

Abstract
Context:
Secondary amenorrhea—the absence of menses for three consecutive cycles—affects approximately 3–4% of reproductive age women, and infertility—the failure to conceive after 12 months of regular intercourse—affects approximately 6–10%. Neuroendocrine causes of amenorrhea and infertility, including functional hypothalamic amenorrhea and hyperprolactinemia, constitute a majority of these cases.
Objective:
In this review, we discuss the physiologic, pathologic, and iatrogenic causes of amenorrhea and infertility arising from perturbations in the hypothalamic-pituitary-adrenal axis, including potential genetic causes. We focus extensively on the hormonal mechanisms involved in disrupting the hypothalamic-pituitary-ovarian axis.
Conclusions:
A thorough understanding of the neuroendocrine causes of amenorrhea and infertility is critical for properly assessing patients presenting with these complaints. Prompt evaluation and treatment are essential to prevent loss of bone mass due to hypoestrogenemia and/or to achieve the time-sensitive treatment goal of conception.
Secondary amenorrhea (the absence of menses for three consecutive cycles) and infertility (the inability to conceive after 12 months of regular intercourse) are common complaints evaluated by primary care physicians, obstetrician/gynecologists, and endocrinologists. Over 50% of cases of secondary amenorrhea result from perturbations in the hypothalamic-pituitary-adrenal (HPA) axis (1). An understanding of the neuroendocrine causes of amenorrhea and infertility is therefore critical when evaluating patients presenting with these complaints in order to implement the most appropriate treatment regimen. We review the physiological, pathological, and iatrogenic causes of neuroendocrine-associated amenorrhea and infertility, including genetic causes, and the hormonal mechanisms responsible for the perturbations in the hypothalamic-pituitary-ovarian (HPO) axis (Figure 1).
Figure 1.

An overview of neuroendocrine causes of amenorrhea and infertility, including physiological, pathophysiological, and iatrogenic etiologies. *, Possible causes of sellar masses and lesions, including infiltrative and infectious processes, are listed in Table 1. **, In humans, high-dose glucocorticoids may also decrease the responsiveness of the pituitary to GnRH stimulation (7).
Physiological Causes of Amenorrhea
Functional hypothalamic amenorrhea (FHA)
FHA, or stress-induced anovulation, is one of the most common causes of secondary amenorrhea (1), and it accounts for the reproductive dysfunction seen in undernutrition, excessive exercise, severe emotional stress, and chronic disease. From a teleological standpoint, in the face of nutritional or physical stress, it is adaptive for an organism to allocate energy resources for its own survival rather than the costly process of reproduction, and therefore FHA is a physiological response to environmental and physical stressors.
Hormonal mediators of FHA
HPA axis.
During periods of physical, nutritional, or extreme emotional stress, the HPA axis is activated and inhibits the HPO axis at multiple levels (Figure 2). At the level of the hypothalamus, CRH suppresses GnRH secretion. In normally cycling women, an infusion of CRH markedly decreases FSH/LH secretion, but GnRH administration prevents this decrease, suggesting that CRH acts by inhibiting GnRH secretion (2). At the level of the pituitary, ACTH has been shown to have negative reproductive effects in mice. In adrenalectomized mice maintained on physiological doses of glucocorticoid, ACTH administration for 10 days results in an absence of corpora lutea (3), demonstrating that ACTH has reproductive effects independent of adrenal steroid production.
Figure 2.
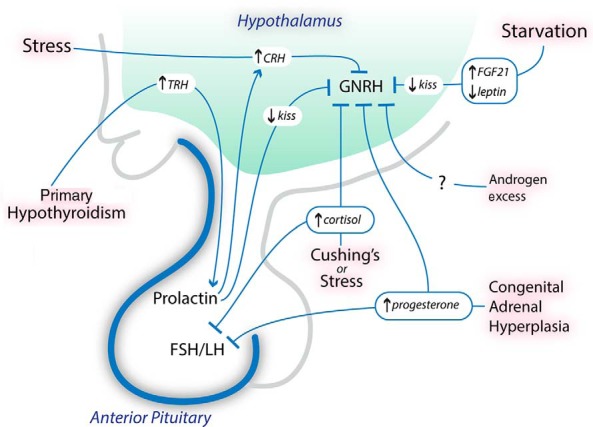
Potential hormonal mediators of amenorrhea and infertility. Physical, emotional, or nutritional stress may increase CRH levels, which suppress GnRH release. Cortisol levels, which increase in the setting of stress and Cushing's syndrome, also suppress GnRH release and likely decrease LH responsiveness to GnRH. Starvation results in low leptin levels and increased FGF-21 levels in animal models; the low leptin levels and increased FGF-21 levels likely suppress GnRH release by decreasing kisspeptin expression. In primary hypothyroidism, TRH levels are increased and stimulate prolactin release. Hyperprolactinemia suppresses GnRH release by decreasing kisspeptin expression, and possibly through a CRH-mediated pathway. High androgen and progesterone levels may be seen in CAH, and these hormones likely suppress GnRH release. kiss, kisspeptin.
Women with FHA also have higher 24-hour mean plasma cortisol levels compared to controls (4), and cortisol, the end-product of the HPA axis, suppresses reproductive function at the hypothalamic, pituitary, and uterine levels. In rhesus monkeys, supraphysiological doses of hydrocortisone suppress gonadotropin secretion, but this suppression is almost completely reversed by intermittent GnRH infusion, suggesting that cortisol suppresses GnRH secretion (5). Women with Cushing's disease and women on long-term supraphysiological prednisolone therapy have a reduced LH response to GnRH (6, 7), suggesting that glucocorticoids also suppress the responsiveness of pituitary gonadotrophs to hypothalamic input. Glucocorticoids also inhibit the effects of estradiol on the uterus. Dexamethasone coadministered with estradiol attenuates the expected increase in uterine weight seen with estradiol alone, at least in part by reducing estrogen receptor concentration (8). Therefore, cross talk between the HPA and HPO axes promotes the development of amenorrhea as a functional adaptation to stress.
Leptin.
Levels of leptin, a hormone secreted by adipocytes, are low in women with FHA (9, 10), which may be a mediator of amenorrhea. Two studies have investigated the effects of recombinant methionyl human leptin (rhleptin) in women with FHA. An open-label study found that LH-pulse frequency increased after 2 weeks of treatment with rhleptin (11). In a randomized placebo-controlled study, significantly more women treated with rhleptin had a menstrual bleed, half of which were associated with ovulation (12). Although the exact mechanism by which decreased leptin levels cause anovulation is not known, low leptin levels may suppress GnRH through a kisspeptin-mediated pathway (13).
Insulin.
Changes in body weight—both weight loss and weight gain—may affect fertility. Significant weight loss due to chronic undernutrition is a cause of FHA and is characterized by low insulin levels, whereas significant weight gain may result in obesity and insulin resistance—a state of functional hypoinsulinemia. Importantly, in animal models, insulin has been shown to modulate the HPO axis, and therefore low or functionally low insulin levels may be a mediator of infertility. In a mouse model, females with neuron-specific disruption of the insulin receptor had a significantly reduced antral follicle count due to a 90% reduction of circulating LH (14), demonstrating the importance of insulin signaling in fertility.
Fibroblast growth factor-21 (FGF-21).
FGF-21 is a liver-derived hormone whose production is up-regulated in response to starvation (15, 16). Recently, FGF-21 was shown to be a potential mediator of starvation-induced amenorrhea (17). FGF-21 transgenic mice are anovulatory, with low LH levels, and administration of GnRH, but not exogenous gonadotropins, elicits an ovulatory LH surge, demonstrating that FGF-21 acts at the level of the hypothalamus to disrupt ovulation (17). FGF-21 transgenic mice have decreased Kiss-1 gene expression in the anteroventral periventricular nuclei of the hypothalamus, and the product of Kiss-1, kisspeptin, is a potent stimulator of GnRH secretion, suggesting a mechanism by which FGF-21 disrupts ovulation (17). Whether FGF-21 is a mediator of FHA in humans is unknown.
