig. 2B). Quantification of HA-eIF4E-expressing ductal/alveolar unit frequency during successive gestations (Fig. 2C) revealed two distinct phases: during the initial phase, encompassing gestations 1 to 5, there were few HA-eIF4E positive ductal/alveolar units (n=2 for each gestation); whereas during the second phase, which included gestations 6 to 9 (from 3 to 7 months of sustained pregnancy/lactation), the number of HA-eIF4E-positive ductal/alveolar units progressively increased with each successive pregnancy indicating a possibility that transgenic eIF4E may confer PI-MECs with an advantage in self-renewal during subsequent gestations.
A cap pull-down assay confirmed participation of HA-eIF4E in the eIF4F complex (representative blot shown in Fig. 2D, right panel); and mammary gland lysates from transgenic mice manifested a subtle shift in their polysome profile towards heavier polysomes (representative tracing shown in Fig. 2E) suggesting activation of global translation. Of note, due to the mosaic pattern of HA-eIF4E expression, quantitative analysis of whole gland lysates underestimates the levels of exogenous eIF4E, total eIF4E, and translational activity in those acini/ducts that expressed the transgene product. Thus, our data indicated that WAP-driven eIF4E overexpression results in physiologically patterned hyperactivation of cap-dependent translation.
Eif4e transgene-ON: overexpressed eIF4E produces neoplastic lesions and mammary tumors
WAP-4E females experienced multiple pregnancies and lactated normally. Histopathological analysis of mammary glands (Fig. 3A and 3B) revealed that during gestations 1 to 5 – the phase characterized by low numbers of HA-eIF4E-positive ductal/alveolar units (Fig. 2C) – the overall rates of cell proliferation in the mammary epithelium of WAP-4E mice was similar to that in wild type females (n=2 for each gestation) (Fig. 3B); however, proliferation was dramatically increased during gestation 6 (100–120 days after beginning joint male/female housing) and later. One hundred percent of ductal/alveolar units in a random sample of mammary glands from transgenic mice (gestations 5 to 9, n=14 mice out of a cohort of 91 mice) manifested signs of diffuse epithelial hyperplasia and anaplasia (Fig. 3A, right panel); and most hyperproliferation was restricted to cells expressing exogenous HA-eIF4E (n=5) (Fig. 3C).
Figure 3.
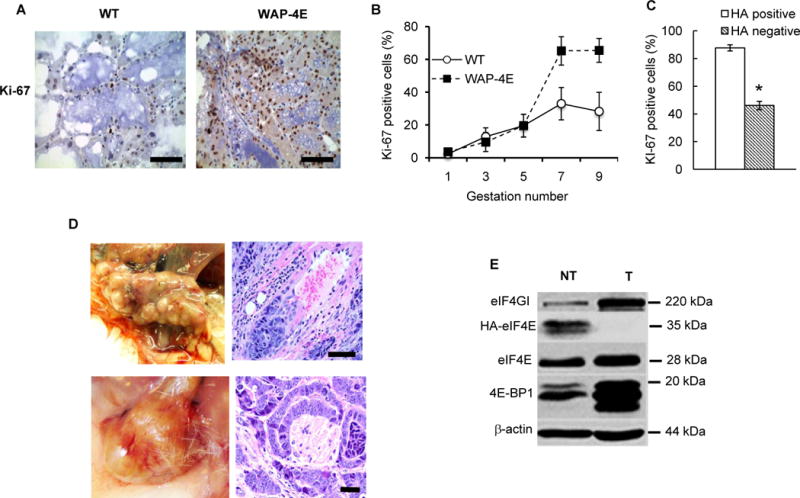
Eif4e transgene-ON: Overexpressed eIF4E produces hyperplastic mammary lesions and stimulates tumorigenesis. A, representative images of mammary tissue from WT and WAP-4E females (gestation 9, day 15 of lactation, n=2) stained for Ki-67 (brown) and counterstained with hematoxylin. B, shown is two-phase kinetics of Ki-67 expression (mean ± SE) during successive pregnancy/lactation cycles (day 15 of lactation) of WT and WAP-4E mice (n=4 for each gestation). C, Ki-67 expression in HA positive and HA-negative mammary epithelial cells (mean ± SE, n=5), *p ≤ 0.001. D, shown are multifocal (top left panel, gestation 5) and unifocal (bottom left panel, gestation 6) breast tumors. Tumor histopathology (hematoxylin/eosin) reveals features of high-grade infiltrating ductal carcinoma including invasion of vessels (top right panel) and perineural invasion (bottom right panel). E, immunoblot showing steady-state levels of eIF4GI, endogenous eIF4E, exogenous eIF4E (HA-eIF4E) and 4E-BP1 in the multifocal breast tumor (T) shown at Figure 3D (top panels) and in the collateral tumor-free mammary gland (NT). Bars = 75 μm.
