

Abstract
The counter-regulatory effects of insulin and catecholamines on carbohydrate and lipid metabolism are well studied, whereas the details of insulin regulation of β adrenergic receptor (βAR) signaling pathway in heart remain unknown. Here, we characterize a novel signaling pathway of insulin receptor (IR) to G protein-coupled receptor kinase 2 (GRK2) in the heart. Insulin stimulates the recruitment of GRK2 to β2AR, which induces β2AR phosphorylation at the GRK sites of serine 355/356 and subsequently β2AR internalization. Insulin thereby suppresses βAR-induced cAMP-PKA activities and contractile response in neonatal and adult mouse cardiomyocytes. Deletion of Insulin receptor substrate 2 (IRS2) disrupts the complex of IR and GRK2, which attenuates insulin-mediated β2AR phosphorylation at GRK sites and β2AR internalization, and the counter-regulation effects of insulin on βAR signaling. These data indicates the requirements of IRS2 and GRK2 for insulin to stimulate counter-regulation of βAR via β2AR phosphorylation and internalization in cardiomyocytes.
Keywords: insulin, adrenergic receptor, GRK2, insulin receptor substrate, internalization, cAMP, PKA, cardiac contractility
1. Introduction
G-protein-coupled receptors and tyrosine-kinase receptors represent two prominent modalities in cell signaling. Cross regulation between members of both receptor super families has been reported, including the counter-regulatory effects of insulin on β-adrenergic action [1]. β2-adrenergic receptor (β2AR) displays acute homologous desensitization in response to βAR agonists as well as counter-regulation by insulin [2]. Insulin stimulates a rapid tyrosine phosphorylation and sequestration of the β2AR [3]. This counter-regulatory effect of insulin on βAR signaling is observed in either DDT1MF-2 smooth muscle cells or Chinese hamster ovary cells (CHO) [4]. Insulin-stimulated internalization of β2AR is dependent upon insulin receptor (IR) kinase-catalyzed phosphorylation of tyrosyl residue at position 350 of the β2AR [4], which creates a docking site for SH2 domains of a variety of proteins, including Grb2 and dynamin. The integrity of Y350 and its phosphorylation in response to insulin are essential for the inhibitory regulation of β2AR functions and β2AR sequestration [5]. These studies largely focus on insulin action in skeletal muscle, liver and adipose tissues, including phosphorylation of the β2AR in HEK293 cells and adipocytes [1,2,6,7]. As a result, insulin induces an acute reduction in the ligand binding capacity of βR in rat adipocytes [8]; and stimulation of fat cells with insulin promotes a marked attenuation of βAR-mediated activation of AC [8,9].
By comparison, little is known how insulin influences βAR trafficking as well as the counter-regulation of βAR signaling in heart tissues. Current literatures report conflict views on cross-regulation between these two distinct classes of receptors in heart tissues [10,11]. Insulin enhances myocardial contractility response to β-adrenergic action in isolated rat cardiac papillary muscle [10]. However, insulin also suppresses β-adrenergic-induced cardiac dysfunction and cell injury in myocardial ischemia and reperfusion [11]. We have recently showed that phosphorylation of β2AR by GRK is required for rapid receptor internalization and desensitization in cardiomyocytes [12]. Disruption of the GRK sites of β2AR prolongs isoprotenolol-induced myocyte contraction response [12] .
A recent study reported that insulin induced membrane translocation of GRK2 in cultured adult rat ventricular cardiomyocytes [13]. In the current work, we probed the role of GRK2 in trafficking of β2AR after insulin stimulation in cardiomyocytes. The results revealed a physical interaction between GRK2 and insulin receptor in heart. Moreover, insulin treatment increased interaction between GRK2 and β2AR, revealing a GRK2-linked pathway between insulin receptor and β-adrenergic signaling. Our data show that a GRK2-mediated β2AR phosphorylation and internalization is necessary for counter-regulation of insulin on β-adrenergic signaling in cardiomyocytes.
2. Material and Methods
2.1 Cell culture
Animal protocols were approved by the IACUC of the University of California at Davis according to NIH regulation. Neonatal cardiomyocytes were isolated from 1- to 2-day-old wild type, β1AR knockout (KO), and β2AR-KO mouse pups. Adult mouse cardiomyocytes were isolated as described previously [14]. H9c2 cardiac myoblasts and Mouse Embryonic Fibroblasts (MEFs) from wild type mice and insulin receptor substrate 2 (IRS2) KO mice (a gift from Dr. Morris White, Harvard University) were cultured in DMEM plus 10% FBS for experiments.