Hyperprolactinemia
Hyperprolactinemia—the most common cause of pituitary-associated amenorrhea—may be due to physiologic or pathophysiologic causes. The exact mechanism by which hyperprolactinemia causes hypogonadotropic hypogonadism is not known, but hyperprolactinemic women have reduced LH-pulse frequency and reduced LH responsiveness to estrogen (18), suggesting that GnRH suppression may be a key factor. This is supported by the fact that in hyperprolactinemic, amenorrheic women, treatment with pulsatile GnRH results in follicular maturation and ovulation when coupled with a human chorionic gonadotropin trigger (18). CRH (19) and kisspeptin may be important mediators of prolactin-induced GnRH suppression. In rodent models, prolactin-receptor mRNA has been localized to kisspeptin neurons in the hypothalamus (20), and kisspeptin administration to hyperprolactinemic female mice increases circulating gonadotropin levels and restores ovulation (21). In vitro, prolactin inhibits granulosa cell function (22), and therefore prolactin may also act by directly disrupting ovarian function.
Although most women with hyperprolactinemia are amenorrheic, some have ovulatory menstrual cycles. These women may still suffer from infertility due to a short luteal-phase defect (23), and therefore treating hyperprolactinemia may be important even in regularly menstruating women. Importantly, a number of physiological causes of hyperprolactinemia and amenorrhea—including pregnancy—do not warrant treatment and therefore should be excluded before initiation of treatment.
Physiological causes of hyperprolactinemia
Pregnancy.
Pregnancy is the most common cause of hyperprolactinemia. Prolactin levels increase during pregnancy and peak at delivery, with levels reaching as high as 600 ng/mL (24). In women who do not nurse, prolactin levels usually decrease during the first 72 hours postpartum (24, 25) and normalize by 3 weeks (25).
Lactation.
In nursing women, suckling increases prolactin levels, although this usually subsides by 12 weeks due to the postpartum drop in estradiol levels resulting in decreased lactotroph hyperplasia (24). Lactational amenorrhea is the result of a number of factors, including maternal nutritional status relative to the frequency/intensity of nursing (26) and hyperprolactinemia. Prolactin levels, their response to suckling, and prolactin bioactivity are all associated with duration of amenorrhea (27–29), and like prolactin-induced amenorrhea, lactational amenorrhea is associated with reduced LH-pulse amplitude and frequency (30) and reduced LH responsiveness to estrogen (31). Moreover, administration of pulsatile GnRH to women with lactational amenorrhea stimulates follicle maturation, ovulation, and luteal-phase function (30), suggesting that GnRH suppression may be a causative factor.
Macroprolactinemia.
Macroprolactinemia is another cause of physiological hyperprolactinemia. The predominant form of prolactin is a 23-kD form, but some individuals may have a significant percentage of circulating prolactin consisting of a higher molecular mass form called macroprolactin. Macroprolactin is thought to be either a complex consisting of the 23-kD form and an immunoglobulin (32) or a complex consisting of glycosylated prolactin aggregates (33). Macroprolactin is cleared from the circulation more slowly than the 23-kD form (34), thereby causing hyperprolactinemia, but it is also less bioactive, supporting the observation that macroprolactinemia is usually asymptomatic (35). Importantly, up to 25% of individuals with hyperprolactinemia may have macroprolactinemia (36, 37), and therefore evaluation for macroprolactinemia in asymptomatic individuals may be warranted to prevent unnecessary treatment. Macroprolactin can be detected through gel-filtration chromatography or polyethylene glycol precipitation (38).
Pathophysiological Causes of Amenorrhea
Pathophysiological causes of hyperprolactinemia
As mentioned above, there are both physiologic and pathophysiologic causes of hyperprolactinemia. Pathophysiologic causes of hyperprolactinemia typically result in amenorrhea and/or infertility in premenopausal women, and therefore an elevated prolactin level requires prompt evaluation (Figure 3). Pathophysiologic causes of hyperprolactinemia include lactotroph adenomas, stalk disruption, primary hypothyroidism, renal failure, and chest wall injury.
Figure 3.

A suggested diagnostic algorithm for evaluating patients with secondary amenorrhea, including non-neuroendocrine causes. Importantly, all patients presenting with secondary amenorrhea should have a history and physical examination performed and a basic laboratory evaluation consisting of serum human chorionic gonadotropin, TSH, FSH, and prolactin levels. *, Individuals who have been prescribed CTLA-4 antibody therapies (ipilimumab or tremelimumab) should have an MRI with pituitary cuts to evaluate for hypophysitis. **, Unless the cause of the hyperprolactinemia can be definitively attributed to the medication (for example, by stopping the medication and repeating the prolactin level), it is difficult to exclude the possibility of a prolactinoma or stalk disruption (due to a sellar lesion) causing the hyperprolactinemia. ***, If the lesion is small (<1 cm) and is found to be nonsecretory, we would recommend following the “No lesion and normal prolactin” pathway because this lesion is unlikely to be the cause of the amenorrhea. Abbreviations: abnl, abnormal; BMI, body mass index; DHEAS, dehydroepiandrosterone sulfate; PCOS, polycystic ovary syndrome; 17-OH progesterone, 17-hydroxyprogesterone; H&P, history and physical.
Lactotroph adenomas
Prolactin-secreting adenomas (lactotroph adenomas) are the most common subtype of secretory pituitary adenoma. These tumors are usually benign, and prolactin levels typically correlate with tumor size. Individuals with large adenomas can have prolactin levels on the order of 104 ng/mL (39), yet with poorly differentiated or cystic lesions, prolactin levels will be lower than expected based on size.
Stalk disruption
Dopamine is produced by neurons in the arcuate nucleus of the hypothalamus and tonically suppresses pituitary prolactin production. Any disruption of the stalk connecting the hypothalamus to the pituitary can prevent the flow of dopamine into the pituitary gland and result in hyperprolactinemia and amenorrhea. Large sellar masses (Table 1) or traumatic injuries are common causes of stalk disruption.
Table 1.
| Benign and malignant neoplastic processes |
| Pituitary adenoma |
| Craniopharyngioma |
| Chordoma |
| Metastatic disease |
| Meningioma |
| Germ cell tumor |
| Ependymoma |
| Granular cell tumor |
| Hemangioma |
| Primary lymphoma |
| Sarcoma |
| Chondrosarcoma |
| Osteosarcoma |
| Fibrosarcoma |
| Carcinoma of sinus origin |
| Pituicytoma |
| Papilloma |
| Schwannoma |
| Cystic lesions |
| Rathke's cleft cyst |
| Arachnoid cyst |
| Dermoid cyst |
| Epidermoid cyst |
| Suprasellar cyst |
| Mucocele |
| Vascular lesions |
| Aneurysm |
| Arteriovenous fistula of cavernous sinus |
| Infiltrative and infectious processes |
| Abscess |
| Hypophysitis |
| Lymphocytic |
| Granulomatous |
| Xanthomatous |
| IgG4-related disease |
| CTLA-4 antibody-related |
| Sarcoidosis |
| Fibrous dysplasia |
| Hemochromatosis |
| Langerhans cell histiocytosis |
| Pituitary hyperplasia |
| Sphenoid sinusitis |
| Tuberculosis |
| Cysticercosis |
Primary hypothyroidism
TRH, secreted by the hypothalamus, not only stimulates release of TSH by the pituitary but also stimulates prolactin release (40). Individuals with primary hypothyroidism have a heightened prolactin response to TRH, resulting in greater prolactin secretion in response to TRH stimulation (41); therefore hyperprolactinemia and resultant amenorrhea can be a consequence of primary hypothyroidism (42). Primary hypothyroidism may also lead to significant enlargement of the pituitary gland due to thyrotroph hyperplasia and likely lactotroph hyperplasia, but treating the hypothyroidism should result in regression of the hyperplasia (43) and normalization of the prolactin level (44).
Chronic renal failure
Chronic renal failure may result in hyperprolactinemia due to both decreased renal clearance and increased secretion of prolactin. The latter may be the result of reduced lactotroph responsiveness to dopamine suppression (45). Although liver disease was once posited as a common cause of hyperprolactinemia (46), recent reports suggest that it is a very rare cause of hyperprolactinemia (47).