One limitation of this model is that the long-term analysis of tumor incidence during continuous pregnancy/lactation cycles in FVB/N females is restricted by their short reproductive period (9–11 gestations during the first 8–10 months of life). During this interval, no multiparous wild type FVB/N mice (n=120) developed mammary tumors. By contrast, among 91 transgenic WAP-4E females followed for 6 to 9 months, one developed a multifocal tumor (Fig. 3D, left upper panels) and another developed a solitary tumor (Fig. 3D, left bottom panels). These lesions were high-grade adenocarcinoma with vascular (Fig. 3D, right upper panel) and neural (Fig. 3D, right bottom panel) invasion. These findings show that overexpressed eIF4E triggered development of breast tumors with a long (minimum of 6 months) latency, which is typical for strong oncogenes driven by the WAP promoter in the tumor resistant FVB/N line of mice (27). Expression of WAP-driven transgenes is not detectable in advanced tumors (28). Accordingly, we did not detect expression of exogenous eIF4E in the tumor tissue (Fig. 3E). Instead, tumors manifested overexpression of endogenous eIF4E and increased expression levels of eIF4GI and 4E-BP1 – alterations that are typically observed in advanced breast cancers (2, 29). Together, our observations provide direct evidence that physiologically patterned overexpression of eIF4E in lactating mammary glands triggered unscheduled proliferation of mammary epithelial cells and development of pre-malignant lesions, some of which progress to invasive mammary tumors.
Eif4e transgene-OFF: eIF4E-induced pre-malignant lesions persist but do not progress
Prior studies indicate that pre-malignant lesions formed during pregnancy can progress to breast cancer only if they escape post-weaning involution (27). When weaning was initiated after gestation 2, lobules and alveoli underwent normal regression in both wild type and transgenic mammary glands (not shown). In contrast, when weaning was delayed until gestations 6 to 10 (high number of HA-eIF4E-positive cells), all transgenic mice (n=28) developed focal neoplastic nodules (range 2 to 4 per gland; representative images of mammary gland whole mounts at gestation 9 are shown in Fig. 4A). At that time, most ductal/alveolar units outside of neoplastic nodules were eliminated in both wild type and transgenic mammary glands, and proliferation rates in areas adjacent to neoplastic nodules returned to a rate that was indistinguishable from rates for mammary glands from wild type mice (Fig. 4B). In marked contrast, proliferation in neoplastic nodules remained increased (Figure 4C and 4D). Apoptosis frequency in areas adjacent to neoplastic nodules was similar to that in glands from wild type mice (Fig. 4E). In sharp contrast, most cells of involution-resistant neoplastic nodules manifested eIF4E-dependent acquisition of resistance to apoptosis (Fig. 4F and 4G). Although neoplastic nodules persisted for at least 45 days after involution, they did not show signs of progression to more advanced stages of neoplasia in the absence of HA-eIF4E expression (Supplementary Fig. S2).
Figure 4.

Eif4e transgene-OFF: eIF4E-Induced pre-malignant lesions persist in post-weaning mammary glands. A, post-weaning mammary glands of multiparous WAP-4E females contain neoplastic foci Whole mounted mammary glands of WT and WAP-4E females (gestation 9, day 10 post-weaning) were stained with carmine (panels 1 and 2). Representative gland pictures and photomicrographs are presented (n=28). Arrows indicate neoplastic nodules. LN designates a lymph node. One nodule (shown in the boxed area, panel 2) was surgically removed, and 5 μm sections were stained with hematoxylin/eosin (panels 3 and 4). Arrows indicate deformed acinar structures. B, shown are the proliferative indices (mean ± SD) in post-weaning WT (n=4) and WAP-4E (n=4) mammary glands (gestation 8, day 20 post-weaning). C, representative image of the focus counterstained with hematoxylin and probed for expression of Ki-67 (brown). D, the relative numbers of Ki-67 positive cells in neoplastic foci and in adjacent areas of mammary glands from WT (n=3) and WAP-4E (n=3) females is shown as mean ± SE, *p ≤ 0.0005. E, time-course of apoptotic activity quantified outside of neoplastic foci in post-weaning WT (n=3) and WAP-4E mice (n=3). F, representative image shows scattered TUNEL positive cells (arrows) in the WAP-4E neoplastic focus. G, shown are the relative numbers of TUNEL positive cells in neoplastic foci and in adjacent areas of post-weaning WAP-4E mammary glands (day 10 post-weaning) (n=3 mice) (mean ± SE), *p ≤ 0.001.
Taken together, results from the eIF4E-ON and eIF4E-OFF states demonstrated that physiologically patterned eIF4E overexpression is sufficient for neoplastic conversion of mammary epithelial cells. We observed two types of neoplastic lesions with distinct fates after weaning: (i) multiple hyperproliferative lesions with cells that were eliminated during involution; and (ii) stochastically-induced involution-resistant neoplastic foci, which remained benign in involuted mammary glands (Eif4e transgene-OFF) or progressed to invasive mammary tumors in continuously lactating glands (Eif4e transgene-ON).