2.2 Adenovirus infection, and plasmid transfection
Neonatal cardiomyocytes were infected with adenoviruses (100 MOI) as previously described to express the cAMP biosensor (ICUE3) [14] or the PKA activity biosensor (AKAR3) [15] as indicated for 24hr. IRS2 mouse shRNA plasmid (Sigma, MO) was used to create recombinant lentiviruses. Neonatal cardiomyocytes were infected with IRS2 shRNA lentivirus for 24hr and cultured for an additional 48 hr.
MEFs or H9C2 cardiac myoblasts were infected with recombinant adenoviruses or transfected as indicated. Experiments were conducted after 48h expression.
2.3 Plasma membrane isolation and immunoprecipitation
H9c2 cardiac myoblasts and MEFs were infected with Flag-tagged wild type β2AR at a multiplicity of infection of 100. After 24 h of expression, cells were serum-starved for 24 h and stimulated with insulin as indicated. For plasma membrane isolation, cells were homogenized in buffer containing 10 mM Tris pH 7.4, 10mM KCl, 1.5mM MgCl2, 1 mM EDTA and protease inhibitor cocktail tablets (Thermo, IL). Intact cells and nuclei were removed by centrifugation at 3,000 g for 10 min. The collected supernatant was further subjected to centrifugation at 100,000 g for 1 hour. The pellets containing membrane fraction were resuspended in RIPA buffer (50 mM Tris pH 7.4, 150mM NaCl, 25 mM sodium pyrophosphate, 2.5 mM EDTA, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS and protease inhibitor cocktail tablets. Whole cell lysates were placed into RIPA lysis buffer rotated for 30 min at 4 °C before centrifugation. For immunoprecipitation, MEFs were stimulated with insulin as indicated before lysed in the coimmunoprecipitaton buffer (150mM NaCl, 20 mM Tris-HCl pH 7.4, 2 mM EDTA, 0.6% NP-40, 10% Glycerol) and Halt Protease/Phosphatase Inhibitor Cocktail (Thermo, IL). Lysates were rotated at 4 °C for 30 min, followed by centrifugation at 16,000×g for 30 min. Heart tissues were lysed with FastPrep®-24 homogenizer for 20 sec in lysis buffer (50mM Tris-HCl (pH 7.5), 150mM NaCl, 1%NP-40, 0.25% deoxycholate, 9.4mg/50ml sodium orthovanadate, 1% sodium dodecyl sulphate). MEF and heart lysates were cleared by centrifugation (40,000 rpm, 30min at 4°C) before immunoprecipitation with antibodies followed with Protein A beads (Repligen, Waltham, MA). Lysates, membrane proteins, and immunoprecipitated complexes were resolved by SDS-PAGE for Western blotting. Insulin receptor (SCBT, Santa Cruz, CA), IRS2 (Millipore), total and phosphorylated β2AR at serine 261/262[12] and serine 355/356, Gi, GFP and GRK2 (SCBT) and γ-tubulin (Sigma) were visualized after incubation with IRDye 680CW goat anti-mouse or with IRDye 800CW goat anti-rabbit secondary antibodies using the Odyssey scanner (LI-COR Biosciences, Lincoln, NE). Signal intensity was quantitated by densitometry of Western blots.
2.4 Receptor internalization
After expressing Flag-β2AR, PKA mutant β2AR or GRK mutant β2AR, H9c2 cardiac myoblasts and MEFs were serum-starved for 24h and stimulated with isoproterenol (10 μM, 5min) or insulin (100 nM, 10 min) to examine receptor internalization. Cells were fixed, permeabilized, and stained with anti-flag antibody (mouse IgG2b, Sigma, MO), which was revealed with an Alexa-fluor-488 conjugated goat anti-mouse IgG2b antibody (Life technologies, CA). Fluorescence images were taken with a CCD camera on a Zeiss microscope with Meta morph software (Molecular Devices, CA). Images were quantified with Image J-based Fiji open source image-processing package as described previously [16]. Briefly, images were rotated to enable optimal selection of areas across the cell body. Plot profile analysis was applied to selected regions for measure of fluorescence intensity. Average of cytosolic fluorescence intensity (CFI) of β2AR staining was divided by that of membrane fluorescence intensity (MFI) for analysis of β2AR internalization. The ratio of CFI/MFI after stimulation was normalized against the ratio of CFI/MFI at resting state.
2.6 Fluorescence Resonance Energy Transfer Recording
Neonatal cardiomyocytes from wild type, β1AR-KO or β2AR-KO pups were infected with viruses to express either PKA activity reporter AKAR3 or cAMP probe ICUE3 as described previously [14]. Living myocytes were imaged on an Axiovert 200M microscope (Carl Zeiss, NY) with a 40×/1.3 numerical aperture oil-immersion objective lens and a charge-coupled device camera controlled by Metafluor software. Dual-emission recording was acquired with a 420DF20 excitation filter, a 450DRLP diachronic mirror, and two emission filters (475DF40 for cyan and 535DF25 for yellow). Exposure time was 200 ms, and recording interval was 20 s. Images in both channels were subjected to background subtraction, and ratios of yellow-to-cyan color were calculated at different time points. After the PKA phosphorylation on the consensus site in AKAR3, the ratio yellow-to-cyan fluorescent protein displayed increases. The binding cAMP to ICUE3 led to decreases in the ratio yellow fluorescent protein/cyan fluorescent protein, which were plotted with inverted y - axis.