Chest wall injury
Traumatic chest wall injury is a rare potential cause of hyperprolactinemia (48). In a patient with hyperprolactinemia after a severe burn to the chest wall, intercostal nerve blockade resulted in normalization of her serum prolactin level (48), suggesting that a neurogenic stimulus at the site of injury was responsible.
Non-lactotroph adenomas and sellar masses
Any sellar mass or lesion (Table 1) can cause amenorrhea. Mechanisms of amenorrhea include hyperprolactinemia due to stalk disruption and/or compression of the pituitary gonadotrophs, particularly when the lesion is ≥ 1 cm. In the case of functioning adenomas, hormonal effects may also play a role in the development of amenorrhea.
Cushing's disease
Amenorrhea is a common finding in women with ACTH-secreting adenomas (Cushing's disease). In a series of 45 women with Cushing's disease, 33% were amenorrheic, and amenorrhea was associated with higher mean serum cortisol levels and lower estradiol and SHBG levels (49). Although adrenal androgen levels are typically elevated in Cushing's disease, amenorrhea was not associated with serum androgen levels or free androgen index (49). Therefore, amenorrhea in Cushing's disease is more likely mediated by suppression of GnRH by cortisol (49) rather than hyperandrogenemia.
Acromegaly
Amenorrhea and infertility are also common findings in women with GH-secreting adenomas (acromegaly). In a study of 47 women with acromegaly, 62% had amenorrhea, which was associated with higher GH levels and lower LH and estradiol levels (50). As with any sellar mass, compression of pituitary gonadotrophs, or hyperprolactinemia—either due to cosecretion of prolactin by the tumor or stalk disruption—may be important in the development of amenorrhea, but another potential cause may be increased androgen bioavailability due to decreased SHBG levels (50). In normally cycling women, T administration reduces LH-pulse frequency, suggesting a mechanism by which increased androgen activity may mediate amenorrhea (51). In men, GH excess may also have a direct effect on gonadal function, suggested by the fact that men with acromegaly have significantly lower semen parameters compared to controls, and post-treatment decreases in IGF-1 levels correlate with increases in total sperm motility (52); therefore, a direct gonadal effect may potentially play a role in the development of amenorrhea as well.
Thyrotroph adenomas
Amenorrhea or oligomenorrhea may also be a presenting finding in women with TSH-secreting adenomas (53), and hyperthyroidism may be a contributing factor. Compared to euthyroid controls, hyperthyroid women with amenorrhea have higher SHBG, FSH, LH, and estradiol levels (54) but do not have a midcycle LH peak, suggesting that amenorrhea results from anovulation due to the failure of estrogen to stimulate LH release (55).
Infiltrative processes
Any inflammatory or infectious disease can infiltrate the hypothalamic-pituitary axis, resulting in anterior pituitary dysfunction and hypogonadotropic hypogonadism. Metastases—most commonly from primary tumors of the breast or lung—may also invade the hypothalamic-pituitary axis and cause amenorrhea (56).
Hypophysitis
An inflammatory infiltrate in the pituitary gland (hypophysitis) typically results in hypopituitarism due to destruction of cells in the anterior pituitary. Hypophysitis may also cause hyperprolactinemia due to stalk disruption, and therefore there are multiple mechanisms by which amenorrhea may occur. The two most common types of hypophysitis are lymphocytic and granulomatous hypophysitis. Lymphocytic hypophysitis, occurring primarily in females, is commonly diagnosed during pregnancy or postpartum, and a majority of patients develop anterior pituitary dysfunction (57). Granulomatous hypophysitis—characterized by granulomas formed by multinucleated giant cells—may occur in the setting of systemic diseases, including Wegener's granulomatosis (58).
IgG4-related disease is a more recently described cause of hypophysitis characterized by an infiltrate consisting of IgG4-positive plasma cells (59). This entity has been associated with hypogonadotropic hypogonadism (59), although it predominantly affects males and individuals >50 years old and therefore is a rare cause of amenorrhea.
Granulomatous diseases
Sarcoidosis and tuberculosis may also cause amenorrhea. In sarcoidosis, hypogonadotropic hypogonadism is usually due to granulomatous infiltration of the hypothalamus, although granulomas may also invade the pituitary and infiltrate gonadotroph cells (60). In addition, both intrasellar tuberculomas and tuberculous meningitis have been associated with amenorrhea (61–63). A study of 49 patients with a history of tuberculous meningitis in childhood found that 10% developed gonadotropin deficiency, likely due to progressive hypothalamic scarring (63).
Hemochromatosis
Although hereditary hemochromatosis is an important cause of hypogonadism in men due to iron deposition in the pituitary gonadotrophs and/or testes, it rarely causes hypogonadism in women (64), likely because menstrual bleeding protects against iron deposition.
Langerhans cell histiocytosis
Langerhans cell histiocytosis is characterized by the inappropriate proliferation and infiltration of a type of dendritic cell into various regions of the body including the hypothalamic-pituitary axis. Although typically associated with diabetes insipidus, hypogonadotropic hypogonadism has been reported (65).
Sheehan's syndrome
During pregnancy, the pituitary gland enlarges due to estrogen stimulation of lactotroph cells (66). This enlarging tissue may compress the superior hypophyseal artery—a major source of blood for the anterior pituitary—making the gland vulnerable to changes in blood supply. Women with significant peripartum blood loss may develop ischemic necrosis of the pituitary gland, resulting in hypopituitarism. This form of hypopituitarism, referred to as Sheehan's syndrome, is usually seen in cases of significant hemorrhage, although rare cases have been reported in individuals with minimal postpartum bleeding (67), and therefore pituitary autoimmunity has been hypothesized to play a role in the development of anterior pituitary dysfunction (68).
Sheehan's syndrome is now less commonly seen in developed countries due to improvements in the management of peripartum hemodynamic complications; however, recent reports suggest that its incidence in the developed world may be greater than previously thought (69). A retrospective study from France demonstrated a mean delay of 9 years before diagnosis (70), suggesting that the diagnosis is often overlooked in developed countries.
Traumatic brain injury
Amenorrhea may also present after traumatic brain injury or subarachnoid hemorrhage. Importantly, even mild head injuries—those not requiring hospitalization—can result in hypogonadism (71). Loss of pituitary function may be delayed and can present 12 months after the initial insult (72).
Adrenal tumors
Amenorrhea is frequently observed in women with glucocorticoid and/or androgen-secreting adrenal neoplasms. As mentioned above, the elevated cortisol levels produced by glucocorticoid-secreting tumors likely suppress GnRH secretion, thereby causing amenorrhea (5). Neoplasms producing androgens alone are exceedingly rare, and approximately 50% are malignant (73). Commonly presenting symptoms include hirsutism, acne, and oligomenorrhea (73). In women with normal menstrual cycles, T reduces LH-pulse frequency (51), suggesting a mechanism by which hyperandrogenemia may cause oligomenorrhea.
Genetic causes of neuroendocrine-associated amenorrhea
Hypothalamus: idiopathic hypogonadotropic hypogonadism (IHH)
IHH is a heterogeneous group of disorders in which individuals have delayed or absent pubertal development—typically due to GnRH deficiency or a GnRH receptor mutation—coupled with normal anatomic findings on hypothalamic/pituitary imaging. Although IHH predominantly affects males, it is a cause of infertility in women, typically presenting as primary amenorrhea.
There are a number of associated phenotypic findings in individuals with IHH, the most common of which is anosmia (Kallman's syndrome). Genetic mutations that impair neuronal migration commonly result in concurrent hypogonadism and anosmia due to the shared embryonic development of GnRH and olfactory neurons. In recent years, a number of genetic mutations that cause Kallman's syndrome have been discovered. These include mutations in the KAL1 gene (74), the FGF-1 receptor (75), FGF-8 (76), the gene encoding semaphorin-3A (77), the CHD7 gene (78), and the genes encoding prokineticin-2 and prokineticin receptor-2 (79).