Exogenous eIF4E is expressed in luminal cells and increases self-renewal
The adult mammary epithelium is maintained by a population of mammary stem/progenitor cells (MaSPCs) that includes bipotent stem cells and committed myoepithelial and luminal progenitors (30). To identify which cell types were targeted by WAP-driven eIF4E for aberrant proliferation, we double-stained mammary glands: i) with anti-HA antibody to identify cells expressing HA-eIF4E; and ii) with either the luminal marker cytokeratin K8 or the basal marker cytokeratin K14. This analysis revealed that only cells expressing luminal K8 expressed HA-eIF4E; whereas cells expressing basal K14 were uniformly HA-negative (Fig. 5A). Further analysis to identify the proliferating pool using Ki-67 revealed that predominantly luminal K8 positive cells manifested increased proliferation compared to their wild type counterparts (Fig. 5B, Supplementary Fig. S3).
Figure 5.
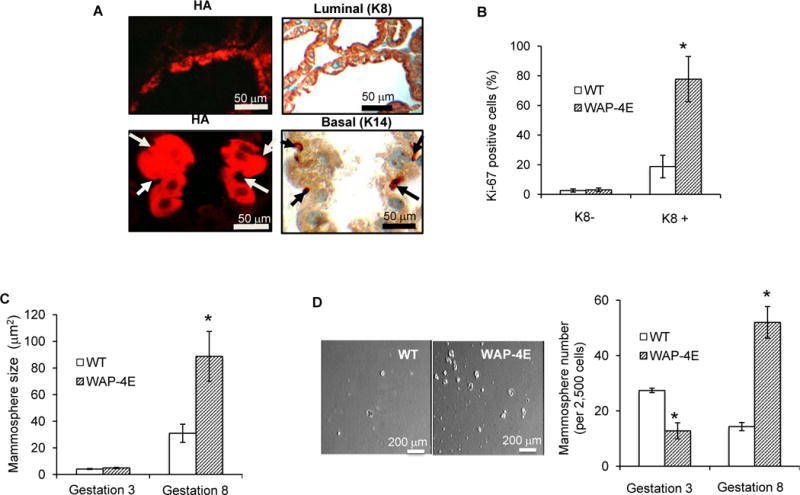
Overexpressed eIF4E increases self-renewal of MaSPC. A, formalin-fixed and paraffin-embedded sections were immunostained for HA (left panels, red) and for cytokeratins (right panels, brown), K8 (luminal marker), and K14 (basal marker). Exogenous HA-eIF4E targets luminal cells (upper panels) and is not expressed in basal cells (arrows at the lower panel). B, quantification of Ki-67 expression in K8 negative and K8 positive mammary cells (mean ± SE, n=3). (C) Primary mammospheres were obtained by plating single cell suspensions of mammary cells (gestations 3 and 8, n=3 for each gestation) on non-adherent substrata. Shown is mammosphere size (mean ± SE), *p ≤ 0.0005. D, secondary mammospheres were formed from a single cell suspension of trypsin-digested primary mammospheres. Shown are the number of mammospheres (left panel) and their appearance by phase contrast microscopy (right panel, bar = 200 μm).
When MaSPCs are targeted by aberrant proliferation-driving stimuli, some combination of self-renewal and differentiation – with or without proliferation of the more differentiated progeny – ensues (31). To evaluate these events in our system, we employed a mammosphere assay (32) in which only MaSPCs from disaggregated mammary glands survive and propagate in vitro without adherence to a substratum. We prepared single-cell suspensions from explants of phenotypically normal (gestation 3, low numbers of HA-eIF4E expressing cells) or hyperplastic (gestation 8, high numbers of HA-eIF4E expressing cells) mammary glands of WAP-4E mice and from pregnancy-matched wild type controls (n=3 in each group). When comparing gestation 8 to gestation 3 on post-weaning day 8 (i.e., expression of HA-eIF4E is turned off), primary mammospheres from WAP-4E mice were 3-fold larger and secondary mammospheres were 4-fold more abundant during gestation 8; whereas those from wild type mice did not vary with parity (Fig. 5C and 5D). Thus, as the number of HA-eIF4E positive cells increased, the mammary epithelium of multiparous WAP-4E females became enriched with MaSPCs displaying increased self-renewal and retained this property at least for several generations when expression of exogenous eIF4E was turned off.
Luminal progenitors can form both luminal and basal-like tumors (33). Histological analysis demonstrated that the epithelial cores of both tumors only expressed the luminal marker K8 (Supplementary Fig. S4). Together, our findings show that the WAP-driven expression of HA-eIF4E in committed luminal progenitors and/or in bipotent mammary stem cells resulted in a population of aberrantly self-renewing cells prone to forming luminal subtypes of pre-malignant lesions and mammary tumors.