2.7 Measurements of myocyte contraction
Spontaneously beating neonatal cardiomyocytes were isolated from newborn pups from WT mice. Measurement of spontaneous neonatal cardiomyocytes contraction rate was carried out as described previously [17]. Briefly, about 3×105 cardiomyocytes were cultured in 35-mm Petri dishes to obtain a uniformly beating syncytium. On day 3, the culture dishes were placed in a temperature-regulation apparatus positioned on the stage of an inverted microscope (Carl Zeiss, NY) connected to a video camera. Cells were equilibrated at 37°C for 10 min before monitoring the contraction rate. Contraction rates of cells within the syncytium were determined at 2 min intervals for 10 min after the addition of isoproterenol as indicated. All assays were recorded and data was analyzed by Meta morph software.
Adult mouse ventricular myocytes were isolated from hearts of 2-to 3-month-old male WT mice. Adult myocytes were placed in a dish with HEPES buffer and electrically stimulated at 30 V/cm at 1 Hz at room temperature. Cell length was recorded with a charge coupled device camera. Cell contraction shortening was analyzed by Meta morph software (Molecular Device, CA) and normalized as the increase over the basal levels after being fitted to a sigmoidal curve. The maximal shortening was normalized to the baseline value.
2.8 Statistical analysis
Student t-test and one or two-way ANOVA followed by post hoc Tukey’s test were performed using Prism (GraphPad, CA). p < 0.05 was considered statistically significant.
3. Results
3.1 Insulin induces β2AR internalization via GRK phosphorylation of β2AR in cardiac myoblasts
We have previously shown that GRK2 is necessary for βAR agonist-induced GRK2 phosphorylation of β2ARs at serine 355/356. Interestingly, we observed that IR formed a complex with GRK2 in mouse hearts and MEFs (Fig. 1A and 1B). Insulin stimulation increased GRK2-IR interaction (Fig 1B). Consistently, when H9c2 cardiac myoblasts were exposed to insulin, GRK2 displayed a membrane translocation (Fig 1C). Next, we directly addressed whether insulin-induced activation of IR leads to GRK2 phosphorylation of β2AR in cardiac muscle cells. Indeed, insulin induced a dose and time-dependent phosphorylation of the β2AR at the GRK sites serine 355/356 in H9c2 cardiac myoblasts (Fig. 1D and 1E). After stimulation with 100 nM of insulin, the GRK-mediated phosphorylation arrived at peak at 5 min and sustained over 60 min (Fig. 1E). To examine whether the insulin-induced phosphorylation of β2AR at serine 355 and 356 is dependent on GRK2, we utilized a well-characterized GRK2 inhibitor (βARKct). We found that inhibition of GRK2 with βARKct significantly reduced S355/S356 phosphorylation after 100 nM insulin stimulation (Supplementary Fig 1). It has been well documented that GRK-mediated phosphorylation of β2AR is involved in endocytosis of β2AR in cardiomyocytes [12], prompting us to explore the role of β2AR phosphorylation in receptor internalization induced by insulin.
Figure 1. Insulin induces GRK2 membrane translocation and GRK-mediated phosphorylation of β2AR.

(A) Mouse heart lysates were subjected to immunoprecipitation with anti-IR antibody. (B) WT MEF cells were lysed for immunoprecipitation with either an anti-IR antibody or an anti-GRK2 antibody upon insulin stimulation (100 nM, 10 min). Immunoprecipitated proteins were detected by Western blot. Data represent at least three independent experiments. (C) H9c2 cardiac myoblasts overexpressed β2AR were stimulated with insulin (100 nM, 10 min). Membrane fractions were blotted for GRK2, IR, β2AR, and Gi. The protein levels on the plasma membrane were normalized to those of Gi. * p < 0.05 by student t-test relative to control group ( n = 3). (D–G) H9c2 cardiac myoblasts were infected with adenovirus expressing mouse β2AR and then stimulated with insulin for 10 min at indicated doses (D and F) or with 100 nM insulin for indicated times (E and G). The levels of phosphorylation of β2AR at serine 261/262 and 355/356 were detected by western blot, and normalized against total β2AR (n = 5). * p < 0.05 and ** p < 0.01 by one-way ANOVA in relative to control followed by Tukey’s test.