Genetic defects in GnRH secretion and function have also been identified in cases of IHH with normosmia. Kisspeptin is a potent regulator of GnRH secretion, and mutations in KISS1 and KISS1R, the genes encoding kisspeptin and its receptor, cause hypogonadism. Mutations of the gene encoding leptin (80), its receptor (81), and mutations in the prohormone convertase 1 gene (82) have all been associated with both hypogonadotropic hypogonadism and severe obesity. GnRH receptor mutations also result in hypogonadotropic hypogonadism (83). Unlike most patients with IHH, individuals with GnRH receptor mutations typically do not respond to conventional doses of pulsatile GnRH administration, although successful conception with high-dose pulsatile GnRH has been reported (84).
Although IHH represents only a small subset of infertility, the underlying mutations may confer susceptibility to the development of FHA. In a study of 55 women with FHA, 13% were found to have heterozygous mutations in genes associated with IHH, as compared to no mutations found in 422 controls (85). Therefore, FHA may stem from interplay between environmental and genetic factors, whereby inherited defects in GnRH biology may lower the threshold at which external stressors suppress the HPO axis (85).
Pituitary
Primary amenorrhea may also result from mutations in genes encoding transcription factors involved in the cellular proliferation and differentiation of the pituitary gland. HESX1 (86), GLI2 (87), and SOX3 (88) mutations have been linked to hypopituitarism, and mutations in SOX2 (89), LHX3 (90), LHX4 (91), and PROP1 (92) may cause gonadotropin deficiency. Alterations in these genes may present with other clinical findings, which may help with the diagnosis. For example, mutations in SOX2 may result in anophthalmia, and mutations in LHX3 have been associated with a rotationally limited cervical spine (90).
Adrenal: congenital adrenal hyperplasia (CAH)
Reduction in the activity of adrenal enzymes responsible for cortisol production may also result in infertility. CAH is a family of inherited disorders characterized by defects in cortisol production, resulting in increased ACTH production due to reduced negative feedback and consequent adrenal gland proliferation. Although there are rare forms of CAH that result in infertility, including 11β-hydroxylase deficiency (93), 17α-hydroxylase deficiency, and 3β-hydroxysteroid dehydrogenase deficiency (94), CAH most commonly arises from autosomal recessive mutations in the gene that encodes 21-hydroxylase (21-OH), CYP21A2, which result in shunting of steroid precursors toward androgen biosynthesis. There is considerable variability in disease phenotype, depending on the severity of the mutation. Classic 21-OH deficiency is typically diagnosed at birth and can present with severe salt wasting and/or prenatal virilization of external female genitalia, whereas nonclassic CAH (nonclassic 21-OH deficiency [NC21OHD]) presents postnatally with signs of hyperandrogenism including hirsutism, acne, frontal balding, and menstrual irregularities (95).
The oligomenorrhea in women with 21-OH deficiency is likely due to elevated androgen and/or progesterone levels. Patients with classic 21-OH deficiency with irregular menstrual cycles have elevated progesterone and androstenedione levels that are associated with reduced LH-pulse amplitude and frequency (96). In vitro, androgens increase GnRH-pulse frequency (97), supporting the observation of elevated LH levels in women with NC21OHD (98); yet in normally cycling women, high-dose T infusion results in reduced LH-pulse frequency, even when blocking estrogen aromatization (51). Therefore, although the exact mechanism by which androgens act to induce oligomenorrhea is unknown, hyperandrogenemia may be a contributing factor in CAH. Elevated levels of progesterone, a substrate of 21-OH, also likely contribute to menstrual irregularity. Administration of progesterone to normally cycling women reduces LH-pulse frequency, and the progesterone receptor has been identified on gonadotroph cells in the pituitary, suggesting that progesterone's gonadotropin-suppressing effects may be mediated by both the hypothalamus and pituitary (99).
Although classic CAH is relatively rare, NC21OHD is one of the most common autosomal recessive diseases known, occurring in 1 in 100 individuals in a mixed ethnic population (100) and at a much higher frequency (1 in 27) in the Ashkenazi Jewish population (101). Given its unique treatment implications, it is important to distinguish NC21OHD from other causes of hyperandrogenism and oligomenorrhea, such as polycystic ovary syndrome. In women with evidence of hyperandrogenism and/or oligomenorrhea, the prevalence of NC21OHD has been reported to range from 1.2–13.8% (102–107). Not detected on newborn screening tests, the most definitive hormonally based diagnostic test for NC21OHD is an ACTH stimulation test measuring serum concentrations of 17-hydroxyprogesterone, a substrate of 21-OH, after ACTH administration (108). Patients with NC21OHD have stimulated 17-hydroxyprogesterone levels that are intermediate between normal subjects and those with classic 21-OH deficiency (108).
Iatrogenic Causes of Amenorrhea
Medication-induced amenorrhea
Various drugs, including hormonal contraceptives and leuprolide, interfere purposefully with normal menses by acting directly on the hypothalamus or pituitary. A number of other medications cause infertility or amenorrhea as a side effect of their use. These medications act by: 1) inhibiting GnRH release; 2) inducing hyperprolactinemia; or 3) inducing hypophysitis.
Inhibition of GnRH release
Opioids.
Opioids are an under-recognized but common cause of hypogonadism (109). The effect of opioids on the gonadal axis has been extensively studied in men, and hypogonadotropic hypogonadism may be a more common side effect in men compared to women (110). In vitro and human studies suggest that opioids act by directly inhibiting GnRH release (111, 112), although they may also act by inducing hyperprolactinemia (113).
Medication-induced hyperprolactinemia
Psychotropic medications.
A number of medications cause hyperprolactinemia by interfering with dopamine's inhibitory effect on prolactin secretion. The medications that most commonly cause hyperprolactinemia are neuroleptics, which act by blocking dopamine receptors. Due to their greater affinity for the D2 receptor (114), typical antipsychotics have a more robust effect on elevating prolactin levels compared to atypical antipsychotics, with the exception of risperidone (115). Data on selective-serotonin reuptake inhibitors are conflicting, but a large open-label study of depressed individuals demonstrated the development of hyperprolactinemia in 22% of women after 12 weeks of fluoxetine use (116). Clomipramine, a tricyclic antidepressant that inhibits serotonin uptake, also increases prolactin levels (117). These serotonin uptake inhibitors may increase prolactin levels by increasing serotonergic inhibition of dopaminergic neurons.
Other medications.
Through a similar antidopaminergic mode of action, certain gastrointestinal motility agents and antihypertensives have also been associated with hyperprolactinemia. Serum prolactin levels increase 6-fold after oral or iv metoclopramide (118), and domperidone also stimulates prolactin release (119). Methyldopa, an α-adrenergic agonist, and verapamil, a calcium-channel blocker, both increase prolactin levels, likely by decreasing dopamine synthesis (120, 121).
Medication-induced hypophysitis
Cytotoxic T-lymphocyte antigen 4 (CTLA-4) antibodies.
Ipilimumab and tremelimumab are human monoclonal antibodies targeting CTLA-4, thereby promoting an immune-mediated response against cancer. Ipilimumab has been approved by the Food and Drug Administration to treat advanced malignant melanoma and is undergoing trials to explore its use in other malignancies. Given its immunostimulatory properties, ipilimumab has been associated with autoimmune reactions including hypophysitis resulting in hypopituitarism. A large randomized controlled trial studying the effects of ipilimumab in metastatic melanoma found the incidence of hypopituitarism to be 2.3% (122).
Pituitary surgery
Although reports in the literature suggest that surgical treatment of a pituitary lesion is rarely associated with hypogonadism (123), this is likely an underestimate of the true prevalence. This is due to the fact that surgeons with higher surgical volumes have fewer postsurgical complications (124), and most of these reports reflect rates of hypogonadism of dedicated pituitary surgeons.
Radiation treatment
Radiation therapy directed at the hypothalamus or pituitary is a well-known cause of hypopituitarism (125). Importantly, individuals who have radiation therapy for nonpituitary brain tumors are also at significant risk of developing hypogonadism (126). The development of hypogonadism and hyperprolactinemia in these individuals is associated with the dose of radiation therapy administered (127).
Conclusion
In conclusion, perturbations of the HPA axis due to physiological, pathological, and iatrogenic causes constitute a majority of cases of amenorrhea and infertility. FHA, a physiologic response to physical, emotional, or nutritional stress is the most common cause of neuroendocrine amenorrhea, and identifying this entity is essential in order to diagnose and treat the underlying disorder. Given the consequences of not treating amenorrhea, namely loss of bone mass, as well as the age constraints in treating female patients with infertility, prompt diagnosis and institution of treatment are critical.