Increased expression of eIF4E orchestrates ATR-mediated signaling and rescues cells from oncogene-induced replication catastrophe
To become malignant, cells harboring oncogenic eIF4E must evade anticancer defenses that are triggered by intense DNA replication stress caused by unrelenting positive proliferation signals from oncogenic proteins. This leads to collapse of replication forks with subsequent cell death (14, 15, 34). Replication stress-induced DSBs are recognized by the meiotic recombination 11 (Mre11)/radiation sensitive 50 (Rad50)/Nijmegen breakage syndrome protein 1 (NBS1) or MRN complex, which triggers the genome surveillance pathway and activates the ataxia-telangiectasia mutated (ATM) kinase. After further processing, vulnerable stretches of single-stranded DNA (ssDNA) are coated with replication protein A (RPA), which orchestrates the activation of the ATM- and Rad3- related (ATR) kinase. In concert, ATM and ATR phosphorylate the downstream effectors histone H2AX (pS139), p53 (pS15) and Chk1 (pS345) (35). During oncogene-driven hyper-replication, however, this system typically proceeds to the induction of premature senescence (36). One exception is hyper-proliferation driven by increases in eIF4E, which stimulates cell proliferation while counteracting premature senescence (20) and apoptosis (10, 19, 37).
In mammalian cells, activation of the DNA damage checkpoint can be detected by immunostaining of the phosphorylated form of chromatin-bound histone H2AX (γH2AX). WAP-4E mice had a moderate increase in the proportion of luminal cells with high expression of γH2AX (Figs. 6A, 6B), indicating that eIF4E-mediated hyper-proliferation of luminal progenitors was associated with replication stress and extensive DSBs. To examine the mechanism and to define whether eIF4E-induced activation of the DDR was associated with rescue of mammary cells from oncogene-induced cell death, we took advantage of an in vitro system, in which overexpression of eIF4E is triggered by peptide growth factors routinely used to maintain hTERT-immortalized human mammary epithelial cells (HMEC/hTERT). Similar to our in vivo model, quiescent HMEC/hTERT harboring the gene encoding HA-eIF4E manifested only traces of exogenous eIF4E, whereas the addition of growth factors dramatically increased expression of HA-eIF4E, which peaked in S and G2/M (Figs. 6C and 6D). Growth factor-induced overexpression of eIF4E accelerated transit of cells into G2 (Fig. 6C), significantly increased expression of the DNA damage sensor Mre11 and intensified ATR/ATM signaling as evidenced by increased phosphorylation of Chk1 and H2AX and increased levels and phosphorylation of p53 (Fig. 6D). Since cells overexpressing eIF4E revealed signs of enhanced replication stress but survived, we examined whether overexpression of eIF4E might aid in preserving cell viability and mediating neoplastic conversion. When we introduced c-Myc or H-RasV12 into HMEC/hTERT cells, we detected massive DNA degradation (Fig. 7A) and profound suppression of colony formation (Fig. 7B). In sharp contrast, HMEC/hTERT/4E cells harboring c-Myc or H-RasV12 displayed predominantly intact DNA and formed multiple colonies (Fig. 7B). Analysis of cells transfected with H-RasV12 (Supplementary Fig. S5A) demonstrated that co-expression of HA-eIF4E decreased Ras-induced accumulation of cells in S/G2/M and efficiently rescued cells from Ras-mediated DNA degradation (Supplementary Fig. S5B).
Figure 6.
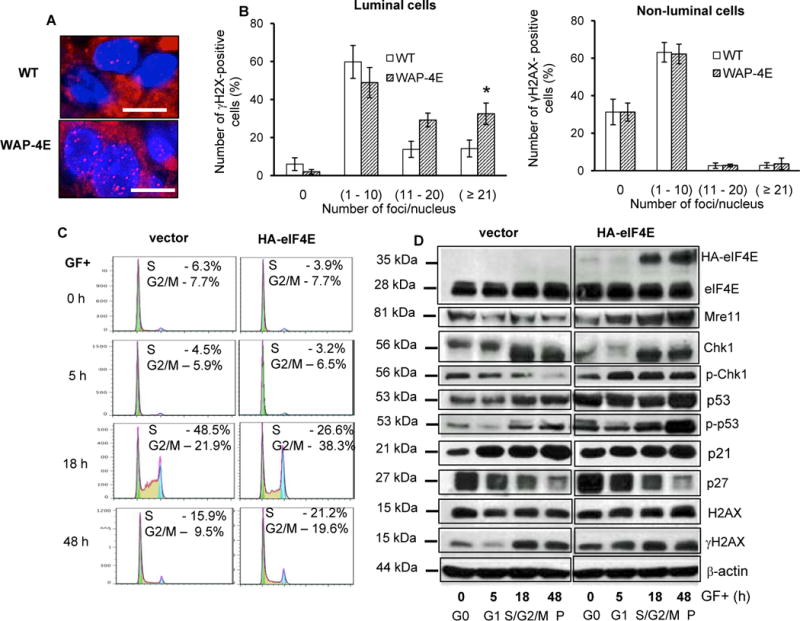
Overexpressed eIF4E accelerates cell cycle progression and stimulates a replication stress response. A and B – analysis in vivo: A, representative images of mammary epithelial cells from wild-type (WT, n=3)) and transgenic (WAP-4E, n=4) mammary glands (gestation 6). Sections were immunostained for γH2AX (red) and DAPI (blue). Bars =10 μm. (B), quantification of γH2AX expression in luminal and non-luminal cells (mean ± SE, n=4), *p ≤ 0.001. C and D – analysis ex vivo: C, representative flow cytometric histograms showing cell-cycle progression of HMEC/hTERT cells harboring HA-eIF4E or an empty vector. Cells were growth factor restricted for 48h, stimulated to cycle with growth factors, and DNA content was analyzed at the indicated times post-growth factor stimulation. D, whole-cell lysates were obtained at the indicated times post-growth factor stimulation of cultured cells and immunoblotted for cell cycle checkpoint regulators (p27, p53, p-p53Ser15 and p21), ATR targets (Chk1, H2AX, γH2AXSer139) and Mre11. β-actin was used as a loading control. P- asynchronously proliferating cells. Shown are represented histograms (C) and blots (D) from 3 independent experiments
Figure 7.