β2AR was distributed throughout the plasma membrane in un-stimulated H9c2 cardiac myoblasts. Both insulin (100 nM, 10 min) and βAR agonist isoproterenol (ISO, 100 nM, 5 min) induced translocation of β2AR from the cell surface to the cytosol (Fig. 2A and 2B). We then directly tested the role of GRK-mediated phosphorylation in receptor internalization by using mutant β2AR lacking GRK phosphorylation sites (GRKmut-β2AR). As expected, GRKmut-β2AR failed to display internalization under insulin stimulation (Fig. 2A and 2B). We also found that insulin induced phosphorylation of β2AR at the PKA sites serine 261/262 in a dose- and time-dependent manner (Fig. 1F an 1G). However, mutation of PKA phosphorylation sites ofx β2AR did not affect insulin-induced receptor internalization (Fig. 2A and 2B), consistent with previous studies showing that PKA phosphorylation is not essential for receptor endocytosis in cardiomyocytes [12,18]. Together, these data indicates a cross talk between IR and βAR mediated by insulin-induced recruitment of GRK2 to β2AR, which promotes phosphorylation and internalization of β2AR.
Figure 2. Insulin and ISO induce internalization of β2AR in H9c2 cardiac myoblasts.

H9c2 myoblasts expressing wild type (WT), mutant β2AR lacking PKA phosphorylation sites (PKAmut), or mutant β2AR lacking GRK phosphorylation sites (GRKmut) were stimulated with insulin (100 nM, 10 min) or ISO (100 nM, 5 min). (A) The distribution of β2AR was examined with immunofluorescence staining. (B) The internalization of β2AR was quantified with Fiji image-processing package. n = 10; *** p < 0.001 when compared to control by one-way ANOVA followed by Tukey’s test.
3.2 Insulin impairs βAR-cAMP-PKA signaling in cardiomyocytes in a β2AR-dependent manner
To determine whether the internalization of β2AR contributes to an insulin counter-regulation of adrenergic signaling in cardiomyocytes, we analyzed the impacts of insulin on βAR-induced cAMP activity through a FRET-based assay using a fluorescent cAMP biosensor ICUE3. Isoproterenol induced a dose-dependent increase in cAMP ICUE3 FRET ratio, which was sustained at 1μM (EC50 at 0.95 nM, Fig. 3A and 3B). Pretreatment with insulin (1–100 nM) dose-dependently inhibited isoproterenol-induced increases in ICUE3 FRET ratio of cAMP activity in cardiomyocytes (with EC50 6.9 nM at 1 nM of insulin, 64.4 nM at 10 nM of insulin, and 811.4 nM at 100 nM of insulin, Fig. 3B). Even at 10 μM of isoproterenol, the increase in ICUE3 FRET ratio of cAMP activity was not sustained (Fig. 3A). Insulin alone (1–100 nM) did not affect the ICUE3 FRET ratio for cAMP activity (Fig. 3C). Furthermore, a specific β2AR antagonist ICI118551, not a specific β1AR antagonist CGP20712A, reversed the inhibitory effect of insulin on isoproterenol-induced response in cAMP activity (Fig. 3D).
Figure 3. Insulin attenuates βAR-induced cAMP signaling in cardiomyocytes.

(A and B) Neonatal cardiomyocytes expressing cAMP biosensor ICUE3 were stimulated with ISO in the absence and presence of pretreatment with insulin (100 nM, 30 min) or as indicated. The changes in cAMP FRET ratio were recorded. (A) Traces show time-courses of changes in FRET ratio in representative cells. (B) The maximal increases in FRET ratio were plotted. Neonatal cardiomyocytes expressing cAMP biosensor ICUE3 were stimulated with insulin, forskolin, or PDE inhibitor IBMX as indicated (C), or were pretreated with insulin together with either 1μM of ICI118551 or 1μM of CGP20712A before stimulation with ISO (D). The changes in cAMP FRET ratio were recorded. The maximal increases in FRET ratio were plotted (C and D). (C) ** p < 0.01, *** p < 0.001 by student t-test in relative to insulin (100nM) group (n = 6~8). (D) * p < 0.05 by one-way ANOVA followed by Tukey’s test (n = 12~14).
In primary WT cardiomyocytes, 10 μM of isoproterenol-induced in a sustained cAMP and PKA FRET response, which was reduced by insulin pretreatment (Fig. 4A and 4D). By activation of the endogenous β1AR in cardiomyocytes lacking β2AR (β2AR-KO), insulin pretreatment didn’t significantly change the isoproterenol-induced sustained cAMP and PKA FRET response (Fig. 4C and 4F). However, in cardiomyocytes lacking β1ARs (β1AR-KO), activation of the β2AR with isoproterenol induced a small transient peak in cAMP and PKA activity, which was attenuated with insulin pretreatment (Fig. 4B and 4E). This finding indicates that there is a β2AR-dependent cross talk between insulin and βAR signaling pathways in cardiomyocytes, which leads to the impairment on βAR-activated cAMP and PKA response.