Acknowledgments
This project was supported by National Institutes of Health grant K23-DK094820 (to P.K.F.) and the Claflin Distinguished Scholar Award (to P.K.F.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- CAH
- congenital adrenal hyperplasia
- CTLA-4
- cytotoxic T-lymphocyte antigen 4
- FGF-21
- fibroblast growth factor-21
- FHA
- functional hypothalamic amenorrhea
- HPA
- hypothalamic-pituitary-adrenal
- HPO
- hypothalamic-pituitary-ovarian
- IHH
- idiopathic hypogonadotropic hypogonadism
- NC21OHD
- nonclassic 21-OH deficiency
- 21-OH
- 21-hydroxylase.
References
- 1. Reindollar RH, Novak M, Tho SP, McDonough PG. Adult-onset amenorrhea: a study of 262 patients. Am J Obstet Gynecol. 1986;155:531–543. [DOI] [PubMed] [Google Scholar]
- 2. Barbarino A, De Marinis L, Tofani A, et al. Corticotropin-releasing hormone inhibition of gonadotropin release and the effect of opioid blockade. J Clin Endocrinol Metab. 1989;68:523–528. [DOI] [PubMed] [Google Scholar]
- 3. Christian JJ. Actions of ACTH in intact and corticoid-maintained adrenalectomized female mice with emphasis on the reproductive tract. Endocrinology. 1964;75:653–669. [DOI] [PubMed] [Google Scholar]
- 4. Biller BM, Federoff HJ, Koenig JI, Klibanski A. Abnormal cortisol secretion and responses to corticotropin-releasing hormone in women with hypothalamic amenorrhea. J Clin Endocrinol Metab. 1990;70:311–317. [DOI] [PubMed] [Google Scholar]
- 5. Dubey AK, Plant TM. A suppression of gonadotropin secretion by cortisol in castrated male rhesus monkeys (Macaca mulatta) mediated by the interruption of hypothalamic gonadotropin-releasing hormone release. Biol Reprod. 1985;33:423–431. [DOI] [PubMed] [Google Scholar]
- 6. Boccuzzi G, Angeli A, Bisbocci D, Fonzo D, Giadano GP, Ceresa F. Effect of synthetic luteinizing hormone releasing hormone (LH-RH) on the release of gonadotropins in Cushing's disease. J Clin Endocrinol Metab. 1975;40:892–895. [DOI] [PubMed] [Google Scholar]
- 7. Sakakura M, Takebe K, Nakagawa S. Inhibition of luteinizing hormone secretion induced by synthetic LRH by long-term treatment with glucocorticoids in human subjects. J Clin Endocrinol Metab. 1975;40:774–779. [DOI] [PubMed] [Google Scholar]
- 8. Rabin DS, Johnson EO, Brandon DD, Liapi C, Chrousos GP. Glucocorticoids inhibit estradiol-mediated uterine growth: possible role of the uterine estradiol receptor. Biol Reprod. 1990;42:74–80. [DOI] [PubMed] [Google Scholar]
- 9. Miller KK, Parulekar MS, Schoenfeld E, et al. Decreased leptin levels in normal weight women with hypothalamic amenorrhea: the effects of body composition and nutritional intake. J Clin Endocrinol Metab. 1998;83:2309–2312. [DOI] [PubMed] [Google Scholar]
- 10. Warren MP, Voussoughian F, Geer EB, Hyle EP, Adberg CL, Ramos RH. Functional hypothalamic amenorrhea: hypoleptinemia and disordered eating. J Clin Endocrinol Metab. 1999;84:873–877. [DOI] [PubMed] [Google Scholar]
- 11. Welt CK, Chan JL, Bullen J, et al. Recombinant human leptin in women with hypothalamic amenorrhea. N Engl J Med. 2004;351:987–997. [DOI] [PubMed] [Google Scholar]
- 12. Chou SH, Chamberland JP, Liu X, et al. Leptin is an effective treatment for hypothalamic amenorrhea. Proc Natl Acad Sci USA. 2011;108:6585–6590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Quennell JH, Howell CS, Roa J, Augustine RA, Grattan DR, Anderson GM. Leptin deficiency and diet-induced obesity reduce hypothalamic kisspeptin expression in mice. Endocrinology. 2011;152:1541–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brüning JC, Gautam D, Burks DJ, et al. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000;289:2122–2125. [DOI] [PubMed] [Google Scholar]
- 15. Inagaki T, Dutchak P, Zhao G, et al. Endocrine regulation of the fasting response by PPARα-mediated induction of fibroblast growth factor 21. Cell Metab. 2007;5:415–425. [DOI] [PubMed] [Google Scholar]
- 16. Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPARα and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007;5:426–437. [DOI] [PubMed] [Google Scholar]
- 17. Owen BM, Bookout AL, Ding X, et al. FGF21 contributes to neuroendocrine control of female reproduction. Nat Med. 2013;19:1153–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Matsuzaki T, Azuma K, Irahara M, Yasui T, Aono T. Mechanism of anovulation in hyperprolactinemic amenorrhea determined by pulsatile gonadotropin-releasing hormone injection combined with human chorionic gonadotropin. Fertil Steril. 1994;62:1143–1149. [DOI] [PubMed] [Google Scholar]
- 19. Kooy A, de Greef WJ, Vreeburg JT, et al. Evidence for the involvement of corticotropin-releasing factor in the inhibition of gonadotropin release induced by hyperprolactinemia. Neuroendocrinology. 1990;51:261–266. [DOI] [PubMed] [Google Scholar]
- 20. Kokay IC, Petersen SL, Grattan DR. Identification of prolactin-sensitive GABA and kisspeptin neurons in regions of the rat hypothalamus involved in the control of fertility. Endocrinology. 2011;152:526–535. [DOI] [PubMed] [Google Scholar]
- 21. Sonigo C, Bouilly J, Carré N, et al. Hyperprolactinemia-induced ovarian acyclicity is reversed by kisspeptin administration. J Clin Invest. 2012;122:3791–3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dorrington J, Gore-Langton RE. Prolactin inhibits oestrogen synthesis in the ovary. Nature. 1981;290:600–602. [DOI] [PubMed] [Google Scholar]
- 23. Corenblum B, Pairaudeau N, Shewchuk AB. Prolactin hypersecretion and short luteal phase defects. Obstet Gynecol. 1976;47:486–488. [PubMed] [Google Scholar]
- 24. Tyson JE, Hwang P, Guyda H, Friesen HG. Studies of prolactin secretion in human pregnancy. Am J Obstet Gynecol. 1972;113:14–20. [DOI] [PubMed] [Google Scholar]
- 25. Bonnar J, Franklin M, Nott PN, McNeilly AS. Effect of breast-feeding on pituitary-ovarian function after childbirth. Br Med J. 1975;4:82–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Valeggia C, Ellison PT. Interactions between metabolic and reproductive functions in the resumption of postpartum fecundity. Am J Hum Biol. 2009;21:559–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Díaz S, Serón-Ferré M, Cárdenas H, Schiappacasse V, Brandeis A, Croxatto HB. Circadian variation of basal plasma prolactin, prolactin response to suckling, and length of amenorrhea in nursing women. J Clin Endocrinol Metab. 1989;68:946–955. [DOI] [PubMed] [Google Scholar]
- 28. Díaz S, Cárdenas H, Brandeis A, et al. Early difference in the endocrine profile of long and short lactational amenorrhea. J Clin Endocrinol Metab. 1991;72:196–201. [DOI] [PubMed] [Google Scholar]
- 29. Campino C, Ampuero S, Díaz S, Serón-Ferré M. Prolactin bioactivity and the duration of lactational amenorrhea. J Clin Endocrinol Metab. 1994;79:970–974. [DOI] [PubMed] [Google Scholar]