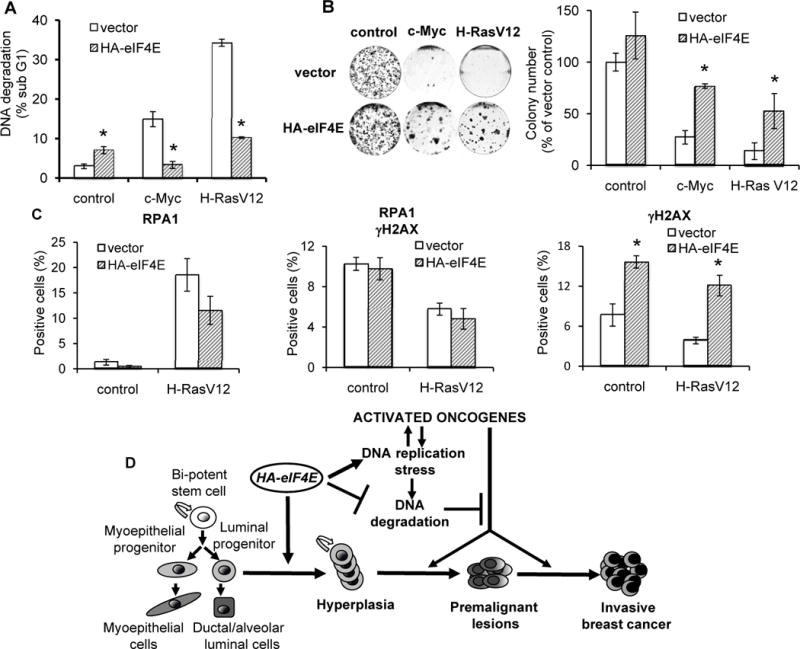
eIF4E-induced rescue from oncogene-induced replication catastrophe is associated with activation of ATR-mediated signaling. A, increased expression of eIF4E protects cells harboring oncogenic c-Myc or Ras from DNA degradation. Shown is flow cytometric quantification of cells with degraded DNA containing sub-diploid (sub-G1) DNA content (mean ± SE, n=4) *p ≤0.001. B, expression of HA-eIF4E in HMEC/hTERT cells harboring oncogenic Myc or Ras enables colony formation. Coomassie stained colonies were photographed (left panel) and scored (right panel) at day 14 post-plating (mean ± SE, two experiments, 3 wells for each experimental point), *p ≤ 0.005. C, increased levels of eIF4E cause replication stress and stimulate the progression of ATR-mediated DDR. Shown are the percentages of HMEC/hTERT cells harboring empty vector (control) or oncogenic Ras which exhibit RPA1-foci alone, overlapping RPA1- and γH2AX-foci, or γH2AX-foci alone (mean ± SE, n=4), *p ≤ 0.001. D. Model of eIF4E-driven development of PABC in transgenic mice (See the text for details).
To determine whether eIF4E-mediated escape from Ras-induced DNA damage was associated with activation of the DDR, we assessed broken DNA by quantification of RPA1 and γH2AX foci in the nucleus (34). The presence of γH2AX foci alone indicates cells harboring DSBs from which RPA has either been displaced with resultant homologous recombination (16), or broken ends have not been resected to ssDNA and have been repaired by other pathways. In the context of eIF4E expression, we observed an increased number of cells expressing γH2AX alone (both control and those harboring H-RasV12) (Fig. 7C and Supplementary Fig. S5C). Thus, overexpressed eIF4E leads to robust DSBs while also activating the repair machinery to promote cell survival, likely at the expense of oncogene-induced neoplastic transformation.