Figure 4. Insulin attenuates βAR-induced cAMP and PKA signaling in cardiomyocytes in a β2AR dependent manner.
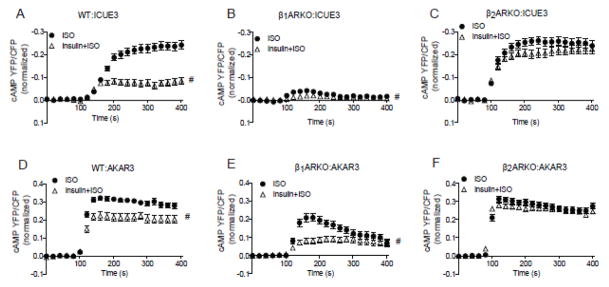
Wild type, β1ARKO, and β2AR-KO neonatal cardiomyocytes expressing cAMP biosensor ICUE3 (A–C) or PKA biosensor AKAR3 (D–F) were treated with insulin (100 nM, 30 min) prior to stimulation with ISO (10 μM). The changes in the FRET ratio were recorded respectively. # p < 0.05 by two-way ANOVA ( n=30~40).
3.3 IRS2 is required for insulin-induced GRK2 phosphorylation of β2AR and β2AR internalization
Consistent with the β2AR phosphorylation data, insulin stimulation promoted recruitment of GRK2 to β2AR in MEF cells (Fig. 5A). Activation of IR induces downstream signaling via recruitment of IRSs. Deletion of the IRS2 gene abolished insulin-induced GRK2 phosphorylation of β2AR as well as β2AR internalization (Fig. 5B–5D). Further examination revealed that IR formed a complex with IRS2 and GRK2, and deletion of IRS2 abolished the complex between IR and GRK2 (Fig. 5E). In agreement, knock down IRS2 in primary wild type cardiomyocytes abolished the inhibitory effect of insulin on isoproterenol-induced cAMP FRET responses (Fig. 5F). Together, these data suggests that IRS2 is necessary for the counter-regulatory effect of insulin on βAR signaling via GRK2-mediated phosphorylation of β2AR and subsequently β2AR internalization.
Figure 5. IRS2 is necessary for insulin-induced phosphorylation and internalization of β2AR and inhibition of adrenergic signaling.

(A) Cell lysates from wild type MEFs were harvested after stimulation with insulin (100 nM, 10 min). Cell lysates were immunoprecipitated with antibody against GRK2. Immunoprecipitated proteins were detected by Western blot. (B) WT or IRS2 KO MEF cells were transfected with mouse β2AR and stimulated with 100 nM insulin for 10 min. GRK-mediated phosphorylation of the β2AR at serine 355/356 was detected by Western blot. Serine 355/356 phosphorylation was normalized against total β2AR. Data represent at least three independent experiments. * p < 0.05 by student t-test in relative to control. (C) Wild type and IRS2-KO MEFs expressing flag-β2AR were stimulated with insulin (100 nM, 10 min) or ISO (100 nM, 5 min). The distribution of β2AR was examined with immunofluorescence staining. (D) The internalization of β2AR was quantified with Fiji image-processing package. n = 10; ** p < 0.01 when compared to control by one-way ANOVA followed by Tukey’s test. (E) WT or IRS2 KO MEF cells were lysed for immunoprecipitation with an anti-IR antibody. Immunoprecipitated proteins were detected by Western blot. (F) Wild type neonatal cardiomyocytes expressing the cAMP biosensor ICUE3 together with scrambled or IRS2-specific shRNA were treated with ISO (10 μM) in the presence or absence of insulin (100 nM, 30min) as indicated. The changes in the cAMP FRET ratio were recorded, and the maximal increases in cAMP FRET ratio were plotted. * p < 0.01 by student t-test in relative to ISO group (n = 8~10).
3.4 Insulin impairs βAR-induced contractile responses in neonatal and adult mouse cardiomyocytes
We further examined the physiological effects of insulin stimulation on βAR signaling in primary cardiomyocytes. Isoproterenol induced a dose-dependent increase in contraction rate in neonatal mouse cardiomyocytes, which was sustained at 1μM (Fig 6A and 6B). Consistent with the cAMP and PKA FRET responses, insulin pretreatment attenuated the isoproterenol-induced contraction rate response and promoted a right shift of the isoproterenol-induced dose response curve of changes in contraction rates (Fig 6A –6C). Insulin alone didn’t affect cardiomyocyte contraction rate.