- 30. Zinaman MJ, Cartledge T, Tomai T, Tippett P, Merriam GR. Pulsatile GnRH stimulates normal cyclic ovarian function in amenorrheic lactating postpartum women. J Clin Endocrinol Metab. 1995;80:2088–2093. [DOI] [PubMed] [Google Scholar]
- 31. Baird DT, McNeilly AS, Sawers RS, Sharpe RM. Failure of estrogen-induced discharge of luteinizing hormone in lactating women. J Clin Endocrinol Metab. 1979;49:500–506. [DOI] [PubMed] [Google Scholar]
- 32. Hattori N, Ishihara T, Ikekubo K, Moridera K, Hino M, Kurahachi H. Autoantibody to human prolactin in patients with idiopathic hyperprolactinemia. J Clin Endocrinol Metab. 1992;75:1226–1229. [DOI] [PubMed] [Google Scholar]
- 33. Hattori N. The frequency of macroprolactinemia in pregnant women and the heterogeneity of its etiologies. J Clin Endocrinol Metab. 1996;81:586–590. [DOI] [PubMed] [Google Scholar]
- 34. Hattori N, Inagaki C. Anti-prolactin (PRL) autoantibodies cause asymptomatic hyperprolactinemia: bioassay and clearance studies of PRL-immunoglobulin G complex. J Clin Endocrinol Metab. 1997;82:3107–3110. [DOI] [PubMed] [Google Scholar]
- 35. Jackson RD, Wortsman J, Malarkey WB. Characterization of a large molecular weight prolactin in women with idiopathic hyperprolactinemia and normal menses. J Clin Endocrinol Metab. 1985;61:258–264. [DOI] [PubMed] [Google Scholar]
- 36. Leslie H, Courtney CH, Bell PM, et al. Laboratory and clinical experience in 55 patients with macroprolactinemia identified by a simple polyethylene glycol precipitation method. J Clin Endocrinol Metab. 2001;86:2743–2746. [DOI] [PubMed] [Google Scholar]
- 37. Vallette-Kasic S, Morange-Ramos I, Selim A, et al. Macroprolactinemia revisited: a study on 106 patients. J Clin Endocrinol Metab. 2002;87:581–588. [DOI] [PubMed] [Google Scholar]
- 38. Olukoga AO, Kane JW. Macroprolactinaemia: validation and application of the polyethylene glycol precipitation test and clinical characterization of the condition. Clin Endocrinol (Oxf). 1999;51:119–126. [DOI] [PubMed] [Google Scholar]
- 39. Delgrange E, Trouillas J, Maiter D, Donckier J, Tourniaire J. Sex-related difference in the growth of prolactinomas: a clinical and proliferation marker study. J Clin Endocrinol Metab. 1997;82:2102–2107. [DOI] [PubMed] [Google Scholar]
- 40. Jacobs LS, Snyder PJ, Wilber JF, Utiger RD, Daughaday WH. Increased serum prolactin after administration of synthetic thyrotropin releasing hormone (TRH) in man. J Clin Endocrinol Metab. 1971;33:996–998. [DOI] [PubMed] [Google Scholar]
- 41. Snyder PJ, Jacobs LS, Utiger RD, Daughaday WH. Thyroid hormone inhibition of the prolactin response to thyrotropin-releasing hormone. J Clin Invest. 1973;52:2324–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Onishi T, Miyai K, Aono T, Shioji T, Yamamoto T. Primary hypothyroidism and galactorrhea. Am J Med. 1977;63:373–378. [DOI] [PubMed] [Google Scholar]
- 43. Jawadi MH, Ballonoff LB, Stears JC, Katz FH. Primary hypothyroidism and pituitary enlargement. Radiological evidence of pituitary regression. Arch Intern Med. 1978;138:1555–1557. [PubMed] [Google Scholar]
- 44. Shahshahani MN, Wong ET. Primary hypothyroidism, amenorrhea, and galactorrhea. Arch Intern Med. 1978;138:1411–1412. [PubMed] [Google Scholar]
- 45. Sievertsen GD, Lim VS, Nakawatase C, Frohman LA. Metabolic clearance and secretion rates of human prolactin in normal subjects and in patients with chronic renal failure. J Clin Endocrinol Metab. 1980;50:846–852. [DOI] [PubMed] [Google Scholar]
- 46. Morgan MY, Jakobovits AW, Gore MB, Wills MR, Sherlock S. Serum prolactin in liver disease and its relationship to gynaecomastia. Gut. 1978;19:170–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ress C, Maeser PA, Tschoner A, et al. Serum prolactin in advanced chronic liver disease. Horm Metab Res. 2014;46:800–803. [DOI] [PubMed] [Google Scholar]
- 48. Morley JE, Dawson M, Hodgkinson H, Kalk WJ. Galactorrhea and hyperprolactinemia associated with chest wall injury. J Clin Endocrinol Metab. 1977;45:931–935. [DOI] [PubMed] [Google Scholar]
- 49. Lado-Abeal J, Rodriguez-Arnao J, Newell-Price JD, et al. Menstrual abnormalities in women with Cushing's disease are correlated with hypercortisolemia rather than raised circulating androgen levels. J Clin Endocrinol Metab. 1998;83:3083–3088. [DOI] [PubMed] [Google Scholar]
- 50. Kaltsas GA, Mukherjee JJ, Jenkins PJ, et al. Menstrual irregularity in women with acromegaly. J Clin Endocrinol Metab. 1999;84:2731–2735. [DOI] [PubMed] [Google Scholar]
- 51. Serafini P, Silva PD, Paulson RJ, Elkind-Hirsch K, Hernandez M, Lobo RA. Acute modulation of the hypothalamic-pituitary axis by intravenous testosterone in normal women. Am J Obstet Gynecol. 1986;155:1288–1292. [DOI] [PubMed] [Google Scholar]
- 52. Colao A, De Rosa M, Pivonello R, et al. Short-term suppression of GH and IGF-I levels improves gonadal function and sperm parameters in men with acromegaly. J Clin Endocrinol Metab. 2002;87:4193–4197. [DOI] [PubMed] [Google Scholar]
- 53. Tritos NA, Eppakayala S, Swearingen B, et al. Pathologic and clinical features of pituitary adenomas showing TSH immunoreactivity. Pituitary. 2013;16:287–293. [DOI] [PubMed] [Google Scholar]
- 54. Akande EO, Anderson DC. Role of sex-hormone-binding globulin in hormonal changes and amenorrhoea in thyrotoxic women. Br J Obstet Gynaecol. 1975;82:557–561. [DOI] [PubMed] [Google Scholar]
- 55. Akande EO, Hockaday TD. Plasma concentration of gonadotrophins, oestrogens and progesterone in thyrotoxic women. Br J Obstet Gynaecol. 1975;82:541–551. [DOI] [PubMed] [Google Scholar]
- 56. Komninos J, Vlassopoulou V, Protopapa D, et al. Tumors metastatic to the pituitary gland: case report and literature review. J Clin Endocrinol Metab. 2004;89:574–580. [DOI] [PubMed] [Google Scholar]
- 57. Thodou E, Asa SL, Kontogeorgos G, Kovacs K, Horvath E, Ezzat S. Clinical case seminar: lymphocytic hypophysitis: clinicopathological findings. J Clin Endocrinol Metab. 1995;80:2302–2311. [DOI] [PubMed] [Google Scholar]
- 58. Hunn BH, Martin WG, Simpson S, Jr, Mclean CA. Idiopathic granulomatous hypophysitis: a systematic review of 82 cases in the literature. Pituitary. 2014;17:357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Leporati P, Landek-Salgado MA, Lupi I, Chiovato L, Caturegli P. IgG4-related hypophysitis: a new addition to the hypophysitis spectrum. J Clin Endocrinol Metab. 2011;96:1971–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Stuart CA, Neelon FA, Lebovitz HE. Hypothalamic insufficiency: the cause of hypopituitarism in sarcoidosis. Ann Intern Med. 1978;88:589–594. [DOI] [PubMed] [Google Scholar]
- 61. Ranjan A, Chandy MJ. Intrasellar tuberculoma. Br J Neurosurg. 1994;8:179–185. [DOI] [PubMed] [Google Scholar]
- 62. Summers VK, Hipkin LJ, Osborne Hughes R, Davis JC. Panhypopituitarism after cured tuberculous meningitis. Br Med J. 1968;1:359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lam KS, Sham MM, Tam SC, Ng MM, Ma HT. Hypopituitarism after tuberculous meningitis in childhood. Ann Intern Med. 1993;118:701–706. [DOI] [PubMed] [Google Scholar]