Discussion
Our findings strongly support the hypothesis that mitogen-driven fluctuations of eIF4E abundance above a safe physiological maximum create a state of vulnerability to subsequent oncogenic events. We show that physiologically patterned eIF4E overexpression in lactating mammary glands leads to aberrant self-renewal of MaSPCs and promotes development of pre-malignant and malignant mammary lesions. We further demonstrate that growth factor-driven overexpression of eIF4E sets the stage for preserving DNA integrity in oncogene-stressed mammary epithelial cells by activating a pro-survival DDR. Our findings support a model in which increased eIF4E abundance above the physiological maximum promotes neoplastic transformation through at least two-steps (Figure 7D). During the first step, overexpressed eIF4E is the sole engine of aberrant self-renewal of MaSPCs. This profoundly disturbs the normal balance of cellular subtypes in the mammary epithelium resulting in aberrantly proliferating MaSPCs, which manifest replicative stress and an increased DDR. Continued hyperproliferation and replicative stress create genomic instability causing some cells to accumulate oncogenic mutations. Such cells serve as a reservoir for the second step in neoplastic transformation in which overexpressed eIF4E mediates escape from oncogene-induced replication catastrophe. This allows cells that may have sustained otherwise lethal oncogenic lesions to survive and continue on the cancer pathway.
During normal cell proliferation, eIF4GI abundance increases, reminiscent of eIF4GI levels in cancer. In contrast, during normal mammary epithelial cell proliferation the intracellular abundance of eIF4E is tightly clamped, whereas relatively modest elevations of eIF4E levels reproducibly resulted in neoplastic lesions. These findings show that distinct threshold levels of eIF4E – within very tight tolerances – mediate either normal or pro-neoplastic cell proliferation. These results support the idea that eIF4E abundance is a crucial metric used by cells to discriminate between normal and oncogenic translational signals, and that normal cells must strongly restrain changes in eIF4E abundance to avoid neoplastic conversion.
When proliferation is enforced by strong oncogenic signals, there is increased DNA damage during S phase, replication fork collapse and cell death (15, 36). Importantly, recent findings establish that genes governing DNA repair and genome integrity are under strong translational control (38). We found that increased eIF4E expression in mammary luminal progenitor cells in vivo induces DNA replication stress reflected by γH2AX focus formation. Because replication stress typically results in genetic instability and oncogene-activating mutations (39), these findings help explain the occurrence of sporadic oncogenic lesions during the second phase of eIF4E-driven oncogenesis. Our in vitro studies using oncogenic Myc or Ras as prototype inducers of the DDR-mediated anticancer barrier (15) documented that overexpressed eIF4E counteracts the adverse effects of oncogene-induced replication stress and rescues mammary epithelial cells from replication catastrophe, enabling them to undergo clonal expansion. This pro-neoplastic activity of eIF4E is associated with activation of the DDR machinery including the MRN complex, which is a major sensor of DSBs and is central to the pro-survival function of the DDR (40). One established mechanism of oncogene-driven replication catastrophe is increased replication initiation or origin firing, a condition that can lead to depletion of intracellular nucleotide pools (16, 41). One possible salvage pathway downstream of overexpressed eIF4E is maintenance of the essential nucleotide pool based upon the recently found ability of eIF4E to stimulate nucleotide synthesis in oncogene-expressing cells by translational activation of phosphoribosyl-pyrophosphate synthetase 2 (PRPS2) (42).
One clinical implication of our study relates to how eIF4E oscillations during pregnancy and lactation might create a high-risk state in which relatively subtle changes in eIF4E-mediated translation could potentially initiate oncogenesis. We speculate that increases in eIF4E abundance above the safe range might occur in expanding cell populations of individuals whose eIF4E levels are at the upper end of the normal eIF4E distribution. Such individuals would represent a potentially identifiable high-risk group for PABC or other malignancies that occur in tissues with actively proliferating stem/progenitor cell populations such as the gastrointestinal tract and bone marrow. Our findings imply that therapeutic interventions to normalize aberrant eIF4E function might not only be promising for advanced cancers (3, 43–45), but might also be productively studied for cancer prevention or interception of the neoplastic process at its early stages.
Supplementary Material
Acknowledgments
We thank Naoko Shima for helpful discussions and advice regarding studies addressing the DNA damage response, David Largaespada for consultations related to gene expression patterns in transgenic mice and for his critical reading of the manuscript. We also thank Sandra Wagner from the University of Minnesota Mouse Genetics Laboratory for her expert assistance in developing transgenic mice.
Grant Support
This work was supported by NIH/NCI grant CA-11338-05, Susan G. Komen Breast Cancer Foundation grant BCTR0402671 and the University of Minnesota MCC/Department of Medicine Bridge Grant to Vitaly Polunovsky; by NIH/NHLBI training grant T32 HL07741 to Jeremy Herrera; by NIH/NCI P30 Masonic Cancer Center support grant 2P30CA077598 to Douglas Yee and Peter Bitterman and NIH GM074917 to Anja-Katrin Bielinsky.