Figure 6. Insulin inhibits βAR-induced contractile shortening in neonatal and adult mouse cardiomyocytes.

Wild type neonatal cardiomyocytes were stimulated with ISO in the presence and absence of pretreatment with insulin (100 nM, 30 min) as indicated (A and B). The changes in spontaneous contraction rates were recorded. (A) Traces show time course of increases in contraction rates over ISO stimulation in representative dishes. The ISO dose-dependent increases (B) and the maximal increases (C) of contraction rates were plotted. # p < 0.05 by two-way ANOVA and * p < 0.05 by one-way ANOVA followed by Tukey’s test between groups. (D) Adult mouse cardiomyocytes paced at 1 Hz were pretreated with insulin (100 nM, 30 min) before stimulation with ISO (100 nM). Contractile shortening was recorded and the maximal increases of contraction rates (E) were plotted. * p < 0.05 by one-way ANOVA followed by Tukey’s test between groups; n = 6~7 from three independent experiments.
Previous studies have shown that insulin stimulates a signal transduction pathway that leads to the activation of phosphoinositide 3-kinase (PI3K) and subsequent phosphorylation of phosphodiesterase 3 (PDE3) in adipocytes [19], which is an important regulator of cellular concentrations of cAMP. Thus, PDE3 inhibitor cilostamide was used to investigate potential role of PDE3 in the inhibitory effect of insulin on βAR signaling. Inhibition of PDE3 with cilostamide only partially rescued insulin-induced impairment of ISO-induced cAMP FRET response and contraction rate in neonatal cardiomyocytes (Supplementary Fig.2A and 2B). These data indicate that the counter-regulatory effect of insulin on βAR signaling is in part due to activation of PDE3.
We further validated the cross talk between IR and βAR signaling cascades in adult cardiomyocytes, a cell model relevant to whole heart contractile function. Isoproterenol induced robust increase in sarcomere shortening in adult mouse cardiomyocytes (Fig. 6D and 6E). While insulin alone minimally affected the baseline shortening, pretreatment with insulin significantly attenuated the isoproterenol-induced myocyte shortening amplitude and slightly extended the isoproterenol-induced shortening duration (Fig. 6D and 6E). These data validate that insulin stimulation inhibits the adrenergic-induced cAMP signal for contraction rate and contractile shortening in primary cardiomyocytes.
4. Discussion
Receptor phosphorylation and internalization are prominent features of agonist-induced desensitization of GRCRs. β2ARs are substrates of several protein kinases [20]. Catecholamines activate both protein kinase A and protein kinase C, as well as GRK2 that phosphorylate serine and threonine residues of β2AR. In addition, β2ARs are phosphorylated on tyrosyl residues by insulin stimulation [1,4]. Activation of both pathways blunts or abolishes β2AR action and promotes sequestration of β2ARs. Our previously study showed that GRK-mediated phosphorylation of the β2AR is necessary for subsequent internalization in cardiomyocytes [12], which results in compartmentalization of the Gs-stimulated cAMP signal, thus selectively affecting cardiac contractile response [21]. Recent studies have described a correlation between insulin resistance and increased levels of GRK2 expression in pathological cardiac conditions [13,22]; importantly, enhanced GRK2 activity negatively affects cardiac contractile function after myocardial injury [13]. Herein, we demonstrate that GRK2 may represent a molecular link between the insulin receptor and βAR signaling in cardiomyocytes for cardiac function.
In the current work, we show that GRK2 forms a complex with IR in an IRS2-dependent manner in the heart. Interestingly, insulin treatment induces GRK2 membrane translocation and increases the association of GRK2 and β2AR. In agreement, Ciccarelli et al have recently found that insulin administration induces GRK2 membrane translocation in adult rat ventricular cardiomyocytes [13]. Moreover, we show that insulin promotes IRS2-dependent β2AR phosphorylation at the GRK2 sites. Similar to isoproterenol stimulation, insulin treatment induces β2AR internalization in H9c2 cardiac myoblasts. In this study, we further reveal that IRS2 and GRK2 are necessary for β2AR internalization after insulin stimulation. Knock down IRS2 in cardiomyocytes also reverses insulin-induced inhibition on cAMP activity after isoproterenol stimulation. These data point out that the IRS2-dependent GRK2-mediated β2AR phosphorylation and internalization are required for insulin-induced counter-regulation of βAR-activated cAMP and PKA activity in cardiomyocytes. Our result also shows that insulin inhibits isoproterenol-induced cAMP and PKA in wild type and β1AR-KO; in comparison, deletion of β2AR (β2AR-KO) in cardiomyocytes abolishes the inhibition of insulin on cAMP and PKA activities induced by isoproterenol. Consequently, insulin attenuates contractile response to βAR stimulation in both neonatal and adult mouse cardiomyocytes.