- 64. Kelly TM, Edwards CQ, Meikle AW, Kushner JP. Hypogonadism in hemochromatosis: reversal with iron depletion. Ann Intern Med. 1984;101:629–632. [DOI] [PubMed] [Google Scholar]
- 65. Kaltsas GA, Powles TB, Evanson J, et al. Hypothalamo-pituitary abnormalities in adult patients with Langerhans cell histiocytosis: clinical, endocrinological, and radiological features and response to treatment. J Clin Endocrinol Metab. 2000;85:1370–1376. [DOI] [PubMed] [Google Scholar]
- 66. Goluboff LG, Ezrin C. Effect of pregnancy on the somatotroph and the prolactin cell of the human adenohypophysis. J Clin Endocrinol Metab. 1969;29:1533–1538. [DOI] [PubMed] [Google Scholar]
- 67. Zargar AH, Singh B, Laway BA, Masoodi SR, Wani AI, Bashir MI. Epidemiologic aspects of postpartum pituitary hypofunction (Sheehan's syndrome). Fertil Steril. 2005;84:523–528. [DOI] [PubMed] [Google Scholar]
- 68. Goswami R, Kochupillai N, Crock PA, Jaleel A, Gupta N. Pituitary autoimmunity in patients with Sheehan's syndrome. J Clin Endocrinol Metab. 2002;87:4137–4141. [DOI] [PubMed] [Google Scholar]
- 69. Kristjansdottir HL, Bodvarsdottir SP, Sigurjonsdottir HA. Sheehan's syndrome in modern times: a nationwide retrospective study in Iceland. Eur J Endocrinol. 2011;164:349–354. [DOI] [PubMed] [Google Scholar]
- 70. Ramiandrasoa C, Castinetti F, Raingeard I, et al. Delayed diagnosis of Sheehan's syndrome in a developed country: a retrospective cohort study. Eur J Endocrinol. 2013;169:431–438. [DOI] [PubMed] [Google Scholar]
- 71. Benvenga S, Campenní A, Ruggeri RM, Trimarchi F. Clinical review 113: hypopituitarism secondary to head trauma. J Clin Endocrinol Metab. 2000;85:1353–1361. [DOI] [PubMed] [Google Scholar]
- 72. Aimaretti G, Ambrosio MR, Di Somma C, et al. Residual pituitary function after brain injury-induced hypopituitarism: a prospective 12-month study. J Clin Endocrinol Metab. 2005;90:6085–6092. [DOI] [PubMed] [Google Scholar]
- 73. Cordera F, Grant C, van Heerden J, Thompson G, Young W. Androgen-secreting adrenal tumors. Surgery. 2003;134:874–880. [DOI] [PubMed] [Google Scholar]
- 74. Franco B, Guioli S, Pragliola A, et al. A gene deleted in Kallmann's syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature. 1991;353:529–536. [DOI] [PubMed] [Google Scholar]
- 75. Dodé C, Levilliers J, Dupont JM, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet. 2003;33:463–465. [DOI] [PubMed] [Google Scholar]
- 76. Falardeau J, Chung WC, Beenken A, et al. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest. 2008;118:2822–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Young J, Metay C, Bouligand J, et al. SEMA3A deletion in a family with Kallmann syndrome validates the role of semaphorin 3A in human puberty and olfactory system development. Hum Reprod. 2012;27:1460–1465. [DOI] [PubMed] [Google Scholar]
- 78. Bergman JE, de Ronde W, Jongmans MC, et al. The results of CHD7 analysis in clinically well-characterized patients with Kallmann syndrome. J Clin Endocrinol Metab. 2012;97:E858–E862. [DOI] [PubMed] [Google Scholar]
- 79. Dodé C, Teixeira L, Levilliers J, et al. Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet. 2006;2:e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Strobel A, Issad T, Camoin L, Ozata M, Strosberg AD. A leptin missense mutation associated with hypogonadism and morbid obesity. Nat Genet. 1998;18:213–215. [DOI] [PubMed] [Google Scholar]
- 81. Clément K, Vaisse C, Lahlou N, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. 1998;392:398–401. [DOI] [PubMed] [Google Scholar]
- 82. Jackson RS, Creemers JW, Ohagi S, et al. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat Genet. 1997;16:303–306. [DOI] [PubMed] [Google Scholar]
- 83. de Roux N, Young J, Misrahi M, et al. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. N Engl J Med. 1997;337:1597–1602. [DOI] [PubMed] [Google Scholar]
- 84. Seminara SB, Beranova M, Oliveira LM, Martin KA, Crowley WF, Jr, Hall JE. Successful use of pulsatile gonadotropin-releasing hormone (GnRH) for ovulation induction and pregnancy in a patient with GnRH receptor mutations. J Clin Endocrinol Metab. 2000;85:556–562. [DOI] [PubMed] [Google Scholar]
- 85. Caronia LM, Martin C, Welt CK, et al. A genetic basis for functional hypothalamic amenorrhea. N Engl J Med. 2011;364:215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Thomas PQ, Dattani MT, Brickman JM, et al. Heterozygous HESX1 mutations associated with isolated congenital pituitary hypoplasia and septo-optic dysplasia. Hum Mol Genet. 2001;10:39–45. [DOI] [PubMed] [Google Scholar]
- 87. França MM, Jorge AA, Carvalho LR, et al. Novel heterozygous nonsense GLI2 mutations in patients with hypopituitarism and ectopic posterior pituitary lobe without holoprosencephaly. J Clin Endocrinol Metab. 2010;95:E384–E391. [DOI] [PubMed] [Google Scholar]
- 88. Alatzoglou KS, Kelberman D, Cowell CT, et al. Increased transactivation associated with SOX3 polyalanine tract deletion in a patient with hypopituitarism. J Clin Endocrinol Metab. 2011;96:E685–E690. [DOI] [PubMed] [Google Scholar]
- 89. Sato N, Kamachi Y, Kondoh H, et al. Hypogonadotropic hypogonadism in an adult female with a heterozygous hypomorphic mutation of SOX2. Eur J Endocrinol. 2007;156:167–171. [DOI] [PubMed] [Google Scholar]
- 90. Netchine I, Sobrier ML, Krude H, et al. Mutations in LHX3 result in a new syndrome revealed by combined pituitary hormone deficiency. Nat Genet. 2000;25:182–186. [DOI] [PubMed] [Google Scholar]
- 91. Pfaeffle RW, Hunter CS, Savage JJ, et al. Three novel missense mutations within the LHX4 gene are associated with variable pituitary hormone deficiencies. J Clin Endocrinol Metab. 2008;93:1062–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wu W, Cogan JD, Pfäffle RW, et al. Mutations in PROP1 cause familial combined pituitary hormone deficiency. Nat Genet. 1998;18:147–149. [DOI] [PubMed] [Google Scholar]
- 93. Zachmann M, Tassinari D, Prader A. Clinical and biochemical variability of congenital adrenal hyperplasia due to 11 β-hydroxylase deficiency. A study of 25 patients. J Clin Endocrinol Metab. 1983;56:222–229. [DOI] [PubMed] [Google Scholar]
- 94. Rosenfield RL, Rich BH, Wolfsdorf JI, et al. Pubertal presentation of congenital δ 5–3 β-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol Metab. 1980;51:345–353. [DOI] [PubMed] [Google Scholar]
- 95. New MI. Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2006;91:4205–4214. [DOI] [PubMed] [Google Scholar]
- 96. Bachelot A, Chakhtoura Z, Plu-Bureau G, et al. Influence of hormonal control on LH pulsatility and secretion in women with classical congenital adrenal hyperplasia. Eur J Endocrinol. 2012;167:499–505. [DOI] [PubMed] [Google Scholar]
- 97. Melrose P, Gross L. Steroid effects on the secretory modalities of gonadotropin-releasing hormone release. Endocrinology. 1987;121:190–199. [DOI] [PubMed] [Google Scholar]