Footnotes
Disclosure of Potential Conflicts of Interest
None
References
- 1.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell. 2009;136:731–45. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Silvera D, Formenti SC, Schneider RJ. Translational control in cancer. Nat Rev Cancer. 2010;10:254–66. doi: 10.1038/nrc2824. [DOI] [PubMed] [Google Scholar]
- 3.Bitterman PB, Polunovsky VA. Translational control of cell fate: From integration of environmental signals to breaching anticancer defense. Cell Cycle. 2012;11:1097–107. doi: 10.4161/cc.11.6.19610. [DOI] [PubMed] [Google Scholar]
- 4.Aitken CE, Lorsch JR. A mechanistic overview of translation initiation in eukaryotes. Nat Struct Mol Biol. 2012;19:568–76. doi: 10.1038/nsmb.2303. [DOI] [PubMed] [Google Scholar]
- 5.Petroulakis E, Parsyan A, Dowling RJ, LeBacquer O, Martineau Y, Bidinosti M, et al. p53-dependent translational control of senescence and transformation via 4E-BPs. Cancer Cell. 2009;16:439–46. doi: 10.1016/j.ccr.2009.09.025. [DOI] [PubMed] [Google Scholar]
- 6.Kim YY, Von Weymarn L, Larsson O, Fan D, Underwood JM, Peterson MS, et al. Eukaryotic initiation factor 4E binding protein family of proteins: Sentinels at a translational control checkpoint in lung tumor defense. Cancer Res. 2009;69:8455–62. doi: 10.1158/0008-5472.CAN-09-1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lazaris-Karatzas A, Montine KS, Sonenberg N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5′ cap. Nature. 1990;345:544–7. doi: 10.1038/345544a0. [DOI] [PubMed] [Google Scholar]
- 8.Rousseau D, Kaspar R, Rosenwald I, Gehrke L, Sonenberg N. Translation initiation of ornithine decarboxylase and nucleocytoplasmic transport of cyclin D1 mRNA are increased in cells overexpressing eukaryotic initiation factor 4E. Proc Natl Acad Sci U S A. 1996;93:1065–70. doi: 10.1073/pnas.93.3.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruggero D, Montanaro L, Ma L, Xu W, Londei P, Cordon-Cardo C, et al. The translation factor eIF-4E promotes tumor formation and cooperates with c-myc in lymphomagenesis. Nat Med. 2004;10:484–6. doi: 10.1038/nm1042. [DOI] [PubMed] [Google Scholar]
- 10.Wendel HG, De Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, et al. Survival signalling by akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004;428:332–7. doi: 10.1038/nature02369. [DOI] [PubMed] [Google Scholar]
- 11.Schedin P. Pregnancy-associated breast cancer and metastasis. Nat Rev Cancer. 2006;6:281–91. doi: 10.1038/nrc1839. [DOI] [PubMed] [Google Scholar]
- 12.Fornetti J, Martinson H, Borges V, Schedin P. Emerging targets for the prevention of pregnancy-associated breast cancer. Cell Cycle. 2012;11:639–40. doi: 10.4161/cc.11.4.19358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar R, Sukumar S, Barbacid M. Activation of ras oncogenes preceding the onset of neoplasia. Science. 1990;248:1101–4. doi: 10.1126/science.2188364. [DOI] [PubMed] [Google Scholar]
- 14.Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature. 2004;432:307–15. doi: 10.1038/nature03098. [DOI] [PubMed] [Google Scholar]
- 15.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–7. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 16.Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 18.Murphy DJ, Junttila MR, Pouyet L, Karnezis A, Shchors K, Bui DA, et al. Distinct thresholds govern myc’s biological output in vivo. Cancer Cell. 2008;14:447–57. doi: 10.1016/j.ccr.2008.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li S, Takasu T, Perlman DM, Peterson MS, Burrichter D, Avdulov S, et al. Translation factor eIF4E rescues cells from myc-dependent apoptosis by inhibiting cytochrome c release. J Biol Chem. 2003;278:3015–22. doi: 10.1074/jbc.M208821200. [DOI] [PubMed] [Google Scholar]
- 20.Larsson O, Li S, Issaenko OA, Avdulov S, Peterson M, Smith K, et al. Eukaryotic translation initiation factor 4E induced progression of primary human mammary epithelial cells along the cancer pathway is associated with targeted translational deregulation of oncogenic drivers and inhibitors. Cancer Res. 2007;67:6814–24. doi: 10.1158/0008-5472.CAN-07-0752. [DOI] [PubMed] [Google Scholar]
- 21.Avdulov S, Li S, Michalek V, Burrichter D, Peterson M, Perlman DM, et al. Activation of translation complex eIF4F is essential for the genesis and maintenance of the malignant phenotype in human mammary epithelial cells. Cancer Cell. 2004;5:553–63. doi: 10.1016/j.ccr.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 22.Duncan R, Hershey JW. Regulation of initiation factors during translational repression caused by serum depletion abundance, synthesis, and turnover rates. J Biol Chem. 1985;260:5486–92. [PubMed] [Google Scholar]