Taken together, these data suggest that insulin enhances IRS2-dependent β2AR phosphorylation at the GRK sites and β2AR internalization by recruiting GRK2 to β2AR. The phosphorylation of β2AR by GRK2 promotes receptor desensitization via internalization and potentially switches the receptor coupling from Gs to inhibitory Gi proteins in cardiomyocytes [12,17], which inhibits βAR-activated AC-cAMP-PKA signaling pathway and reduces cardiac contraction. Further efforts with transgenic mice of either expressing the mutant β2AR lacking the GRK sites or deletion of the GRK2 gene in heart tissues will yield evidence of the GRK2-mediated cross talk between IR and βAR in cardiac contractile response in vivo.
5. Conclusion
This study reveals the counter-regulation of insulin on βAR signaling in cardiomyocytes, highlighting the role of IRS2 and GRK2 in insulin-induced β2AR phosphorylation and internalization in cardiomyocytes. Thus our study elucidates a distinct mechanism on insulin regulation of βAR signaling in cardiomyocytes in comparison to other tissues, providing a basis to understand cross talk between these two regulatory signaling pathways in heart.
Supplementary Material
Highlights.
Insulin receptor and GRK2 form a complex in the heart.
Insulin stimulation promotes recruitment of GRK2 to β2AR.
IRS2 mediates insulin-induced β2AR phosphorylation at GRK sites and internalization.
IRS2 and GRK2 are required for the counter regulation of insulin on βAR signaling in cardiomyocytes.
Acknowledgments
This study was supported by a National Natural Science Foundation of China grant 81473212 and a Central Authorities of an Institution of Higher Learning of Scientific Research Special Fund of China 2014QN031 to QF, a NIH grant RO1 HL082846 and a National Natural Science Foundation of China grant 81428022 to YKX. YKX is an AHA established investigator.
Abbreviations
- AR
Adrenergic receptor
- IR
Insulin receptor
- IRS
Insulin receptor substrate
- GRK
G protein-coupled receptor kinase
- GPCR
G-protein coupled receptor
- KO
knockout
- MEFs
Mouse Embryonic Fibroblasts
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hadcock JR, Port JD, Gelman MS, Malbon CC. Cross-talk between tyrosine kinase and G-protein-linked receptors. Phosphorylation of beta 2-adrenergic receptors in response to insulin. J Biol Chem. 1992;267:26017–26022. [PubMed] [Google Scholar]
- 2.Morris AJ, Malbon CC. Physiological regulation of G protein-linked signaling. Physiol Rev. 1999;79:1373–1430. doi: 10.1152/physrev.1999.79.4.1373. [DOI] [PubMed] [Google Scholar]
- 3.Karoor V, Malbon CC. G-protein-linked receptors as substrates for tyrosine kinases: cross-talk in signaling. Adv Pharmacol. 1998;42:425–428. doi: 10.1016/s1054-3589(08)60779-6. [DOI] [PubMed] [Google Scholar]
- 4.Karoor V, Wang L, Wang HY, Malbon CC. Insulin stimulates sequestration of beta-adrenergic receptors and enhanced association of beta-adrenergic receptors with Grb2 via tyrosine 350. J Biol Chem. 1998;273:33035–33041. doi: 10.1074/jbc.273.49.33035. [DOI] [PubMed] [Google Scholar]
- 5.Shih M, Malbon CC. Serum and insulin induce a Grb2-dependent shift in agonist affinity of beta-adrenergic receptors. Cell Signal. 1998;10:575–582. doi: 10.1016/s0898-6568(97)00195-2. [DOI] [PubMed] [Google Scholar]
- 6.Doronin S, Wang HY, Malbon CC. Insulin stimulates phosphorylation of the beta 2-adrenergic receptor by the insulin receptor, creating a potent feedback inhibitor of its tyrosine kinase. J Biol Chem. 2002;277:10698–10703. doi: 10.1074/jbc.M109432200. [DOI] [PubMed] [Google Scholar]
- 7.Shumay E, Song X, Wang HY, Malbon CC. pp60Src mediates insulin-stimulated sequestration of the beta(2)-adrenergic receptor: insulin stimulates pp60Src phosphorylation and activation. Mol Biol Cell. 2002;13:3943–3954. doi: 10.1091/mbc.E02-03-0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Engfeldt P, Hellmer J, Wahrenberg H, Arner P. Effects of insulin on adrenoceptor binding and the rate of catecholamine-induced lipolysis in isolated human fat cells. J Biol Chem. 1988;263:15553–15560. [PubMed] [Google Scholar]