- 98. Levin JH, Carmina E, Lobo RA. Is the inappropriate gonadotropin secretion of patients with polycystic ovary syndrome similar to that of patients with adult-onset congenital adrenal hyperplasia? Fertil Steril. 1991;56:635–640. [DOI] [PubMed] [Google Scholar]
- 99. Couzinet B, Young J, Kujas M, et al. The antigonadotropic activity of a 19-nor-progesterone derivative is exerted both at the hypothalamic and pituitary levels in women. J Clin Endocrinol Metab. 1999;84:4191–4196. [DOI] [PubMed] [Google Scholar]
- 100. Zerah M, Ueshiba H, Wood E, et al. Prevalence of nonclassical steroid 21-hydroxylase deficiency based on a morning salivary 17-hydroxyprogesterone screening test: a small sample study. J Clin Endocrinol Metab. 1990;70:1662–1667. [DOI] [PubMed] [Google Scholar]
- 101. Speiser PW, Dupont B, Rubinstein P, Piazza A, Kastelan A, New MI. High frequency of nonclassical steroid 21-hydroxylase deficiency. Am J Hum Genet. 1985;37:650–667. [PMC free article] [PubMed] [Google Scholar]
- 102. Chetkowski RJ, DeFazio J, Shamonki I, Judd HL, Chang RJ. The incidence of late-onset congenital adrenal hyperplasia due to 21-hydroxylase deficiency among hirsute women. J Clin Endocrinol Metab. 1984;58:595–598. [DOI] [PubMed] [Google Scholar]
- 103. Kuttenn F, Couillin P, Girard F, et al. Late-onset adrenal hyperplasia in hirsutism. N Engl J Med. 1985;313:224–231. [DOI] [PubMed] [Google Scholar]
- 104. Pang SY, Lerner AJ, Stoner E, et al. Late-onset adrenal steroid 3 β-hydroxysteroid dehydrogenase deficiency. I. A cause of hirsutism in pubertal and postpubertal women. J Clin Endocrinol Metab. 1985;60:428–439. [DOI] [PubMed] [Google Scholar]
- 105. Baskin HJ. Screening for late-onset congenital adrenal hyperplasia in hirsutism or amenorrhea. Arch Intern Med. 1987;147:847–848. [PubMed] [Google Scholar]
- 106. Azziz R, Sanchez LA, Knochenhauer ES, et al. Androgen excess in women: experience with over 1000 consecutive patients. J Clin Endocrinol Metab. 2004;89:453–462. [DOI] [PubMed] [Google Scholar]
- 107. Escobar-Morreale HF, Sanchón R, San Millán JL. A prospective study of the prevalence of nonclassical congenital adrenal hyperplasia among women presenting with hyperandrogenic symptoms and signs. J Clin Endocrinol Metab. 2008;93:527–533. [DOI] [PubMed] [Google Scholar]
- 108. New MI, Lorenzen F, Lerner AJ, et al. Genotyping steroid 21-hydroxylase deficiency: hormonal reference data. J Clin Endocrinol Metab. 1983;57:320–326. [DOI] [PubMed] [Google Scholar]
- 109. Abs R, Verhelst J, Maeyaert J, et al. Endocrine consequences of long-term intrathecal administration of opioids. J Clin Endocrinol Metab. 2000;85:2215–2222. [DOI] [PubMed] [Google Scholar]
- 110. Fraser LA, Morrison D, Morley-Forster P, et al. Oral opioids for chronic non-cancer pain: higher prevalence of hypogonadism in men than in women. Exp Clin Endocrinol Diabetes. 2009;117:38–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Rasmussen DD, Liu JH, Wolf PL, Yen SS. Endogenous opioid regulation of gonadotropin-releasing hormone release from the human fetal hypothalamus in vitro. J Clin Endocrinol Metab. 1983;57:881–884. [DOI] [PubMed] [Google Scholar]
- 112. Stubbs WA, Delitala G, Jones A, et al. Hormonal and metabolic responses to an enkephalin analogue in normal man. Lancet. 1978;2:1225–1227. [DOI] [PubMed] [Google Scholar]
- 113. Tolis G, Dent R, Guyda H. Opiates, prolactin, and the dopamine receptor. J Clin Endocrinol Metab. 1978;47:200–203. [DOI] [PubMed] [Google Scholar]
- 114. Meltzer HY, Matsubara S, Lee JC. Classification of typical and atypical antipsychotic drugs on the basis of dopamine D-1, D-2 and serotonin2 pKi values. J Pharmacol Exp Ther. 1989;251:238–246. [PubMed] [Google Scholar]
- 115. David SR, Taylor CC, Kinon BJ, Breier A. The effects of olanzapine, risperidone, and haloperidol on plasma prolactin levels in patients with schizophrenia. Clin Ther. 2000;22:1085–1096. [DOI] [PubMed] [Google Scholar]
- 116. Papakostas GI, Miller KK, Petersen T, et al. Serum prolactin levels among outpatients with major depressive disorder during the acute phase of treatment with fluoxetine. J Clin Psychiatry. 2006;67:952–957. [DOI] [PubMed] [Google Scholar]
- 117. Golden RN, Hsiao J, Lane E, Hicks R, Rogers S, Potter WZ. The effects of intravenous clomipramine on neurohormones in normal subjects. J Clin Endocrinol Metab. 1989;68:632–637. [DOI] [PubMed] [Google Scholar]
- 118. McCallum RW, Sowers JR, Hershman JM, Sturdevant RA. Metoclopramide stimulates prolactin secretion in man. J Clin Endocrinol Metab. 1976;42:1148–1152. [DOI] [PubMed] [Google Scholar]
- 119. Sowers JR, Sharp B, McCallum RW. Effect of domperidone, an extracerebral inhibitor of dopamine receptors, on thyrotropin, prolactin, renin, aldosterone, and 18-hydroxycorticosterone secretion in man. J Clin Endocrinol Metab. 1982;54:869–871. [DOI] [PubMed] [Google Scholar]
- 120. Steiner J, Cassar J, Mashiter K, Dawes I, Fraser TR, Breckenridge A. Effects of methyldopa on prolactin and growth hormone. Br Med J. 1976;1:1186–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Kelley SR, Kamal TJ, Molitch ME. Mechanism of verapamil calcium channel blockade-induced hyperprolactinemia. Am J Physiol. 1996;270:E96–E100. [DOI] [PubMed] [Google Scholar]
- 122. Hodi FS, O'Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Fatemi N, Dusick JR, Mattozo C, et al. Pituitary hormonal loss and recovery after transsphenoidal adenoma removal. Neurosurgery. 2008;63:709–718; discussion 718–719. [DOI] [PubMed] [Google Scholar]
- 124. Barker FG, 2nd, Klibanski A, Swearingen B. Transsphenoidal surgery for pituitary tumors in the United States, 1996–2000: mortality, morbidity, and the effects of hospital and surgeon volume. J Clin Endocrinol Metab. 2003;88:4709–4719. [DOI] [PubMed] [Google Scholar]
- 125. Snyder PJ, Fowble BF, Schatz NJ, Savino PJ, Gennarelli TA. Hypopituitarism following radiation therapy of pituitary adenomas. Am J Med. 1986;81:457–462. [DOI] [PubMed] [Google Scholar]
- 126. Constine LS, Woolf PD, Cann D, et al. Hypothalamic-pituitary dysfunction after radiation for brain tumors. N Engl J Med. 1993;328:87–94. [DOI] [PubMed] [Google Scholar]
- 127. Agha A, Sherlock M, Brennan S, et al. Hypothalamic-pituitary dysfunction after irradiation of nonpituitary brain tumors in adults. J Clin Endocrinol Metab. 2005;90:6355–6360. [DOI] [PubMed] [Google Scholar]
- 128. Freda PU, Wardlaw SL, Post KD. Unusual causes of sellar/parasellar masses in a large transsphenoidal surgical series. J Clin Endocrinol Metab. 1996;81:3455–3459. [DOI] [PubMed] [Google Scholar]
- 129. Valassi E, Biller BM, Klibanski A, Swearingen B. Clinical features of nonpituitary sellar lesions in a large surgical series. Clin Endocrinol (Oxf). 2010;73:798–807. [DOI] [PMC free article] [PubMed] [Google Scholar]