- 23.Pyronnet S, Sonenberg N. Cell-cycle-dependent translational control. Curr Opin Genet Dev. 2001;11:13–8. doi: 10.1016/s0959-437x(00)00150-7. [DOI] [PubMed] [Google Scholar]
- 24.Wagner KU, Boulanger CA, Henry MD, Sgagias M, Hennighausen L, Smith GH. An adjunct mammary epithelial cell population in parous females: Its role in functional adaptation and tissue renewal. Development. 2002;129:1377–86. doi: 10.1242/dev.129.6.1377. [DOI] [PubMed] [Google Scholar]
- 25.Henry MD, Triplett AA, Oh KB, Smith GH, Wagner KU. Parity-induced mammary epithelial cells facilitate tumorigenesis in MMTV-neu transgenic mice. Oncogene. 2004;23:6980–5. doi: 10.1038/sj.onc.1207827. [DOI] [PubMed] [Google Scholar]
- 26.Krepulat F, Lohler J, Heinlein C, Hermannstadter A, Tolstonog GV, Deppert W. Epigenetic mechanisms affect mutant p53 transgene expression in WAP-mutp53 transgenic mice. Oncogene. 2005;24:4645–59. doi: 10.1038/sj.onc.1208557. [DOI] [PubMed] [Google Scholar]
- 27.Mack DL, Boulanger CA, Callahan R, Smith GH. Expression of truncated Int6/eIF3e in mammary alveolar epithelium leads to persistent hyperplasia and tumorigenesis. Breast Cancer Res. 2007;9:R42. doi: 10.1186/bcr1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schulze-Garg C, Lohler J, Gocht A, Deppert W. A transgenic mouse model for the ductal carcinoma in situ (DCIS) of the mammary gland. Oncogene. 2000;19:1028–37. doi: 10.1038/sj.onc.1203281. [DOI] [PubMed] [Google Scholar]
- 29.Braunstein S, Karpisheva K, Pola C, Goldberg J, Hochman T, Yee H, et al. A hypoxia-controlled cap-dependent to cap-independent translation switch in breast cancer. Mol Cell. 2007;28:501–12. doi: 10.1016/j.molcel.2007.10.019. [DOI] [PubMed] [Google Scholar]
- 30.Rios AC, Fu NY, Lindeman GJ, Visvader JE. In situ identification of bipotent stem cells in the mammary gland. Nature. 2014;506:322–7. doi: 10.1038/nature12948. [DOI] [PubMed] [Google Scholar]
- 31.Li Q, Bohin N, Wen T, Ng V, Magee J, Chen SC, et al. Oncogenic nras has bimodal effects on stem cells that sustainably increase competitiveness. Nature. 2013;504:143–7. doi: 10.1038/nature12830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253–70. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Molyneux G, Geyer FC, Magnay FA, McCarthy A, Kendrick H, Natrajan R, et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell. 2010;7:403–17. doi: 10.1016/j.stem.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 34.Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, et al. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell. 2013;155:1088–103. doi: 10.1016/j.cell.2013.10.043. [DOI] [PubMed] [Google Scholar]
- 35.Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer. 2012;12:801–17. doi: 10.1038/nrc3399. [DOI] [PubMed] [Google Scholar]
- 36.Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–42. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 37.Polunovsky VA, Rosenwald IB, Tan AT, White J, Chiang L, Sonenberg N, et al. Translational control of programmed cell death: Eukaryotic translation initiation factor 4E blocks apoptosis in growth-factor-restricted fibroblasts with physiologically expressed or deregulated myc. Mol Cell Biol. 1996;16:6573–81. doi: 10.1128/mcb.16.11.6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stumpf CR, Moreno MV, Olshen AB, Taylor BS, Ruggero D. The translational landscape of the mammalian cell cycle. Mol Cell. 2013;52:574–82. doi: 10.1016/j.molcel.2013.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–13. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 40.Stracker TH, Petrini JH. The MRE11 complex: Starting from the ends. Nat Rev Mol Cell Biol. 2011;12:90–103. doi: 10.1038/nrm3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–5. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- 42.Cunningham JT, Moreno MV, Lodi A, Ronen SM, Ruggero D. Protein and nucleotide biosynthesis are coupled by a single rate-limiting enzyme, PRPS2, to drive cancer. Cell. 2014;157:1088–103. doi: 10.1016/j.cell.2014.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bitterman PB, Polunovsky VA. Attacking a nexus of the oncogenic circuitry by reversing aberrant eIF4F-mediated translation. Mol Cancer Ther. 2012;11:1051–61. doi: 10.1158/1535-7163.MCT-11-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hsieh AC, Truitt ML, Ruggero D. Oncogenic AKTivation of translation as a therapeutic target. Br J Cancer. 2011;105:329–36. doi: 10.1038/bjc.2011.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nasr Z, Robert F, Porco JA, Jr, Muller WJ, Pelletier J. eIF4F suppression in breast cancer affects maintenance and progression. Oncogene. 2013;32:861–71. doi: 10.1038/onc.2012.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.