- 9.Martiny L, Dib K, Haye B, Correze C, Jacquemin C, Lambert B. The effect of glycosyl inositol-phosphate on cAMP production in isolated rat fat-cells is transduced by a pertussis toxin sensitive G-protein. FEBS Lett. 1991;286:105–109. doi: 10.1016/0014-5793(91)80951-x. [DOI] [PubMed] [Google Scholar]
- 10.Ferrara N, Abete P, Corbi G, Paolisso G, Longobardi G, Calabrese C, Cacciatore F, Scarpa D, Iaccarino G, Trimarco B, Leosco D, Rengo F. Insulin-induced changes in beta-adrenergic response: an experimental study in the isolated rat papillary muscle. Am J Hypertens. 2005;18:348–353. doi: 10.1016/j.amjhyper.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 11.Yu QJ, Si R, Zhou N, Zhang HF, Guo WY, Wang HC, Gao F. Insulin inhibits beta-adrenergic action in ischemic/reperfused heart: a novel mechanism of insulin in cardioprotection. Apoptosis. 2008;13:305–317. doi: 10.1007/s10495-007-0169-2. [DOI] [PubMed] [Google Scholar]
- 12.Liu R, Ramani B, Soto D, De Arcangelis V, Xiang Y. Agonist dose-dependent phosphorylation by protein kinase A and G protein-coupled receptor kinase regulates beta2 adrenoceptor coupling to G(i) proteins in cardiomyocytes. J Biol Chem. 2009;284:32279–32287. doi: 10.1074/jbc.M109.021428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ciccarelli M, Chuprun JK, Rengo G, Gao E, Wei Z, Peroutka RJ, Gold JI, Gumpert A, Chen M, Otis NJ, Dorn GW, 2nd, Trimarco B, Iaccarino G, Koch WJ. G protein-coupled receptor kinase 2 activity impairs cardiac glucose uptake and promotes insulin resistance after myocardial ischemia. Circulation. 2011;123:1953–1962. doi: 10.1161/CIRCULATIONAHA.110.988642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soto D, De Arcangelis V, Zhang J, Xiang Y. Dynamic protein kinase a activities induced by beta-adrenoceptors dictate signaling propagation for substrate phosphorylation and myocyte contraction. Circ Res. 2009;104:770–779. doi: 10.1161/CIRCRESAHA.108.187880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu S, Li Y, Kim S, Fu Q, Parikh D, Sridhar B, Shi Q, Zhang X, Guan Y, Chen X, Xiang YK. Phosphodiesterases coordinate cAMP propagation induced by two stimulatory G protein-coupled receptors in hearts. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:6578–6583. doi: 10.1073/pnas.1117862109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang D, Yuen EY, Zhou Y, Yan Z, Xiang YK. Amyloid beta peptide-(1–42) induces internalization and degradation of beta2 adrenergic receptors in prefrontal cortical neurons. J Biol Chem. 2011;286:31852–31863. doi: 10.1074/jbc.M111.244335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiang Y, Kobilka B. The PDZ-binding motif of the beta2-adrenoceptor is essential for physiologic signaling and trafficking in cardiac myocytes. Proc Natl Acad Sci U S A. 2003;100:10776–10781. doi: 10.1073/pnas.1831718100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu R, Wang D, Shi Q, Fu Q, Hizon S, Xiang YK. Palmitoylation regulates intracellular trafficking of beta2 adrenergic receptor/arrestin/phosphodiesterase 4D omplexes in cardiomyocytes. PLoS One. 2012;7:e42658. doi: 10.1371/journal.pone.0042658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitamura T, Kitamura Y, Kuroda S, Hino Y, Ando M, Kotani K, Konishi H, Matsuzaki H, Kikkawa U, Ogawa W, Kasuga M. Insulin-induced phosphorylation and activation of cyclic nucleotide phosphodiesterase 3B by the serine-threonine kinase Akt. Mol Cell Biol. 1999;19:6286–6296. doi: 10.1128/mcb.19.9.6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malbon CC, Karoor V. G-protein-linked receptors as tyrosine kinase substrates: new paradigms in signal integration. Cell Signal. 1998;10:523–527. doi: 10.1016/s0898-6568(97)00194-0. [DOI] [PubMed] [Google Scholar]
- 21.Xiao RP. Beta-adrenergic signaling in the heart: dual coupling of the beta2-adrenergic receptor to G(s) and G(i) proteins. Sci STKE. 2001;2001:re15. doi: 10.1126/stke.2001.104.re15. [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Guerra L, Nieto-Vazquez I, Vila-Bedmar R, Jurado-Pueyo M, Zalba G, Diez J, Murga C, Fernandez-Veledo S, Mayor F, Jr, Lorenzo M. G protein-coupled receptor kinase 2 plays a relevant role in insulin resistance and obesity. Diabetes. 2010;59:2407–2417. doi: 10.2337/db10-0771. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.